 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(phosphinoselenoic amides) as versatile chelating ligands for alkaline earth metal (Mg, Ca, Sr and Ba) complexes: syntheses, structure and ε-caprolactone polymerisation†
Ravi K.
Kottalanka
a,
Harinath
Adimulam
a,
Jayeeta
Bhattacharjee
a,
H.
Vignesh Babu
b and
Tarun K.
Panda
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Ordnance Factory Estate, Yeddumailaram 502205, Andhra Pradesh, India. E-mail: tpanda@iith.ac.in; Fax: +91(40)2301 6032; Tel: +91(40)2301 6036
bSchool of Chemistry, University of Hyderabad, Hyderabad, India
First published on 3rd April 2014
Abstract
We report here a series of heavier alkaline earth metal complexes with N,N′-(ethane-1,2-diyl)bis(P,P-diphenylphosphinoselenoic amide) using two synthetic routes. In the first route, the heavier alkaline earth metal bis(trimethylsilyl)amides [M{N(SiMe3)2}2(THF)n] (M = Ca, Sr, Ba), when treated with phosphinoselenoic amine [Ph2P(Se)NHCH2CH2NHPPh2(Se)] (1), afforded the corresponding alkaline earth metal complexes of the composition [(THF)3M{Ph2P(Se)NCH2CH2NPPh2(Se)}] [M = Ca (2), Sr (3), Ba (4)]. The metal complexes 2–4 were also obtained from a one-pot reaction, where potassium phosphinoselenoic amide was generated in situ by the reaction of compound 1 and [KN(SiMe3)2], followed by the addition of the respective metal diiodides in THF at room temperature. The magnesium complex [(THF)3Mg{Ph2P(Se)NCH2CH2NPPh2(Se)}] (5) was also prepared. The solid-state structures of alkaline earth metal complexes 2–5 were established by single crystal X-ray diffraction analysis. In the solid state, all the metal complexes are monomeric but in complexes 2–4, ligand 1 is chelated in a tetra-dentate fashion to each metal ion but in complex 5, ligand 1 behaves as a bidentate ligand. Complexes 2–4 were tested as catalysts for the ring-opening polymerisation of ε-caprolactone and a high level of activity for the barium complex 4 was observed, with narrow polydispersity. We also report the synthesis and structure of the bis(amidophosphino borane) ligand [Ph2P(BH3)NHCH2CH2NHPPh2(BH3)] (6) and the corresponding barium complex [(THF)2Ba{Ph2P(BH3)NCH2CH2NPPh2(BH3)}]2 (7).
Introduction
Aliphatic polyesters are currently considered as alternatives to synthetic petrochemical-based polymers and are attractive to researchers, since the starting materials for their synthesis can be obtained from annually renewable resources. Their biodegradable and biocompatible nature along with their mechanical and physical properties make them prospective thermoplastics with broad commercial applications (e.g., single-use packaging materials, medical sutures and drug delivery systems).1,2 It is well established that polymer properties are highly dependent on their intrinsic structural parameters, such as polymer composition, molecular weight, polydispersity, tacticity, and polymer chain ends.3,4 Ring-opening polymerisation (ROP) of cyclic esters promoted, for example, by metal initiators proved to be the most efficient way for preparing polyesters with controlled molecular weight and microstructure and narrow molecular-weight distribution.5–7 This leads to the design and synthesis of new, well-defined, single-site catalysts that exhibit good activity, productivity and selectivity for cyclic ester polymerisation and allow crucial polymer architecture control.Group 2 metal complexes have attracted considerable attention as initiators for the ROP of cyclic esters, and some of them have demonstrated impressive results.8 Recently, we reported the synthesis of alkaline earth metal complexes with iminopyrrolyl and amidopyrrolyl ligands and they proved to be highly active for the ROP of ε-caprolactone, affording high-molecular-weight poly(ε-caprolactone)s.9 We have continuously studied the complexation reaction of amidophosphine-chalcogen-based ligands to alkaline earth metal precursors, for example synthesis of homoleptic alkaline earth metal complexes of the composition [M(THF)2{Ph2P(Se)N(CHPh2)}2] (M = Ca, Sr, Ba).10 We also reported the amido-phosphine borane complexes of [M(THF)2{Ph2P(BH3)N(CHPh2)}2] (M = Ca, Sr, Ba).11 All these ligands are monoanionic and form homoleptic mono-nuclear complexes. To explore the chemistry of heavier alkaline earth metals and their application in ε-caprolactone polymerisation, we focused on the dianionic system N,N′-(ethane-1,2-diyl)bis(P,P-diphenylphosphinoselenoic amide) [Ph2P(Se)NCH2CH2NPPh2(Se)}]2− which was recently prepared by Woollins et al.12 However, their detailed study of alkaline earth metal chemistry has not been available so far. In this context, the synthetic and structural details of bis(phosphinoselenoic amide) alkaline earth metal complexes with the composition [(THF)3M{Ph2P(Se)NCH2CH2NPPh2(Se)}] [M = Ca (2), Sr (3), Ba (4), Mg (5)] are presented. We also report the ROP study of ε-caprolactone using complexes 2–4 as catalysts. In addition, we present the synthesis and structure of the bis(amidophosphino borane) ligand [Ph2P(BH3)NHCH2CH2NHPPh2(BH3)}] and the corresponding dimeric barium complex [(THF)2Ba{Ph2P(BH3)NCH2CH2NPPh2(BH3)}]2.
Results and discussion
Ligands
N,N′-(Ethane-1,2-diyl)bis(P,P-diphenylphosphinoselenoic amide) was prepared according to the method described by Woollins et al. by the reaction of bis(diphenylphosphino)ethane-1,2-diamine, [Ph2PNHCH2CH2NHPPh2] and elemental selenium in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio at room temperature in THF.12 The spectroscopic data for compound 1 were in full agreement with the reported values. The solid-state structure of compound 1 was established by single crystal X-ray diffraction analysis. When compound 1 was re-crystallised from a mixture of THF–pentane (1:2) at −35 °C, a trans product (1-trans) was obtained. However, crystallisation from dichloromethane at room temperature afforded a cis product (1-cis). This indicates that there is equilibrium between cis and trans forms in solution (see Scheme 1). 1-trans crystallises in the orthorhombic space group Pbca, with one molecule each of 1-trans and THF in the asymmetric unit. In contrast, 1-cis crystallises in the monoclinic space group C2/c, with four isolated molecules in the unit cell. The molecular structures of 1-cis and 1-trans are shown in Fig. 1. The details of the structural parameters are given in Table TS1 in the ESI.†
:2 molar ratio at room temperature in THF.12 The spectroscopic data for compound 1 were in full agreement with the reported values. The solid-state structure of compound 1 was established by single crystal X-ray diffraction analysis. When compound 1 was re-crystallised from a mixture of THF–pentane (1:2) at −35 °C, a trans product (1-trans) was obtained. However, crystallisation from dichloromethane at room temperature afforded a cis product (1-cis). This indicates that there is equilibrium between cis and trans forms in solution (see Scheme 1). 1-trans crystallises in the orthorhombic space group Pbca, with one molecule each of 1-trans and THF in the asymmetric unit. In contrast, 1-cis crystallises in the monoclinic space group C2/c, with four isolated molecules in the unit cell. The molecular structures of 1-cis and 1-trans are shown in Fig. 1. The details of the structural parameters are given in Table TS1 in the ESI.†
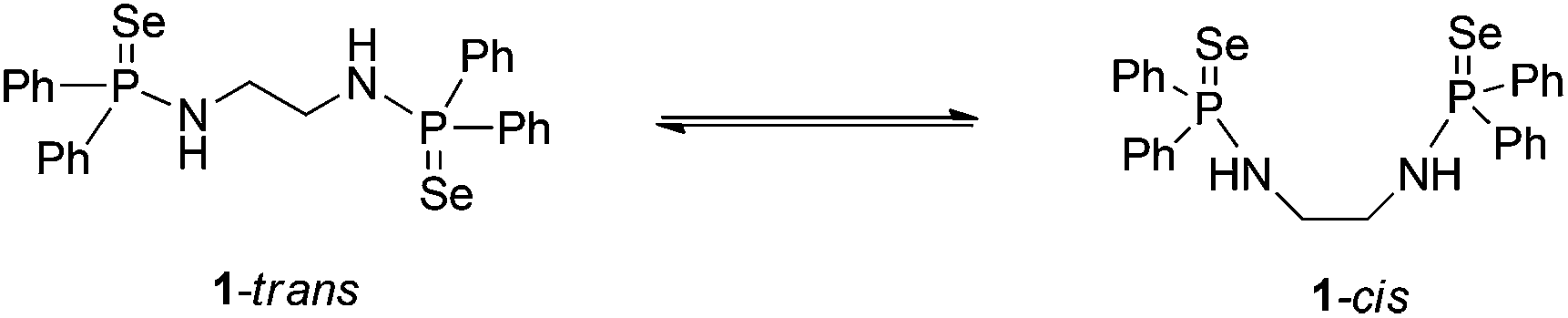 | ||
| Scheme 1 trans and cis forms of ligand 1. | ||
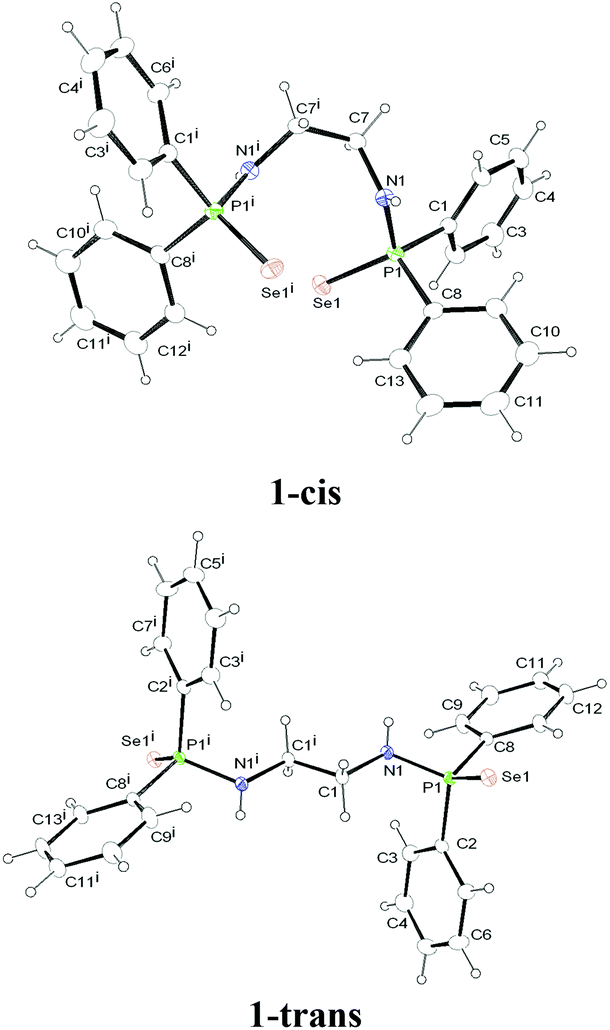 | ||
| Fig. 1 Solid-state structures of compounds 1-cis and 1-trans, ellipsoids are drawn to encompass 30 per cent probability. Selected bond lengths [Å] and bond angles [°]: 1-cis: P1–N1 1.665(2), P1–Se1 2.1194(7), P1–C1 1.810(3), P1–C8 1.810(3), N1–C7 1.468(3), C7–C7i 1.512(5), C7i–C7–N1 112.9(2), N1–P1–C8 102.55(12), N1–P1–Se1 116.97(8), N1–P1–C1 104.95(12), P1–N1–C7 120.73(18), Se1–P1–C8 113.29(10), Se1–P1–C1 111.36(9). 1-trans: P1–N1 1.6660(18), P1–Se1 2.1057(5), P1–C2 1.817(2), P1–C8 1.816(2), N1–C1 1.468(3), C1–C1i 1.507(4), C1i–C1–N1 109.4(2), N1–P1–C8 101.65(9), N1–P1–Se1 118.10(7), N1–P1–C2 102.72(10), P1–N1–C1 117.58(14), Se1–P1–C8 112.19(7), Se1–P1–C2 112.45(7). | ||
The P–Se bond distances [2.1194(7) Å for 1-cis and 2.1057(5) Å for 1-trans] are in the range similar to that of [Ph2P(Se)NH(CHPh2)] [2.1086(12) Å], [Ph2P(Se)NH(CPh3)] [2.1166(8) Å] and [Ph2P(Se)NH(2,6-Me2C6H3)] [2.1019(8) Å], previously we reported to consider the phosphorus–selenium bond as a double bond.10,13b P1–N1 bond distances of 1.665(2) Å and 1.116(2) Å for 1-cis and 1-trans respectively are in the expected range, as reported for other phosphinoselenoic amido compounds in the literature.13 In the centro-symmetric 1-trans form, C1–N1 and C1i–N1i bonds are trans to each other. Compound 1-cis also possesses a centre of inversion i, the middle of the C7–C7i bond. C7–N1 and C7i–N1i bonds are cis to each other and a dihedral angle of 67.09° is formed by the planes containing C7, N1, P1 and C7i, N1i, P1i atoms. Thus, planes containing the N1, P1, Se1 and N1i, P1i, Se1i atoms are not coplanar, but almost orthogonal (86.90°) to each other.
Alkaline earth metal complexes
The title compounds [(THF)3M{Ph2P(Se)NCH2CH2NPPh2(Se)}] [M = Ca (2), Sr (3), Ba (4)] can be obtained by two different synthetic approaches. Both the approaches were used for the calcium complex 2, whereas the other complexes were obtained using a one-pot reaction only. In the first approach, the well-established compound [(THF)2Ca{N(SiMe3)2}2]14,15 was made to react with compound 1 in a 1:1 molar ratio in THF at room temperature to afford the corresponding bis(diphenylphosphinoselenoic amide) complex 2via the elimination of volatile bis(trimethylsilyl)amine (see Scheme 2). However, the most convenient approach for obtaining complexes 2–4 is a one-pot reaction, in which the ligand, [Ph2P(Se)NHCH2CH2NHP(Se)Ph2], is made to react with anhydrous potassium bis(trimethylsilyl)amide in a 1:2 molar ratio in THF to generate in situ potassium salt of ligand 1, followed by the addition of anhydrous alkaline earth metal diiodide to the reaction mixture (see Scheme 2).16
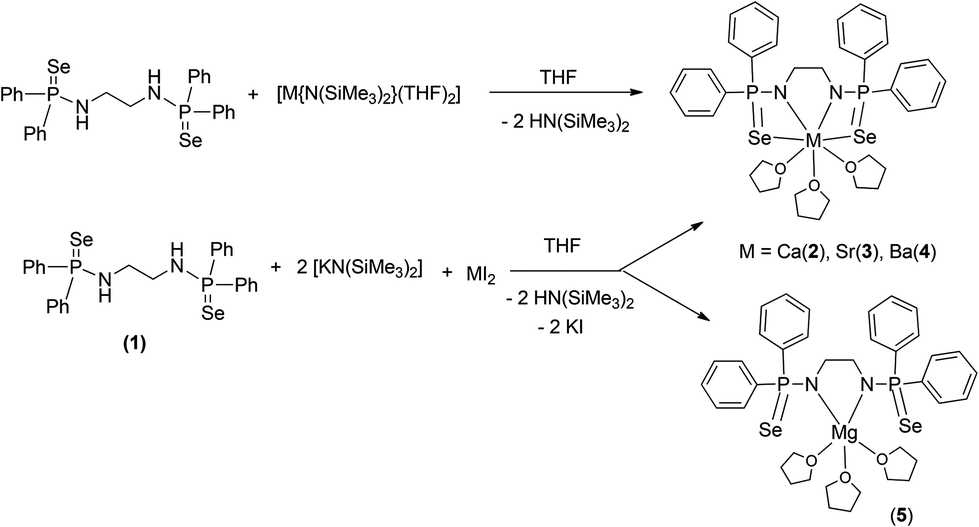 | ||
| Scheme 2 Syntheses of alkaline earth metal phosphinoselenoic amide complexes. | ||
The corresponding magnesium complex [(THF)3Mg{Ph2P(Se)NCH2CH2NPPh2(Se)}] (5) was obtained by the second route, using ligand 1 and [KN(SiMe3)2], followed by addition of magnesium diiodide in THF solvent (see Scheme 2).
The new complexes were characterised using standard analytical and spectroscopic techniques, and the solid-state structures of all four alkaline earth metal complexes were established by single crystal X-ray diffraction analysis. A strong absorption band at 550 cm−1 (for complex 2), 552 cm−1 (for complex 3), 555 cm−1 (for complex 4) and 551 cm−1 (for complex 5) in FT-IR spectra indicates a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se bond in each complex. The PSe stretching frequencies are within the range reported by us.10,13b The 1H NMR spectra of the diamagnetic compounds 2–5 show a multiplet signal [δ 3.39 ppm (complex 2), 2.87 ppm (complex 3), 3.14 ppm (complex 4) and 3.09 ppm (complex 5)] for the four methylene protons and this is very close to the resonance signal (δ 3.16 ppm) of the analogous methylene protons present in free ligand 1. Each of the complexes 2–5 shows a sharp signal in the 31P{1H} NMR spectra [δ 71.8 (complex 2), 71.9 (complex 3), 73.3 ppm (complex 4) and 43.7 ppm (complex 5)], which is significantly low field shifted for complexes 2–4 and high field shifted for complex 5 to free ligand 1 (δ 59.6 ppm),12 showing that both the phosphorous atoms in each complex are chemically equivalent in solution. All three complexes are coordinated to THF molecules, as is evident from the typical multiplet signals at 3.65–3.55 ppm and 1.35–1.33 ppm observed in 1H NMR spectra.
Se bond in each complex. The PSe stretching frequencies are within the range reported by us.10,13b The 1H NMR spectra of the diamagnetic compounds 2–5 show a multiplet signal [δ 3.39 ppm (complex 2), 2.87 ppm (complex 3), 3.14 ppm (complex 4) and 3.09 ppm (complex 5)] for the four methylene protons and this is very close to the resonance signal (δ 3.16 ppm) of the analogous methylene protons present in free ligand 1. Each of the complexes 2–5 shows a sharp signal in the 31P{1H} NMR spectra [δ 71.8 (complex 2), 71.9 (complex 3), 73.3 ppm (complex 4) and 43.7 ppm (complex 5)], which is significantly low field shifted for complexes 2–4 and high field shifted for complex 5 to free ligand 1 (δ 59.6 ppm),12 showing that both the phosphorous atoms in each complex are chemically equivalent in solution. All three complexes are coordinated to THF molecules, as is evident from the typical multiplet signals at 3.65–3.55 ppm and 1.35–1.33 ppm observed in 1H NMR spectra.
Although there is ongoing interest in alkaline earth organometallics17 and particularly in the cyclopentadienyl chemistry of these elements,18 complexes 2–5 represent, to the best of our knowledge, the first alkaline earth metal complexes containing a bis(diphenylphosphinoselenoic amide) ligand having two sets of three heteroatoms, N, P and Se, adjacent to each other in the ligand. Therefore, their molecular structures in the solid state were determined by X-ray diffraction analysis.
The calcium, strontium bis(diphenylphosphinoselenoic amido) complexes 2 and 3 crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two molecules of 2 and 3 in the unit cell respectively. The slightly larger barium compound 4 also crystallises in the triclinic space group P, with two independent molecules of complex 4 in the asymmetric unit. The details of the structural parameters are given in Table TS1 in ESI.† The solid-state structures of complexes 3 and 4 are shown in Fig. 2 and 3 respectively. Complexes 2–4 are isostructural to each other due to the similar ionic radii of the metal ions (1.00 Å, 1.18 Å and 1.35 Å respectively) for a coordination number of six.19
, with two molecules of 2 and 3 in the unit cell respectively. The slightly larger barium compound 4 also crystallises in the triclinic space group P, with two independent molecules of complex 4 in the asymmetric unit. The details of the structural parameters are given in Table TS1 in ESI.† The solid-state structures of complexes 3 and 4 are shown in Fig. 2 and 3 respectively. Complexes 2–4 are isostructural to each other due to the similar ionic radii of the metal ions (1.00 Å, 1.18 Å and 1.35 Å respectively) for a coordination number of six.19
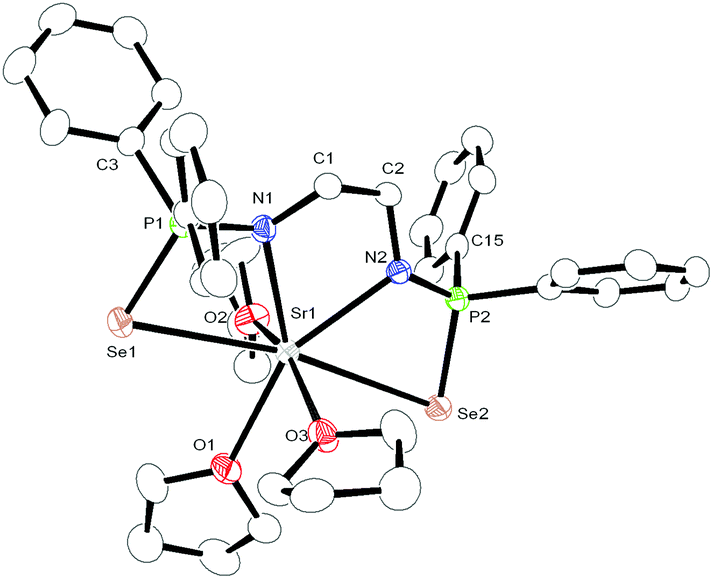 | ||
| Fig. 2 Solid-state structure of compound 3, ellipsoids are drawn to encompass 30 per cent probability, omitting hydrogen atoms for clarity. Selected bond lengths [Å] and bond angles [°]: complex 2 (Ca): P1–N1 1.592(8), P1–Se1 2.148(3), P1–C9 1.824(10), P1–C3 1.832(10), N1–C1 1.450(13), P2–N2 1.595(8), P2–Se2 2.155(3), P2–C15 1.833(9), P2–C21 1.824(10), N2–C2 1.433(13), C1–C2 1.526(13), Ca1–N1 2.386(8), Ca1–Se1 3.252(2), Ca1–P1 3.359(3), Ca1–N2 2.418(8), Ca1–Se2 3.300(2), Ca1–P2 3.364(3), Ca1–O1 2.374(7), Ca1–O2 2.429(7), Ca1–O3 2.385(7), C2–C1–N1 108.0(8), N1–P1–C9 112.3(4), N1–P1–Se1 108.4(3), N1–P1–C3 111.0(4), P1–N1–C1 123.0(7), Se1–P1–C9 110.3(3), Se1–P1–C3 111.3(3), C1–C2–N2 109.2(8), N2–P2–C15 111.9(4), N2–P2–Se2 108.8(3), N2–P2–C21 112.4(5), P2–N2–C2 123.6(7), Se2–P2–C15 111.8(3), Se2–P2–C21 110.1(3), N1–Ca1–Se1 63.3(2), N1–Ca1–N2 68.6(3), Se1–Ca1–Se2 165.42(7), O1–Ca1–O2 79.6(3), O1–Ca1–O3 159.1(3), O2–Ca1–O3 80.4(3), P1–Se1–Ca1 73.76(8), P2–Se2–Ca1 72.76(8). Complex 3 (Sr): P1–N1 1.592(5), P1–Se1 2.1548(17), P1–C9 1.823(7), P1–C3 1.826(7), N1–C1 1.465(7), P2–N2 1.589(5), P2–Se2 2.1559(17), P2–C15 1.837(6), P2–C21 1.819(6), N2–C2 1.463(7), C1–C2 1.524(8), Sr1–N1 2.517(5), Sr1–Se1 3.2788(10), Sr1–P1 3.4456(18), Sr1–N2 2.540(5), Sr1–Se2 3.3259(10), Sr1–P2 3.4521(16), Sr1–O1 2.568(5), Sr1–O2 2.529(5), Sr1–O3 2.537(5), C2–C1–N1 108.4(5), N1–P1–C9 111.3(3), N1–P1–Se1 109.13(19), N1–P1–C3 112.7(3), P1–N1–C1 122.7(4), Se1–P1–C9 110.3(2), Se1–P1–C3 109.1(2), C1–C2–N2 109.3(5), N2–P2–C15 112.2(3), N2–P2–Se2 108.72(19), N2–P2–C21 112.6(3), P2–N2–C2 123.4(4), Se2–P2–C15 111.2(2), Se2–P2–C21 109.8(2), N1–Sr1–Se1 62.35(11), N1–Sr1–N2 65.80(15), Se1–Sr1–Se2 170.54(2), O1–Sr1–O2 80.67(18), O1–Sr1–O3 80.37(17), O2–Sr1–O3 159.22(17), P1–Se1–Sr1 75.57(5), P2–Se2–Sr1 74.67(5). | ||
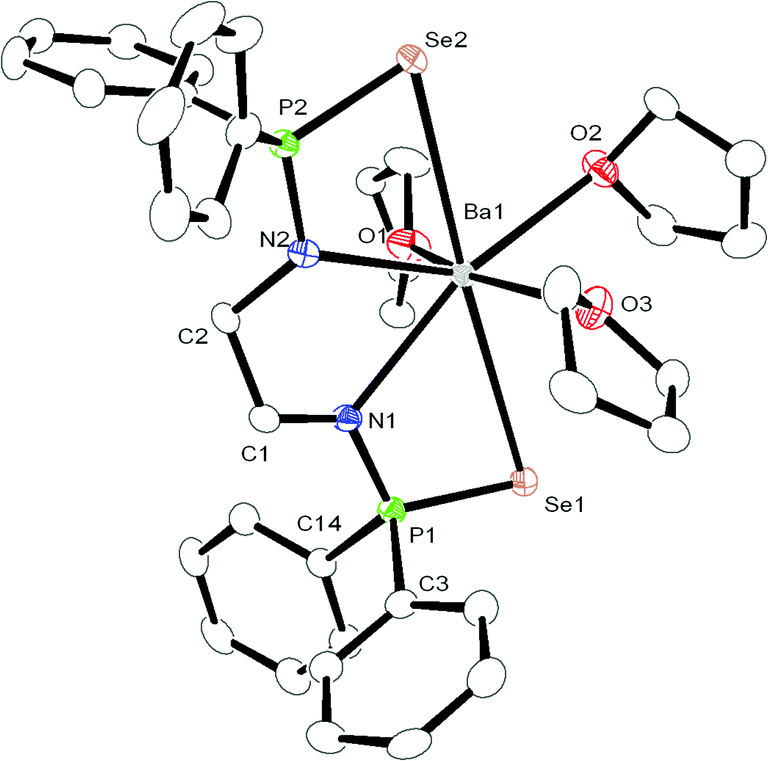 | ||
| Fig. 3 Solid-state structure of compound 4, ellipsoids are drawn to encompass 30 per cent probability, omitting hydrogen atoms for clarity. Selected bond lengths [Å] and bond angles [°]: (Molecule 1) P1–N1 1.585(6), P1–Se1 2.1583(19), P1–C14 1.834(7), P1–C3 1.835(7), N1–C1 1.458(9), P2–N2 1.591(6), P2–Se2 2.1490(18), P2–C15 1.833(7), P2–C21 1.837(7), N2–C2 1.478(8), C1–C2 1.517(9), Ba1–N1 2.657(5), Ba1–Se1 3.4706(9), Ba1–P1 3.6129(17), Ba1–N2 2.654(6), Ba1–Se2 3.4071(9), Ba1–P2 3.5929(18), Ba1–O1 2.716(5), Ba1–O2 2.812(5), Ba1–O3 2.721(5), C2–C1–N1 110.6(6), N1–P1–C14 112.6(3), N1–P1–Se1 110.1(2), N1–P1–C3 111.6(3), P1–N1–C1 122.3(5), Se1–P1–C14 109.4(2), Se1–P1–C3 110.0(2), C1–C2–N2 108.8(6), N2–P2–C15 111.8(3), N2–P2–Se2 109.5(2), N2–P2–C21 112.3(3), P2–N2–C2 121.9(5), Se2–P2–C15 110.4(2), Se2–P2–C21 110.0(3), N1–Ba1–Se1 58.71(12), N1–Ba1–N2 63.45(17), Se1–Ba1–Se2 175.44(2), O1–Ba1–O2 74.26(18), O1–Ba1–O3 155.90(19), O2–Ba1–O3 85.91(18), P1–Se1–Ba1 75.90(5), P2–Se2–Ba1 76.91(5). | ||
In all three complexes, the coordination polyhedron is formed by dianionic bis(diphenylphosphinoselenoic amide) [Ph2P(Se)NCH2CH2NP(Se)Ph2]2− ligands, and three THF molecules which are present as solvates to provide the metal ion seven-fold coordination. The ligand 1 coordinates to the alkaline earth metal ion via chelation of two amido nitrogen atoms and two selenium atoms attached to the phosphorus atoms. The phosphorus–metal distances (3.365 and 3.359 Å for complex 2, 3.452 and 3.446 Å for complex 3, and 3.593 and 3.613 Å for complex 4) are significantly greater than the sum of the covalent radii of the respective metal ion and phosphorus atom (3.07 Å for complex 2, 3.25 Å for complex 3 and 3.34 Å for complex 4). This indicates that the metal ion and phosphorous have no interaction between themselves. Thus, in each case, the central metal ion adopts a distorted pentagonal bi-pyramidal geometry around it, with two selenium atoms, two nitrogen atoms of ligand 1, along with one oxygen atom from the THF molecule, which is in the basal plane, whereas two remaining THF molecules occupy the apical positions. In complex 2, the M–N distances [2.386(8) Å and 2.418(8) Å for complex 2, 2.517(5) Å and 2.540(5) Å for complex 3, and 2.657(5) Å and 2.654(6) Å for complex 4] and M–Se distances [3.252(2) Å and 3.300(2) Å for complex 2, 3.2788(1) Å and 3.3259(1) Å for complex 3 and 3.4706(9) Å and 3.4071(9) Å for complex 4] indicate a slight asymmetrical attachment of the tetra-dentate ligand 1 to the alkaline earth metal ion. This is due to the presence of four phenyl rings attached to two phosphorus atoms. However, similar M–N distances and M–Se distances were observed in our previously reported complexes [(THF)2M{Ph2P(Se)N(CHPh2)}2] (M = Ca, Sr, Ba)10 and heavier alkaline earth metal complexes reported by other groups.20 Thus, we observe that bis(diphenylphosphinoselenoic amide) 1 behaves as a tetra-dentate chelating ligand to form a five-membered metallacycle M1–N1–C1–C2–N2, where two four-membered metallacycles M1–Se1–P1–N1 and M1–Se2–P2–N2 are fused together to construct a polymetallacyclic motif tricyclometalla[5.2.0.01,4]nonane structure. To the best of our knowledge, this is the first example of such a structural motif in alkaline earth metal complexes, with three adjacent hetero donor atoms—selenium, phosphorus and nitrogen. Among the three M–O distances for each complex [M–O distance is 2.429(7) Å for complex 2, 2.568(5) Å for complex 3, 2.812(5) Å for complex 4], the THF molecule resides in the basal plane of the distorted pentagonal bi-pyramidal structure and is slightly elongated compared to the remaining M–O distances [2.374(7) Å and 2.385(7) Å for complex 2, 2.537(5) Å and 2.568(5) Å for complex 3, 2.716(5) Å and 2.721(5) Å for complex 4] measured for the THF molecules placed in the apical position. This slight elongation can be explained by the extensive electron release from the two anionic basal nitrogen atoms opposite the THF molecule in the metal complex.
The magnesium ion has the smallest ionic radii among the Mg2+ to Ba2+ ions and different coordination behaviours can be anticipated.21 As we have observed that ligand 1 is acting as a tetra-dentate chelating ligand towards moderately larger ions (Ca2+ to Ba2+) it would be interesting to study its solid-state structure to learn more about its flexible nature. The magnesium compound 5 was crystallised from the THF–pentane mixture as a colourless solid. Compound 5 crystallises in the triclinic space group P, with two molecules in the unit cell. Table TS1 in ESI† contains details of the structural refinement parameters for compound 5 and its solid-state structure is shown in Fig. 4. In contrast to compounds 2–4, it is observed that for complex 5, a five magnesa-metallacycle Mg1–N1–C1–C2–N2 is formed by the chelation of two amido nitrogen atoms of ligand 1. Two selenium atoms, which are coordinated to Ca–Ba to make two four-membered rings in complexes 2–4, are unable to interact with the smaller magnesium ion (Mg1–Se1 3.661 Å and Mg1–Se2 3.769 Å). This is an instance of flexibility of the chelating ligand 1, switching from tetra-dentate to bi-dentate fashion depending upon the metal ion. As three THF molecules are chelated to the magnesium ion, the geometry around it is best described as a distorted trigonal bi-pyramidal geometry, having the amido nitrogens and one THF at the equatorial position and two THF molecules in the apical position. As expected, the Mg–N bond distances [2.066(3) Å and 2.083(3) Å] are among the shortest with respect to M–N distances [2.386(8) and 2.418(8) Å for Ca, 2.517(5) and 2.540(5) Å for Sr, and 2.657(5) and 2.654(6) Å for Ba]. Five-membered magnesium metallacycles are reported in the literature.22
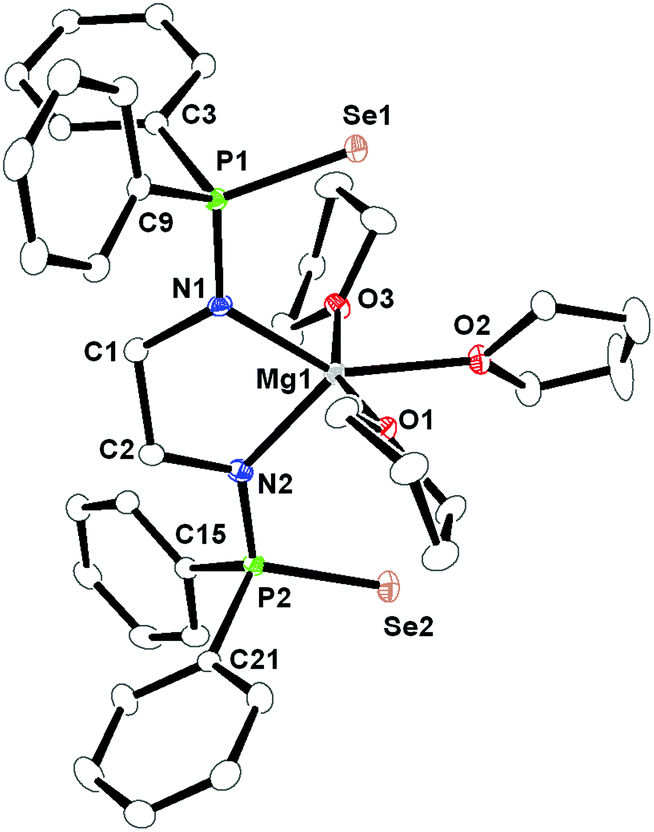 | ||
| Fig. 4 Solid-state structure of compound 5, ellipsoids are drawn to encompass 30 per cent probability, omitting hydrogen atoms for clarity. Selected bond lengths [Å] and bond angles [°]: P1–N1 1.606(3), P1–Se1 2.1308(10), P1–C3 1.831(3), P1–C9 1.825(3), N1–C1 1.477(4), P2–N2 1.612(3), P2–Se2 2.1240(10), P2–C15 1.833(3), P2–C21 1.834(3), N2–C2 1.482(4), C1–C2 1.524(4), Mg1–N1 2.083(3), Mg1–N2 2.066(3), Mg1–O1 2.124(3), Mg1–O2 2.137(2), Mg1–O3 2.106(2), C2–C1–N1 108.9(3), N1–P1–C9 112.57(15), N1–P1–Se1 110.69(10), N1–P1–C3 111.46(15), P1–N1–C1 121.0(2), Se1–P1–C9 109.18(11), Se1–P1–C3 111.65(11), C1–C2–N2 109.0(3), N2–P2–C15 111.99(15), N2–P2–Se2 111.98(11), N2–P2–C21 110.05(15), P2–N2–C2 118.6(2), Se2–P2–C15 109.14(11), Se2–P2–C21 110.70(11), N1–Mg1–N2 83.40(12), O1–Mg1–O2 80.49(10), O1–Mg1–O3 160.13(11), O2–Mg1–O3 80.11(10), N1–Mg1–O1 94.85(11), N2–Mg1–O1 99.30(11), N1–Mg1–O3 97.76(11), N2–Mg1–O2 134.70(12), N2–Mg1–O1 99.30(11), N2–Mg1–O3 97.36(11). | ||
Bis(amidodiphenylphosphine borane) ligand
Recently we introduced an amidophosphine–borane adduct as a ligand and exploited its chelating behaviour in alkali metal and alkaline earth metal chemistry.11 The amidophosphine borane [Ph2P(BH3)NHR] acts as a monoanionic ligand and coordinates to the metal ions through amido nitrogen and borane hydrogen. We intended to extend the idea of amidophosphine–borane to bis(amidodiphenylphosphine borane) by introducing a spacer of the ethylene bridge. Thus, bis(amidodiphenylphosphine borane) would form a dianion and act as a tetra-dentate ligand towards metal ions. In this paper we present the synthesis and structure of the bis(amidophosphine borane) ligand [Ph2P(BH3)NHCH2CH2NHPPh2(BH3)] and the corresponding dimeric barium complex [(THF)2Ba{Ph2P(BH3)NCH2CH2NPPh2(BH3)}]2 to demonstrate our concept of versatility of the amidophosphine backbone. The bis(amidodiphenylphosphine borane) ligand [Ph2P(BH3)NHCH2CH2NHPPh2(BH3)] (6) was isolated as a white precipitate from the reaction between bis(phosphineamine) [Ph2PNHCH2CH2NHPPh2] and the borane adduct [BH3·SMe2] at room temperature in a 1:2 molar ratio in toluene as the solvent (see Scheme 3).
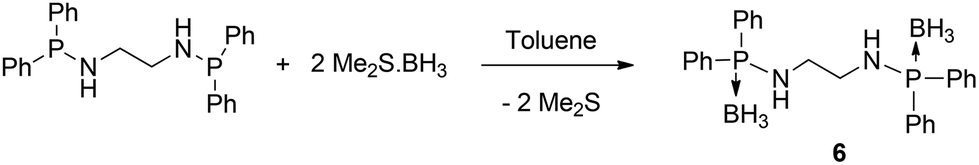 | ||
| Scheme 3 Synthesis of the bis(amidodiphenylphosphine borane) ligand 6. | ||
The formation of the amidophosphine borane ligand 6 from [Ph2PNHCH2CH2NHPPh2] can easily be followed by 1H NMR spectroscopy measured in CDCl3, since additional resonances for the two chemically equivalent borane (BH3) groups attached to the phosphorus atoms appear as a broad signal at δ 1.4 ppm. In the 1H NMR spectra, the resonances of the amidophosphine moiety in ligand 6 are only slightly shifted in comparison to the starting material with those reported for the phosphine amines.23 The multiplet signals at 2.78 ppm can be assigned to the four methylene protons of ligand 6 in which both the hydrogen atoms are diastereotopic to each other. This indicates that methylene signals are slightly high field shifted compared to the selenium analogue 1 (3.16 ppm). Another broad signal at 3.02 ppm corresponding to the two NH protons of ligand 6 is observed and is also shifted to the higher field (3.24 ppm) compared to 1. Ligand 6 shows a doublet signal in the 31P{1H} NMR spectrum at 58.8 ppm with a coupling constant of 67.9 Hz due to coupling with the adjacent boron atom. In 11B{1H} NMR spectrum, the signal at −38.1 ppm can be assigned to the BH3 group attached with phosphorus. This observation is in agreement with our previously reported values.11 In the FT-IR spectra, a characteristic signal for P–B bond stretching at 606 cm−1 was observed along with another characteristic signal at 2380 cm−1 assigned to the B–H stretching frequency. These values are in agreement with those reported in the literature.24
The molecular structure of ligand 6 was established using single crystal X-ray diffraction analysis. It crystallises in the monoclinic space group Cc, with four independent molecules in the unit cell (see Fig. 5). The details of the structural parameters are given in Table TS1 in the ESI.† The P1–B1 bond distances in 6 [1.9091(2) Å and 1.916(1) Å] are almost similar and in agreement with reported values—1.918(6) Å for [Ph2P(BH3)NH(CHPh2)], 2.1019(8) Å for [{Ph2P(BH3)}2CH2] and 1.921(3) Å for [(CH2-o-CF3C6H4)-(Ph)P(BH3)C4H8P(BH3)(Ph)(CH2-o-CF3C6H4)] to be considered as the phosphorus–boron dative bond reported by us and others.19,25,26 The P1–N1 bond ranges from 1.659(1) Å to 1.660(9) Å and the C1–N1 bond distances of 1.443(1) Å and 1.480(1) are also similar to those reported by us previously: P1–N1 1.673(6) Å and C1–N1 1.453(8) Å for Ph2PNH(CHPh2) and P1–N1 1.638(3) Å and C1–N1 1.468(5) Å for Ph2P(BH3)NH(CHPh2).11,13
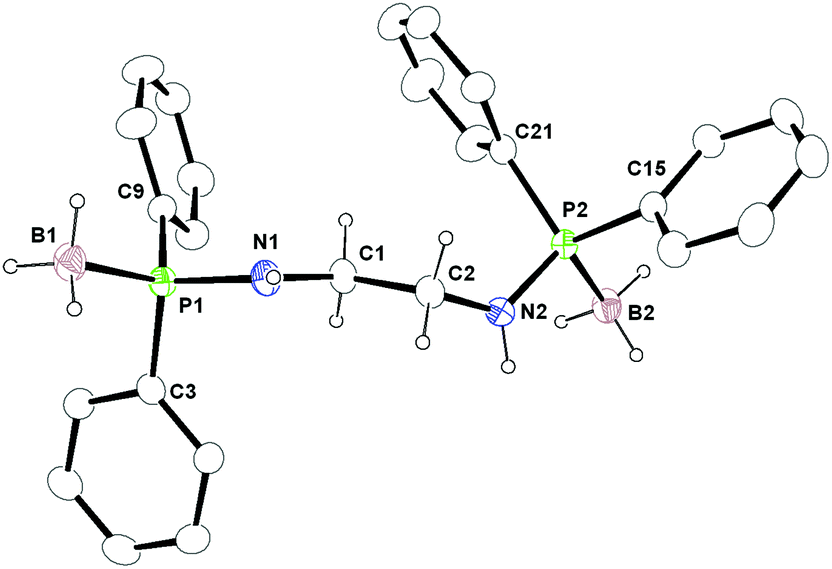 | ||
| Fig. 5 Solid-state structure of compound 6, ellipsoids are drawn to encompass 30 per cent probability, omitting aromatic ring hydrogen atoms for clarity. Selected bond lengths [Å] and bond angles [°]: P1–N1 1.668(3), P1–B1 1.920(5), P1–C3 1.805(4), P1–C9 1.815(4), N1–C1 1.463(5), C1–C2 1.523(5), C2–N2 1.437(5), P2–N2 1.660(3), P2–B2 1.896(5), P2–C15 1.810(4), P2–C21 1.817(4), B1–H1c 0.9600, B2–H2c 0.9600, B1–P1–N1 110.0(2), C3–P1–B1 110.3(2), C9–P1–B1 117.1(2), N1–P1–C3 106.20(19), N1–P1–C9 107.62(17), P1–N1–C1 121.4(3), N1–C1–C2 110.3(3), C1–C2–N2 111.9(3), C2–N2–P2 128.8(3), B2–P2–N2 106.7(2), B2–P2–C15 111.7(2), B2–P2–C21 116.8(2), N2–P2–C15 108.20(18), N2–P2–C21 107.85(17). | ||
Bis(amidodiphenylphosphine borane) barium complex
Ligand 6 was made to react with [K{N(SiMe3)2}] in THF at an ambient temperature in a 1:2 molar ratio followed by addition of barium diiodide to afford the dimeric barium bis(amidodiphenyl-phosphine borane) complex [(THF)2Ba{Ph2P(BH3)NCH2CH2N-PPh2(BH3)}]2 (7) through the elimination of KI and volatile tetramethylsilane (see Scheme 4).
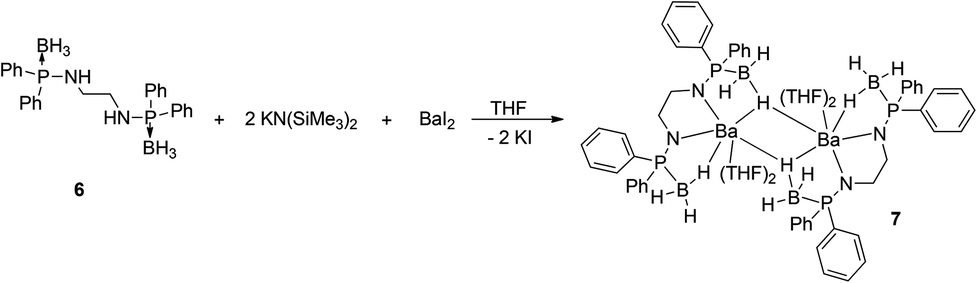 | ||
| Scheme 4 Synthesis of barium complex 7. | ||
In the FT-IR spectra, the strong absorption band at 605 cm−1 is assigned to the P–B bond of complex 7, which is in the range of that of ligand 6 (606 cm−1). The 1H NMR spectrum of complex 7 in C6D6 is very similar to the spectra recorded for compound 6 and reveals time-averaged Cs-symmetry in solution. The four methylene protons in the ligand backbone appear as a multiplet at 2.69 ppm. The resonances of the three protons attached to the boron atom appear as a multiplet at 1.38 ppm in the 1H NMR spectra. In the proton decoupled 31P NMR spectra, complex 7 shows only one doublet signal at 70.8 ppm and these values are significantly low-field shifted compared to the value for compound 6 (58.8 ppm) upon the coordination of barium atoms to the bis(amidophosphine-borane) ligand. The phosphorus atoms present in the [Ph2P(BH3)NCH2CH2NPPh2(BH3)]2− moieties are chemically equivalent. A broad signal at −37.6 ppm was observed in the 11B{1H} NMR spectra of complex 7.
Compound 7 was re-crystallised from THF and n-pentane (1:2 ratio) and was found to crystallise in the triclinic space group P, which has two molecules in the unit cell. The solid-state structure of complex 7 is given in Fig. 6. The details of the structural parameters are given in Table TS1 in the ESI.† Compound 7 is dimeric and two barium ions are coordinated by four amido nitrogen atoms and four BH3 groups of two ligands 6. Out of four borane groups, two are in mode to coordinate to the two barium ions. Each of the borane (BH3) group coordinates through the hydrogen atoms in a η1 fashion and has a Ba1–B1 bond length of 3.332(6) and Ba1–B2 is 3.251(7) Å. Thus, ligand 6 can be considered a tetra-dentate ligand, similar to what was observed for ligand 1 in the Ca–Ba complexes (see above). Additionally, two THF molecules are coordinated to each barium ion and the geometry around each barium ion is best described as distorted pentagonal bi-pyramidal. It is noteworthy that the P–B distances [1.938(7) and 1.943(8) Å] are slightly elongated compared to that of the ligand 6 [1.920(5) and 1.896(5) Å] even after the coordination of the BH3 group to the barium centre. The Ba–N [2.671(5) and 2.647(5) Å] and Ba1–O1 [2.730(5) and 2.744(5) Å] distances are in the range similar to that of the reported complexes.11
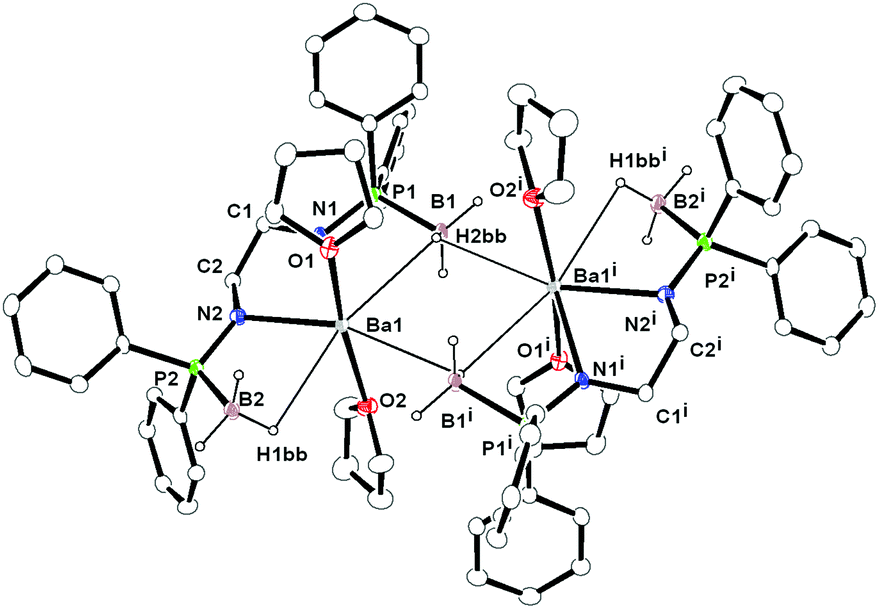 | ||
| Fig. 6 Solid state structure of compound 7, ellipsoids are drawn to encompass 30 per cent probability, omitting hydrogen atoms (except in the borane groups) for clarity. Selected bond lengths [Å] and bond angles [°]: P1–N1 1.595(5), P1–B1 1.938(7), C1–N1 1.468(7), C1–C2 1.531(8), C2–N2 1.475(7), P2–N2 1.596(5), P2–B2 1.943(8), B1–Ba1i 3.245(7), B1–H2aa 1.16(6), B1–H2bb 1.07(7), B1–H2cc 1.10(5), B2–H1aa 1.10(5), B2–H1bb 1.12(6), B1–H1cc 1.11(6), Ba1–N1 2.671(5), Ba1–N2 2.647(5), Ba1–B1 3.332(6), Ba1–B2 3.251(7), Ba1–P1 3.6169(16), Ba1–P2 3.5540(16), Ba1–O1 2.730(5), Ba1–O2 2.744(5), Ba1–B1i 3.245(7), Ba1–H1bb 2.90(6), Ba1–H2bb 2.95(7), B1–P1–N1 107.2(3), P1–N1–C1 125.3(4), N1–Ba1–N2 62.32(15), O1–Ba1–O2 169.80(14), N1–C1–C2 109.1(5), C1–C2–N2 109.2(5), N2–P2–B2 108.1(3). | ||
Ring opening polymerization study
Catalytic activities of the calcium, strontium and barium complexes 2, 3 and 4 were performed (see Scheme 5). Polymerisation studies were typically conducted in toluene, with various monomer/catalyst ratios at 25 °C. Selected data obtained with respect to complexes 2, 3 and 4 are given in Table 1. | ||
| Scheme 5 ROP of ε-CL in toluene with calcium, strontium and barium complexes 2–4 respectively. | ||
Se)2(THF)3] (where M = Ca, Sr, Ba)a
| Entry | [M] | [ε-CL]0/[M]0 | Solvent | Reac. temp. [°C] | Reac. timeb [min] | Conv.c [%] | M n(theo) [g mol−1] | M n(GPC) [g mol−1] | M w(GPC) [g mol−1] | M w/Mnf |
|---|---|---|---|---|---|---|---|---|---|---|
| a Results are representative of at least two experiments. b Reaction times were not necessarily optimized. c Monomer conversions were determined by 1H NMR spectroscopy. d Theoretical molar mass values calculated from the relation: [monomer]0/[M]0 × monomer conversion where [M]0 = 8.76 × 10−3 mmol and monomer weight of ε-CL = 114 g mol−1. e Experimental molar masses were determined by GPC versus polyethylene glycol standards. f Molar mass distributions were calculated from GPC. | ||||||||||
| 1 | Ca | 200 | Toluene | 25 | 10 | 75 | 15000 |
17065 |
11658 |
1.473 |
| 2 | Ca | 300 | Toluene | 25 | 15 | 96 | 28872 |
31027 |
17769 |
1.746 |
| 3 | Ca | 400 | Toluene | 25 | 20 | 89 | 35689 |
27238 |
19883 |
1.370 |
| 4 | Ca | 500 | Toluene | 25 | 20 | 64 | 32080 |
33618 |
24744 |
1.359 |
| 5 | Sr | 100 | Toluene | 25 | 10 | 99 | 17759 |
28789 |
17338 |
1.660 |
| 6 | Sr | 200 | Toluene | 25 | 10 | 98 | 35159 |
50353 |
44198 |
1.139 |
| 7 | Sr | 300 | Toluene | 25 | 10 | 95 | 51125 |
60384 |
53266 |
1.134 |
| 8 | Sr | 400 | Toluene | 25 | 10 | 72 | 28872 |
29925 |
22051 |
1.357 |
| 9 | Sr | 500 | Toluene | 25 | 15 | 80 | 40100 |
31590 |
22461 |
1.406 |
| 10 | Ba | 200 | Toluene | 25 | 5 | 82 | 16441 |
24947 |
18278 |
1.365 |
| 11 | Ba | 300 | Toluene | 25 | 5 | 94 | 28270 |
29965 |
20262 |
1.479 |
| 12 | Ba | 400 | Toluene | 25 | 5 | 85 | 34085 |
35033 |
25045 |
1.399 |
| 13 | Ba | 500 | Toluene | 25 | 5 | 70 | 35087 |
43215 |
34650 |
1.247 |
The catalytic ability of the newly synthesised mono-nuclear calcium complex 2 to promote the ROP of ε-CL was first evaluated (Table 1, entries 1–4). Indeed, the sluggish reactivity of the calcium complexes is very similar to that observed in some previously reported studies using other calcium complexes for ROP of ε-caprolactone.8 Since the larger strontium derivatives have been reported to be more active than the calcium congeners in ROP,27,28 we tested compound 3 as a catalyst and observed an enhanced rate of polymerisation (Table 1, entries 5–8). In both cases, higher reactivity was observed for conversion of ε-caprolactone to poly-caprolactone and up to 500 ε-CL units were successfully converted in high yields (90 per cent and 80 per cent) within 15 and 10 minutes respectively at 25 °C. The control over the ROP process was rather good, affording PCLs, featuring a good match between the observed (as determined by GPC) and calculated molar mass values, as well as moderate dispersity data (PDI = Mw/Mn < 1.80). The overall efficiency of the calcium initiator 2 towards the ROP of ε-CL was weaker than that of the strontium analogue 3. Being the largest ionic radius of the barium atom, it was anticipated that complex 4 would show the highest reactivity among all the three alkaline earth metal complexes.29,30 In reality we observed that up to 500 ε-CL units were successfully converted in good yields (70 per cent) within 5 minutes at 25 °C (Table 1, entries 10–13). The poly-caprolactone produced by the use of the barium catalyst was a good match between the observed and calculated molar mass values, and we observed a relatively narrow poly-dispersity data (PDI up to 1.25, entry 13 in Table 1). Thus, among three metal complexes 2, 3 and 4, the barium complex showed the highest activity for ROP of ε-caprolactone.
Experimental
General consideration
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−4 torr) line, or in an argon-filled M. Braun glove box. THF was pre-dried over Na wire and distilled under nitrogen from sodium and benzophenone ketyl prior to use. Hydrocarbon solvents (toluene and n-pentane) were distilled under nitrogen from LiAlH4 and stored in the glove box. 1H NMR (400 MHz), 13C{1H} and 31P{1H} NMR (161.9 MHz) spectra were recorded on a BRUKER AVANCE III-400 spectrometer. BRUKER ALPHA FT-IR was used for FT-IR measurement. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad. Metal iodides (MgI2, CaI2, SrI2 and BaI2), KN(SiMe3)2, Me2S·BH3 and ε-caprolactone were purchased from Sigma Aldrich and used as such. The bis(phosphineamine) [Ph2PNHCH2CH2NHPPh2] and bis(phosphinoselenoic amine) [Ph2P(Se)NHCH2CH2NHPPh2(Se)] (1) were prepared according to procedure prescribed in the literature.12,23 The NMR solvent C6D6 was purchased from Sigma Aldrich and dried under Na/K alloy prior to use.Preparation of [(THF)3Ca{Ph2P(Se)NCH2CH2NPPh2(Se)}] (2)
In a 50 mL dry Schlenk flask ligand 1 (200 mg, 0.34 mmol), KN(SiMe3)2 (136 mg, 0.68 mmol) and CaI2 (100 mg, 0.34 mmol) were mixed together in 10 mL of THF at an ambient temperature and stirred for 14 hours. The precipitate of KI was filtered using a filter dropper and the filtrate was dried in vacuo. The resulting white compound was further purified by washing with pentane and crystals suitable for X-ray analysis were grown from a THF–pentane (1:2 ratio) mixture solvent at −40 °C.
Yield 202.0 mg (70.6%).
1H NMR (400 MHz, C6D6): δ = 8.04–7.99 (m, 8H, ArH), 7.10–7.05 (m, 12H, ArH), 3.77–3.74 (m, 12H, THF), 3.39 (m, 4H, CH2), 1.36–1.33 (m, 12H, THF) ppm; 13C NMR (100 MHz, C6D6): δ = 133.1 (P–ArC), 131.9 (o-ArC), 131.8 (m-ArC), 129.7 (p-ArC), 68.5 (THF), 42.9 (CH2), 25.3 (THF) ppm; 31P NMR (161.9 MHz, C6D6): δ = 71.8 ppm; FT-IR (selected frequencies): 3052 (ArC–H), 2920 (C–H), 1435 (P–C), 969 (P–N), 550 (PSe) cm−1. (C38H48CaN2O3P2Se2) (840.74) Calc. C 54.29, H 5.75, N 3.33; found C 53.83, H 5.39, N 2.98.
Preparation of [(THF)3Sr{Ph2P(Se)NCH2CH2NPPh2(Se)}] (3)
In a 50 mL dry Schlenk flask ligand 1 (130 mg, 0.23 mmol), KN(SiMe3)2 (89 mg, 0.45 mmol) and SrI2 (100 mg, 0.23 mmol) were mixed together in 10 mL of THF at an ambient temperature and stirred for 14 hours. The precipitate of KI was filtered using a filter dropper and the filtrate was dried in vacuo. The resulting white compound was further purified by washing with pentane and crystals suitable for X-ray analysis were grown from a THF–pentane (1:2 ratio) mixture solvent at −40 °C.
Yield 176.5 mg (87.8%).
1H NMR (400 MHz, C6D6): δ = 8.05–7.99 (m, 8H, ArH), 7.12–6.97 (m, 12H, ArH), 3.57–3.53 (m, 12H, THF), 2.87 (m, 4H, CH2), 1.41–1.37 (m, 12H, THF) ppm; 13C NMR (100 MHz, C6D6): δ = 133.8 (P–ArC), 132.9 (P–ArC), 131.0 (o-ArC), 130.8 (m-ArC), 130.2 (p-ArC), 66.4 (THF), 40.9 (CH2), 24.4 (THF) ppm; 31P NMR (161.9 MHz, C6D6): δ = 71.9 ppm; FT-IR (selected frequencies): 3052 (ARC–H), 2922 (C–H), 1435 (P–C), 998 (P–N), 552 (PSe) cm−1. (C38H48N2O3P2Se2Sr) (888.26) Calc. C 51.38, H 5.45, N 3.15; found C 50.65, H 5.08, N 3.01.
Preparation of [(THF)3Ba{Ph2P(Se)NCH2CH2NPPh2(Se)}] (4)
In a 50 mL dry Schlenk flask ligand 1 (150 mg, 0.256 mmol), KN(SiMe3)2 (102 mg, 0.512 mmol) and BaI2 (100 mg, 0.256 mmol) were mixed together in 10 mL of THF at an ambient temperature and stirred for 14 hours. The precipitate of KI was filtered using a filter dropper and the filtrate was dried in vacuo. The resulting white compound was further purified by washing with pentane and crystals suitable for X-ray analysis were grown from a THF–pentane (1:2 ratio) mixture solvent at −40 °C.
Yield 210.0 mg (87.5%).
1H NMR (400 MHz, C6D6): δ = 8.14–8.02 (m, 8H, ArH), 7.11–7.03 (m, 12H, ArH), 3.57–3.53 (m, 12H, THF), 3.14 (m, 4H, CH2), 1.41–1.38 (m, 12H, THF) ppm; 13C NMR (100 MHz, C6D6): δ = 133.1 (P–ArC), 132.8 (P–ArC), 131.5 (o-ArC), 131.3 (m-ArC), 130.7 (p-ArC), 67.5 (THF), 48.5 (CH2), 25.5 (THF) ppm; 31P NMR (161.9 MHz, C6D6): δ = 73.3 ppm; FT-IR (selected frequencies): 3052 (ArC–H), 2951 (C–H), 1434 (P–C), 997 (P–N), 555 (PSe) cm−1. (C38H48BaN2O3P2Se2) (937.97) Calc. C 48.66, H 5.16, N 2.99; found C 47.88, H 4.72, N 2.69.
Preparation of [(THF)3Mg{Ph2P(Se)NCH2CH2NPPh2(Se)}] (5)
In a 50 mL dry Schlenk flask ligand 1 (210 mg, 0.36 mmol), KN(SiMe3)2 (143 mg, 0.72 mmol) and MgI2 (100 mg, 0.36 mmol) were mixed together in 10 mL of THF at an ambient temperature and stirred for 14 hours. The precipitate of KI was filtered using a filter dropper and the filtrate was dried in vacuo. The resulting white compound was further purified by washing with pentane and crystals suitable for X-ray analysis were grown from a THF–pentane (1:2 ratio) mixture solvent at −40 °C.
Yield 202.0 mg (70.6%).
1H NMR (400 MHz, C6D6): δ = 7.92–7.87 (m, 8H, ArH), 7.01–6.90 (m, 12H, ArH), 3.61–3.58 (m, 12H, THF), 3.09 (m, 4H, CH2), 1.32–1.26 (m, 12H, THF) ppm; 13C NMR (100 MHz, C6D6): δ = 132.1 (P–ArC), 131.9 (P–ArC), 131.7 (o-ArC), 131.6 (m-ArC), 129.6 (p-ArC), 68.1 (THF), 29.9 (CH2), 25.4 (THF) ppm; 31P NMR (161.1 MHz, C6D6): δ = 43.7 ppm; FT-IR (selected frequencies): 3052 (ArC–H), 2917 (C–H), 1435 (P–C), 997 (P–N), 551 (PSe) cm−1. (C38H48MgN2O3P2Se2) (824.95) Calc. C 55.32, H 5.86, N 3.40; found C 54.83, H 5.49, N 3.18.
Preparation of [Ph2P(BH3)NHCH2CH2NHPPh2(BH3)] (6)
To a stirring solution of ethylenediamine (325.2 mg, 5.4 mmol) and triethylamine (1.09 g, 1.5 mL, 10.8 mmol) in a THF–CH2Cl2 mixture solvent (10 mL), was added a solution of chlorodiphenylphosphine (2.39 g, 10.8 mmol) in THF (5 mL), drop-wise, and stirred for 3 hours. The precipitate formed was filtered and the solvent was removed in vacuo. To this, 20 mL of dry toluene and two equivalents of borane-dimethyl sulphide (822.2 mg, 10.8 mmol) were added and stirred for another 12 hours. The title compound was formed as a white precipitate, which further purified by washing several times with n-hexane. Crystals suitable for X-ray diffraction analysis were obtained from THF–n-pentane combination in a 1:2 ratio.
Yield 58.7% (1.45 g).
1H NMR (400 MHz, CDCl3): δ = 7.66–7.57 (m, 8H, ArH), 7.55–7.32 (m 12H, ArH), 3.03–3.01 (m, 2H, P(BH3)NH), 2.81–2.75 (m, 4H, CH2), 1.87–0.95 (m, 6H, BH3) ppm; 13C NMR (100 MHz, C6D6): δ = 134.3 (P–ArC), 134.2 (P–ArC), 132.9 (o-ArC), 132.8 (o-ArC), 132.2 (p-ArC), 132.1 (p-ArC), 131.4 (m-ArC), 131.2 (m-ArC), 44.2 (CH2) ppm; 31P–{1H} NMR (161.9 MHz, CDCl3): δ = 58.8 (d, JP–B = 67.9 Hz) ppm; 11B–{1H} NMR (128.4 MHz, CDCl3): δ −38.1 ppm. FT-IR (selected frequencies): 3366 (N–H), 3056 (ArC–H), 2960 (C–H), 2380 (B–H), 1436 (P–C), 935 (P–N), 606 (P–B) cm−1. (C26H32B2N2P2) (456.10) Calc. C 68.46, H 7.07, N 6.14; found C 67.98, H 6.79, N 5.88.
Preparation of [(THF)2Ba{Ph2P(BH3)NCH2CH2NPPh2(BH3)}]2 (7)
In a 50 mL dry Schlenk flask, ligand 6 (116.8 mg, 0.256 mmol), KN(SiMe3)2 (102 mg, 0.512 mmol) and BaI2 (100 mg, 0.256 mmol) were mixed together in 10 mL of THF at an ambient temperature and stirred for 14 hours. The precipitate of KI was filtered using a filter dropper and the filtrate was dried in vacuo. The resulting white compound was further purified by washing with pentane and crystals suitable for X-ray analysis were grown from a THF–pentane (1:2 ratio) mixture solvent at −40 °C.
Yield 153.0 mg (85.0%).
1H NMR (400 MHz, C6D6): δ = 7.59–7.54 (m, 8H, ArH), 7.15–7.01 (m, 12H, ArH), 2.71–2.67 (m, 4H, CH2), 1.96–0.80 (m, 6H, BH3) ppm; 13C NMR (100 MHz, C6D6): δ = 133.3 (P–ArC), 132.6 (P–ArC), 132.2 (o-ArC), 132.1 (o-ArC), 131.3 (p-ArC), 131.2 (p-ArC), 128.8 (m-ArC), 128.7 (m-ArC), 44.4 (CH2) ppm; 31P NMR (161.9 MHz, C6D6): δ = 70.8 (d, JP–B = 62.4 MHz) ppm; 11B–{1H} NMR (128.4 MHz, C6D6): δ −37.6 ppm. FT-IR (selected frequencies): 3056 (ArC–H), 2957 (C–H), 2375 (B–H), 1434 (P–C), 996 (P–N), 605 (P–B) cm−1. (C34H46B2BaN2O2P2) (735.62) Calc. C 55.51, H 6.30, N 3.81; found C 54.92, H 6.02, N 3.31.
Typical polymerisation experiment
In a glove box under an argon atmosphere, the catalyst was dissolved in the appropriate amount (1.0 mL) of dry toluene. ε-Caprolactone in 1.0 mL of toluene was then added under vigorous stirring. The reaction mixture was stirred at room temperature for 5–20 minutes, after which the reaction mixture was quenched by addition of a small amount of (1.0 mL) methanol and then added acidified methanol in little excess. The polymer was precipitated in excess methanol and it was filtered and dried under vacuum. The final polymer was then analysed by NMR and GPC.X-Ray crystallographic studies of 2–6
Single crystals of compounds 2–5 and 7 were grown from a THF and pentane mixture at −40 °C under an inert atmosphere. The single crystals of 1-cis, 1-trans and 6 suitable for X-ray measurement were grown at room temperature. For compounds 2–5 and 7, a crystal of suitable dimensions was mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed in a nitrogen stream at 150(2) K. However, for compounds 1-cis, 1-trans and 6, the data were collected at 293 K. All measurements were made on an Oxford Supernova Xcalibur Eos CCD detector with graphite-monochromatic Cu-Kα (1.54184 Å) radiation. Crystal data and structure refinement parameters are summarised in Table TS1 in the ESI.† The structures were solved by direct methods (SIR92)31 and refined on F2 by full-matrix least-squares methods using SHELXL-97.32 Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. No restraint was made for all the compounds. For compounds 2 and 5, two carbon atoms which are part of the two coordinated THF molecules are slightly thermally disorder and treated anisotropically. The function minimised was [∑w(Fo2 − Fc2)2] (w = 1/[σ2(Fo2) + (aP)2 + bP]), where P = (Max(Fo2,0) + 2Fc2)/3 with σ2(Fo2) from counting statistics. The functions R1 and wR2 were (∑||Fo| − |Fc||)/∑|Fo| and [∑w(Fo2 − Fc2)2/∑(wFo4)]1/2, respectively. The ORTEP-3 program was used to draw the molecule. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 987282–987289.Conclusion
We have demonstrated a series of alkaline earth metal complexes with the bis(phosphinoselenoic amine) ligand via two synthetic routes. In the solid-state structures of Ca–Ba complexes, the bis(phosphinoselenoic amine) acts as a tetra-dentate ligand by the chelation of two amido nitrogen and two selenium atoms, whereas due to the smaller size of magnesium, the same ligand behaves as a bidentate ligand through chelation of two amido-nitrogen atoms only, showing its flexible nature. We have also introduced another poly-dentate ligand bis(amidodiphenylphosphine borane) in barium chemistry to prepare the barium dimeric complex. We have tested complexes 2–4 as catalysts for the ROP of ε-caprolactone and observed that the barium complex, being the largest ionic radius, acts as the best catalyst among the three analogous complexes.Acknowledgements
This work was supported by the Department of Science and Technology India (DST) under the SERC Fast Track Scheme (SR/FT/CS-74/2010) and start-up grant from IIT Hyderabad. R. K. K., A. H. and J. B. thank the University Grants Commission, India for their PhD fellowship. We thank Dr K. Muralidharan of University of Hyderabad for providing the facility to measure the polymers.Notes and references
- (a) M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed; (b) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS PubMed; (c) K. Sudehsh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef; (d) Y. Ikada and H. Tsuji, Macromol. Rapid Cummun., 2000, 21, 117–132 CrossRef CAS; (e) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- (a) K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 42–65 CrossRef; (b) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2002, 3, 1–41 CrossRef PubMed; (c) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS; (d) A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- D. Meimaroglou and C. Kiparissides, Macromolecules, 2010, 43, 5820–5832 CrossRef CAS.
- R. W. Nunes, J. R. Martin and J. F. Johnson, Polym. Eng. Sci., 1982, 2, 205–228 Search PubMed.
- (a) H. Yasuda, J. Organomet. Chem., 2002, 647, 128–138 CrossRef CAS; (b) Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22 CrossRef CAS; (c) H. Yasuda, Prog. Polym. Sci., 2000, 25, 573–626 CrossRef CAS; (d) S. Agarwal, C. Mast, K. Denicke and A. Greiner, Macromol. Rapid Commun., 2000, 21, 195–212 CrossRef CAS; (e) J. Wu, T. L. Yu, C. T. Chen and C. C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS PubMed; (f) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- (a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (b) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (c) C. A. Wheaton, P. G. Hayes and B. Ireland, Dalton Trans., 2009, 4832–4846 RSC; (d) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- (a) S. M. Li, I. Rashkov, J. L. Espartero, N. Manolova and M. Vert, Macromolecules, 1996, 29, 57–62 CrossRef CAS; (b) P. Dobrzyński, J. Kasperczyk and M. Bero, Macromolecules, 1999, 32, 4735–4737 CrossRef; (c) Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS; (d) M. Westerhausen, S. Schneiderbauer, A. N. Kneifel, Y. Söltl, P. Mayer, H. Nöth, Z. Zhong, P. J. Dijkstra and J. Feijen, Eur. J. Inorg. Chem., 2003, 3432–3439 CrossRef CAS; (e) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (f) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2003, 1758–1759 RSC; (g) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed; (h) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC.
- M. Kuzdrowska, L. Annunziata, S. Marks, M. Schmid, C. G. Jaffredo, P. W. Roesky, S. M. Guillaume and L. Maron, Dalton Trans., 2013, 42, 9352–9360 RSC.
- T. K. Panda, K. Yamamoto, K. Yamamoto, H. Kaneko, Y. Yang, H. Tsurugi and K. Mashima, Organometallics, 2012, 31, 2268–2274 CrossRef CAS.
- R. K. Kottalanka, K. Naktode, S. Anga, H. P. Nayek and T. K. Panda, Dalton Trans., 2013, 42, 4947–4956 RSC.
- R. K. Kottalanka, S. Anga, K. Naktode, P. Laskar, H. P. Nayek and T. K. Panda, Organometallics, 2013, 32, 4473–4482 CrossRef CAS.
- R. Ahmed, A. Altieri, D. M. D'Souza, D. A. Leigh, K. M. Mullen, M. Papmeyer, A. M. Z. Slawin, J. K. Y. Wong and J. D. Woollins, J. Am. Chem. Soc., 2011, 133, 12304–12310 CrossRef CAS PubMed.
- (a) R. K. Kottalanka, K. Naktode and T. K. Panda, J. Mol. Struct., 2013, 1036, 188–195 CrossRef CAS PubMed; (b) K. Naktode, R. K. Kottalanka and T. K. Panda, New J. Chem., 2012, 36, 2280–2285 RSC.
- J. M. Boncella, C. J. Coston and J. K. Cammack, Polyhedron, 1991, 10, 769–770 CrossRef CAS.
- P. S. Tanner, D. J. Burkey and T. P. Hanusa, Polyhedron, 1995, 14, 331–333 CrossRef CAS.
- The bonding situation in the drawing of the ligand system is simplified for clarity.
- T. P. Hanusa, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and M. P. Mingos, Elsevier, Oxford, 2007, vol. 2, p. 67 Search PubMed.
- (a) T. P. Hanusa, Organometallics, 2002, 21, 2559–2571 CrossRef CAS; (b) T. P. Hanusa, Chem. Rev., 2000, 100, 1023–1036 Search PubMed; (c) T. P. Hanusa, Coord. Chem. Rev., 2000, 210, 329–367 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press, Oxford, U.K., 1984 Search PubMed.
- (a) T. K. Panda, H. Kaneko, O. Michael, H. Tsurugi, K. Pal, W. Tömroos, R. Anwander and K. Mashima, Organometallics, 2012, 31, 3178–3184 CrossRef CAS; (b) A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock, G. Kociok-Köhn and P. A. Procopiou, Inorg. Chem., 2008, 47, 7366–7376 CrossRef CAS PubMed.
- (a) V. C. R. Grignard, Acad. Sci., 1900, 130, 1322–1324 CAS; (b) V. Grignard, Ann. Chim., 1901, 24, 433–490 CAS; (c) D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS; (d) F. Bickelhaupt, J. Organomet. Chem., 1994, 475, 1–14 CrossRef CAS.
- (a) D. C. Bradley, D. M. Frigo, I. S. Harding and M. Motevalli, Eur. J. Solid State Inorg. Chem., 1993, 30, 241–258 CAS; (b) A. Xia, M. J. Heeg and C. H. Winter, Organometallics, 2002, 21, 4718–4725 CrossRef CAS; (c) O. Michel, K. Yamamoto, H. Tsurugi, C. Maichle-Möossmer, K. W. Tömroos, K. Mashima and R. Anwander, Organometallics, 2011, 30, 3818–3825 CrossRef CAS.
- M. C. B. Dolinskya, W. O. Lina and M. L. Dias, J. Mol. Catal. A: Chem., 2006, 258, 267–274 CrossRef PubMed.
- J. D. Odom, B. A. Hudgens and J. R. Durig, J. Phys. Chem., 1973, 77, 1972 CrossRef CAS.
- R. K. Kottalanka, P. Laskar, K. Naktode, B. S. Mallik and T. K. Panda, J. Mol. Struct., 2013, 1047, 302–309 CrossRef CAS PubMed.
- B. J. Anderson, D. S. Glueck, A. G. DiPasquale and A. L. Rheingoldb, Organometallics, 2008, 27, 4992–5001 CrossRef CAS.
- B. Liu, T. Roisnel, J.-P. Guegan, J.-F. Carpentier and Y. Sarazin, Chem. – Eur J., 2012, 18, 6289–6301 CrossRef CAS PubMed.
- Y. Sarazin, B. Liu, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- M. G. Davidson, C. T. O'Hara, M. D. Jones, C. G. Keir, M. F. Mahon and G. Kociok-Köhn, Inorg. Chem., 2007, 46, 7686–7688 CrossRef CAS PubMed.
- (a) I. Palard, A. Soum and S. M. Guillaume, Chem. – Eur J., 2004, 10, 4054–4062 CrossRef CAS PubMed; (b) J. Jenter, P. W. Roesky, N. Ajellal, S. M. Guillaume, N. Susperregui and L. Maron, Chem. – Eur J., 2010, 16, 4629–4638 CrossRef CAS PubMed.
- M. Sheldrick, SHELXS-97, Program of Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program of Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For crystallographic details in CIF. CCDC 987282–987289. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00669k |
| This journal is © The Royal Society of Chemistry 2014 |