DOI:
10.1039/C4DT00629A
(Paper)
Dalton Trans., 2014,
43, 6819-6827
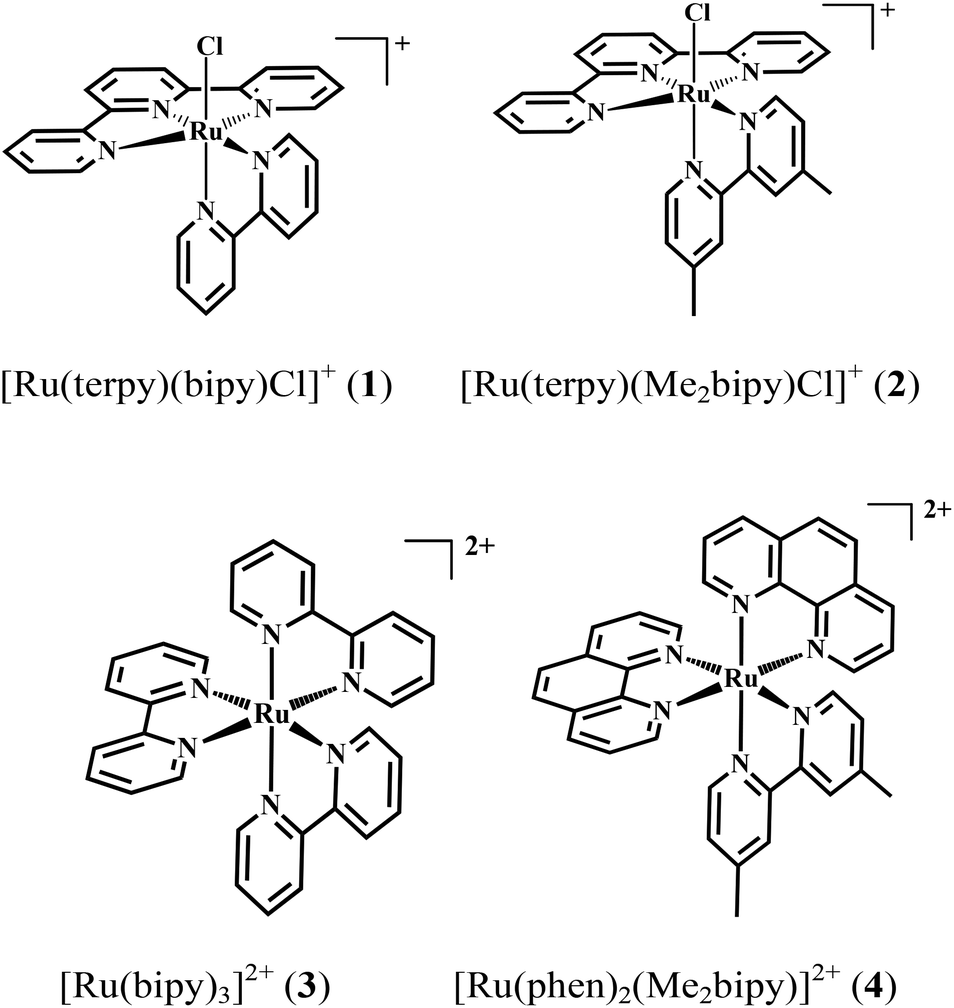
Cooperative effects in homogenous water oxidation catalysis by mononuclear ruthenium complexes†
Received
28th February 2014
, Accepted 7th March 2014
First published on 10th March 2014
Abstract
The homogenous water oxidation catalysis by [Ru(terpy)(bipy)Cl]+ (1) and [Ru(terpy)(Me2bipy)Cl]+ (2) (terpy = 2,2′:6′,2′′-terpyridine, bipy = 2,2′-bipyridine, Me2bipy = 4,4′-dimethyl-2,2′-bipyridine) under the influence of two redox mediators [Ru(bipy)3]2+ (3) and [Ru(phen)2(Me2bipy)]2+ (4) (phen = 1,10-phenanthroline) was investigated using Ce4+ as sacrificial oxidant. Oxygen evolution experiments revealed that mixtures of both 2–4 and 2–3 produced more molecular oxygen than catalyst 2 alone. In contrast, the combination of mediator 4 and catalyst 1 resulted in a lower catalytic performance of 1. Measurements of the temporal change in the intensity of a UV transition at 261 nm caused by the addition of four equivalents of Ce4+ to 2 revealed three distinctive regions-suggested to correspond to the stepwise processes: (i) [RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+ → [RuVO]3+; (ii) [RuVO]3+ → [RuIII–(OOH)]2+; and (iii) [RuIII–(OOH)]2+ → [RuII–OH2]2+. UV-Visible spectrophotometric experiments on the 1–4 and 2–4 mixtures, also carried out with four equivalents of Ce4+, demonstrated a faster [Ru(phen)2(Me2bipy)]3+ → [Ru(phen)2(Me2bipy)]2+ reduction rate in 2–4 than that observed for the 1–4 combination. Cyclic voltammetry data measured for the catalysts and the mixtures revealed a coincidence in the potentials of the RuII/RuIII redox process of mediators 3 and 4 and the predicted [RuIVO]2+/[RuVO]3+ potential of catalyst 2. In contrast, the [RuIVO]2+/[RuVO]3+ process for catalyst 1 was found to occur at a higher potential than the RuII/RuIII redox process for 4. Both the spectroscopic and electrochemical experiments provide evidence that the interplay between the mediator and the catalyst is an important determinant of the catalytic activity.
O]2+ → [RuVO]3+; (ii) [RuVO]3+ → [RuIII–(OOH)]2+; and (iii) [RuIII–(OOH)]2+ → [RuII–OH2]2+. UV-Visible spectrophotometric experiments on the 1–4 and 2–4 mixtures, also carried out with four equivalents of Ce4+, demonstrated a faster [Ru(phen)2(Me2bipy)]3+ → [Ru(phen)2(Me2bipy)]2+ reduction rate in 2–4 than that observed for the 1–4 combination. Cyclic voltammetry data measured for the catalysts and the mixtures revealed a coincidence in the potentials of the RuII/RuIII redox process of mediators 3 and 4 and the predicted [RuIVO]2+/[RuVO]3+ potential of catalyst 2. In contrast, the [RuIVO]2+/[RuVO]3+ process for catalyst 1 was found to occur at a higher potential than the RuII/RuIII redox process for 4. Both the spectroscopic and electrochemical experiments provide evidence that the interplay between the mediator and the catalyst is an important determinant of the catalytic activity.
Introduction
The splitting of water into oxygen and hydrogen using sunlight has a great potential for developing an alternative renewable energy source that will help to overcome the looming shortfall in fossil fuels. However, despite the promise, to achieve water splitting (2H2O → O2 + 2H2) a substantial energy input additional to the thermodynamic requirement (1.23 V or 267 kJ mol−1 per mole of H2 produced) is needed to compensate for losses arising from, for example, cell resistance and various activation barriers. In this regard, the mechanistically complex water oxidation half reaction (2H2O → O2 + 4H+ + 4e−) presents particular challenges as it involves the extraction of four electrons and four protons from two water molecules and the formation of a stable diatomic O–O bond. Thus, efficient catalysts need to be developed such that the energy required to drive the water-splitting reaction is as close as possible to the thermodynamic value (1.23 V). There has been much interest in this field of research and efforts have been made to develop heterogeneous photo-catalysts as well as photo-electrocatalysts for water splitting.1 Single semiconductors or combinations of semiconductors with appropriate valence and conduction band positioning (Z-scheme) have been used to split water directly and more active catalysts have been introduced to enhance performance (these include metals and a variety of transition metal oxides).1,2 In parallel with these studies, there has been a long-standing interest in molecular catalysts that oxidise water or reduce water, with a focus on the former being driven in part by the fact that a tetramanganese cluster in water oxidation complex (WOC) of photosystem II is the only catalyst known to catalyse water oxidation in nature.3 Amongst the range of molecular water oxidation catalysts that have been reported,4 however ruthenium complexes have been investigated for several decades now and, in contrast to some purported molecular catalysts incorporating other transition metals, there is clear evidence that such complexes are indeed molecular catalysts operating in homogeneous solution and when attached to surfaces.5–9 Initially, the focus was on dinuclear complexes owing to the discovery that the ruthenium blue dimer (cis,cis-[(bipy)2(H2O)RuIII(μ-O)RuIII(OH2)(bipy)2]4+; bipy = 2,2′-bipyridine) catalysed this reaction,6 and a mechanistic understanding of the pathways leading to oxygen formation was developed from these studies.7 More recently, mononuclear ruthenium complexes have also been shown to catalyse water oxidation.5,8–11 They do so by forming a reactive [RuVO] intermediate, generated through successive oxidation processes [RuII–OH2] → [RuIII–OH] → [RuIVO] → [RuVO].5,8 The formation of the [RuVO] moiety is central to the catalytic evolution of oxygen, which has been proposed to occur through either the attack of [RuVO] by water or, alternatively, through intra- or inter-molecular coupling of two [RuVO] species.5,8 These mononuclear ruthenium complexes have largely been investigated using Ce4+ as a sacrificial oxidant,5,8,9,11 and although investigations of the interactions between potential photosensitisers (or dyes) and the ruthenium catalysts is very important for the development of a light-induced water-splitting system, to date only a handful of reports have focussed on this aspect in homogenous systems.9,10 For example, the catalytic rate of Ce4+-assisted water oxidation by the blue dimer in homogenous solution was enhanced by a factor of ∼30 on addition of mediators such as [Ru(bipy)2L]2+, where L = 2,2′-bipyridine, 2,2′-bipyrimidine and 2,2′-bipyrazine.9 In a more recent report, a covalently-linked mediator/catalyst dinuclear complex [(bipy)2Ru(4-Mebpy-4′-bimpy)Ru(terpy)(OH2)]4+ or [RuaII–RubII–OH2]4+ (4-Mebpy-4′-bimpy = 4-(methylbipyridin-4′-yl)-N-benzimid-N′-pyridine; terpy = 2,2′:6′,2′′-terpyridine) displayed a faster catalytic rate than the individual mononuclear complex [Ru(terpy)(Mebim-py)(OH2)]2+ (Mebim-py = 2-pyridyl-N-methylbenzimidazole).10
The relative lack of investigations focussing on cooperativity between ruthenium catalysts and redox-active ruthenium polypyridyl complexes has led us to study the interplay between two tris(diimine)ruthenium complexes {[Ru(bipy)3]2+ (3) and [Ru(phen)2(Me2bipy)]2+ (4) (phen = 1,10-phenanthroline, Me2bipy = 4,4′-dimethyl-2,2′-bipyridine)} as redox mediators and two closely-related mononuclear catalysts [Ru(terpy)(bipy)Cl]+ (1) and [Ru(terpy)(Me2bipy)Cl]+ (2) in Ce4+-activated homogenous water oxidation catalysis. Building on previous related investigations of mononuclear complexes of type [Ru(terpy)(xbipy)(H2O)]2+ (xbipy = derivatives of 2,2′-bipyridines with various substitutions on the 4,4′ position),11 we report an investigation of the influence of redox mediators 3 and 4 on water oxidation mononuclear catalysis by 1 and 2 (Scheme 1).
 |
| | Scheme 1 | |
Results and discussion
Following our reports describing the synthesis of the stereochemically-resolved Δ-[Ru(phen)2(Me2bipy)]2+ and the non-chiral [Ru(terpy)(Me2bipy)Cl]+ for the study of their biological activity,12,13 in the present work we adopted the same synthetic pathway to obtain complexes 1 and 2. These compounds were prepared by refluxing mixtures of [Ru(terpy)Cl3] and 2,2′-bipyridine or 4,4′-dimethyl-2,2′-bipyridine in ethanol–water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), while 4 was obtained from the reaction between [Ru(phen)2Cl2] and 4,4′-dimethyl-2,2′-bipyridine in ethanol–water (1:1). Size-exclusion chromatography was used to separate each complex from the unreacted starting material and any oxidised ruthenium by-products. The purity of the compounds was confirmed by 1H NMR spectroscopy.
:1), while 4 was obtained from the reaction between [Ru(phen)2Cl2] and 4,4′-dimethyl-2,2′-bipyridine in ethanol–water (1:1). Size-exclusion chromatography was used to separate each complex from the unreacted starting material and any oxidised ruthenium by-products. The purity of the compounds was confirmed by 1H NMR spectroscopy.
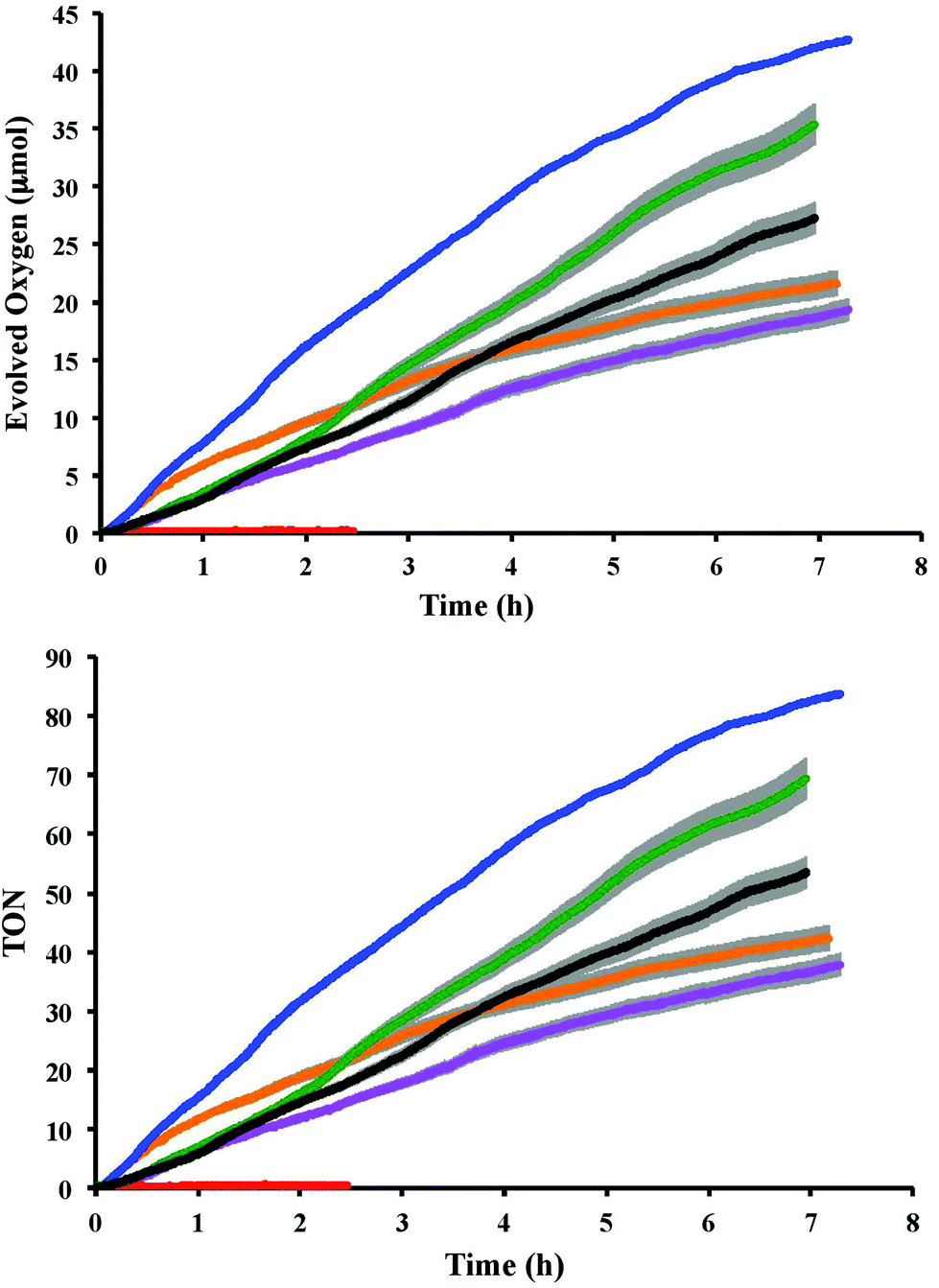
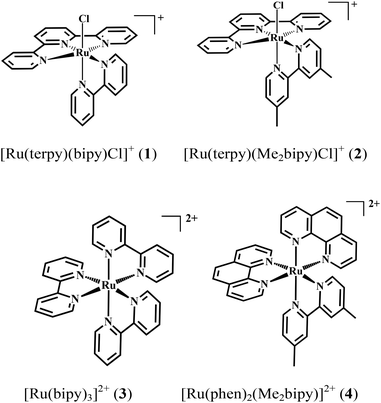
A series of oxygen evolution experiments using catalysts 1 and 2 as a 1:1 mixture with either 3 or 4 at the concentration 73 μM was carried out in an air-tight vessel using 0.1 M HClO4 solution as the reaction medium and (NH4)2[Ce(NO3)6] (0.073 M or 1000 equivalents) as the oxidant. The molecular oxygen evolved from the reaction was detected in the headspace over the course of several hours by a Clark-type micro-sensor. The oxygen measurement for the individual catalysts revealed that 2 produced approximately 16 μmol oxygen after six hours whereas 1 produced twice as much (30 μmol). In order to test the effect of the redox mediators 3 and 4 on the catalytic performance of 1 and 2, the oxygen evolution of each catalyst in the presence of the mediators was investigated. Upon mixing with 4 in a 1:1 ratio, oxygen production catalysed by 1 was found to decrease slightly to 23 μmol over the six hours of testing. In contrast, the addition of mediator 4 to catalyst 2 resulted in a significant increase in oxygen production from 16 μmol to 38 μmol over the same period. No molecular oxygen could be detected from a control experiment using only mediator 4 (Fig. 1). The enhancement of the catalytic properties of 2 affected by the redox mediator prompted us to perform further analyses in order to elucidate the mode of interaction between the tris(diimine)ruthenium(II) complexes and the catalyst. Our initial investigations using UV-Visible, 1H NMR and electrochemical techniques are discussed in the following sections.
 |
| | Fig. 1 (Top) Oxygen evolution from the reaction between the complexes and Ce4+ in 0.1 M HClO4 detected in the headspace. (Bottom) Turnover numbers (TONs). Blue = 2–4 mixture; green = 1 only; black = 1–4 mixture; orange = 2–3 mixture; purple = 2 only; red = 4 only. The error bars of the full and crossed lines are in grey. In all cases, the concentration of each complex and Ce4+ were 73 μM and 0.073 M, respectively. | |
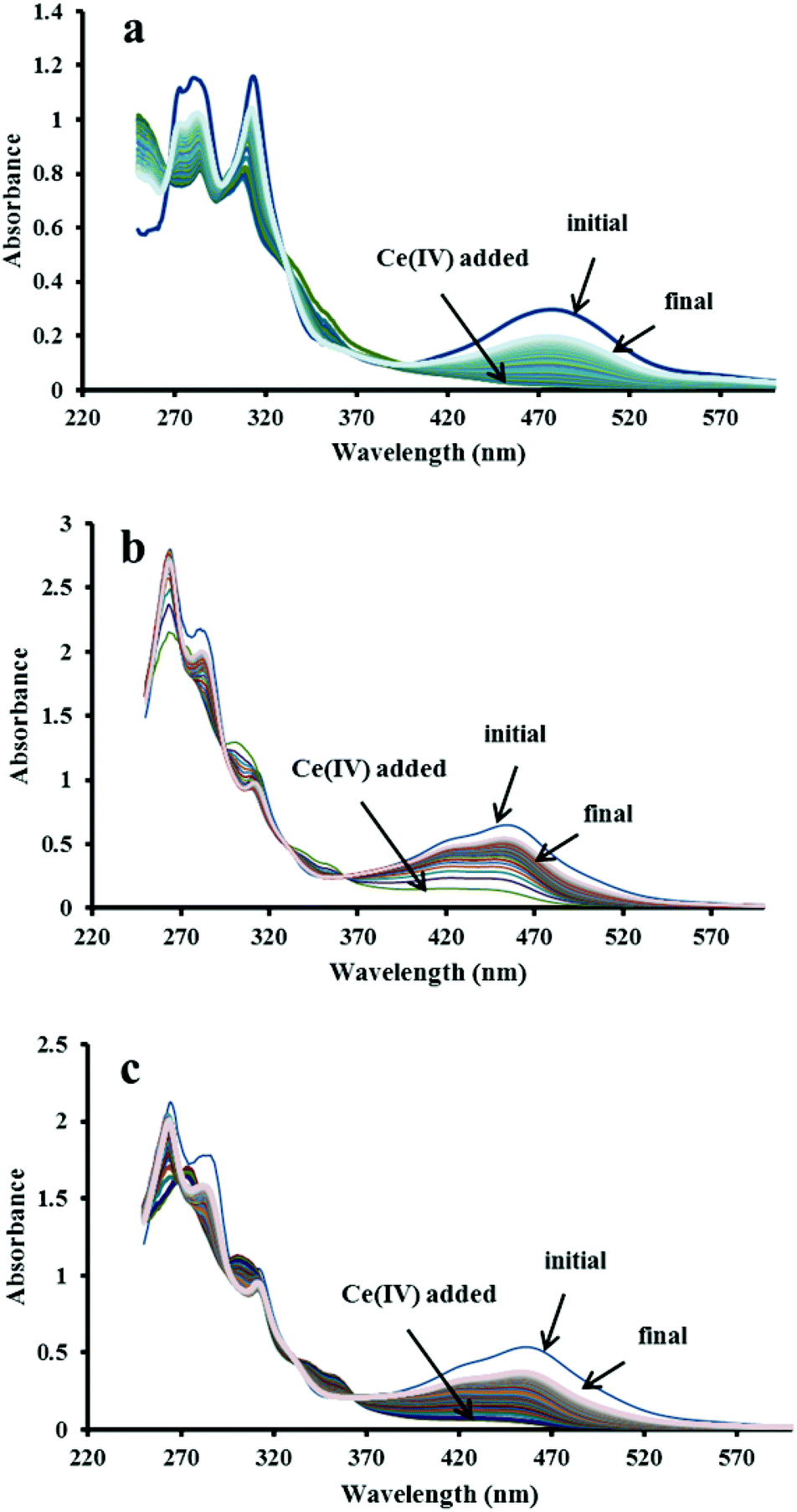
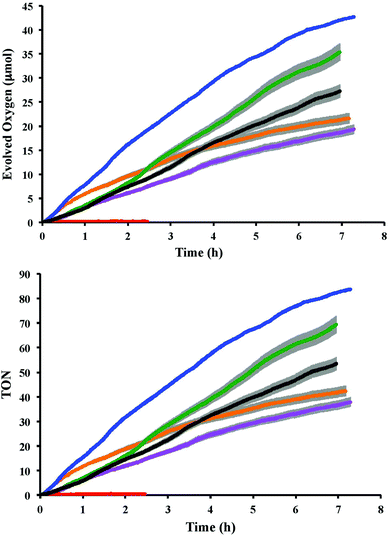
The changes in the UV-Visible spectrum of the individual catalysts and the mixtures, before and after the addition of four equivalents of Ce4+, were studied in 0.1 M HClO4. The initial UV-Visible spectrum of catalyst 2 (before Ce4+ was added) showed three absorption bands (480 nm, 313 nm and 281 nm). The absorption at 480 nm, assigned as MLCT (metal-to-ligand charge transfer) band, was found to disappear upon addition of four equivalents of Ce4+ (Fig. 2, panel a). This spectral assignment has been reported earlier for the reaction of the closely-related [Ru(terpy)(bpm)(OH2)]2+ complex with Ce4+ in 0.1 M HClO4.8 The addition of two equivalents of Ce4+ to the solution of [Ru(terpy)(bpm)(OH2)]2+ resulted in the two oxidation steps that could be followed spectroscopically:
| | | [RuII–OH2]2+ + Ce4+ → [RuIII–OH]2+ + Ce3+ | (1) |
| | | [RuIII–OH]+ + Ce4+ → [RuIVO]2+ + Ce3+ | (2) |
 |
| | Fig. 2 Spectral changes before and after addition of four equivalent Ce4+ in (a) 2 only; (b) 2–4 mixture and (c) 1–4 mixture. In all cases the complex concentration was 24 μM. | |
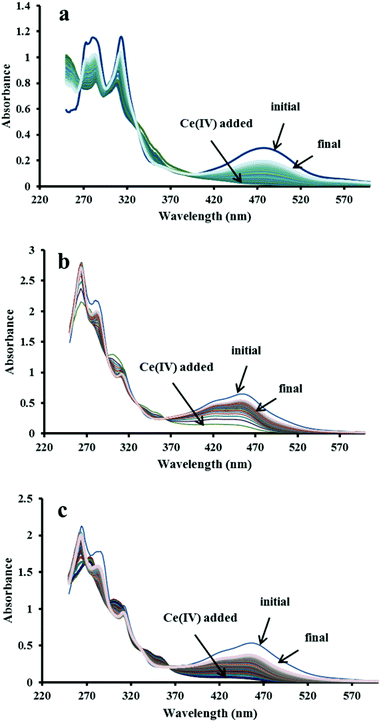
Further addition of one equivalent of Ce4+ to the [RuIVO]2+ species (or three equivalents to [RuII–OH2]2+) triggered three stepwise redox processes. Three distinctive regions based on the absorbance changes at 283 nm were observed and attributed to the reactions given in eqn (3)–(5).8
| | | [RuIVO]2+ + Ce4+ → [RuVO]3+ + Ce3+ | (3) |
| | | [RuVO]3+ + H2O → [RuIII–(OOH)]2+ + H+ | (4) |
| | | [RuIII–(OOH)]2+ → [RuII–OH2]2+ | (5) |
The transient [RuVO]3+ ion has been proposed to react further with water to give what is tentatively assigned as the [RuIII–OOH]2+ species which slowly decays to [RuII–OH2]2+ possibly by releasing O2.8 In the present investigation, three regions were indeed observed at the slightly lower wavelength (261 nm), compared with 283 nm in the previous study, suggesting that catalyst 2 also undergoes the stepwise redox processes described by eqn (3)–(5) after the addition in this case of four equivalents of Ce4+ (see Fig. 3, top panel). Oxygen can be released if further Ce4+ is added to the peroxido species [RuIII–OOH]2+ according to eqn (6) and (7).8 This was demonstrated in the present study by using an excess (1000 equivalents) of Ce4+ in the oxygen evolution experiment mentioned earlier.
| | | [RuIII–(OOH)]2+ + Ce4+ → [RuIV–(O2)]2+ + Ce3+ + H+ | (6) |
| | | [RuIV–(O2)]2+ + H2O → [RuII–(OH2)] + O2 | (7) |
 |
| | Fig. 3 (Top) Absorbance changes at 261 nm in 2 after the addition of four equivalent of Ce4+ showing three distinctive changes with time. (Bottom) Absorbance changes at 261 nm measured on addition of four equivalents of Ce4+ to a mixture of catalyst 2 and 4. No clear evidence was found for the redox processes identified for 2 alone. | |
The UV-Visible spectrum of the mixture of catalyst 2 and 4 before the Ce4+ addition showed the MLCT band at 455 nm and three other bands in the UV regions (313, 281 and 264 nm). As observed for the individual catalyst, the MLCT band also disappeared upon adding the oxidant (Fig. 2, panel b). In addition to the three redox processes observed for the individual catalyst, oxidation of the mixture with four equivalents of Ce4+ is also expected to trigger the formation of [Ru(phen)2(Me2bipy)]3+ according to eqn (8).
| | | [Ru(phen)2(Me2bipy)]2+ + Ce4+ → [Ru(phen)2(Me2bipy)]3+ + Ce3+ | (8) |
As in the study with 2 alone, the absorbance changes at 261 nm was also monitored for the 2–4 mixture to see whether the features associated with (3), (4) and (5) could be identified on the same time scale. These features were absent (Fig. 3, bottom) partly because of the coincidental rise in a strong ligand-centred absorption at 264 nm as is typically observed in this region for similar tris(diimine)ruthenium(II) complexes.14 Alternatively, it is possible that the oxidation step (3) is greatly accelerated by [Ru(phen)2(Me2bipy)]3+ such that it could not be detected on the minute time scale. The acceleration of reaction (3) by [Ru(phen)2(Me2bipy)]3+ (see eqn (9)) during the oxidation of 2 by Ce4+ is consistent with the enhanced oxygen production by 2 in the presence of 4, as shown in Fig. 1.
| | | [Ru(phen)2(Me2bipy)]3+ + [RuIVO]2+ → [RuVO]3+ + [Ru(phen)2(Me2bipy)]2+ | (9) |
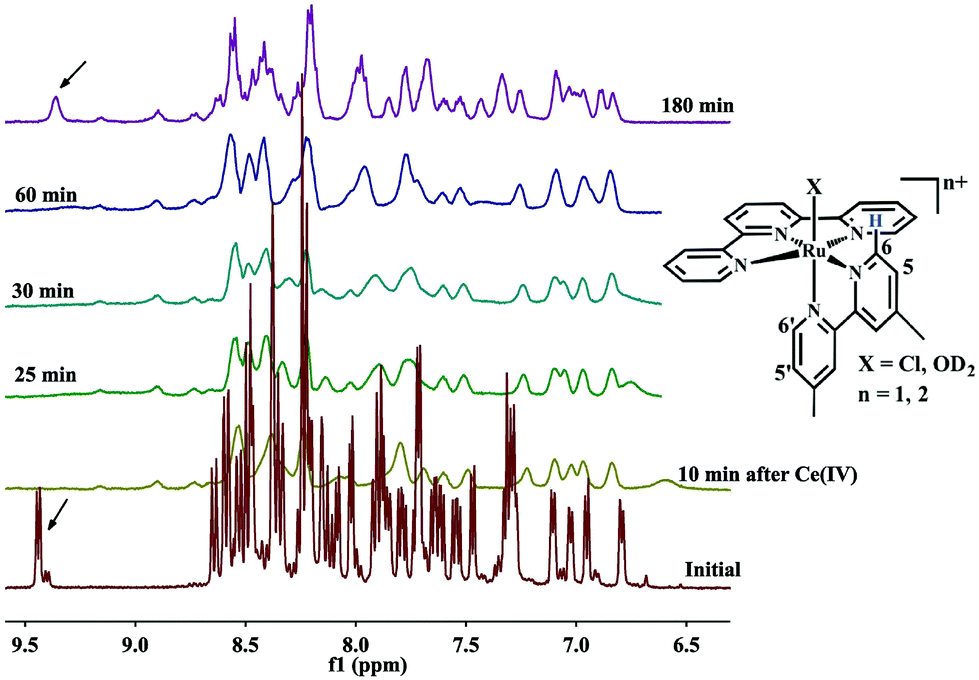
A 1H NMR experiment was carried out on the mixture of 2–4 in D2O–CD3CN to further probe the processes occurring on addition of Ce4+ (Fig. 4). The characteristic C6 proton resonance of the bipy ligand, located between 9 to 10 ppm, is affected by the adjacent chlorido or aqua ligand in [Ru(terpy)(xbipy)Cl]+ or [Ru(terpy)(xbipy)(OH2)]2+ is a useful tool to identify the presence of [RuII–Cl]+ or [RuII–OH2]2+ species.11,13 As shown in Fig. 4, the C6 proton at 9.45 ppm of 2 disappears on addition of Ce4+ to the mixture because of the formation of paramagnetic higher-valency ruthenium species. Over a period of 180 minutes, [RuII–OD2]2+ was clearly generated as indicated by the reappearance of the C6 proton signal.
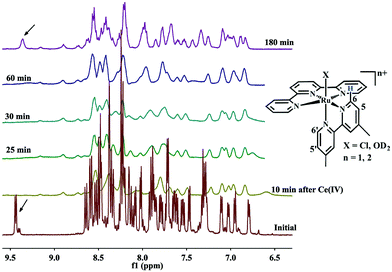 |
| | Fig. 4 Time dependence of the evolution of the 1H NMR spectrum of a 1:1 mixture of 2 and 4 in CD3CN–0.1 M HClO4 in D2O (1:4) (4.22 mM) before and after addition of four equivalent Ce4+. | |
The spectrum of the mixture before Ce4+ addition shows a proton resonance at 9.45 ppm, attributed to the C6 Me2bipy proton adjacent to the chloro ligand in [Ru(terpy)(Me2bipy)Cl]+ (2). The proton resonance located nearby (9.4 ppm) is attributed to the corresponding C6 proton of Me2bipy of the solvation product, [Ru(terpy)(Me2bipy)(OD)2)]2+.11,13 Integration of the two proton resonances reveals that the mixture contains approximately 90% [Ru(terpy)(Me2bipy)Cl]+ and 10% [Ru(terpy)(Me2bipy)(OD2)]2+. Both proton resonances disappear following the addition of Ce4+. The spectrum measured at 180 minutes after the addition of Ce4+ shows the appearance of the C6 proton at 9.4 ppm, suggesting that the [Ru(terpy)(Me2bipy)(OD2)]2+ was regenerated but not [Ru(terpy)(Me2bipy)Cl]+.
Although the stepwise processes (3) to (5) in the 2–4 mixture could not be identified from the UV-Visible spectral data, process (5) or the regeneration of [RuII–OH2]2+ (or the deuterated forms) was evident from the time-course 1H NMR data.
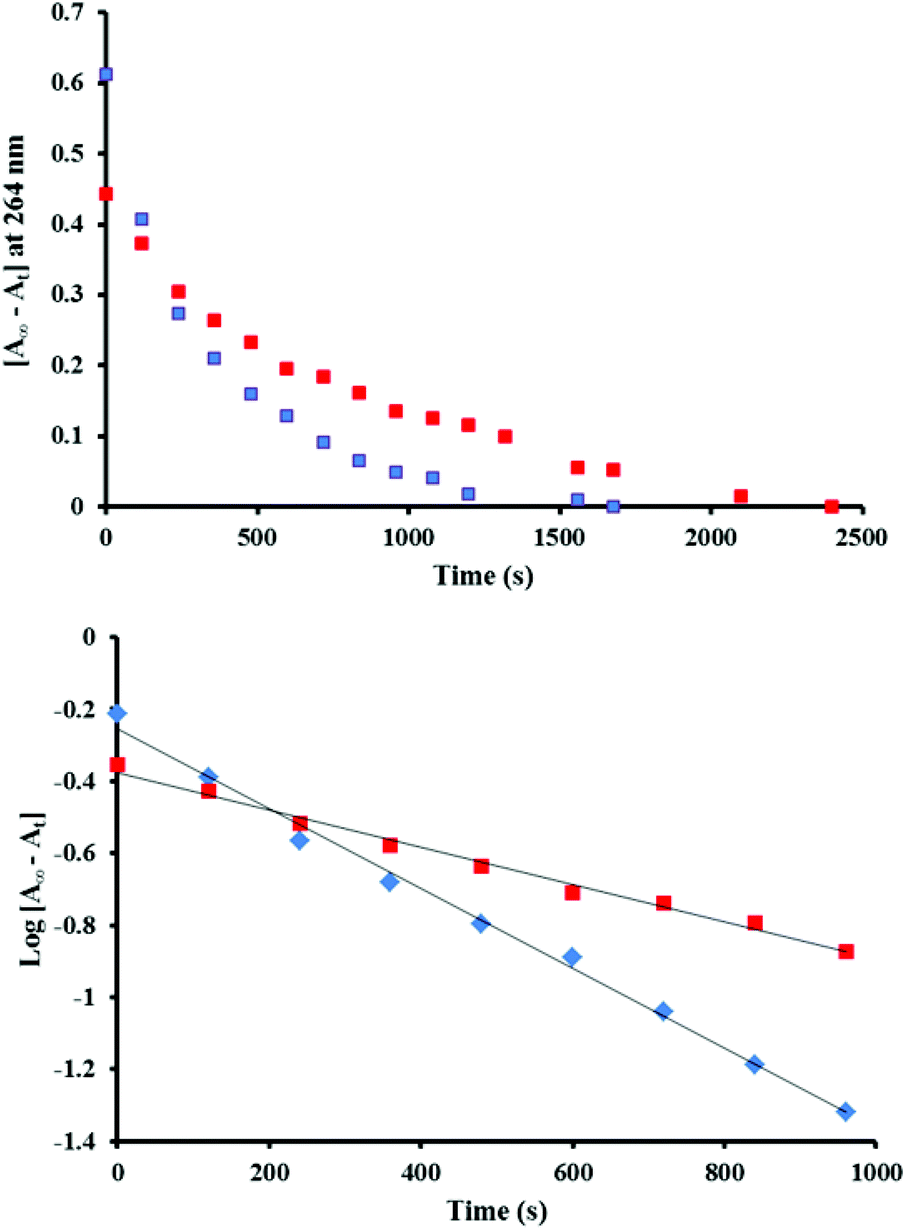
In order to investigate the kinetics of the reduction of [Ru(phen)2(Me2bipy)]3+ to the initial [Ru(phen)2(Me2bipy)]2+ state with respect to catalyst 1 and 2, the absorbance changes at 264 nm in 1–4 and 2–4 mixtures after Ce4+ addition were monitored. The UV-Visible spectrum of catalyst 2 before Ce4+ addition showed no absorption below 270 nm (Fig. 2), and thus the strong 264 nm band is more specific for [Ru(phen)2(Me2bipy)]2+ and was chosen for the kinetic evaluation. As can be seen from Fig. 5 (top panel), the absorbance changes at 264 nm reflecting the regeneration of [Ru(phen)2(Me2bipy)]2+ follow first-order exponential kinetics and the rate constants for the [Ru(phen)2(Me2bipy)]3+ to [Ru(phen)2(Me2bipy)]2+ reduction in the 2–4 and 1–4 combinations were calculated to be 1.11 (±0.03) × 10−3 s−1 and 5.2 (±0.2) × 10−4 s−1, respectively (Fig. 5, bottom panel). The faster rate of reduction of [Ru(phen)2(Me2bipy)]3+ to [Ru(phen)2(Me2bipy)]2+ in the 2–4 combination in comparison to 1–4 suggests that the redox processes of 2 and 4 under Ce4+ activation may be inter-dependent, thus supporting the cooperative interaction given in eqn (9). In contrast to the ‘cooperative’ effect in the 2–4 mixture, 1 and 4 appear to consume Ce4+ independently, such that a ‘competitive’ effect leads to a slight decrease in the oxygen evolution performance of 1 (Fig. 1). Accordingly, in this case 4 merely reacts with some of the available Ce4+ but the [Ru(phen)2(Me2bipy)]3+ complex produced is not capable of oxidising the catalyst 1 from [RuIVO]2+ to [RuVO]3+ state.
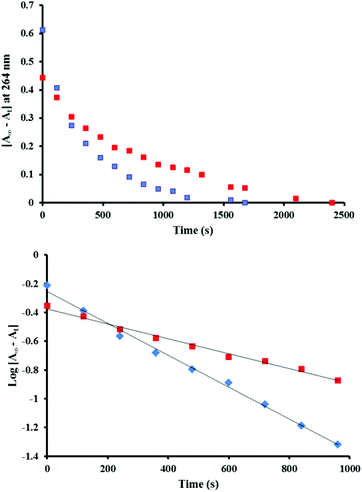 |
| | Fig. 5 (Top) Absorbance changes reflecting the regeneration of the [Ru(phen)2(Me2bipy)]2+, measured over time at 264 nm after four equivalents of Ce4+ were added to mixtures of 2–4 (blue dotted line) and 1–4 (red dotted line). The concentration of each complex was 24 μM. The final absorbance values are denoted as A∞ and the absorbance at a time t as At. (Bottom) Analysis of the linear plots of log[A∞ − At] vs. time in 2–4 (blue dotted line) and in 1–4 (red dotted line), assumed to correspond to the decay of [Ru(phen)2(Me2bipy)]3+, gave first-order rate constants of 1.11 (±0.03) × 10−3 s−1 and 5.2 (±0.2) × 10−4 s−1 for the [Ru(phen)2(Me2bipy)]2+ recovery in the 2–4 and in 1–4 combinations, respectively. | |
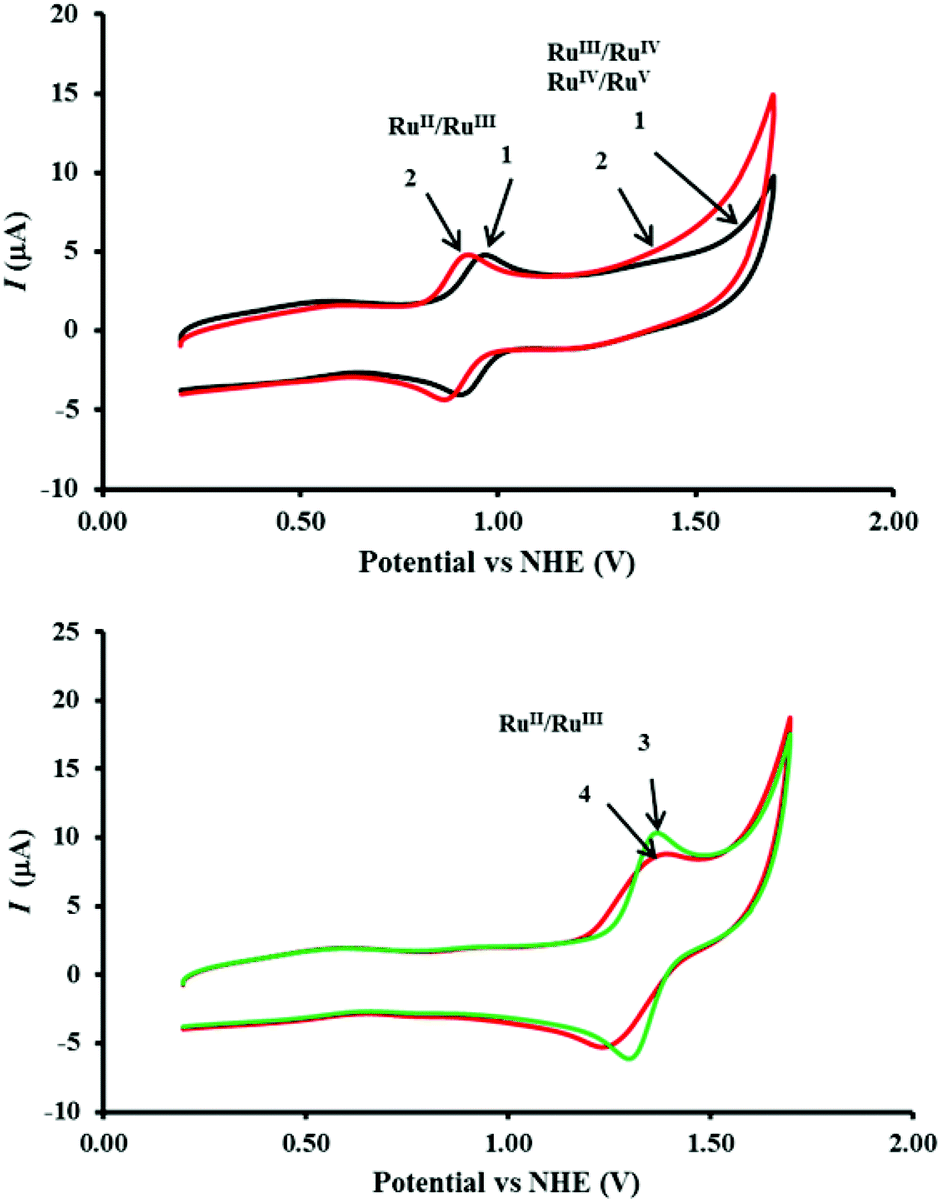
The cyclic voltammograms of 1, 2, 3 and 4 as individual complexes and in mixtures were measured in CH3CN–0.1 M HClO4 (1:6) at the concentration 0.48 mM and the data are presented in Fig. 6, 7 and Table 1. The RuII/RuIII oxidation potentials (Epavs. NHE) of 1 and 2 of 0.97 V and 0.93 V, respectively, lie within the range of those reported for closely-related complexes.11 The RuII/RuIII oxidation potentials (Epavs. NHE) for 3 and 4 were observed at 1.37 V and 1.39 V, respectively. There were no significant differences in these potentials when measured as mixtures of complexes.
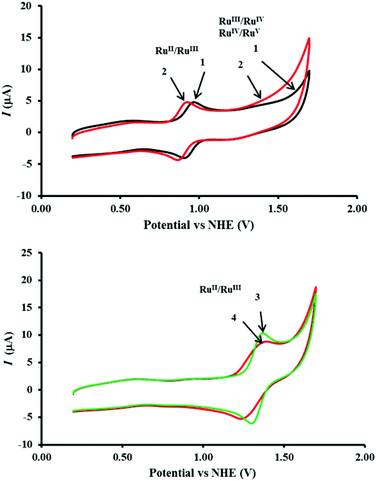 |
| | Fig. 6 (Top) Comparison of the cyclic voltammetry (CV) of 1 (black) and 2 (red) showing the RuII/RuIII redox wave and the predicted RuIII to RuIV and RuIV to RuV oxidations leading to the onset of water oxidation. (Bottom) Comparison of the CV traces showing the [Ru(bipy)3]2+/3+ redox wave for 3 (green) and [Ru(phen)2(Me2bipy)]2+/3+ in 4 (red). The traces were recorded at 0.48 mM complex concentrations in CH3CN–0.1 M HClO4 (1:6) using a three electrode system comprising a glassy carbon working electrode, platinum auxiliary electrode and Ag/AgCl reference electrode at the scan rate 50 mV s−1 at 23 °C. | |
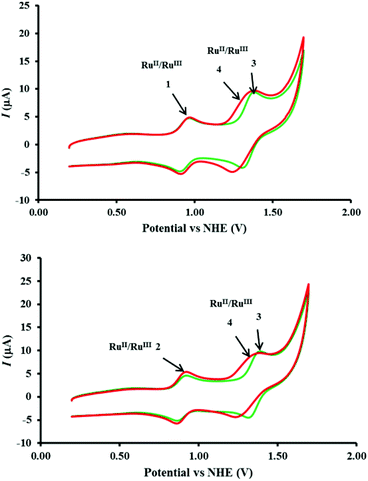 |
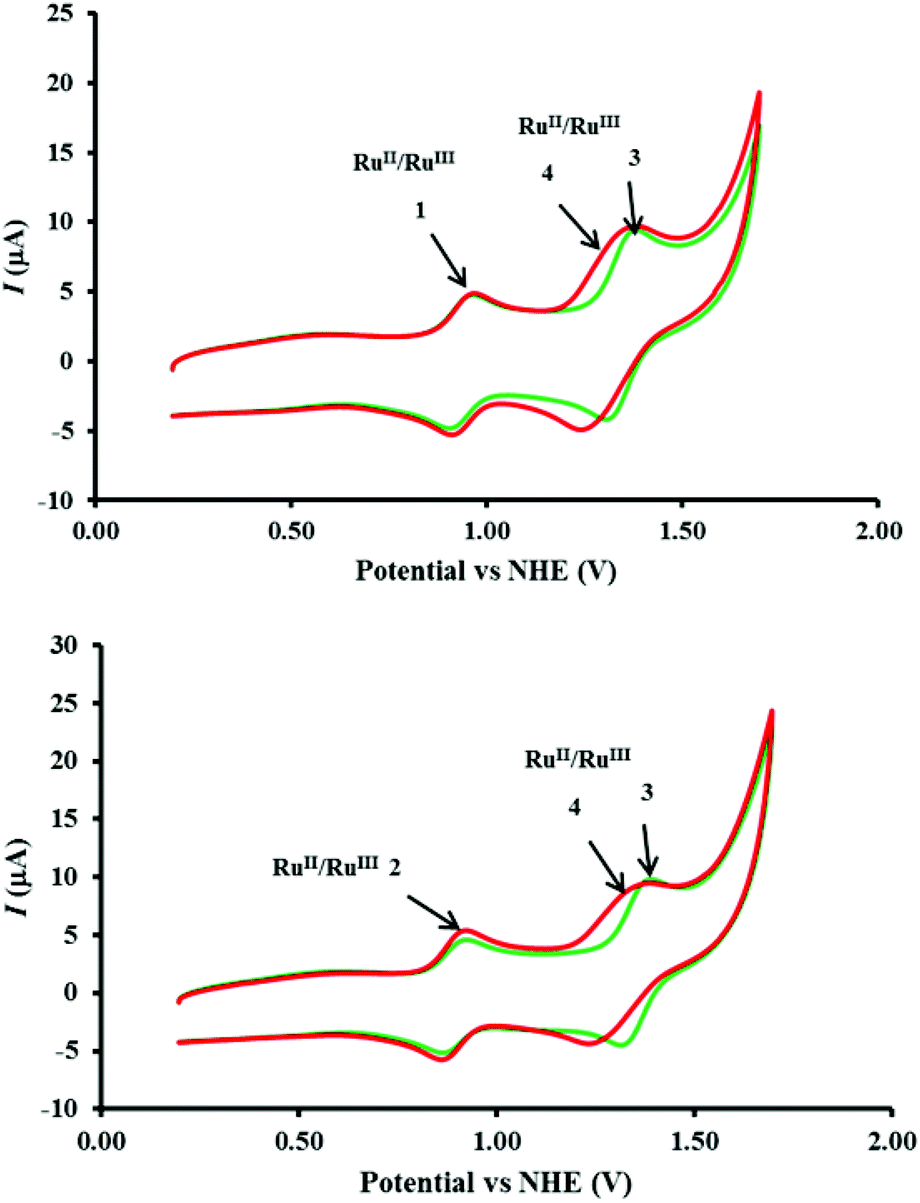
| | Fig. 7 (Top) Comparison of the cyclic voltammetry of mixtures of 1–3 (green) and 1–4 (red) in top panel and of 2–3 (green) and 2–4 (red) in bottom panel. Complex concentrations, electrode configuration and CV measurement conditions as indicated in caption to Fig. 6. | |
Table 1 Assignment of the redox potentials obtained from the CV experiments of the individual complexes and the mixtures, at 0.48 mM concentrations, carried out in CH3CN–0.1 M HClO4 (1:6) on a glassy carbon electrode at the scan rate 50 mV s−1. E1/2 = (Epa − Epc)/2 in volts relative to NHE; ΔE = |Epa − Epc| in mV
| Complexes and mixtures |
Assignment |
E
pa (V) |
E
pc (V) |
E
1/2 (V) |
ΔE (mV) |
|
An increase in current starting from 1.37 V for catalyst 2 (Fig. 6, top) reflects the RuIII/RuIV and RuIV/RuV oxidation processes.
RuIII/RuIV and RuIV/RuV oxidation processes in 1 predicted to start at 1.55 V.
|
|
4 only |
RuII/RuIII (4) |
1.39 |
1.23 |
1.31 |
160 |
|
2–4 |
RuII/RuIII (4) |
1.39 |
1.23 |
1.31 |
160 |
|
|
RuII/RuIII (2) |
0.92 |
0.86 |
0.89 |
60 |
|
1–4 |
RuII/RuIII (4) |
1.39 |
1.24 |
1.32 |
150 |
|
|
RuII/RuIII (1) |
0.97 |
0.91 |
0.94 |
60 |
|
3 only |
RuII/RuIII (3) |
1.37 |
1.30 |
1.34 |
70 |
|
2–3 |
RuII/RuIII (3) |
1.39 |
1.32 |
1.36 |
70 |
|
|
RuII/RuIII (2) |
0.92 |
0.86 |
0.89 |
60 |
|
1–3 |
RuII/RuIII (3) |
1.38 |
1.30 |
1.34 |
80 |
|
|
RuII/RuIII (1) |
0.96 |
0.90 |
0.93 |
60 |
|
2 onlya |
RuII/RuIII (2) |
0.93 |
0.86 |
0.90 |
70 |
|
1 onlyb |
RuII/RuIII (1) |
0.97 |
0.90 |
0.94 |
70 |
A close analysis of the cyclic voltammetry of the individual catalysts reveals a significant increase in current starting from 1.37 V (Fig. 6, bottom) in 2 which, as described in eqn (2) and (3), may be attributed to the formation of [RuIVO]2+ and [RuVO]3+ leading to the onset of water oxidation. The redox waves of the [RuIVO]2+ and [RuVO]3+ are generally not well-defined as they coincide with the onset of water oxidation. On the other hand, the cyclic voltammetry of 1 reveals that the oxidation processes occurs at much higher potential (1.55 V). The predicted [RuIVO]2+ and [RuVO]3+ species that are accessible at a reasonably lower potential in 2 may be ‘switched on’ by both redox mediators 3 and 4 as the RuII/RuIII potentials of the two complexes occur in the same range. In contrast, this may be more difficult for catalyst 1 because the higher potential results in a slight increase in the reaction driving force. This postulate is consistent with both the oxygen evolution and the UV-Visible spectrophotometric data presented earlier.
The significant difference in the oxygen evolution performance of 2 in the presence of 3 and 4 is interesting (Fig. 1), and worthy of further comment. From the electrochemistry point of view, although the anodic potentials (Epa) of both 3 and 4 are identical, the cathodic potential (Epc) of 4 was significantly lower (see Table 1). When compared with [Ru(bipy)3]2+, it is feasible that the [Ru(phen)2(Me2bipy)]2+/3+ redox process in the acidified, and predominantly aqueous, solution may be accompanied by structural changes that lead to electrochemical irreversibility which manifests itself in a larger ΔE. In addition to the potential matching described earlier, the acceleration of the oxidation of the [RuIVO]2+ conversion to [RuV=O]3+ by [Ru(phen)2(Me2bipy)]3+ (eqn (9)), we tentatively propose that this may be due to a greater thermodynamic stability of [Ru(phen)2(Me2bipy)]2+, resulting in faster reduction of [Ru(phen)2(Me2bipy)]3+ (compared with [Ru(bipy)3]3+ to [Ru(bipy)3]2+), and a concomitantly faster oxidation of [RuIVO]2+ to [RuVO]3+.
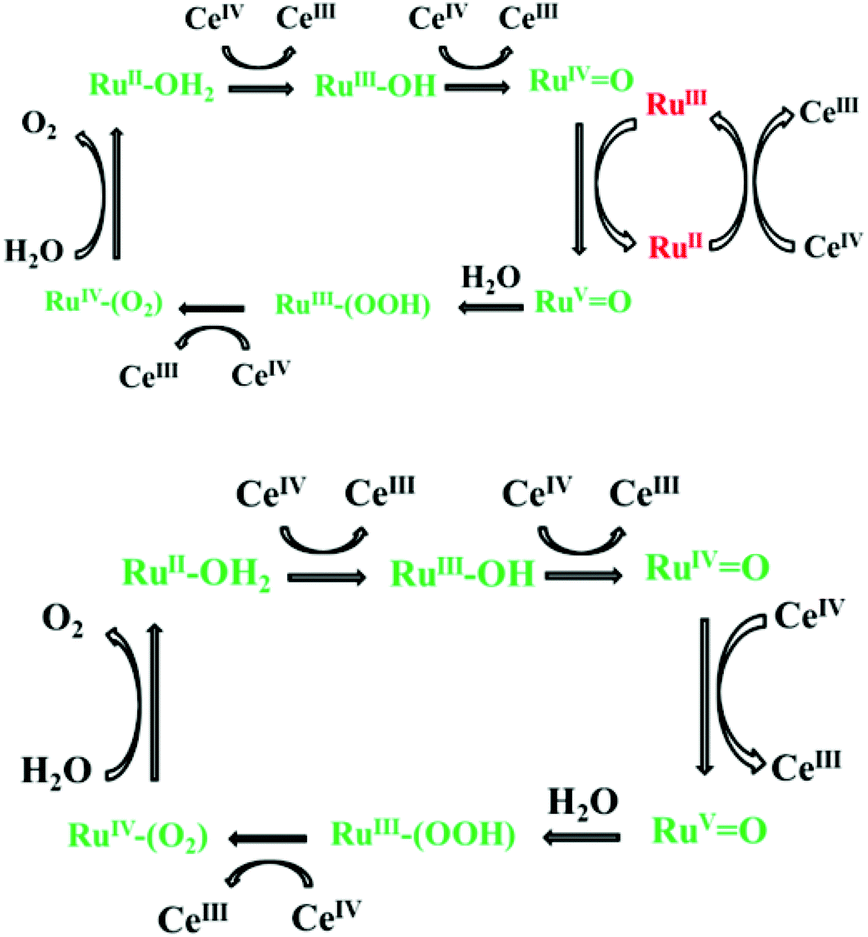
The proposed mechanism for the homogenous catalysis by the recently-reported dinuclear complex [(bipy)2Ru(4-Mebpy-4′-bimpy)Ru(terpy)(OH2)]4+, referred to as [RuaII–RubII–OH2]4+ (a = mediator and b = catalyst) highlighted the role of the redox mediator in accelerating the catalytic process.10 Following the oxidation of [RuaII–RubII–OH2]4+ to [RuaIII–RubIVO]5+ by Ce4+ (eqn (10)), an intra-molecular electron transfer between the two ruthenium centres leads to a redox equilibrium described in eqn (11).10 Further reactions (eqn (12)–(14)) follow what was described earlier in eqn (4), (6) and (7).10
| | | [RuaII–RubII–OH2]4+ + 3Ce4+ → [RuaIII–RubIVO]5+ + 3Ce3+ + 2H+ | (10) |
| | | [RuaIII–RubIVO]5+ ⇄ [RuaII–RubVO]5+ | (11) |
| | | [RuaII–RubVO]5+ + H2O → [RuaII–RubIII(OOH)]4+ + H+ | (12) |
| | | [RuaII–RubIII(OOH)]4+ + Ce4+ → [RuaII–RubIV(O2)]4+ + Ce3+ + H+ | (13) |
| | | [RuaII–RubIV(O2)]4+ + H2O → [RuaII–RubII–OH2]4+ + O2 | (14) |
In keeping with this study, an (inter-molecular) electron transfer between [Ru(phen)2(Me2bipy)]3+ in 4 and [RuIVO]2+ in 2 was demonstrated in the present study suggesting a similar mechanism of the cooperative effect proposed in Fig. 8.
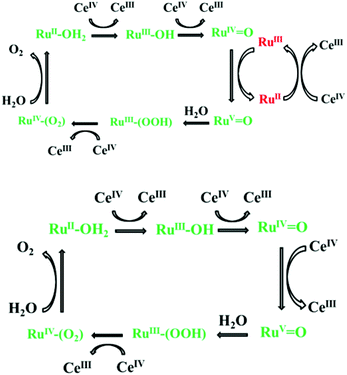 |
| | Fig. 8 (Top) Proposed mechanism of the mediator/catalyst cooperative effect observed in the 2–4 mixture.9,10 The catalytic mechanism involving only Ce4+ is given in the bottom diagram.8 The green colour indicates the catalyst and the red indicates the redox mediator. | |
Conclusions
This study has provided evidence of the enhancement of the water oxidation catalytic properties of 2 under the influence of the redox mediator 4, [Ru(phen)2(Me2bipy)]3+. The ability of 4 to accelerate the critical oxidation of the catalyst, [RuIVO]2+ → [RuVO]3+, may be due in part to the slightly better potential match between the two redox events when compared to [Ru(bpy)3]2+ (3). Work is currently underway to further probe such electron transfer processes using ruthenium catalysts and mediators appropriate for immobilisation on electrode surfaces. This accessibility of the electron transfer between the redox mediator (dye) and the catalyst is significant for the design of catalytic assemblies for the photo-induced oxidation of water. For example, mediator-catalyst assemblies can be attached to, for example, dyed-titania semiconductor films to construct photoanodes capable of splitting water with visible light.15
Experimental section
Materials and synthesis
[Ru(bipy)3]Cl2 and (NH4)2[Ce(NO3)6] were purchased from Sigma Aldrich and used as received. All other chemicals were sourced from commercial suppliers and used without further purification except where otherwise indicated.
The synthesis and characterisation of the two precursors [Ru(terpy)Cl3] and [Ru(phen)2Cl2]16,17 and the [Ru(phen)2(Me2bipy)]Cl2,12 [Ru(terpy)(Me2bipy)Cl]Cl and [Ru(terpy)(bipy)Cl]Cl complexes13 have been reported previously and the same methods were used in the present work. In a typical synthesis, solid [Ru(terpy)Cl3] (0.20 g, 0.45 mmol) and appropriate amounts of the ligands (0.084 g, 0.45 mmol for Me2bpy and 0.071 g, 045 mmol for bipy) were refluxed in EtOH–H2O (4:1; 40 ml) for 4 h. After cooling, the solvent mixture was evaporated to dryness. The crude product was dissolved in methanol and loaded onto a Sephadex LH-20 exclusion column and the pure compounds were separated from the impurities using methanol as the eluent. Yield of [Ru(terpy)(Me2bipy)Cl]Cl 0.16 g (60%). To prepare [Ru(phen)2(Me2bipy)]Cl2, [Ru(phen)2Cl2] (0.20 g, 0.38 mmol) and Me2bipy (0.069 g, 0.38 mmol) were refluxed in EtOH–H2O (1:1, 30 ml) for 6 h, after which the bright orange solution was evaporated to dryness and loaded onto a Sephadex LH-20 column. The pure [Ru(phen)2(Me2bipy)]Cl2 compound was separated from the impurities using methanol as the eluent. The yield was 0.24 g (90%). The 1H NMR data of the compounds were consistent with those reported in the literature.12,13
Methods
Oxygen evolution
Oxygen measurements were performed using a Unisense OXY-500 microsensor connected to a Unisense OXY Meter. The sensor was calibrated using air and argon for 100% O2 and 0% O2, respectively. The signal was processed using Sensor Trace software. To measure oxygen evolution by the individual complexes and the mixtures (1:1 complex to redox mediator), each complex and redox mediator (0.51 μmol) was suspended in 0.1 M HClO4 (6 ml) in an air-tight vessel. Argon was then purged to obtain zero oxygen reading. A solution of (NH4)2[Ce(NO3)6] (0.28 g, 0.51 mmol) in 0.1 M HClO4 (1 ml) was charged through a septum after the reading had stabilised for 20 min. The final concentrations of the complexes and the Ce4+ in the vessel were 73 μM and 0.073 M, respectively. The oxygen sensor was measured in the headspace and the signal was collected for 6 to 10 h.
UV-Visible spectrophotometry
UV-Visible spectra were recorded on a Varian Cary 300 spectrophotometer. To measure the spectra of either individual complexes or the mixture of complexes, each compound (0.51 μmol) was dissolved in distilled water (0.5 ml) and 0.1 M HClO4 (6.5 ml) was then added. A 1 ml aliquot was taken from this stock into a 1 cm path length cuvette and 1.95 ml of 0.1 M HClO4 was added. The initial spectrum was recorded prior to adding 0.05 ml (NH4)2[Ce(NO3)6] in 0.1 M HClO4. The concentration of the complex and (NH4)2[Ce(NO3)6] in the cuvette were 24 μM and 97 μM, respectively. The spectra were then recorded in 2, 5, 10 and 15 min intervals for 15 h.
1H NMR experiment
1H NMR experiments were performed on a Bruker Avance 400 MHz. Solid [Ru(terpy)(Me2bipy)Cl]Cl (2.0 mg, 3.4 μmol) and [Ru(phen)(Me2bipy)]Cl2 (2.4 mg, 3.4 μmol) were dissolved in CD3CN (0.2 ml) in an NMR tube and was topped up by 0.1 M HClO4 in D2O (0.6 ml). The concentration of each complex was 4.22 mM. The initial spectrum was measured prior to adding the solid (NH4)2[Ce(NO3)6] (7.4 mg, 14 μmol). The spectrum was then recorded every 10 min for the first 30 min then every 30 min for additional 2.5 h.
Electrochemistry
Electrochemical measurements were performed on a VSP Bio-Logic potentiostat connected to a three-electrode system. Cyclic voltammetry of the individual complexes and the mixtures were measured in CH3CN–0.1 M HClO4 (1:6) at the scan rate 50 mV s−1 on a glassy carbon working electrode. A platinum wire was used as an auxiliary electrode and Ag/AgCl electrode was used the reference. The concentration of each complex in all cases was 0.48 mM. The observed potentials were corrected relative to NHE.
Acknowledgements
The authors thank the Australian Research Council for financial support received through the Australian Centre of Excellence for Electromaterials Science (CE0561616).
Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991 CrossRef CAS; A. Currao, Chimia, 2007, 61, 815 CrossRef; A. Heller, Science, 1984, 223, 1141 Search PubMed; R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892 CrossRef PubMed; W. J. Younpblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef PubMed; S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645 CrossRef PubMed; D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS PubMed; J. Barber and J. W. Murray, Coord. Chem. Rev., 2008, 252, 233 CrossRef PubMed; C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444 CrossRef PubMed; R. Brimblecombe, D. R. J. Kolling, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Inorg. Chem., 2009, 48, 7269 CrossRef PubMed; R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335 CrossRef PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21 CrossRef CAS PubMed; R. Manchanda, G. W. Brudvig and R. H. Crabtree, Coord. Chem. Rev., 1995, 144, 1 CrossRef; W. Ruettinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1 CrossRef PubMed; S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981 CrossRef PubMed; M. Yagi, A. Syouji, S. Yamada, M. Komi, H. Yamazaki and S. Tajima, Photochem. Photobiol. Sci., 2009, 8, 139 Search PubMed; H. Yamazaki, A. Shouji, M. Kajita and M. Yagi, Coord. Chem. Rev., 2010, 254, 2483 CrossRef PubMed.
- W. Song, A. Ito, R. A. Binstead, K. Hanson, H. Luo, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 1158 Search PubMed; K. Kobayashi, H. Ohtsu, T. Wada, T. Kato and K. Tanaka, J. Am. Chem. Soc., 2003, 125, 6729 CrossRef CAS PubMed; J. T. Muckerman, D. E. Polyansky, T. Wada, K. Tanaka and E. Fujita, Inorg. Chem., 2008, 47, 1787 CrossRef PubMed; Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845 CrossRef PubMed; J. Nyhlen, L. Duan, B. Akermark, L. Sun and T. Privalov, Angew. Chem., Int. Ed., 2010, 49, 1773 CrossRef PubMed; L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397 CrossRef PubMed; D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto and C. P. Berlinguette, Eur. J. Inorg. Chem., 2010, 3135 CrossRef PubMed; L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS; J. A. Gilbert, D. S. Eggleston, W. R. Murphy, D. A. Geselowitz, S. W. Gersten, D. K. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855 CrossRef.
- X. Yang and M.-H. Baik, J. Am. Chem. Soc., 2006, 128, 7476 CrossRef CAS PubMed; D. Moonshiram, J. W. Jurss, J. J. Concepcion, T. Zakharova, I. Alperovich, T. J. Meyer and Y. Pushkar, J. Am. Chem. Soc., 2012, 134, 4625 CrossRef PubMed; D. Moonshiram, V. Purohit, J. J. Concepcion, T. J. Meyer and Y. Pushkar, Materials, 2013, 6, 392 CrossRef.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462 CrossRef CAS; J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277 CrossRef PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17632 CrossRef CAS PubMed.
- M. R. Norris, J. J. Concepcion, D. P. Harrison, R. A. Binstead, D. L. Ashford, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2080 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202 CrossRef CAS PubMed.
- Y. Mulyana, D. K. Weber, D. P. Buck, C. A. Motti, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 1510 RSC; F. Li, Y. Mulyana, M. Feterl, J. M. Warner, J. G. Collins and F. R. Keene, Dalton Trans., 2012, 40, 5032 RSC; M. J. Pisani, P. D. Fromm, Y. Mulyana, R. J. Clarke, K. Heimann, J. G. Collins and F. R. Keene, ChemMedChem, 2011, 6, 848 CrossRef CAS PubMed.
- Y. Mulyana, J. G. Collins and F. R. Keene, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 371 CrossRef CAS PubMed.
- D. Hesek, Y. Inoue, S. R. L. Everitt, H. Ishida, M. Kunieda and M. G. B. Drew, Inorg. Chem., 2000, 39, 308 CrossRef CAS.
- W. Song, C. R. K. Glasson, H. Luo, K. Hanson, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Phys. Chem. Lett., 2011, 2, 1808 CrossRef CAS; L. Wang, D. L. Ashford, D. W. Thompson, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. C, 2013, 117, 24250 Search PubMed.
- P. A. Adcock, F. R. Keene, R. S. Smythe and M. R. Snow, Inorg. Chem., 1984, 23, 2336 CrossRef CAS.
- T. Togano, N. Nagao, M. Tsuchida, H. Kumakura, K. Hisamatsu, F. S. Howell and M. Mukaida, Inorg. Chim. Acta, 1992, 195, 221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00629a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a