 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and thermal decomposition of a pyridylene-bridged bis-β-diketiminate magnesium hydride cluster†
Sjoerd
Harder
*a,
Jan
Spielmann
b and
Julia
Intemann
ac
aInorganic and Organometallic Chemistry, University Erlangen-Nürnberg, Egerlandstrasse 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
bInorganic Chemistry, University Duisburg-Essen, Universitätsstrasse 5, 45117 Essen, Germany
cStratingh Institute of Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, Netherlands
First published on 14th April 2014
Abstract
Reaction of PYR-(MgnBu)2, in which PYR is 2,6-[(DIPP)NC(Me)CHC(Me)N-]2-pyridine and DIPP is 2,6-iPr2-phenyl, with (DIPP)NH2BH3 gave PYR-[MgNH(DIPP)BH3]2 (56%) which was characterized by crystal structure determination. Addition of THF resulted in β-H elimination and formation of PYR-[MgNH(DIPP)BH3](MgH)·THF (57%), likewise characterized by crystal structure determination. Conversion of the second amidoborane anion in H− could not be achieved. Reaction of PYR-(MgnBu)2 with PhSiH3 gave PYR-(MgH)2, which crystallized as a dimer. The structure of [PYR-(MgH)2]2 shows an 8-membered ring of Mg2+ and H− ions. Thermal decomposition at 130 °C releases one equivalent of H2, i.e. 50% of the expected value. Nucleophilic attack at the para-position and reduction of the pyridylene bridge might explain reduced H2 release.
Introduction
Transition metal hydride complexes play an important role in the broader context of organometallic chemistry and have paved the way for the early development of homogeneous transition metal catalysis.1 The extended class of late main group metal hydride compounds (e.g. alanes, boranes, stannanes) is also well-established and heavily used as reducing agents in organic synthesis.2 The early main group metal hydrides, however, suffer from their highly polar character, which results in ionic crystal structures, (MH)∞ and (MH2)∞, with very high lattice energies.2,3 The latter are an obstacle in the synthesis of well-defined early main group metal hydride complexes like L0⋯M–H or L−1–M–H (in which L0 and L−1 represent neutral and −1 charged ligand systems, respectively). The challenge of selectively synthesizing early main group metal hydride complexes was recently taken on, and the field is rapidly expanding.4–15 This is especially due to rewarding contributions of the field to early main group metal catalysis16–19 and hydrogen storage.11,14In light of this, β-diketiminate complexes of magnesium hydride (Scheme 1) have been shown to function as molecular models for the hydrogen storage system: MgH2 ⇄ Mg + H2.11,14,20 Apart from the magnesium hydride dimer [(DIPPnacnac)MgH]2 (Scheme 1),9 larger magnesium hydride clusters based on bridged β-diketiminate ligands were introduced.11,14 The ligand with direct connection of β-diketiminate units gave the tetranuclear magnesium hydride cluster [NN-(MgH)2]2.14 Using a para-phenylene spacer gave a larger octanuclear cluster in which MgH2 is incorporated: [PARA-(MgH)2]3[MgH2]2 (Scheme 1).11 It was shown that these magnesium hydride complexes are stable towards thermal decomposition when dissolved in aromatic solvents. However, in the solid state, thermal decomposition with release of the expected amount of H2 is observed. As predicted by theoretical calculation,21 the hydrogen release temperatures (120–200 °C) are much lower than that for bulk (MgH2)∞ (>300 °C) and increase with the cluster size (Scheme 1).14 Hitherto, the low-valent products after H2 elimination could not be characterized. Also, reversibility has so far not been observed.
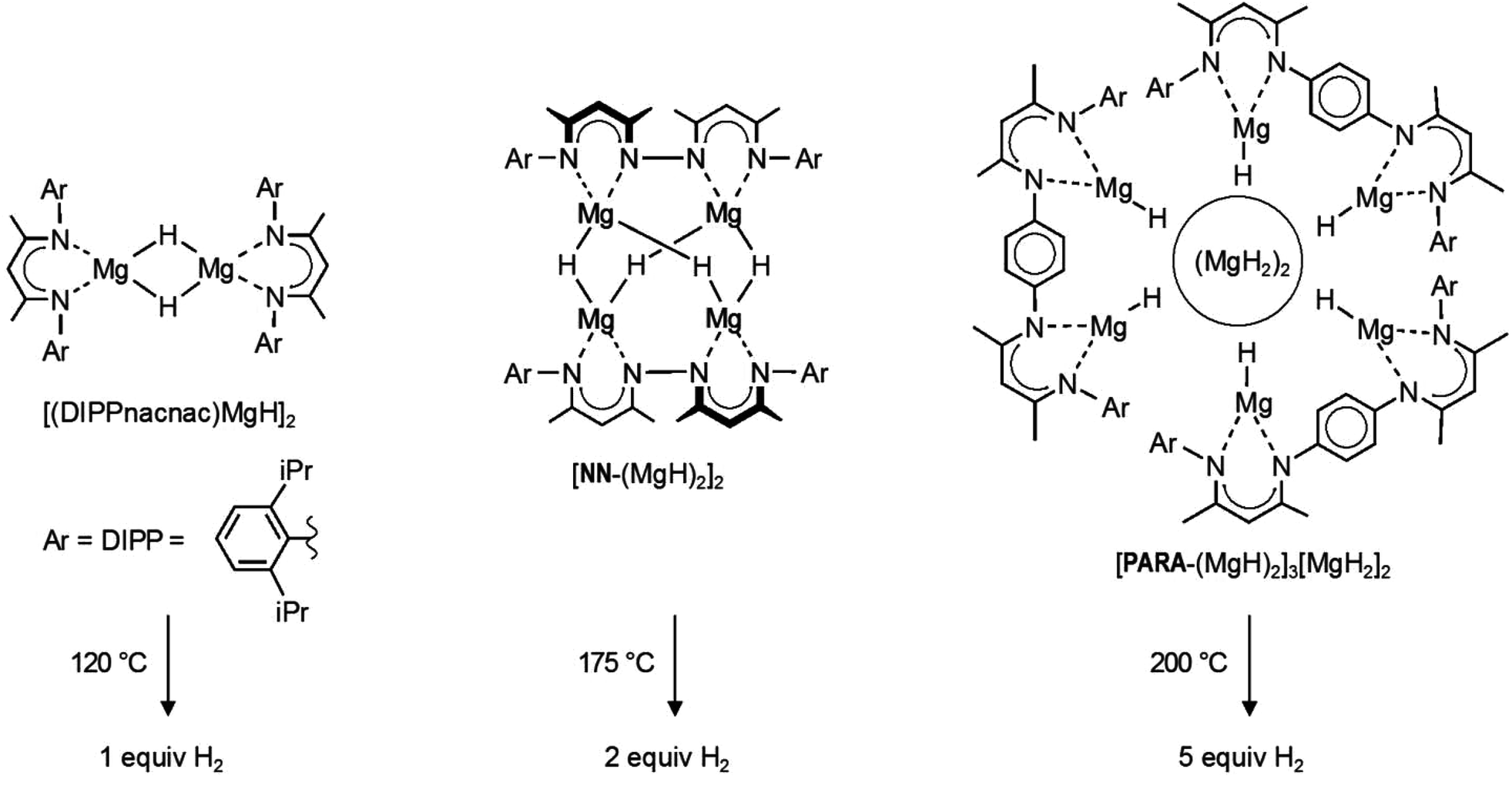 | ||
| Scheme 1 Magnesium hydride complexes. | ||
In continuation, we extend our investigations with the 2,6-pyridylene bridged β-diketiminate ligand (PYR). This ligand has been shown to have unusual coordination chemistry.22–24 It can bridge a dimeric unit symmetrically with long, but not negligible pyridine–metal contacts (i) in Scheme 2. This leads to significant strain: the N–C–N angles in PYR-(MgnBu)2 of circa 109° are much smaller than the idealized 120° value for sp2 hybridization. Also asymmetric coordination modes (ii) have been observed. Due to essential free rotation of the pyridylene unit in respect of the β-diketiminate NCCCN planes, another geometry with non-classic hydrogen C–H⋯N bonding is feasible (iii). As the bimetallic complexes PYR-(ZnR)2 were found to be completely inactive in CO2/epoxide copolymerization, it was reasoned that such a geometry might block metal–metal communication and synergistic effects.
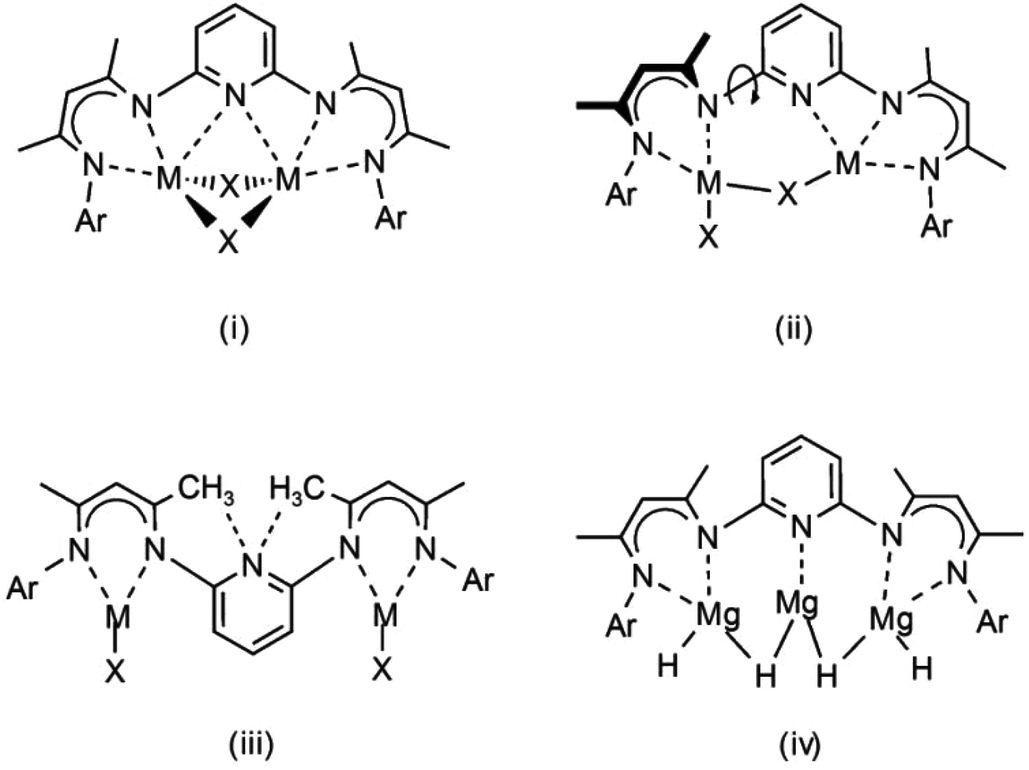 | ||
| Scheme 2 Coordination modes of the PYR ligand. | ||
The pyridylene bridge in PYR is potentially a strongly coordinating donor that also may assist in incorporation of additional metal units: a magnesium hydride complex like (iv) could be envisioned. Here, we evaluate the magnesium hydride chemistry of the PYR ligand.
Results and discussion
We recently reported a convenient high-yield method for the synthesis of [(DIPPnacnac)MgH]2 by thermally induced β-H elimination in the magnesium amidoborane precursor (DIPPnacnac)MgNH(iPr)BH3.23 The analogue bimetallic complex PYR-[MgNH(iPr)BH3]2 can be prepared according to Scheme 3, but thermal decomposition led to formation of a BNBN-product, presumably through an hydride intermediate.23 We found that, in case of a 2,6-iPr2C6H3 substituent (DIPP) on N, the fate of thermal decomposition is different. | ||
| Scheme 3 Synthesis and decomposition of [PYR-(MgH)2]2. | ||
PYR-[MgNH(DIPP)BH3]2 was prepared by careful deprotonation of H2N(DIPP)BH3 by PYR-(MgnBu)2 at −78 °C. The low temperature is essential and prevents catalytic decomposition of H2N(DIPP)BH3 in [(DIPP)NH]2BH and BH3, as reported earlier.25 The product, which could be isolated in the form of yellow block-like crystals (56%), has been characterized by crystal structure determination (Fig. 1a, Table 1). Its structure is similar to that of PYR-[MgNH(iPr)BH3]2 and shows a near C2-symmetric complex with two B,N-bridging amidoborane anions. Their Mg⋯H, Mg⋯B and Mg–N distances average 2.10(2) Å, 2.546(2) Å and 2.126(1) Å, respectively, and are similar to those in the iPr-substituted complex.23
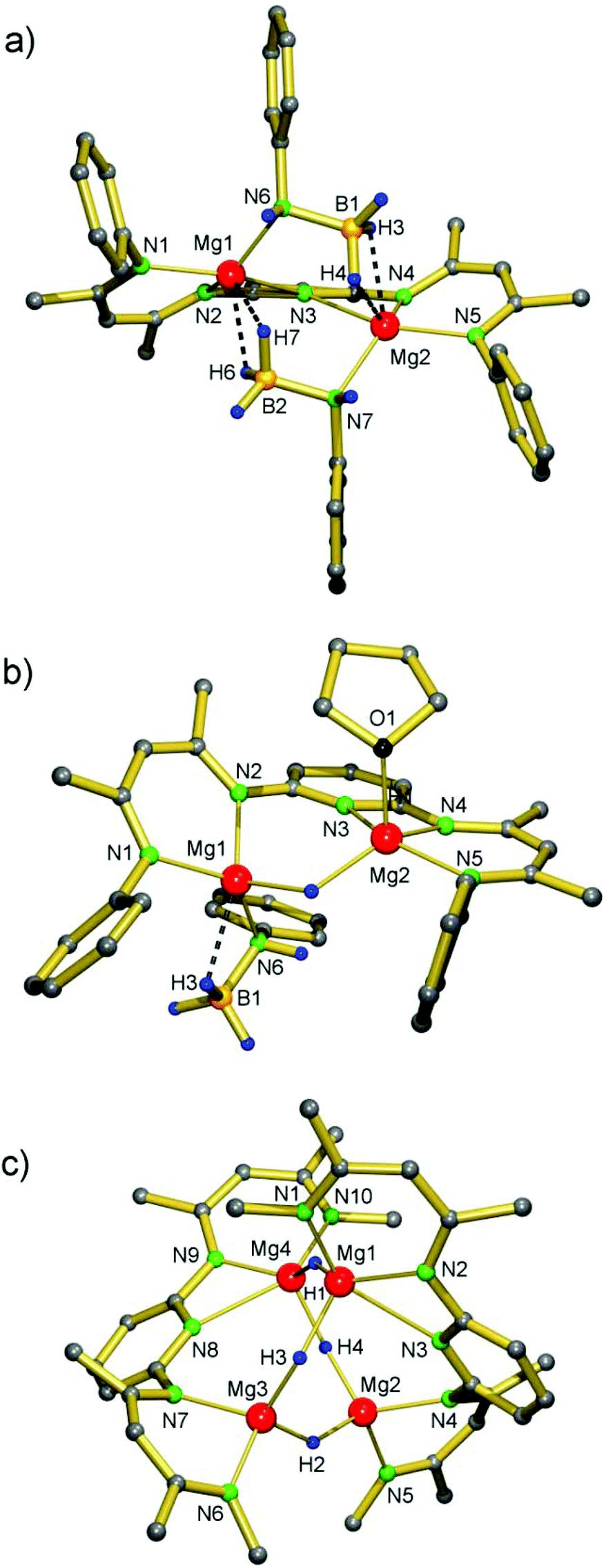 | ||
| Fig. 1 Crystal structures of (a) PYR-[MgNH(DIPP)BH3]2, (b) PYR-[MgNH(DIPP)BH3](MgH)·THF and (c) [PYR-(MgH)2]2. In all cases, the iPr substituents of the DIPP are not shown for clarity (for [PYR-(MgH)2]2 only the Cipso atoms of DIPP are shown). | ||
| PYR-[MgNH(DIPP)BH3]2 | |||
| Mg1–N1 | 2.085(1) | Mg2⋯N3 | 2.701(1) |
| Mg1–N2 | 2.031(1) | Mg2–N4 | 2.039(1) |
| Mg1⋯N3 | 2.780(1) | Mg2–N5 | 2.093(3) |
| Mg1–N6 | 2.121(1) | Mg2–N7 | 2.130(1) |
| Mg1⋯B2 | 2.538(2) | Mg2⋯B1 | 2.554(2) |
| Mg1⋯H6 | 2.21(2) | Mg2⋯H3 | 2.22(2) |
| Mg1⋯H7 | 1.98(2) | Mg2⋯H4 | 1.97(1) |
| PYR-[Mg2(H)NH(DIPP)BH3]2(THF) | |||
| Mg1–H1 | 1.95(2) | Mg2–H1 | 1.93(3) |
| Mg1–N1 | 2.081(2) | Mg2–N3 | 2.199(2) |
| Mg1–N2 | 2.093(2) | Mg2–N4 | 2.068(2) |
| Mg1–N6 | 2.093(2) | Mg2–N5 | 2.119(2) |
| Mg1⋯B1 | 2.649(3) | Mg2–O1 | 2.029(2) |
| [PYR-(MgH)2]2 | |||
| Mg1–H1 | 1.83(2) | Mg1–N1 | 2.121(2) |
| Mg1–H3 | 1.84(2) | Mg1–N2 | 2.055(2) |
| Mg2–H2 | 1.79(2) | Mg1⋯N3 | 2.614(2) |
| Mg2–H4 | 1.82(2) | Mg2–N4 | 2.031(2) |
| Mg3–H2 | 1.81(2) | Mg2–N5 | 2.062(2) |
| Mg3–H3 | 1.79(2) | Mg3–N6 | 2.055(2) |
| Mg4–H1 | 1.86(2) | Mg3–N7 | 2.040(2) |
| Mg4–H4 | 1.81(2) | Mg4⋯N8 | 2.542(2) |
| Mg4–N9 | 2.052(2) | ||
| Mg4–N10 | 2.139(2) | ||
The pyridylene bridge is symmetrically bridging the two Mg ions with rather long distances of 2.7–2.8 Å.
1H NMR signals for PYR-[MgNH(DIPP)BH3]2 in toluene-d8 are, at room temperature, rather broad. Cooling the sample to −50 °C gives a set of sharp signals which allow for unambiguous NMR characterization. The NH and BH3 groups show broad 1H NMR signals at 3.13 ppm and 2.28 ppm, respectively. The iPr groups in the ligand give rise to four doublet and two septet resonances and the same was found for the iPr groups in the NH(DIPP)BH3− ion.
In contrast to a toluene solution of (DIPPnacnac)MgNH(DIPP)BH3, which decomposes into (DIPPnacnac)MgN(DIPP) = BH2 and H2 at forced conditions (120 °C, 24 hours),23 a toluene solution of PYR-[MgNH(DIPP)BH3]2 already decomposes slowly at room temperature. A singlet 1H NMR signal at 4.45 ppm indicates H2 formation. 11B NMR spectra suggest formation of [(DIPP)NH]2BH and the BH4− ion, but well-defined products could not be isolated.
Interestingly, addition of minor amounts of THF to a benzene solution of PYR-[MgNH(DIPP)BH3]2 initiated a fast decomposition reaction and resulted in precipitation of yellow crystals (Scheme 3). Structural characterization of the product revealed the occurrence of a single β-H elimination: PYR-[MgNH(DIPP)BH3](MgH)·THF. The crystal structure (Fig. 1b, Table 1) shows a binuclear molecule with symmetrical bridging of H− between the Mg2+ centers: 193(3) Å and 1.95(2) Å. The remaining amidoborane anion binds only to Mg1. This unsymmetrical charge distribution of anions is balanced by strong coordination of the pyridylene bridge to Mg2 (2.199(2) Å) and an additional THF ligand.
1H NMR signals can be assigned unambiguously: the amidoborane part shows a broad BH3 signal at 1.58 ppm and a quartet for NH at 2.61 ppm (3J(H,H) = 3.4 Hz) whereas the bridging H− gives a singlet at 3.21 ppm.
The mechanism for the here observed THF-induced β-H elimination is subject of speculation. In light of the fact that the preliminary step for β-hydrogen elimination generally requires a low-coordinate metal center with an agostic β-H⋯metal contact,26 addition of THF should rather prevent such process. Lewis-base induced β-H eliminations are rare.27
We propose the following reaction sequence (Scheme 4): (i) coordination of THF results in slippage of a bridging amidoborane ion to terminal coordination and a BH3 group bridging two Mg2+ centers (in fact this intermediate has been observed earlier in the form of the NN-[MgNH(iPr)BH3]2·THF complex),23 (ii) this is followed by β-H elimination and formation of the product, (iii) the remaining DIPPN(H) = BH2 decomposes to [(DIPP)NH]2BH (detected by 11B NMR) and BH3 (detected in the form of BH4− by 11B NMR). The last step might go through an intermediate [DIPPN(H)BH2(DIPP)NHBH3]− anion. Similar intermediates have been isolated in early main group metal and transition metal chemistry.25,28–31
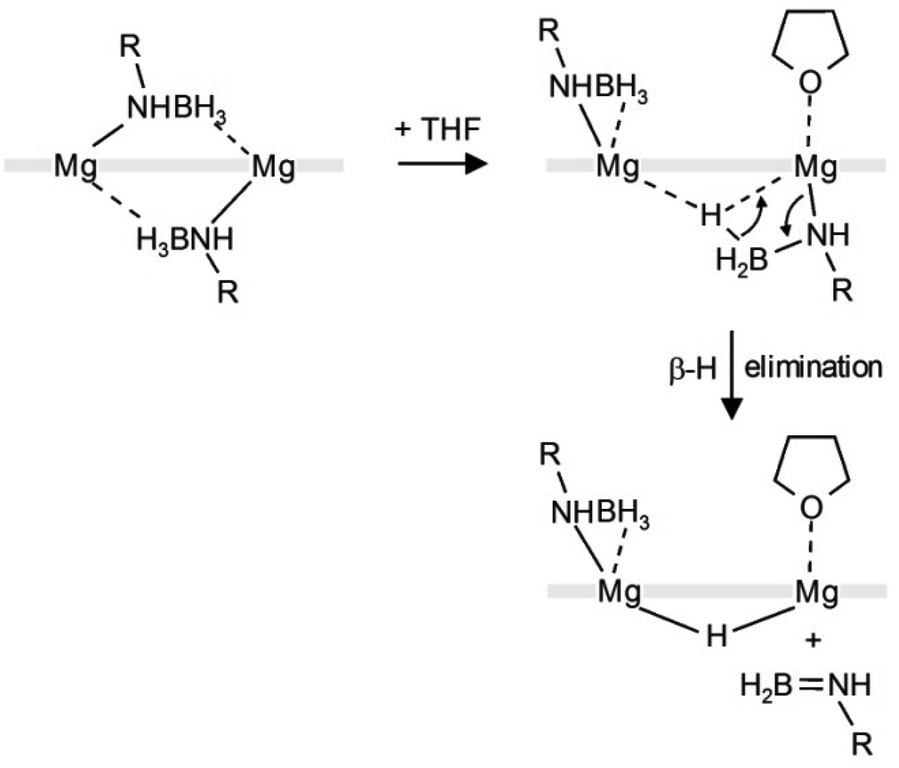 | ||
| Scheme 4 Proposed mechanism for THF-induced hydride formation; front view (the thick grey line represents PYR). | ||
The very mild nature of this Lewis-base induced hydride formation, which is smooth at room temperature, posed the question whether a second H− elimination might open a new route for preparation of magnesium hydride complexes. Unfortunately, addition of larger amounts of THF (or using THF as the solvent) did not trigger further β-H elimination. Slowly increasing the temperature of the THF solution led to H2 formation, presumably by reaction of the Mg–H function with [(DIPP)NH]2BH, and PYR-(MgH)2 could not be isolated.
Therefore, we chose the slower but more effective phenylsilane route:8,9 reaction of a PYR-(MgnBu)2 solution in benzene with two equivalents of PhSiH3 gave the product at 60 °C after three days, which could be crystallized from hexane (54%). The crystal structure of the dimer [PYR-(MgH)2]2 shows a near C2-symmetric tetranuclear cluster without exact crystallographic symmetry (Fig. 1c, Table 1) but with some similarity to [NN-(MgH)2]2.14 Although composition and connectivity are similar, the arrangement of Mg ions in [PYR-(MgH)2]2 is strongly distorted from the Mg4 tetrahedron found in [NN-(MgH)2]2. Whereas in the latter Mg⋯Mg distances vary from 3.030(1) to 3.586(1) Å, those in [PYR-(MgH)2]2 are longer and show larger variation: 3.410(1)–4.293(1) Å. This flattened Mg4 geometry is undoubtedly due to the large span width of the pyridylene bridged PYR ligand and results in an 8-membered ring of alternating Mg2+ and H− ions. However, the average Mg–H distances (H atoms have been located and were refined) are very similar in both structures: [NN-(MgH)2]2 1.808(9) and [PYR-(MgH)2]2 1.82(2), but are naturally subject to a large error margin. The pyridylene N atoms are asymmetrically bridging the Mg2+ ions with shorter weakly-bound (2.614(2)/2.542(2) Å) and longer essentially non-bonding (3.353(2)/3.407(2) Å) distances. Attempts to incorporate additional MgH2 into the cluster (cf. (iv) in Scheme 2) by reaction of PYR-(MgnBu)2/(MgnBu)2 mixtures with excess PhSiH3 failed.
The cluster [PYR-(MgH)2]2 contains three different hydride positions (H1, H3/H4 and H2). In case fast pyridyl-Mg1/Mg2 and pyridyl-Mg4/Mg3 switching takes place, at least two different hydride positions are present (H1/H2 and H3/H4). 1H NMR spectra of [PYR-(MgH)2]2 in toluene-d8, however, only show one singlet for the H− ligands. Cooling of the solution to −75 °C did not result in decoalescence. This strongly contrasts with the dynamic behaviour observed for [NN-(MgH)2]2: below room temperature the singlet hydride signal decoalesces in two triplets.14 We presume that for [PYR-(MgH)2]2 the solid state structure is maintained in solution but fast dynamics of ligand coordination geometries result in equal hydride positions: a time-averaged structure with symmetrically bridging pyridylene units and flat PYR ligands would be S4-symmetric. This is in agreement with the observation of two iPr doublet signals and one iPr heptet signal. Although a monomeric PYR-(MgH)2 structure with bridging H− ligands (type i in Scheme 2) cannot be ruled out, cf.PYR-(MgnBu)2,23 in apolar solvents it seems the least stable option (especially if one considers the short Mg–H bond distances of 1.8 Å).
The thermal decomposition of yellow crystals of [PYR-(MgH)2]2 was investigated by stepwise heating in a thermostated air-bath. At 130 °C the color changed via yellow-orange to dark red. Gas quantification by pumping the gas quantitatively into a calibrated burette of a Töpler pump system indicated the release of 1.1 ± 0.1 mol-equivalents of gas per [PYR-(MgH)2]2. Extended heating did not result in more gas elimination. The gas was proven to be H2: (i) it does not condense in liquid N2 but is fully converted to condensable water after leading it over CuO of 300 °C; (ii) after leading the gas in deuterated THF, a clear 1H NMR resonance could be observed at 4.55 ppm which is the chemical shift for H2 in this solvent.18
Thermal H2 desorption from the [PYR-(MgH)2]2 cluster differs from the earlier reported results for [NN-(MgH)2]2: hydrogen is released at the significantly lower temperature of 130 °C (vs. 175 °C) and only 50% of the theoretical hydrogen content could be detected (vs. full conversion). This is likely due to the fact that pyridine is susceptible towards nucleophilic addition of H− in ortho- or para-positions to form a dearomatized amide. Proposed nucleophilic attack at the para-position (Scheme 3) explains reduced H2 release. In contrast to [NN-(MgH)2]2, which when dissolved in toluene is stable even after prolonged heating at 150 °C, a toluene solution of [PYR-(MgH)2]2 decomposes by a colour change from yellow to dark-red after one hour at this temperature. 1H NMR analysis indicates dearomatization of the pyridyl ring, but due to the presence of several different species, no well-defined products could be isolated. We propose initial formation of a dihydropyridide bridged β-diketiminate-Mg(I) complex (Scheme 3) which decomposes into various undefined species.
Conclusions
The magnesium hydride chemistry with the 2,6-pridylene bridged bis-β-diketiminate ligand, PYR, has been investigated. Although one of the amidoborane anions in PYR-[MgNH(DIPP)BH3]2 could be converted into H− by a THF-induced β-H elimination, such a transformation could not be achieved for the second amidoborane anion. Reaction of PYR-(MgnBu)2 with PhSiH3 gave the desired magnesium hydride complex in the form of the dimer [PYR-(MgH)2]2. Attempts to incorporate additional MgH2 into the cluster failed. The structure of [PYR-(MgH)2]2 is different from that reported previously for [NN-(MgH)2]2, a complex with directly coupled β-diketiminate units and four Mg2+ ions at the corners of a tetrahedron. Instead, a flattened Mg4 geometry is found in which alternating Mg2+ and H− ions form an 8-membered ring. [PYR-(MgH)2]2 decomposes at 130 °C releasing one equivalent of H2. In contrast, [NN-(MgH)2]2 decomposes at 175 °C and releases two equivalents of H2. It is proposed that nucleophilic attack at the para-position explains reduced H2 formation (Scheme 3). Although the fate of the magnesium is unclear, it is likely that low-valent magnesium species were formed initially. We continue our investigations with the challenging isolation of larger well-defined Mg(I) clusters.Experimental
General
All experiments were carried out using standard Schlenk-techniques and freshly dried solvents. The following compounds were prepared according to the literature: PYR-H2,22PYR-[Mg(nBu)]2,23 (DIPP)NH2BH3.32 NMR spectra were measured using a Bruker DPX300 and DRX500 spectrometer. Crystals were measured using a Siemens Smart diffractometer with APEXII area detector system.Syntheses
1H{11B} NMR (500 MHz, toluene-d8, −40 °C): δ = 0.19 (d, 3J(H,H) = 6.3 Hz, 6H, iPr), 0.62 (d, 3J(H,H) = 6.2 Hz, 6H, iPr), 0.86 (d, 3J(H,H) = 6.5 Hz, 6H, iPr), 0.95 (d, 3J(H,H) = 6.3 Hz, 6H, iPr), 1.19 (d, 3J(H,H) = 6.0 Hz, 6H, iPr), 1.25 (d, 3J(H,H) = 6.5 Hz, 6H, iPr), 1.42 (d, 3J(H,H) = 6.2 Hz, 6H, iPr), 1.44 (s, 6H, Me backbone), 1.49 (d, 3J(H,H) = 6.0 Hz, 6H, iPr), 1.98 (s, 6H, Me backbone), 2.28 (br, 6H, BH3), 2.64 (sept, 3J(H,H) = 6.2 Hz, 2H, iPr), 2.81 (sept, 3J(H,H) = 6.3 Hz, 2H, iPr), 3.13 (br, 2H, NH), 3.17 (sept, 3J(H,H) = 6.5 Hz, 2H, iPr), 3.59 (sept, 3J(H,H) = 6.0 Hz, 2H, iPr), 4.79 (s, 2H, H backbone), 6.34 (d, 3J(H,H) = 8.0 Hz, 2H, aryl), 6.78 (d, 3J(H,H) = 6.4 Hz, 2H, aryl), 6.94 (t, 3J(H,H) = 8.0 Hz, 1H, aryl), 6.97–7.07 (m, 8H, aryl), 7.12 (m, 2H, aryl). 11B NMR (160 MHz, toluene-d8, 20 °C): δ = –18.0 (br). 13C NMR (data taken from 2D-spectra, 75 MHz, toluene-d8, −40 °C): δ = 23.2 (iPr), 24.6 (iPr), 24.6 (Me backbone), 24.6 (iPr), 24.8 (iPr), 25.0 (iPr), 25.3 (iPr), 25.3 (Me Backbone), 25.9 (iPr), 27.3 (iPr), 27.4 (iPr), 28.3 (iPr), 28.4 (iPr), 29.1 (iPr), 100.0 (backbone), 110.7 (aryl), 122.9 (aryl), 123.7 (aryl), 124.1 (aryl), 124.2 (aryl), 125.2 (aryl), 126.4 (aryl), 138.4 (aryl), 139.7 (aryl), 140.9 (aryl), 142.3 (aryl), 142.3 (aryl), 145.5 (aryl), 145.6 (aryl), 161.1 (aryl), 162.5 (backbone), 173.1 (backbone).
Crystal structure determinations
The structures were solved by direct methods (SHELXS-97)33 and refined with SHELXL-97.34 All geometry calculations and graphics were performed with PLATON.35![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, ρcalc = 1.103 g cm−3, μ(MoKα) = 0.078 mm−1, 53
, Z = 2, ρcalc = 1.103 g cm−3, μ(MoKα) = 0.078 mm−1, 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 697 measured reflections, 15620 independent reflections (Rint = 0.029), 12390 reflections observed with I > 2σ(I), θmax = 26.4°, R = 0.0401, wR2 = 0.1043, GOF = 1.03, 1128 parameter, min/max residual electron density −0.30/+0.44 e Å−3. Cocrystallized solvent molecules, 3.5 molecules of benzene, were relatively well-ordered and refined anisotropically. All hydrogen atoms, except those of cocrystallized benzene molecules, were found in the difference-Fourier map and were refined isotropically. Hydrogen atoms on the benzene molecules were placed on idealized calculated positions.
850 measured reflections, 10889 independent reflections (Rint = 0.038), 9207 reflections observed with I > 2σ(I), θmax = 26.5°, R = 0.0480, wR2 = 0.1260, GOF = 1.06, 621 parameter, min/max residual electron density −0.24/+0.61 e Å−3. Two cocrystallized molecules of heavily disordered benzene were treated by the SQUEEZE procedure incorporated in PLATON (214 Å3, 98 e).15 The hydride and hydrogens on N and B were found in the difference-Fourier map and refined isotropically. All other hydrogen atoms have been placed on calculated positions and were refined in a riding mode. The correct handedness of the chiral unit cell has been checked by refinement of the Flack parameter to 0.01 with esd 0.18.
576 measured reflections, 15303 independent reflections (Rint = 0.077), 15303 reflections observed with I > 2σ(I), θmax = 25.8°, R = 0.0487, wR2 = 0.1165, GOF = 0.94, 1228 parameter, min/max residual electron density −0.24/+0.43 e Å−3. Heavily disordered cocrystallized solvent was treated by the SQUEEZE procedure incorporated in PLATON (290 Å3, 52 e).15 One of the iPr groups shows rotational disorder over two positions that were refined isotropically. Hydrogen atoms were found in the difference-Fourier map and were refined isotropically, except for those on the disordered iPr group. The latter were placed on idealized positions and refined in a riding mode.
697 measured reflections, 15620 independent reflections (Rint = 0.029), 12390 reflections observed with I > 2σ(I), θmax = 26.4°, R = 0.0401, wR2 = 0.1043, GOF = 1.03, 1128 parameter, min/max residual electron density −0.30/+0.44 e Å−3. Cocrystallized solvent molecules, 3.5 molecules of benzene, were relatively well-ordered and refined anisotropically. All hydrogen atoms, except those of cocrystallized benzene molecules, were found in the difference-Fourier map and were refined isotropically. Hydrogen atoms on the benzene molecules were placed on idealized calculated positions.
850 measured reflections, 10889 independent reflections (Rint = 0.038), 9207 reflections observed with I > 2σ(I), θmax = 26.5°, R = 0.0480, wR2 = 0.1260, GOF = 1.06, 621 parameter, min/max residual electron density −0.24/+0.61 e Å−3. Two cocrystallized molecules of heavily disordered benzene were treated by the SQUEEZE procedure incorporated in PLATON (214 Å3, 98 e).15 The hydride and hydrogens on N and B were found in the difference-Fourier map and refined isotropically. All other hydrogen atoms have been placed on calculated positions and were refined in a riding mode. The correct handedness of the chiral unit cell has been checked by refinement of the Flack parameter to 0.01 with esd 0.18.
576 measured reflections, 15303 independent reflections (Rint = 0.077), 15303 reflections observed with I > 2σ(I), θmax = 25.8°, R = 0.0487, wR2 = 0.1165, GOF = 0.94, 1228 parameter, min/max residual electron density −0.24/+0.43 e Å−3. Heavily disordered cocrystallized solvent was treated by the SQUEEZE procedure incorporated in PLATON (290 Å3, 52 e).15 One of the iPr groups shows rotational disorder over two positions that were refined isotropically. Hydrogen atoms were found in the difference-Fourier map and were refined isotropically, except for those on the disordered iPr group. The latter were placed on idealized positions and refined in a riding mode.
Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 992444–992446 for PYR-[MgNH(DIPP)BH3](MgH)·THF, PYR-[MgNH(DIPP)BH3]2, and [PYR-(MgH)2]2, respectively.
Acknowledgements
The DFG is gratefully acknowledged for financial support. Prof. R. Boese and D. Bläser are thanked for measuring X-ray data and H. Bandmann for measuring the 500 MHz NMR spectra.Notes and references
- G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305 CrossRef CAS PubMed.
- P. Rittmeyer and U. Wietelmann, Hydrides, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012, p. 103 Search PubMed.
- S. Harder, Chem. Commun., 2012, 48, 11165 RSC.
- D. Hoffmann, T. Kottke, R. J. Lagow and R. D. Thomas, Angew. Chem., Int. Ed., 1998, 37, 1537 CrossRef.
- D. R. Armstrong, W. Clegg, R. P. Davies, S. T. Liddle, D. J. Linton, P. R. Raithby, R. Snaith and A. E. H. Wheatley, Angew. Chem., Int. Ed., 1999, 38, 3367 CrossRef CAS.
- D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O'Hara, R. E. Mulvey and R. B. Rowlings, Chem. Commun., 2002, 376 RSC.
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2009, 48, 4013 CrossRef CAS PubMed.
- S. Harder, J. Spielmann, J. Intemann and H. Bandmann, Angew. Chem., Int. Ed., 2011, 50, 4156 CrossRef CAS PubMed.
- (a) A. Stasch, Angew. Chem., Int. Ed., 2012, 51, 1930 CrossRef CAS PubMed; (b) C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2012, 51, 5911 CrossRef CAS PubMed.
- P. Jochmann, J. P. Davin, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 2098 CrossRef PubMed.
- J. Intemann, J. Spielmann, P. Sirsch and S. Harder, Chem. – Eur. J., 2013, 19, 8478 CrossRef CAS PubMed.
- A. Stasch, Angew. Chem., Int. Ed., 2014, 53, 1338 CrossRef CAS PubMed.
- F. Buch, J. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741 CrossRef CAS PubMed.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS PubMed.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A., 2010, 466, 927 CrossRef CAS.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865 CrossRef CAS PubMed.
- R. W. P. Wagemans, J. H. van Lenthe, P. E. de Jongh, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2005, 127, 16675 CrossRef CAS PubMed.
- D. F.-J. Piesik, S. Range and S. Harder, Organometallics, 2008, 27, 6178 CrossRef CAS.
- J. Spielmann, D. F.-J. Piesik and S. Harder, Chem. – Eur. J., 2010, 16, 8307 CrossRef CAS PubMed.
- H. Gehring, R. Metzinger, C. Hertwig, J. Intemann, S. Harder and C. Limberg, Chem. – Eur. J., 2013, 19, 1629 CrossRef CAS PubMed.
- J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934 RSC.
- (a) P. S. Braterman and R. J. Cross, Chem. Soc. Rev., 1973, 2, 271 RSC; (b) G. F. Schmidt and M. Brookhart, J. Am. Chem. Soc., 1985, 107, 1443 CrossRef CAS; (c) B. J. Burger, M. E. Thompson, W. D. Cotter and J. E. Bercaw, J. Am. Chem. Soc., 1990, 112, 1566 CrossRef CAS.
- (a) K. Yan, J. J. D. Heredia, A. Ellern, M. S. Gordon and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 15225 CrossRef CAS PubMed; (b) D. Mukherjee, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 7582 CrossRef CAS PubMed.
- D. J. Liprot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem. – Eur. J., 2010, 16, 8508 CrossRef PubMed.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS PubMed.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 27 RSC.
- D. Y. Kim, N. J. Singh, H. M. Lee and K. S. Kim, Chem. – Eur. J., 2009, 15, 5598 CrossRef CAS PubMed.
- J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, Universität Göttingen, Göttingen (Germany), 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, Universität Göttingen, Göttingen (Germany), 1997 Search PubMed.
- A. L. Spek, Platon, A Multipurpose Crystallographic Tool, Utrecht (The Netherlands), 2000 Search PubMed.
Footnote |
| † CCDC 992444–992446. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00835a |
| This journal is © The Royal Society of Chemistry 2014 |