 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational development of iron catalysts for asymmetric transfer hydrogenation
Peter E.
Sues
,
Karl Z.
Demmans
and
Robert H.
Morris
*
Department of Chemistry, University of Toronto, 80 Saint George St., Toronto, Ontario M5S 3H6, Canada. E-mail: rmorris@chem.utoronto.ca
First published on 15th April 2014
Abstract
The asymmetric reduction of ketones and imines by transfer of hydrogen from isopropanol as the solvent catalyzed by metal complexes is a very useful method for preparing valuable enantioenriched alcohols and amines. Described here is the development of three generations of progressively more active iron catalysts for this transformation. Key features of this process of discovery involved the realization that one carbonyl ligand was needed (as in hydrogenases), the synthesis of modular ligands templated by iron, the elucidation of the mechanisms of catalyst activation and action, as well as the rational synthesis of precursors that lead directly and easily to the species in the catalytic cycle. The discovery that iron, an abundant element that is essential to life, can form catalysts of these hydrogenation reactions is a contribution to green chemistry.
 Peter E. Sues | Peter E. Sues was born in Toronto, Canada, in 1987. He received his H. B. Sc. in Chemistry and Biochemistry at the University of Toronto in 2009 and he is currently a Ph. D. candidate in the Department of Chemistry at the University of Toronto under the guidance of Prof. Robert H. Morris. His research interests include the development of phosphine containing ligands for iron group metals with the aim of developing transition metal catalysts for a variety of chemical transformations. |
 Karl Z. Demmans | Karl Z. Demmans was born in Sarnia, Ontario, Canada in 1990. He obtained a bachelor's degree with an Honours in Chemistry from McGill University in 2012, and then spent a year under the guidance of Dr Audrey Moores and Dr Tomislav Friščić where he explored his interests in green chemistry and catalysis. He is presently in his first year of doctoral studies at the University of Toronto investigating the heterogenization of homogeneous iron catalysts. |
 Robert H. Morris | Robert H. Morris was born in Ottawa, Canada, in 1952. He received his PhD from the University of British Columbia in 1978 under the guidance of Prof. Brian James. After postdoctoral work at the Nitrogen Fixation Laboratory, University of Sussex and the Pennsylvania State University he joined the faculty of the University of Toronto in 1980. He was appointed full Professor there in 1989. He is a Fellow of the Chemical Institute of Canada and of the Royal Society of Canada. His research interests include the organometallic, bioinorganic and catalytic chemistry of transition metal complexes, with a focus on iron. |
Introduction
Green chemistry
Homogeneous catalyst development has traditionally focused on the more active second- and third-row transition metals in combination with simple ligands.1 As a rule, these catalysts provided high turnover frequencies (TOF) and product selectivities which could not be easily matched by the less active first-row transition metals.2–5 However, with new environmental constraints being imposed on industry, there has been a push towards more sustainable practices or “Green Chemistry.”6 The basic tenets of this movement challenge chemists to develop new alternatives, focusing on transformations that use more abundant, cheap, and less toxic materials. In addition there is an emphasis on producing less waste overall by reducing the amount of by-products formed during synthesis, while reducing the need for auxiliary chemicals.6,7 These demands have pushed industry and academia to shift their focus towards the first-row transition metals, which are much more sustainable and “green” than their second- or third-row analogues.An important process in the fragrance and pharmaceutical industries is the asymmetric hydrogenation (AH) of ketones and imines to enantiopure alcohols and amines.8 As opposed to direct hydrogenation, asymmetric transfer hydrogenation (ATH) does not require pressurized hydrogen gas and often employs isopropanol as the solvent as well as the reducing agent, usually with base added. This provides safer, moderate reaction conditions while producing acetone as the major side product which can be recycled.7,9 Other hydrogen sources have also been employed such as formic acid–triethylamine mixtures or primary/secondary alcohols other than isopropanol. ATH typically employs second- or third-row transition metals such as Ru, Rh or Pd, Os, Ir or Pt, in order to achieve high catalyst TOF and product selectivities.10–12 An excellent candidate for a “greener” alternative is iron since it is significantly more abundant and is essential to life; thus trace impurities would be less harmful when these catalysts are applied in drug synthesis.7,13,14 For comparison, ruthenium costs $3 g−1, only 10 ppm is acceptable in medicinal products, and the maximum intake for a 50 kg human is 100 μg per day, whereas iron costs $0.0003 g−1, and the FDA does not require testing for trace amounts of iron in pharmaceutical products.15,16 With this in mind, chemists have successfully developed iron catalysts for use in many reactions including cross-coupling, reduction, oxidation, cycloaddition and polymerization.17–23 Herein we present our group's contribution to Green Chemistry and ATH, detailing an in-depth mechanistic study which led to a rational and systematic approach to ligand design in order to activate iron(II).
The idea of using iron came from our studies of corresponding ruthenium hydride complexes that were very active for ketone hydrogenation.24 Noyori et al. first explained why the presence of primary amino groups in ruthenium hydride complexes resulted in very active ketone transfer hydrogenation catalysts, later referred to as the “N–H effect.”9,11,39 The ketone is attacked in the outer sphere by the metal hydride and amine proton, simultaneously as shown in Fig. 1. Catalysis involving such an intermediate is commonly referred to as bifunctional catalysis.25
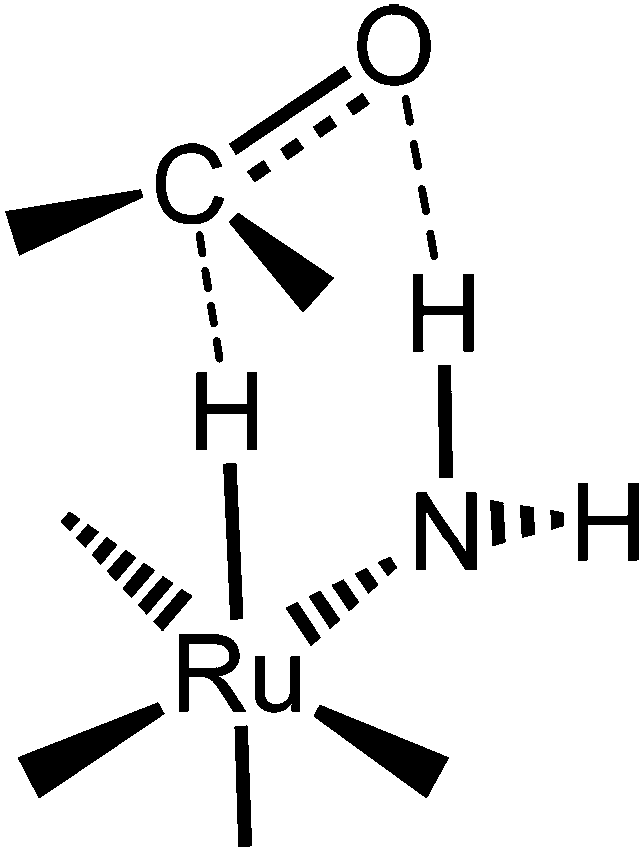 | ||
| Fig. 1 Representation of the metal hydride and amino proton interaction demonstrated by the dashed line.24 | ||
Noyori and coworkers showed that a trans-dichloro(6,5,6-PNNP)ruthenium(II) complex (where 6,5,6 refers to the sizes of the rings that the tetradentate ligand makes with the ruthenium) when activated with KOtBu in isopropanol had good activity and enantioselectivity for the ATH of ketones.26 Our group's interest in mechanistic studies of ruthenium hydride and dihydrogen chemistry as well as protonic–hydridic NH–HM bonds led us to study such systems in order to characterize the hydride complexes responsible for catalysis.27–30 Our group synthesized hydride complexes by refluxing chlorohydridotris(triphenylphosphine)ruthenium(II) with the corresponding tetradentate 6,5,6-PNNP ligand in THF, which produced a pair of diastereomers with the N–H groups syn or anti (Fig. 2).24,31,32 The syn complex isomerized upon addition of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) or KOtBu to obtain pure anti isomer. When tested for the ATH and AH of acetophenone in isopropanol with KOtBu at 45 °C, we observed much higher activity with pure anti isomer than with a mixture of the two isomers. The trans-dihydride that forms from the anti isomer will have the parallel HRu–NH motif on each side of the complex. The high activity of these tetradentate systems encouraged us to find out whether related iron complexes would also be hydrogenation catalysts.
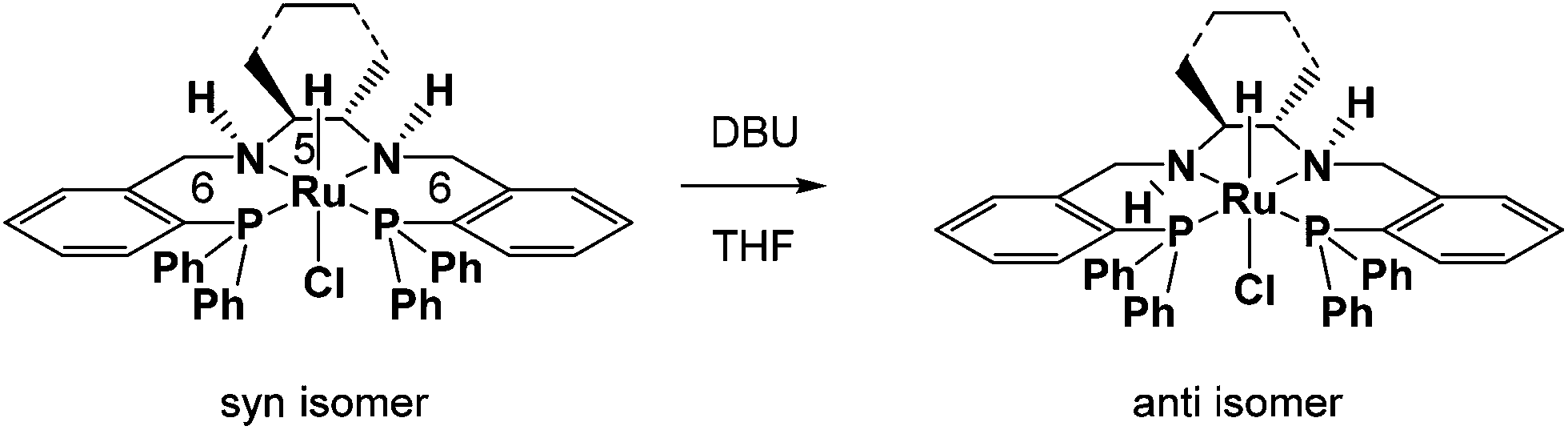 | ||
| Fig. 2 Ruthenium complexes used to investigate the effect of metal hydride-amino proton stereoisomerism on catalytic activity. | ||
Brief overview of the work summarized in the 2009 Chem. Soc. Rev. Article33
In 2008–9 the Morris group reported that a series of iron complexes bearing tetradentate ligands with two phosphorus donors and two nitrogen donors were active for ATH and AH of ketones once activated with KOtBu in isopropanol.34,35 The ligands were formed from Schiff base condensation reactions between an air stable, commercially available phosphine aldehyde, 2-(diphenylphosphino)benzaldehyde, and various diamines. This type of ligand had been reported previously by Noyori and coworkers as mentioned above26 and Gao and coworkers36 for ruthenium-based ATH catalysts, as well as for iron systems generated in situ. The complexes described by the Morris group were the first well defined-iron species capable of effecting AH reactions (Fig. 3).34,35,37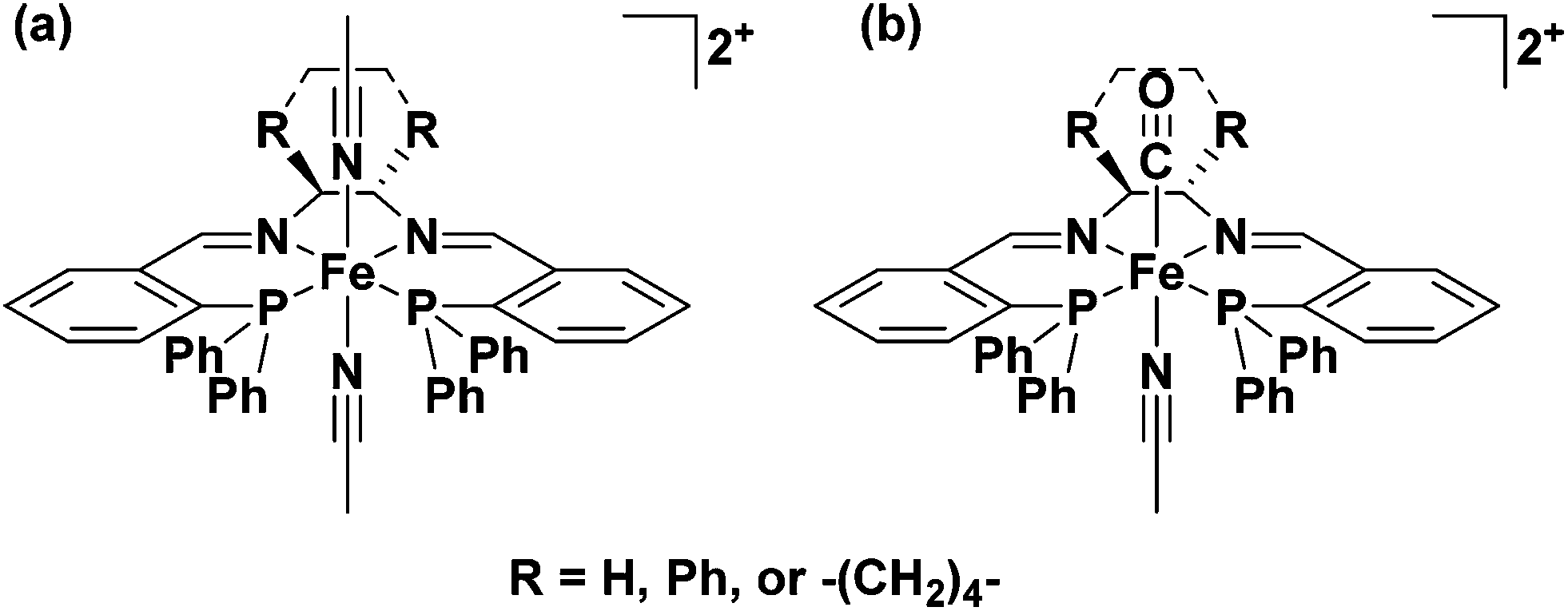 | ||
| Fig. 3 (a) trans-Bis(acetonitrile)(6,5,6-PNNP)iron(II) complexes developed for the AH of polar double bonds; (b) trans-carbonyl(acetonitrile)(6,5,6-PNNP)iron(II) complexes developed for the ATH of polar double bonds.34,35,37,38 | ||
A crucial point in the development of these catalysts was the realization that, much like hydrogenase enzymes in nature,39–42 a CO ligand was needed to activate the iron complexes for ATH using iPrOH as the solvent and sacrificial reductant; without this ligand they were completely inactive for this type of transformation (Fig. 4).33,37 This first generation of ‘6,5,6’ iron carbonyl catalysts were fairly active for the reduction of acetophenone in iPrOH with 8 equiv. KOtBu (TOF of 2600 h−1 at room temperature), but their selectivity was only moderate (up to 63% enantioselectivity for acetophenone, up to 96% for tBuCOPh→tBuHCOHPh).37 It was thought that removal of the o-phenylene linkers between the phosphorus and imine moieties, to shrink the metallocyclic ring sizes down from six to five, would make the ligand more suitable for iron as the original ligand was designed for the larger second row transition metal ruthenium.
 | ||
| Fig. 4 A simplified general schematic outlining ATH using iPrOH as the sacrificial reductant. | ||
Further investigation of the 6,5,6 systems revealed that the active catalytic species were iron(0) nanoparticles.43 The flexibility of this tetradentate ligand provided a pathway to the formation of iron(0).38 Upon reaction with base in iPrOH, one of the ligand imines of the carbonyl precatalyst becomes reduced and deprotonated. The ligand arm subsequently folds upwards to form a ferraaziridine species and eventually, iron nanoparticles. This result prompted the development of a smaller ring system which would be significantly more rigid.
One difficulty with removing the o-phenylene linker of the tetradentate ligand in favour of a methylene linker, however, is the inherent instability of the free phosphine aldehyde species, which is prone to oxidation and oligomerization. A way to avoid this unwanted decomposition is to protect the aldehyde such that the reactive phosphine species can be generated in situ. Fortunately, Matt et al.44 had reported a phosphonium salt that fit this criterion: a protonated dimer of two phosphine aldehydes that could be deprotected upon exposure to base. Moreover, the phosphonium dimer was a completely air- and moisture-stable solid, making it an ideal ligand precursor. With the phosphonium salt in hand, attempts to generate a free PNNP ligand were carried out, but these reactions invariably gave a mixture of unidentified products, most likely due to the reactivity of the product imines and the phosphorus lone pairs.
A breakthrough in the synthesis of the 5,5,5-PNNP ligand came with the use of a metal template approach.45 In the presence of iron, the phosphonium dimer 1a (the a refers to the PPh2 moiety) was deprotected with base and the phosphine aldehyde was produced in situ (Fig. 5). Subsequent addition of a diamine resulted in a metal templated Schiff base condensation between the coordinated phosphine aldehyde and the diamine (1,2-ethylenediamine (en) or (S,S)-NH2CHPhCHPhNH2 ((S,S)-dpen)) to generate a kinetic product, a bis(tridentate) iron complex with two PNN ligands 2a or 3a. The kinetic product could then be converted to the thermodynamic tetradentate product 4a or 5a through reflux, or by increasing the polarity of the reaction by using a protic solvent (Fig. 5).
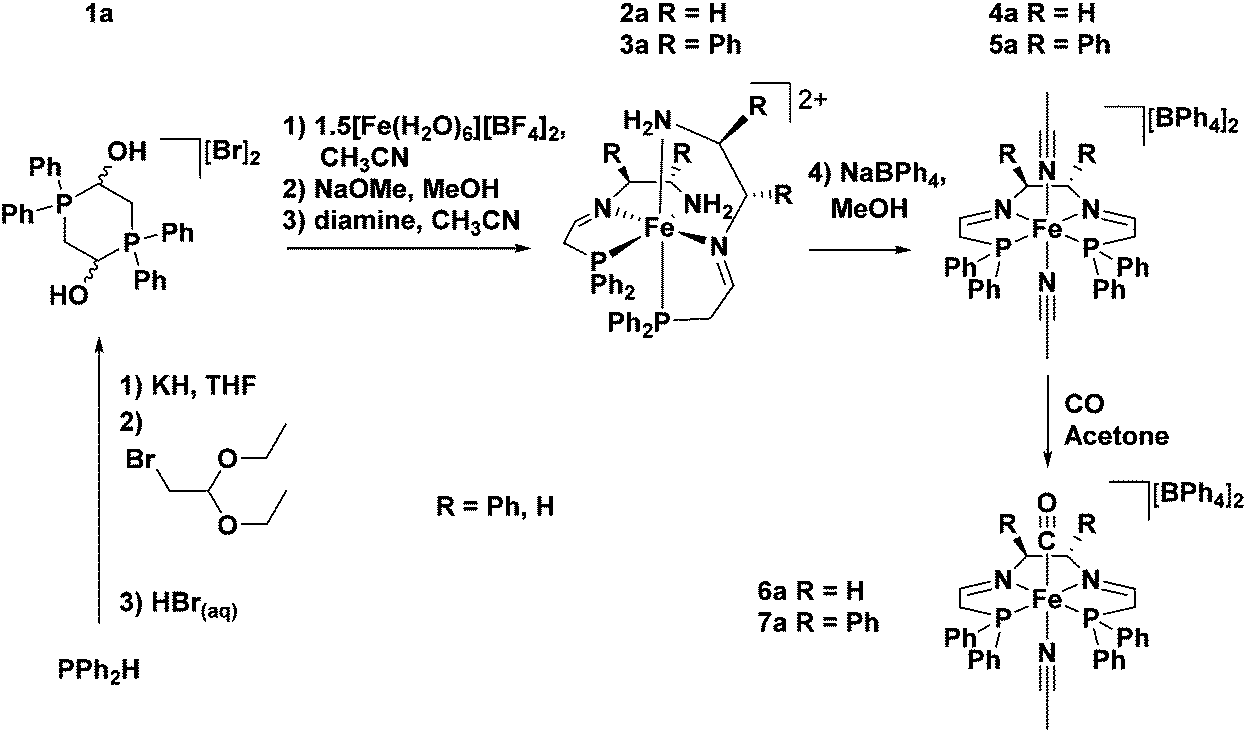 | ||
| Fig. 5 Synthesis of the phosphonium dimeric salt precursor, the intermediate with tridentate ligands and the bis(acetonitrile) and carbonyl complexes with a 5,5,5 tetradentate ligand.45 | ||
As seen previously for the 6,5,6-PNNP systems, the second generation 5,5,5-PNNP bis(acetonitrile) iron(II) complexes 4a and 5a were completely inactive for ATH.45 Replacement of one acetonitrile ligand with CO, however, gave precursors (6a and 7a) to extremely active ATH catalysts.46 This second generation of catalysts showed marked improvements over the original 6,5,6 systems in both activity (TOF of 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 at 28 °C) and selectivity (up to 82% enantioselectivity for acetophenone reduction to (S)-1-phenylethanol) when the (R,R)-catalyst 7a derived from the diamine (R,R)-dpen was used.46 It should be noted that for both generations of catalyst, the addition of base was needed for catalytic activity (Fig. 6).37,46
000 h−1 at 28 °C) and selectivity (up to 82% enantioselectivity for acetophenone reduction to (S)-1-phenylethanol) when the (R,R)-catalyst 7a derived from the diamine (R,R)-dpen was used.46 It should be noted that for both generations of catalyst, the addition of base was needed for catalytic activity (Fig. 6).37,46
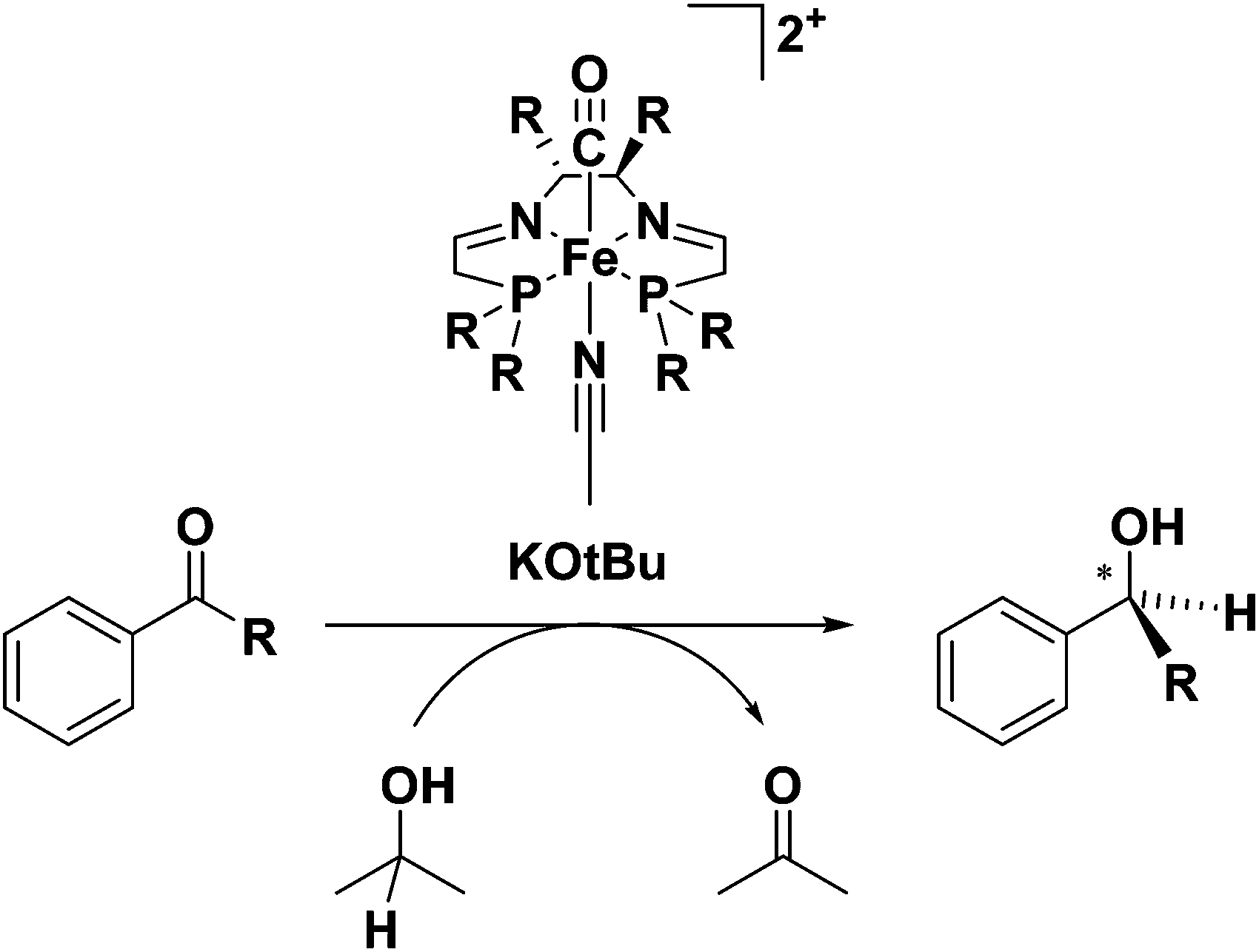 | ||
| Fig. 6 The ATH of ketones catalyzed by the second generation 5,5,5 systems 6a or 7a using iPrOH as the sacrificial reductant and solvent and KOtBu as the base.46 | ||
Development of the second generation 5,5,5 systems
Stereoelectronic effects
After the initial synthesis and screening of the 5,5,5 iron complexes in 2009, further modifications were made to the system to elucidate more about the catalytic mechanism, as well as to increase the activity and selectivity of the catalyst. It was thought that the role of the acetonitrile ligand was very minor in the catalytic cycle, and that its main function was to act as a labile ligand and decoordinate during precatalyst activation to generate a vacant site. This assumption was verified in that replacement of the acetonitrile ligand with a bromide did not impact the activity nor the selectivity of the 5,5,5 system (Table 1).47,48 Moreover, the iron complexes with a bromide trans to the carbonyl ligand were synthetically much easier and cheaper to make because isolation of the bis(acetonitrile) intermediate using salt metathesis was unnecessary, and fewer equivalents of NaBPh4 were needed to isolate the final monocationic product (Fig. 7).47,48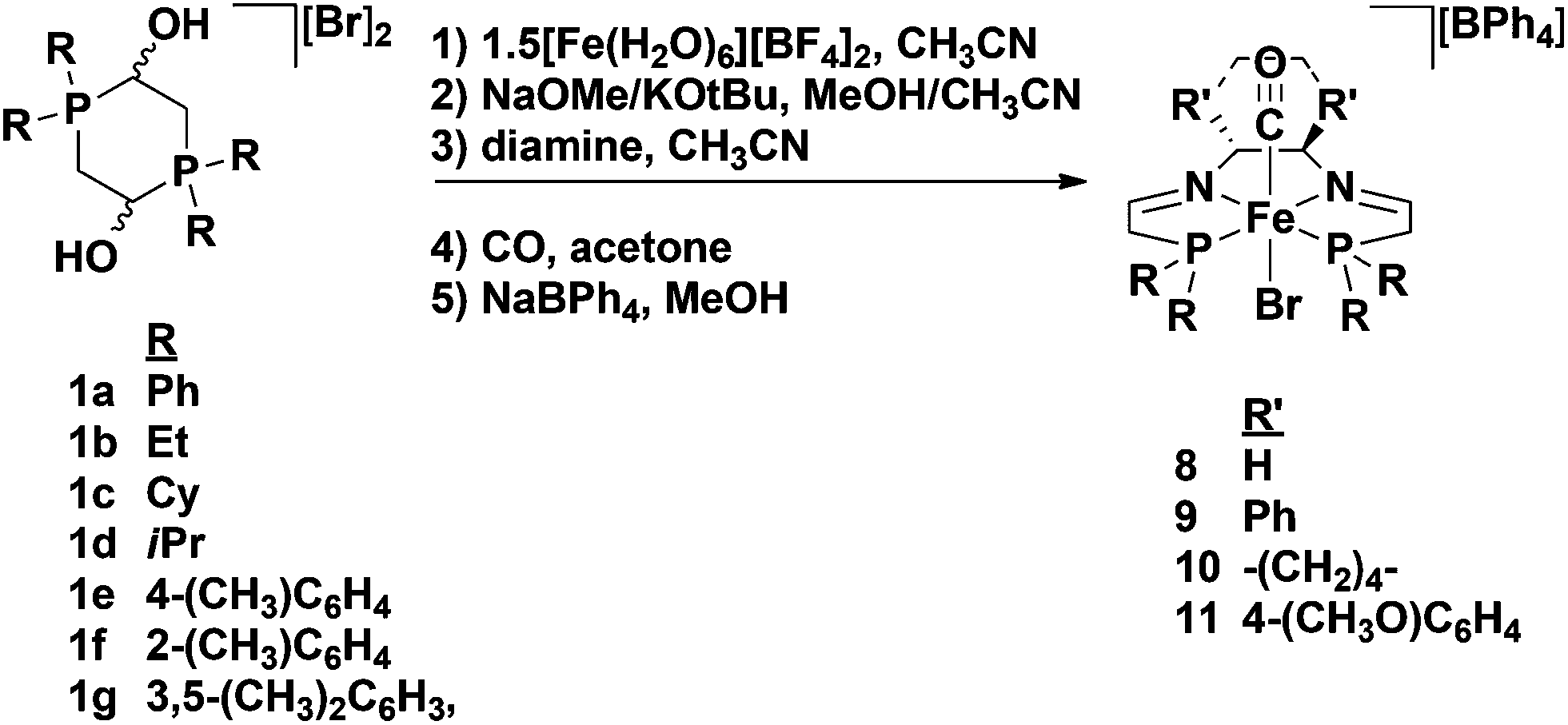 | ||
| Fig. 7 General synthesis of substituted 5,5,5 precatalysts with a bromide trans to the carbonyl ligand. The synthesis works for both the (R,R)-diamines (shown here) or the (S,S)-diamines. For the cyclohexyl and isopropyl analogues KOtBu and CH3CN were used in the second step; for all other substituents NaOMe and MeOH were used instead.47–49 | ||
|
|
|||
|---|---|---|---|
| Diamine precursor | Precatalyst structure | TOFa (h−1) | Enantiomeric excess (ee %) |
| a TOF is defined as the slope of the linear portion of the conversion (between 15% and 50%) plotted against time. The conversion was determined by GC after exposing the reaction solution to oxygen to destroy the catalyst. | |||
| (R,R)-dpen | (R,R)-7a | 21000 |
82 (S) |
| (R,R)-dpen | (R,R)-9a | 20000 |
81 (S) |
| (R,R)-dach | (R,R)-10a | 4300 | 63 (S) |
| en | 8a | 2100 | 0 |
| (R,R)-dpen-OMe | (R,R)-11a | 20000 |
82 (S) |
The impact of the diamine backbone on catalytic performance was also investigated. The synthesis of a variety of 5,5,5-PNNP carbonyl precatalysts with different NN linkers was facile due to the modular nature of the ligand framework. The range of structures possible was limited only by the number of easily accessible diamine precursors with the substituents R′ as listed in Fig. 7. The (R,R)-dpen backbone gave catalysts that were significantly more active and selective than those with the (R,R)-diaminocyclohexane (dach) or ethylenediamine (en) backbones (Table 1).47 Additionally, the electronic effects of the aryl groups on the diamine backbone were explored by comparing the activity of the dpen-derived complex (R,R)-9a with that of (R,R)-1,2-di(p-methoxyphenyl)ethylenediamine (dpen-OMe), complex 11a. The catalytic performance was unaffected by the change indicating that although the activity of the 5,5,5 systems for ATH was optimized with aromatic groups in the diamine backbone, the specific electronic characteristics of those aryl groups were not very important.47
Our group was also interested in varying the substituents on the phosphorus donors of the 5,5,5-PNNP ligand. Aliphatic groups were chosen in order to greatly increase the sigma donation from the phosphine arms and thus increase the electron density of the entire system. As such, phosphonium salt precursors with cyclohexyl, isopropyl, and ethyl substituents needed to be synthesized. The dialkylphosphine starting materials, however, could not be deprotonated with KH in the same way as diphenylphosphine. Due to the increased electron density on the phosphorus centres, the use of butyl lithium is usually necessary to generate dialkylphosphide nucleophiles. This same electron richness of the dialkylphosphines, on the other hand, makes the neutral species much more nucleophilic; so much so that they are strong enough nucleophiles to displace the bromide in bromoacetaldehyde diethyl acetal under reflux conditions thus avoiding the use of a lithium reagent (Fig. 8). In addition, because water instead of base was added to the reaction mixture, an acidic work-up was not needed to generate the phosphonium dimer salts 1b–d.50,51
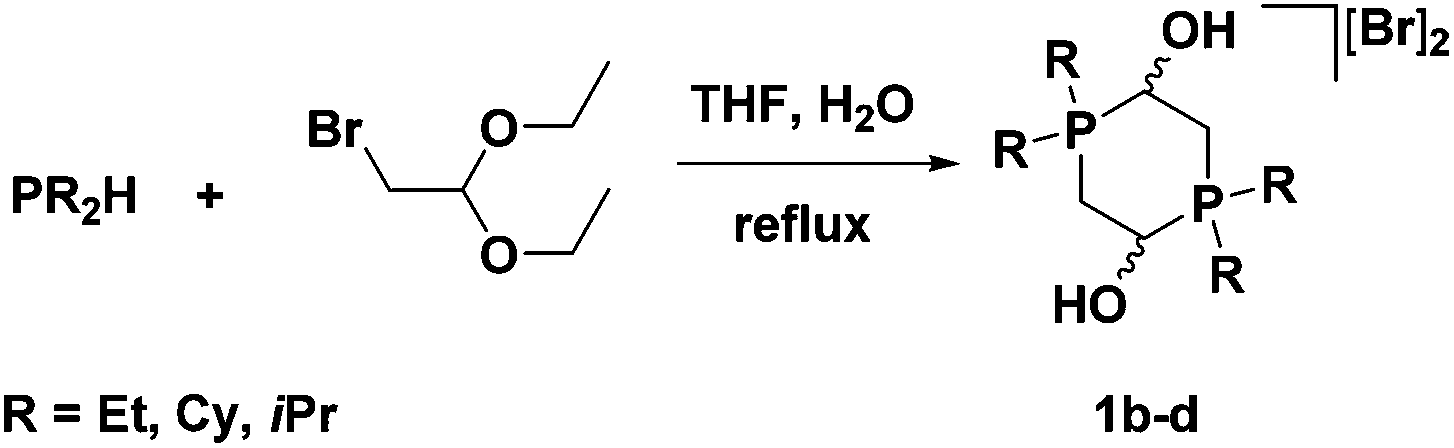 | ||
| Fig. 8 Synthesis of alkyl-substituted phosphonium salt precursors in THF at reflux in the presence of water.50,51 | ||
With the phosphonium salts readily available, the synthesis of the corresponding iron complexes was carried out. Much like the original phenyl-substituted species derived from 1a, the alkyl variants of the 5,5,5-PNNP ligand could only be generated in a one-pot template reaction in the presence of iron.48,51 Unlike the diphenylphosphino compounds, the cyclohexyl (1c) and isopropyl (1d) analogues gave the tetradentate complexes 5c and 5d directly without going through a bis(tridentate) intermediate analogous to 3a (Fig. 5), even in the absence of MeOH.48,51 In the case of ethyl substituents, on the other hand, the kinetic product 3b similar to 3a was seen, and either reflux in acetonitrile or the addition of MeOH was necessary to facilitate conversion to the desired 5,5,5-PNNP bis(acetonitrile) complex 5b.48 These observations suggested that the steric demands of the dialkylphosphino moieties played a role in determining the stability of the bis(tridentate) complex. With bulky cyclohexyl or isopropyl groups, the iron centre was too crowded to accommodate two PNN tridentate ligands, whereas with ethyl groups there was enough space. Therefore the bulkier groups likely initially formed a tridentate PNN tris-acetonitrile complex that quickly converted to the desired product, while the less sterically encumbered substituents formed the bis(tridentate) intermediate first.48
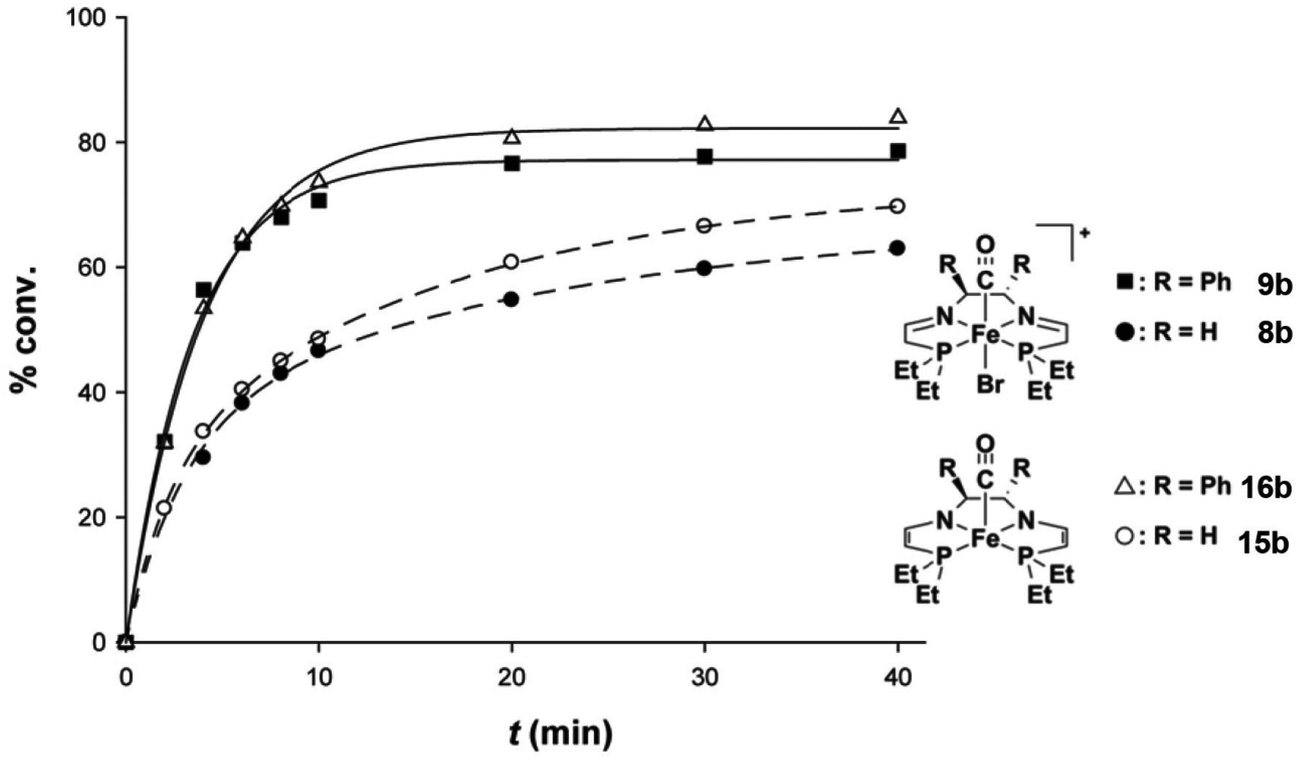
The bis(acetonitrile) alkyl complexes were not active catalysts for ATH or AH, as seen with the phenyl 5,5,5 system; therefore a ligand exchange reaction was performed to install a carbonyl ligand trans to a bromide on the metal centre in complexes 8b–d and 9b–d (Fig. 7).48 When the carbonyl complexes were tested for ATH, however, only the diethylphosphino-substituted variants 8b and 9b generated an active catalyst at elevated temperatures; the cyclohexyl (8c, 9c) and isopropyl (8d, 9d) analogues were completely inactive.48 In addition, 9b was significantly less active and selective than the diphenylphosphino catalyst 9a: TOF of 2400 h−1 at 50 °C and 47% enantioselectivity.48 Moreover, the catalyst system using the ethyl precatalyst 9b decomposed much more rapidly than the one from the phenyl precatalyst 9a, as shown by the fact that additional equivalents of catalyst were needed to push the ATH of acetophenone in isopropanol to equilibrium, whereas for the phenyl-substituted catalyst, additional substrate could be added to the reaction mixture once equilibrium was reached, and catalysis continued (Fig. 9).48
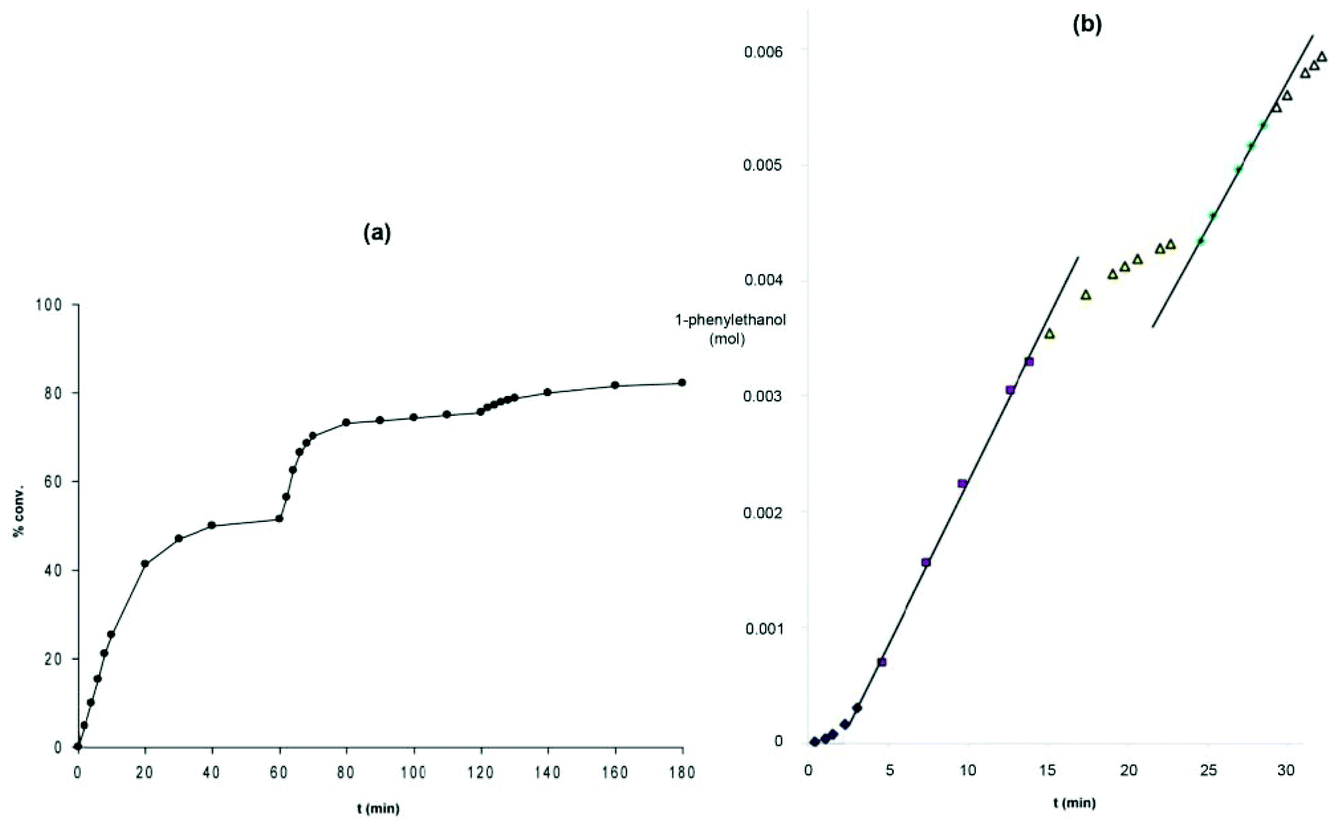 | ||
| Fig. 9 Reaction profiles for (a) the diethylphosphino-substituted catalyst (S,S)-9b and (b) the diphenylphosphino-substituted catalyst (R,R)-9a. The sharp increases in activity in (a) corresponds to the addition of more catalyst (S,S)-9b, while the second sharp increase in activity seen for (R,R)-9a represents the addition of more substrate. Adapted with permission from ref. 47 and 48. Copyright (2014) American Chemical Society. | ||
Although there seemed to be a correlation between the steric bulk at the phosphorus donors and catalyst activity, with respect to the alkyl-substituted 5,5,5 systems, the extreme electronic difference that accompanied replacing the phenyl groups also appeared to be a factor in the observed poor catalytic performance. In order to separate the steric parameters from the electronic parameters, a series of iron carbonyl complexes with aryl-substituted phosphorus donors were targeted. By utilizing the same electron-donating or electron-withdrawing groups in different positions on the aryl ring, (in this case in the ortho and para positions) the same electronic characteristics of the diarylphosphino functionalities would be maintained, while drastically altering the steric demands of the tetradentate ligand. In this way, our group endeavoured to increase the activity of our 5,5,5 systems while concurrently elucidating more about the catalytic mechanism. Moreover, Noyori and Ohkuma52 and our group53 reported an increase in selectivity for ruthenium based asymmetric hydrogenation catalysts when the phenyl groups of a bidentate phosphine ligand were replaced by m-xylyl groups, and as such, substitutions in the meta positions were also targeted in an attempt to increase the selectivity of the second generation catalysts.
The electron-donating aryl groups o-tolyl, p-tolyl, and m-xylyl were chosen as substituents on phosphorus for the investigation of their effect on catalysis. The syntheses of the corresponding phosphonium salts (1e–g) were straightforward and much like the original diphenylphosphino phosphonium dimer when employing the appropriate diarylphosphino starting material (Fig. 10, route (a)).49,50 A drawback of this conventional route for synthesizing phosphonium dimers, however, is that it relies on the use of expensive, as well as highly air-sensitive phosphine precursors. The literature procedure for synthesizing the required electron-donating diarylphosphines involves the use of Grignard reagents and diethylphosphite to produce diarylphosphine oxides, which can, in turn, be reduced to diarylphosphines by diisobutylaluminum hydride (DIBAL).54 The reduction step in particular is extremely tedious and time-consuming, as well as low yielding in many cases. A more convenient method was therefore developed which bypassed the DIBAL reduction and generated the phosphonium salts from diarylphosphinoacetaldehyde diethyl acetals (Fig. 10, route (b)).49 This alternative method also had the added benefit of removing the bromide ions generated from the substitution reaction in a previous step, which allowed for the synthesis of the phosphonium dimers with any desired counter-ion.
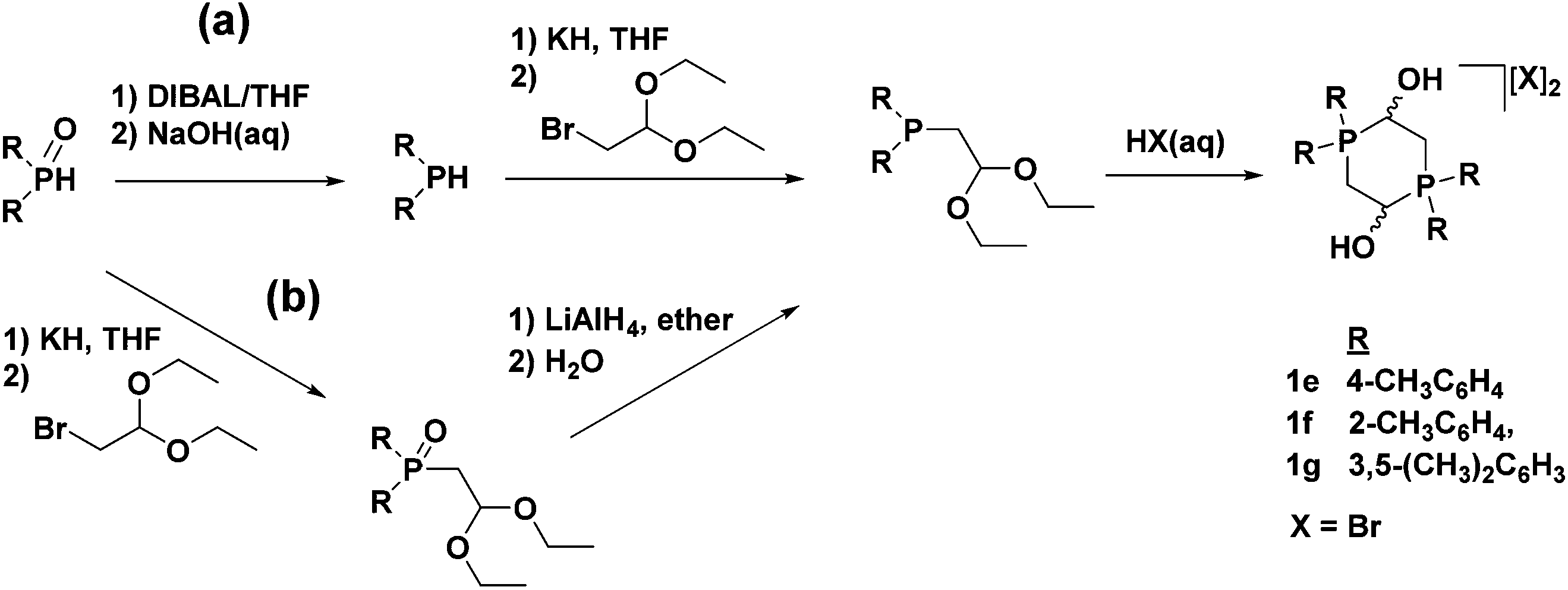 | ||
| Fig. 10 The conventional method (a) and alternative method (b) of synthesizing the electron-donating aryl-substituted phosphonium dimer salts. Compounds with X = BF4− were also made. Adapted with permission from ref. 49. Copyright (2014) American Chemical Society. | ||
With the phosphonium dimers in hand, a series of bis(acetonitrile) 5,5,5-PNNP complexes were synthesized utilizing (S,S)-dpen as the diamine backbone. The p-tolyl and m-xylyl variants behaved much like the parent phenyl 5,5,5 complex in that the templating reaction first gave bis(tridentate) complexes 3e and 3g, which then converted to the desired products in the presence of MeOH.49 The synthesis of the o-tolyl-substituted system, on the other hand, was more similar to that of the bulky alkyl-substituted complexes (cyclohexyl and isopropyl); the 5,5,5-PNNP product 5f was directly obtained and the kinetic product 3f was never observed.48 A ligand exchange reaction was then carried out to generate monocationic carbonyl complexes to test as ATH catalysts 9e–g (Fig. 7). A modest increase in catalytic activity was seen for the p-tolyl-substituted species 9e (TOF of 30000 h−1versus a TOF of 28000 h−1 for the parent diphenylphosphino analogue 9a under identical reaction conditions), while the m-xylyl version 9g was slightly slower (TOF of 26000 h−1 under identical reaction conditions).49 The m-xylyl catalyst also displayed an increase in selectivity over the original second generation system (90% enantioselectivity for the reduction of acetophenone versus an enantioselectivity of 82% (R) using (S,S)-9a).49 The o-tolyl-substituted carbonyl complex 9f, conversely, was completely inactive for ATH, thus supporting the hypothesis that increased steric bulk around the phosphorus donors completely shuts down catalytic activity.49 Moreover, this result ruled out de-coordination of a phosphine arm during catalysis in a Meerwein–Ponndorf–Verley type mechanism.
For electron-withdrawing aryl groups, p-trifluoromethylphenyl and 3,5-bis(trifluoromethyl)phenyl substituents were chosen for investigation, given that a substitution in the ortho position had already been determined to prevent catalysis. Attempted syntheses of phosphonium salt precursors for the trifluoromethyl-substituted aryl groups, however, invariably lead to multiple unidentifiable products. This was most likely due to the electron-withdrawing nature of the substituents which deactivated the lone pair of the phosphorus centre to such an extent that it was unable to attack an aldehyde after deprotection with acid (and thus prevented formation of a dimeric phosphonium species). Fortunately, the electron poor nature of the phosphine functionality also made it possible to isolate the protected form of the phosphine aldehyde 12h and 12i; the diarylphosphinoacetaldehyde diethyl acetal was only mildly air sensitive (Fig. 11).49
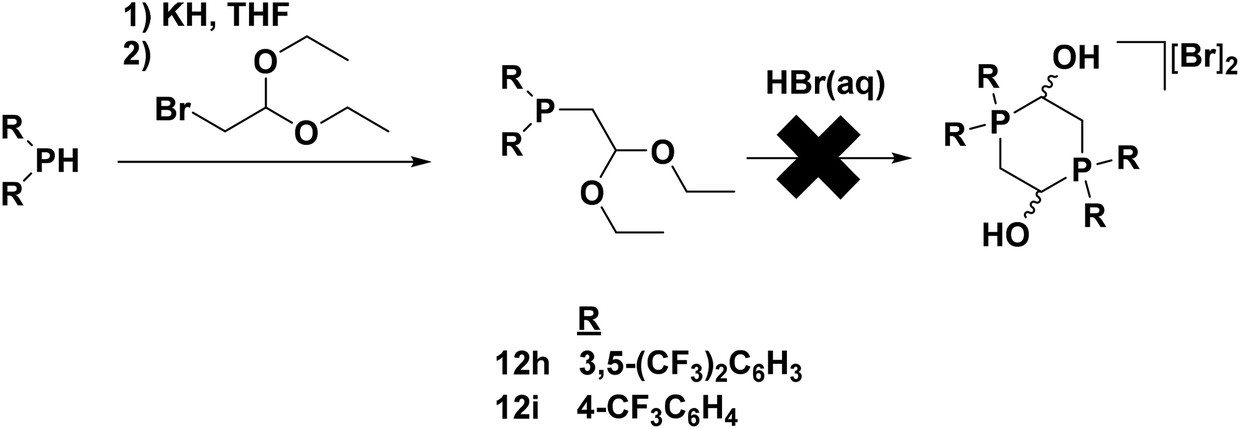 | ||
| Fig. 11 Synthesis of electron-withdrawing aryl-substituted phosphinoacetaldehyde diethyl acetal ligand precursors 12h and 12i. Adapted with permission from ref. 49. Copyright (2014) American Chemical Society. | ||
Unlike the phosphonium dimers, which are stable with respect to acid and are deprotected in the presence of base, diethyl acetals are stable with respect to base and are deprotected by acid. As such an acidic template procedure was developed in order to synthesize the 5,5,5-PNNP bis(acetonitrile) iron complexes 13h and 13i (Fig. 12).49 These compounds were predictably inactive for ATH, and therefore a ligand exchange reaction with KBr under a CO atmosphere was subsequently performed to remove the acetonitrile ligands and install a carbonyl ligand trans to a bromide in 14h and 14i (Fig. 12).49 The iron carbonyl species thus generated were tested for ATH, but they were found to be completely inactive for this transformation.
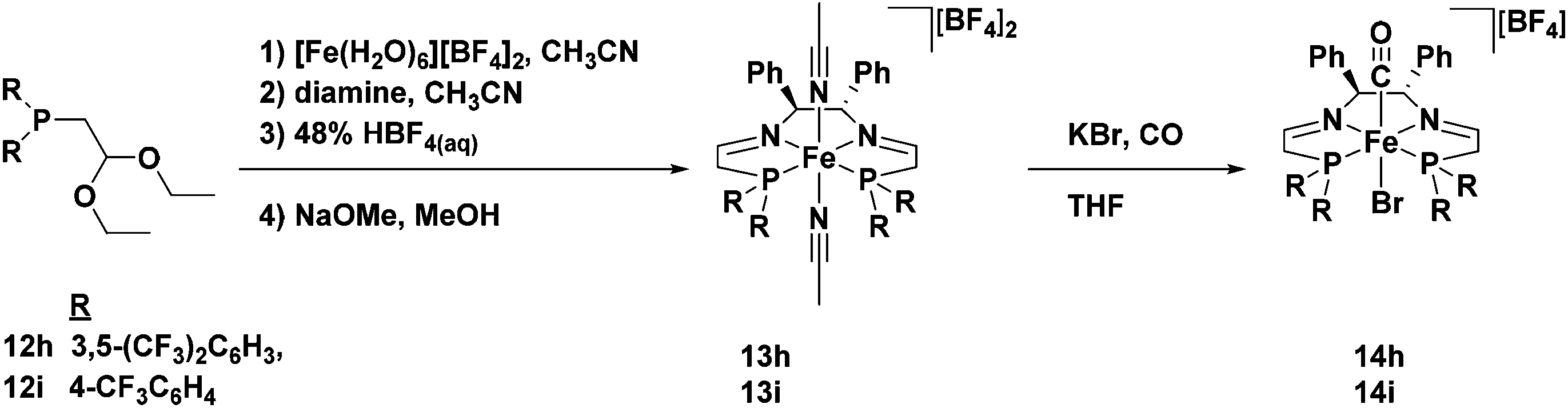 | ||
| Fig. 12 The acid-promoted template synthesis of 5,5,5-PNNP iron compounds with electron-withdrawing substituents on the phosphorus donors, as well as the ligand substitution reaction to generate carbonyl complexes. Adapted with permission from ref. 49. Copyright (2014) American Chemical Society. | ||
With all the various substitutions of the phosphorus donors for the second generation 5,5,5 systems that had been carried out by our group, it was observed that the iron catalysts required a delicate balance of electronic and steric factors in order to be active as well as selective for ATH reactions.49 If the phosphine arms were too electron-donating then catalytic activity dropped significantly. On the other hand, if the phosphorus functionalities were not donating enough then catalyst activity was completely shut down. Moreover, the catalyst had low activity with small ethyl groups and were completely inactive for sterically demanding substituents on phosphorus. This balance of stereoelectronic parameters could be visually depicted using a three-dimensional volcano plot, which compared catalyst activity to the Tolman cone angles of the phosphorus donors, as well as the v(CO) stretching frequency of the carbonyl precatalysts (Fig. 13).49 From the graph it is evident that a narrow region of electronic and steric parameters defines active catalysts, and that even relatively small deviations from these values is followed either by a large drop in activity or by complete catalyst deactivation.
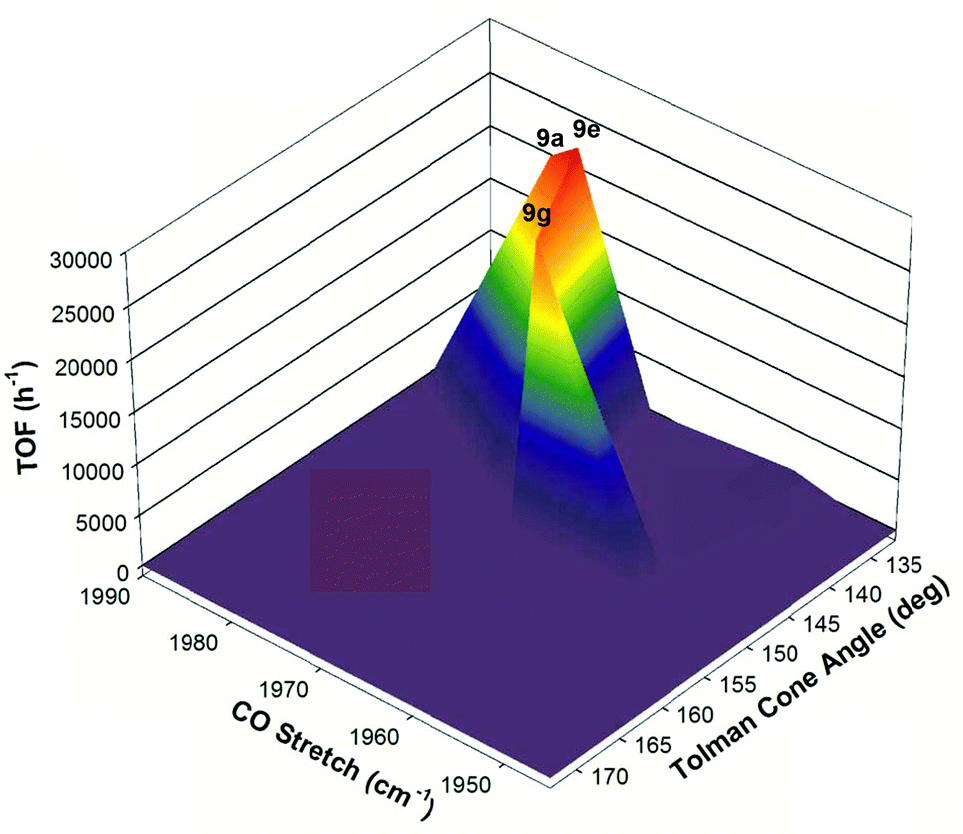 | ||
| Fig. 13 A three-dimensional volcano plot correlating the activity of the second generation 5,5,5 systems (TOF h−1) with respect to electronic (CO stretching frequency, cm−1) and steric (Tolman cone angles, deg.) parameters. Adapted with permission from ref. 49. Copyright (2014) American Chemical Society. | ||
Role of base in the activation of the iron carbonyl precatalysts
It should be noted that the iron carbonyl complexes reported by our group are not active for ATH by themselves; strong base, such as KOtBu, is needed to generate active catalytic species.34,35,37,43,46,48 This is in fact quite typical for ATH reactions and there are many similar reports in the literature.55 It is not always known why this is the case, but for bifunctional catalysts the base is commonly thought to deprotonate an amino group on the ligand and thus generate an amido intermediate which can then enter the catalytic cycle.10 For our 5,5,5 systems, on the other hand, the role of the base was not immediately apparent because there were no acidic amino groups to deprotonate in the ligand architecture. When conducting catalysis, however, upon addition of base to the catalytic mixture, a dramatic colour change from yellow to green was observed.56,57 Investigations into the nature of this green compound were therefore conducted in order to elucidate the role of base in activating the iron carbonyl species.The majority of the studies on the role of base were carried out on the alkyl-substituted analogues because of the relative stability of the corresponding deprotonated species.57 Initial studies were performed mimicking reaction conditions, in iPrOH, but without any ketone substrate. A deep green solution was obtained upon addition of KOtBu to complex 8c, but the compound was unstable and rapidly decomposed. The solvent was subsequently changed to benzene, which greatly improved the stability of the deprotonated species and lead to their isolation as a sticky green residue. The 1H NMR spectrum of the oily products indicated that a methylene proton alpha to each of the phosphorus donors had been removed, doubly-deprotonating the tetradentate ligand and generating two eneamido ligand arms in complex 15c (Fig. 14).57 This result was further supported by deuterium labelling by reaction of the green compound with DCl. This demonstrated that the only deuterated sites were the positions alpha to the phosphorus donors.57 Moreover, ESI+ MS studies indicated that the product of the deuteration had also incorporated a chloride ligand from the DCl.
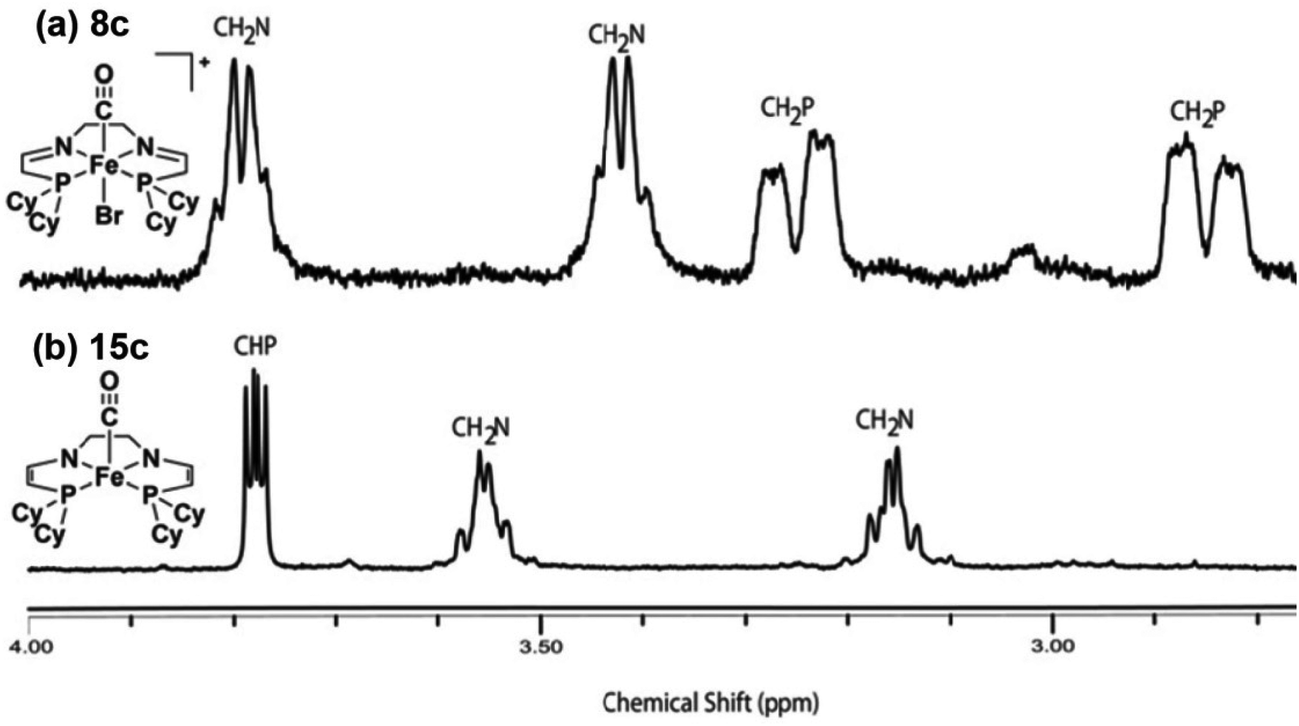 | ||
| Fig. 14 1H NMR spectra of the cyclohexyl-substituted iron carbonyl complex 8c (top) and the doubly-deprotonated eneamido species 15c (bottom). Adapted with permission from ref. 57. Copyright (2014) American Chemical Society. | ||
Definitive proof of the exact structure of the doubly-deprotonated species was obtained via X-ray diffraction studies when crystals of an isopropyl analogue with a 1,2-diamino-benzene diamine backbone were grown from cold pentane. The molecular structure showed that the iron centre adopted a distorted square pyramidal geometry with the carbonyl ligand in the axial position trans to a vacant site (Fig. 15).57 The C–C bond lengths of the phosphine arms contracted upon deprotonation, whereas the C–N bond lengths increased, which suggested extensive delocalization over the entire ligand arm.57
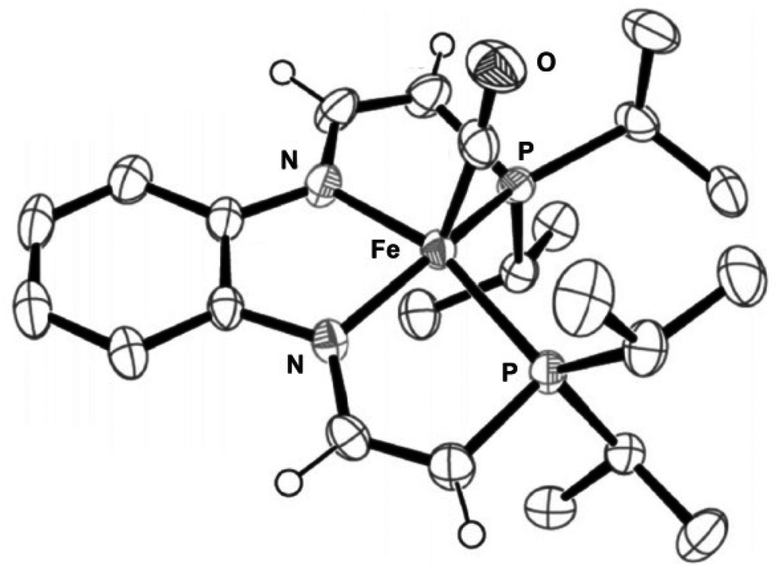 | ||
Fig. 15 ORTEP plot (thermal ellipsoids at 50% probability) of the crystallized doubly-deprotonated eneamido species Fe(CO){PiPr2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN(o-C6H4)NCHCHPiPr2} (most of the H atoms are removed for clarity). Adapted with permission from ref. 57. Copyright (2014) American Chemical Society. CHN(o-C6H4)NCHCHPiPr2} (most of the H atoms are removed for clarity). Adapted with permission from ref. 57. Copyright (2014) American Chemical Society. | ||
With the structure of the doubly-deprotonated species elucidated, ethyl-substituted green compounds 15b (derived from ethylenediamine) and 16b (derived from (S,S)-dpen) were tested for TH and ATH in the absence of added base. The isolated eneamido species 16b displayed catalytic activity and enantioselectivity (of 52% (R)-1-phenylethanol) similar to that of the in situ activation ATH procedure (addition of 8 equivalents of base to the iron carbonyl precatalyst 9b), indicating that the base is only needed to deprotonate the tetradentate ligand and does not play any further role during catalysis (Fig. 16).57 Moreover, this result also suggested that the eneamido species and the in situ activated precatalyst lead to the same active catalytic species. Unfortunately, attempts to hydrogenate the base sensitive substrate 4-acetylbenzoate ethyl ester using the eneamido species in the absence of added KOtBu led to transesterification, demonstrating that although no extra base was needed for catalysis, the deprotonated complexes were basic enough to generate iPrO− in solution.57
 | ||
| Fig. 16 Reaction profiles for the ATH of acetophenone in iPrOH at 50 °C catalyzed by the in situ activated ethyl-substituted iron precatalyst (S,S)-9b (■, catalyst/base/substrate = 1/8/500), the isolated ethyl-substituted eneamido species (S,S)-16b (△, catalyst/substrate = 1/500), the in situ activated ethyl-substituted iron precatalyst 8b (●, catalyst/base/substrate = 1/8/200), and the isolated ethyl-substituted eneamido species 15b (○, catalyst/substrate = 1/200). Adapted with permission from ref. 57. Copyright (2014) American Chemical Society. | ||
Following the characterization of the alkyl-substituted eneamido species, identical deprotonation studies were carried out on the original 5,5,5 system (S,S)-9a. Although the phenyl variants were significantly more sensitive, they could be partially characterized by 1H and 31P NMR spectroscopy.56 The results showed that deprotonation occurred in exactly same position as the alkyl analogues and that eneamido species were being formed. In addition, the isolated eneamido species could be used for ATH in the absence of additional base with similar activity and selectivity as the in situ activated precatalyst, but the induction period evident for the original second generation catalyst was prolonged when the additional equivalents of base were not used.56
Kinetic and mechanistic investigations
One of the most intriguing observations made about the second generation 5,5,5 precatalysts, with respect to catalysis, was that the reaction profile for ATH displayed a marked induction period, followed by rapid catalysis and then a rapid decrease in activity as the system reached a plateau (Fig. 17).47,56 Such sigmoidal reaction profiles often indicate the presence of heterogeneous catalysis,58 but nanoparticles were ruled out early on in mechanistic investigations via electron microscopy and poisoning studies.43 In this case, the induction period is representative of a high barrier activation process which occurs throughout catalysis. The steep linear portion of the curve is indicative of rapid catalysis, but because the activity does not decay exponentially, the amount of new catalyst being activated must counterbalance the decrease in the concentration of substrate. Lastly, the sharp decline and plateau at the end of catalysis is typical for ATH as the system approaches equilibrium (ATH is an equilibrium process driven by the large excess of iPrOH).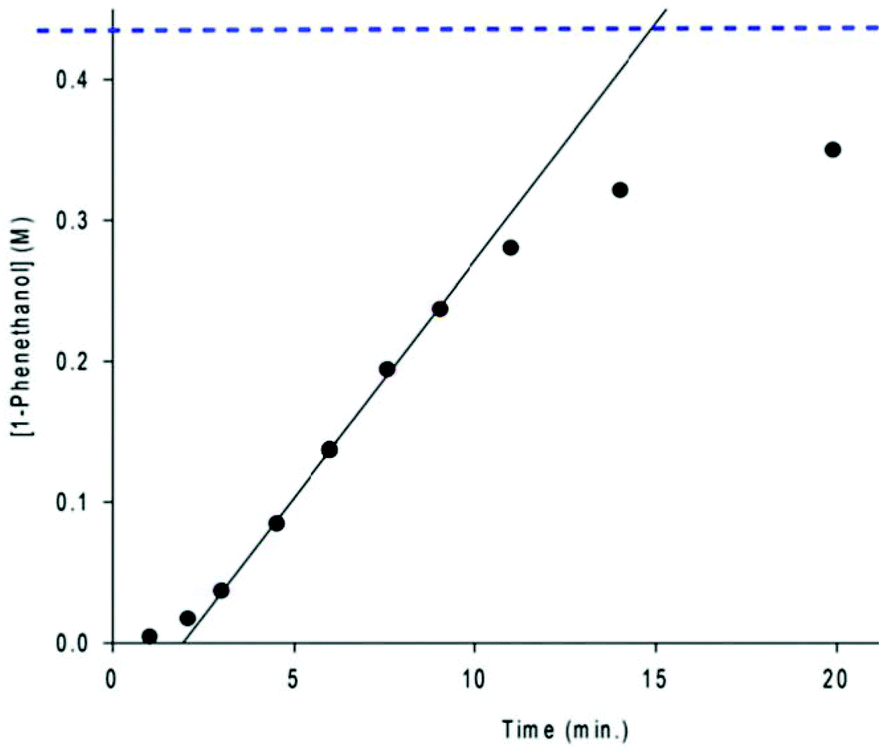 | ||
| Fig. 17 A typical reaction profile for the ATH of acetophenone using the precatalyst (R,R)-9a or (S,S)-9a. The dotted blue line represents the equilibrium concentration of 1-phenethanol.56 | ||
In order to obtain more detailed information on the activation process as well as the catalytic mechanism, a detailed kinetic analysis was conducted on the second generation catalyst (R,R)-9a using acetophenone as the model substrate. Using a maximum rate method for determining rate laws (a kinetic method using initial rates would not be suitable given the long induction period) a systematic variation of the reaction conditions was carried out.56 It was determined that there was a positive correlation between rate and the concentration of precatalyst, but the dependence was not linear. With respect to the amount of time required for activation, on the other hand, there was an inverse relationship versus the concentration of precatalyst when the amount of base was in excess. When the concentration of precatalyst was increased beyond a certain point, however, the activation time became almost independent of the precatalyst concentration and the base became the rate-limiting reagent. Attempts to activate the precatalyst before the addition of substrate revealed that although the catalyst could be activated in the presence of just basic iPrOH, the induction period was shortened if the precatalyst was treated with base in iPrOH for several minutes with similar activity to that of a standard run, but if the precatalyst was left in basic iPrOH too long then the catalyst decomposed.56 These observations showed that the activation period required the reaction of the precatalyst with basic iPrOH, but in the absence of ketone, the catalytic species decomposed under strongly basic conditions.
The concentration of acetophenone was also varied with respect to the reaction conditions during the kinetic investigations.56 It was discovered that there was a positive rate dependence on the concentration of substrate, while the induction period, conversely, was lengthened with increased ketone concentration indicating that it interfered with the activation of the catalyst. The interference of the ketone substrate on the activation process can be attributed to the formation and coordination of an enolate form of acetophenone, a result that was supported by previous experiments where benzophenone, a non-enolizable ketone, was used as the ketone substrate.49 This hypothesis was further tested through the addition of acetone, a byproduct of ATH in iPrOH, to the catalytic mixture. This ketone can also form an enolate, and it was found that it inhibited the activation step as well.
The effect of base on the maximum rate of catalysis was explored as well as its effect on the activation period. As long as the concentration of base was above a threshold value, 6 equiv. of base per iron carbonyl precatalyst, the rate of catalysis was independent of the base concentration.56 The activation period, on the other hand, was inversely proportional to the amount of base, indicating that the more basic the solution the faster the catalyst was activated.
As mentioned previously, the initial formation of a bis(eneamido) species (15, 16) was discovered upon reaction of the precatalyst with base, but this was shown to need further activation to become catalytically active. If the bis(eneamido) complex was pretreated with iPrOH before the addition of substrate, then the induction period was not seen, but if the bis(eneamido) was treated with substrate before the addition of iPrOH, on the other hand, then the induction period was more pronounced (thus supporting enolate inhibition of the activation process).56 Initially, it was thought that reduction of the tetradentate ligand imine functionalities to produce two amino groups was a plausible activation pathway for these systems, as was seen for related ruthenium ATH catalysts with 5,5,5-PNNP ligands. An independently synthesized iron carbonyl complex with two amino groups, however, was shown to be significantly less active.56 A solution of the bis(eneamido) species which had reacted with iPrOH for 20 min provided a clue to the structure of the active catalyst. The reaction mixture was quenched with HCl in ether, and yellow solid was isolated. Spectroscopic studies indicated that the tetradentate ligand had been partially reduced and contained both imine and amine functionalities.56 When the partially reduced iron carbonyl complex was used as a precatalyst and treated with base, no induction period was seen and catalysis began immediately.56 The conversion versus time plot for the catalytic reduction of acetophenone had a shape similar to that of the (S,S)-9a system but there was no induction period and the initial slope was much greater (TOF of 55000 h−1).56 In addition the enantioselectivity in the reduction of acetophenone (82% (R)) was the same as that of the (S,S)-9a system. It was hypothesized that the partially reduced species rapidly reacts with base to produce an amido eneamido species, which can then enter the catalytic cycle.
With this information in hand along with DFT calculations as discussed later, a kinetic model was developed for the system that treated the activation and ATH steps separately.56 The overall scheme of the process modelled is shown in Fig. 18.56 In the activation step, the carbonyl precatalyst reacts with base and forms a set of rapid equilibria between the neutral bis(eneamido) complex 16a, the cationic eneamido complex 17a, and two enolate complexes (from acetone and the ketone substrate). Only the reaction of 17a with iPrO− in a slow activation step (kact, Fig. 18) leads to the catalytic cycle. Once formed, the product amido eneamido species 18a reacts in the catalytic cycle by abstracting a proton and hydride from iPrOH in an outer sphere step-wise process. The resulting amino hydride complex 19a can then deliver the hydrogen equivalent to the substrate, once again in an outer-sphere step-wise process, thus completing the catalytic cycle generating the product alcohol and regenerating 18a.
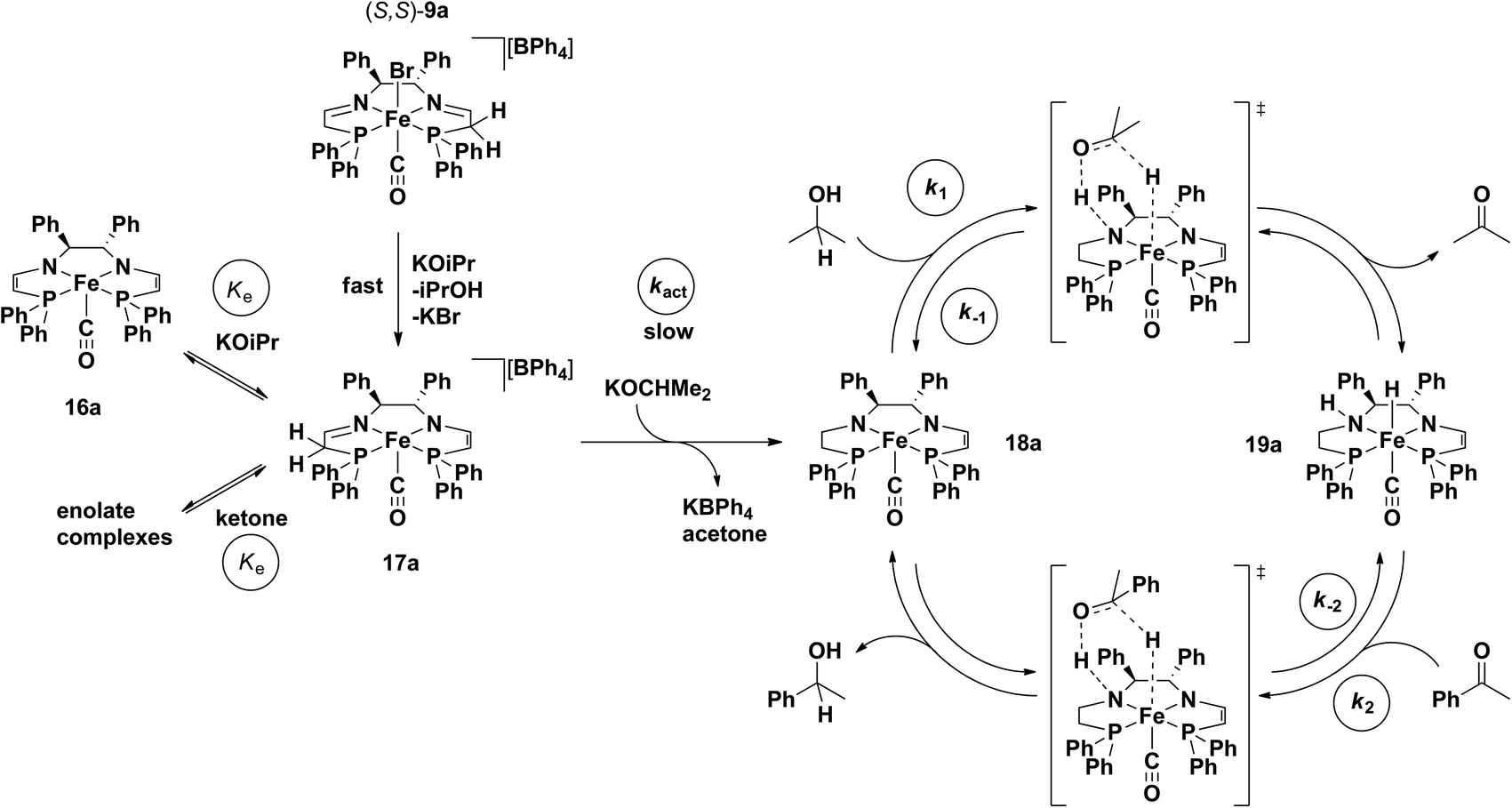 | ||
| Fig. 18 The proposed mechanism of action for the 5,5,5 systems, including the activation and propagation steps. Adapted with permission from ref. 56. Copyright (2014) American Chemical Society. | ||
In the context of the actual catalytic transformation, the rate of acetophenone reduction was dependent on the concentration of active catalyst, the concentration of substrate as well as the concentration of iPrOH. The actual catalytic cycle could be broken down into two dependent equilibria: the first where a postulated iron amido complex 18a with a saturated side arm reacts with iPrOH to give an intermediate species with a proposed iron hydride 19a as well as acetone (k1/k−1), and the second where the iron hydride reacts with the substrate acetophenone to generate the product alcohol and regenerate 18a (k2/k−2).56 Unfortunately, due to the fact that the activation process was relatively slow and the total amount of active iron species was constantly changing during catalysis, very few simplifying assumptions could be made and an in depth numerical simulation was needed to discern the rate laws from the kinetic data.
Using equations that can be derived from the kinetic model, the change in concentration of all the components in the catalytic mixture can be modelled over very small time intervals (Δt = 0.00025 s), assuming that the concentration of base and iPrOH are constant throughout the reaction and equal to their initial values.56 The experimental data (up to 18 catalytic runs) were thus simulated with a consistent set of estimated rate constants, which were determined through trial and error (Fig. 18).56 The extracted rate constants were the values that gave the best agreement with the observed reaction profiles (see Table 2, example in Fig. 19a).
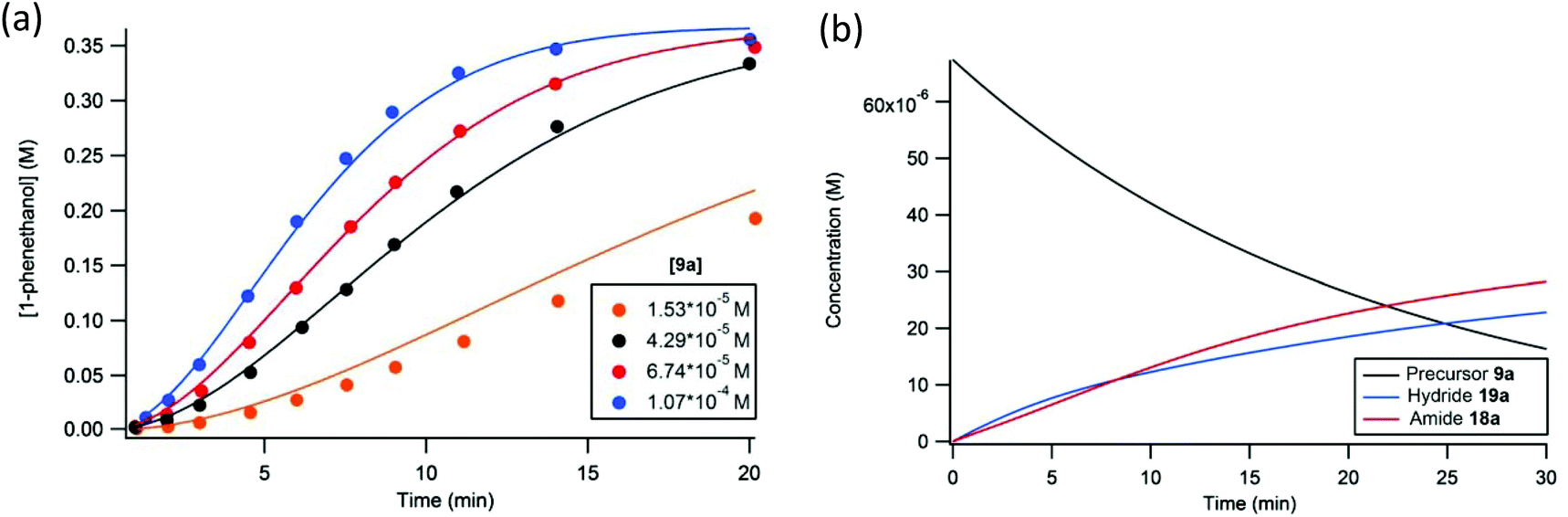 | ||
| Fig. 19 The simulated (solid line) and experimentally observed (circles) reaction profiles used to determine the rate constants for the proposed kinetic model. Plot (a) is representative of the kinetic data where the concentration precatalyst was varied. Plot (b) depicts the concentration of iron species in the catalytic mixture under standard conditions. Adapted with permission from ref. 56. Copyright (2014) American Chemical Society. | ||
| Rate constants | (M−1 min−1) |
|---|---|
| k 1 | 6.5 × 102 |
| k −1 | 2.8 × 104 |
| k 2 | 1.4 × 104 |
| k −2 | 1.4 × 103 |
| k act | 2.8 × 102 |
Another important result of this investigation was the confirmation that the unique features of the catalytic profile were consistent with a slowly activating catalyst that was constantly increasing in concentration throughout the reaction. This behaviour was modelled using a plot of the concentration of the precatalyst and active iron species in solution over time (Fig. 19b).56
In addition to varying the concentrations of the reaction components, the temperature dependence of the catalytic system was also explored. Due to the fact that the maximum rates of the reaction results from the combined effects of multiple rate constants (the activation process as well as the ATH steps), they could not be used to determine activation energy for the rate determining step. Therefore, in much the same way as the previous investigations, the experimentally determined reaction profiles were simulated and the rate constants extracted from the data (Fig. 20a).56 Subsequently, the various rate constants thus obtained were then used to calculate the activation energies of the various steps (Fig. 20b).56 Kinetic isotope experiments were also conducted and showed that a hydride transfer was likely involved in the rate limiting step for both the activation as well as the ATH steps.56 Thus the use of (CD3)2CDOD resulted in a retardation of the rate representing a kinetic isotope effect (KIE) of 2.5 ± 0.1 whereas the use of (CH3)2CHOD gave a KIE of 1.3 ± 0.1.
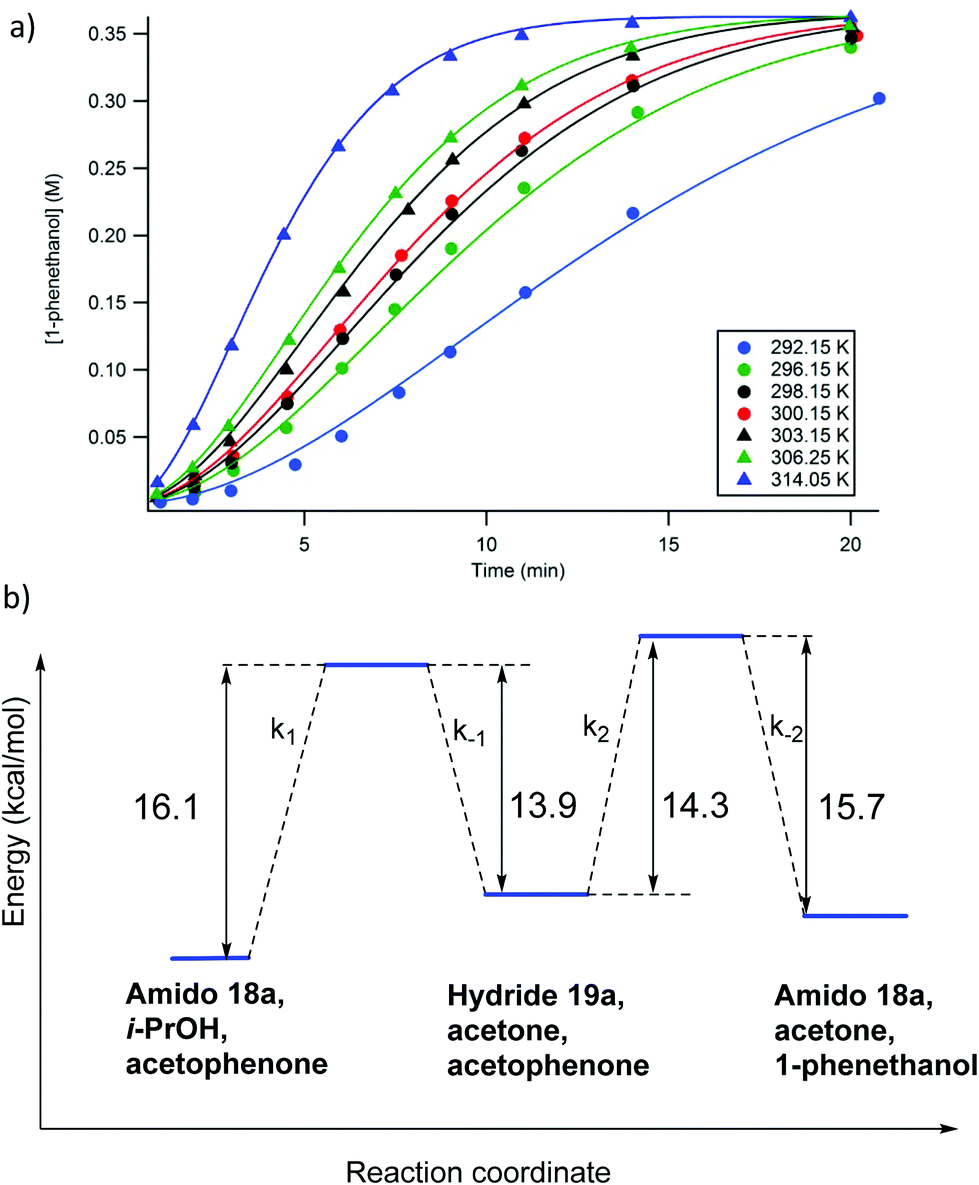 | ||
| Fig. 20 (a) The simulated (solid line) and experimentally observed (circles) reaction profiles used to determine the rate constants for the proposed kinetic model (Fig. 18) at various temperatures; and (b) the calculated activation barriers for the catalytic steps. Adapted with permission from ref. 56. Copyright (2014) American Chemical Society. | ||
While the kinetic studies and experimental mechanistic investigations were being conducted, concurrent DFT calculations were performed to elucidate the mechanism of activation and catalysis for the second generation iron carbonyl catalysts. An additional objective was to determine whether outer-sphere hydride and proton transfer between the iron and the substrates occurs in a concerted or stepwise fashion. DFT calculations can more easily distinguish between these pathways than experiment.
With respect to the activation period, the neutral bis(eneamido) structure (20) was used as a starting/reference point, and the phenyl rings on the diamine backbone as well as the phosphorus donors were replaced by hydrogens to give practical computation times. Various reactions of alcohol and substrate were found to be possible energetically including the enolate equilibria shown in Fig. 18, however only the pathway that leads to catalyst activation is discussed here. Isopropanol coordinates to iron (20iPrOH) and then protonates the carbon alpha to the phosphorus (Fig. 21). Although protonation at the amido group had a lower activation energy, the product alkoxide was significantly higher in energy, 9.2 kcal mol−1, than the alkoxide product from carbon protonation (alkoxide complex 21) at −2.4 kcal mol−1 relative to 20 and isopropanol, indicating that the first process most likely led to a kinetic product, while the second led to the thermodynamic product 21. The iPrO− ligand then transfers a hydride to reduce the imine moiety. The reduction activation barrier was quite high, 22.8 kcal mol−1, but not unreasonable for a room temperature process and the product, an amido eneamido species 22, was very low in energy, −8.1 kcal mol−1 compared to 20 and isopropanol (Fig. 21). Thus, the reduction of the imine functionality was found to be the rate-limiting step for the activation process, which was consistent with the experimental observation that the use of isopropanol-d8 as the solvent resulted in a much longer induction period before catalysis.
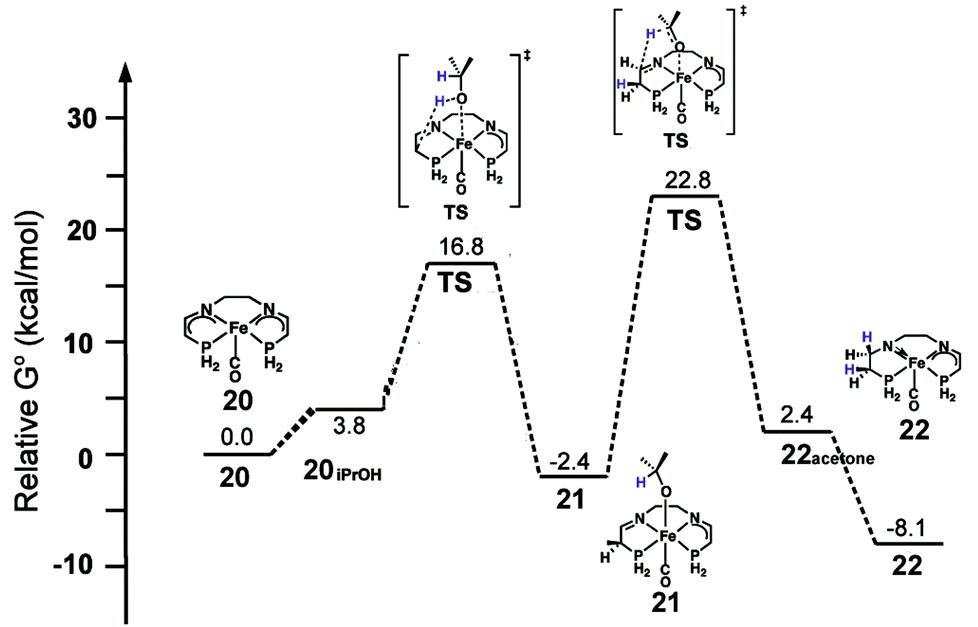 | ||
| Fig. 21 The energy profile for the activation of a simplified model of the 5,5,5 system. All energies are relative to the bis(eneamido) complex 20, iPrOH, and acetophenone. Adapted with permission from ref. 59. Copyright (2014) American Chemical Society. | ||
The catalytic mechanism for transfer hydrogenation was explored as well by DFT using the amido eneamido intermediate 22 calculated from the investigation of the activation process as a starting point, while still using 20 as a reference point for relative energies. Binding of iPrOH producing 22iPrOH caused a small increase in energy, up to −4.2 kcal mol−1, followed by a barrierless proton transfer from the alcohol to the amido nitrogen (Fig. 22). This generated a high energy intermediate at −1.9 kcal mol−1, an intimate ion-pair 24 between an iPrO− and the protonated eneamido complex. The proton transfer was then followed by hydride transfer from the alkoxide anion, which produced acetone and an amino iron hydride intermediate 25 with a relatively low barrier, −0.6 kcal mol−1. The hydride transfer was calculated to be the rate-limiting step of the entire catalytic cycle, which was consistent with the experimentally determined KIE values (2.5 ± 1 for kiPrOH/kiPrOH-d8 and 1.3 ± 1 for kiPrOH/kiPrOD), but was also found to be substantially lower in energy than the 22.8 kcal mol−1 needed for the activation process.56 It was crucial to use a solvent continuum model to locate the ion pairs and discrete proton/hydride transfer transition states. Then, in a very similar process, but in the reverse direction, acetophenone could be reduced to 1-phenethanol and 22 regenerated. It is interesting to note that the DFT calculations suggest that the bifunctional transfer of a proton and hydride to and from the iron complex and the reductant as well as the substrate occurs in a step-wise fashion.
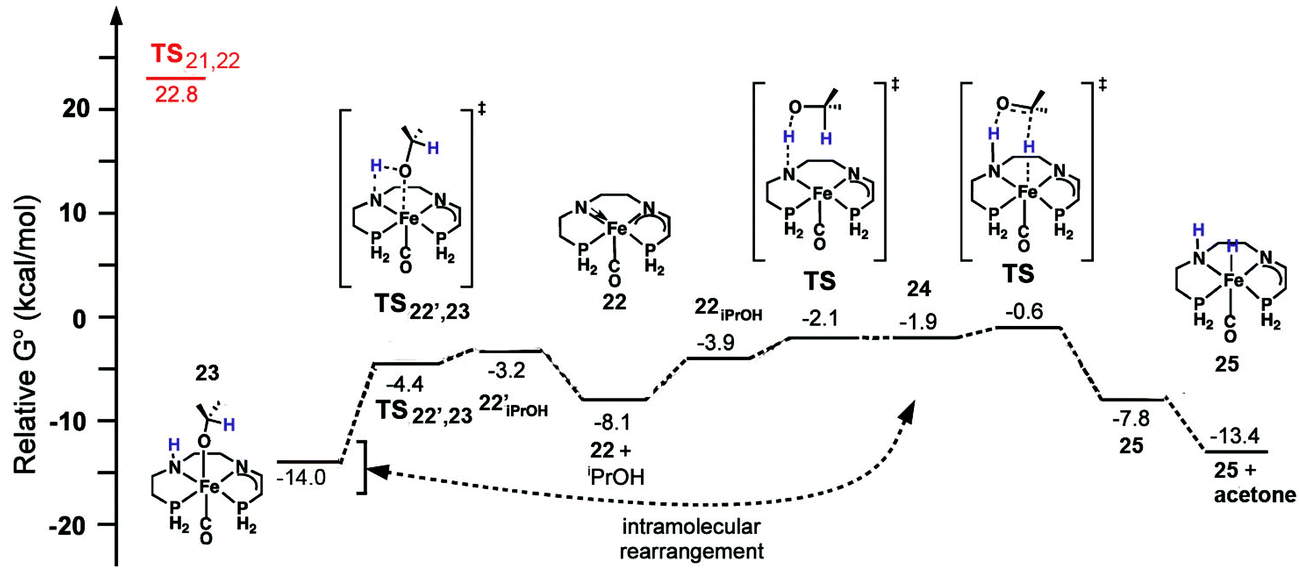 | ||
| Fig. 22 The energy profile for the oxidation of iPrOH as well as a possible pathway to an alkoxide resting state 23. All energies are relative to the bis(eneamido) complex 20 and iPrOH. The activation energy for the rate-limiting step of the activation process is shown at left at 22.8 kcal mol−1. Adapted with permission from ref. 59. Copyright (2014) American Chemical Society. | ||
Another possible pathway for protonation of the amido eneamido species 22 was also calculated, using both iPrOH and the 1-phenethanol product as the proton sources. Coordination of an alcohol caused a small increase in energy, −3.2 and −3.0 kcal mol−1 for iPrOH and 1-phenethanol, respectively, and was followed by a barrierless proton transfer to the amido nitrogen, producing the alkoxide adduct 23 in the case of iPrOH (Fig. 22). The corresponding alkoxide complexes were very low in energy, −14.0 and −16.0 kcal mol−1 for iPrOH and 1-phenethanol, respectively, and represented thermodynamic sinks for the catalytic cycle. The binding of the product alkoxide has significant implications for catalysis, because, as the reaction progresses, this equilibrium becomes more and more prevalent, potentially slowing down catalysis. The alkoxide complexes may not represent a deactivation pathway, however, as calculations have shown that these types of compounds can rearrange in a reversible process to give intimate ion pairs similar to the high energy intermediates discussed previously.60 In this way, these alkoxide species could act as an alternative, productive pathway in the catalytic cycle.
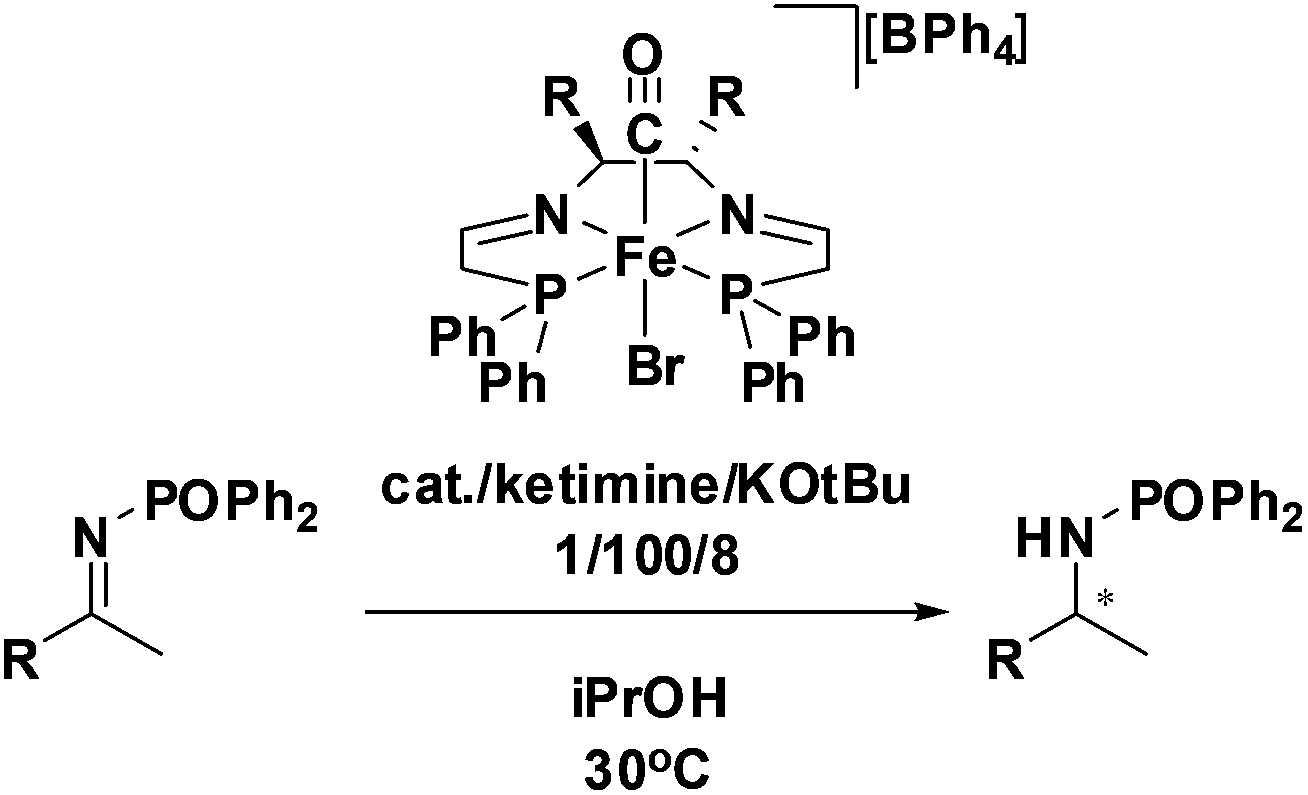
Ketimine hydrogenation
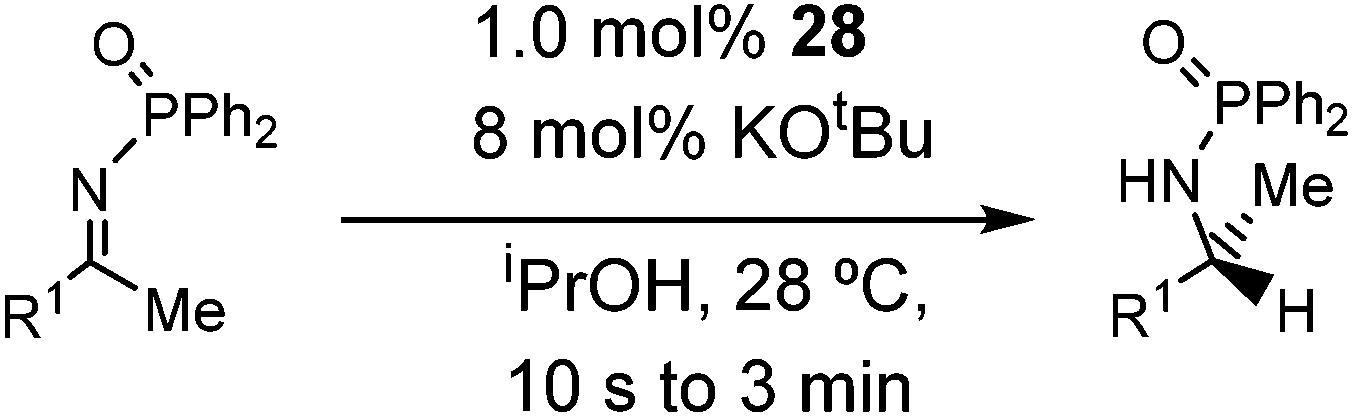
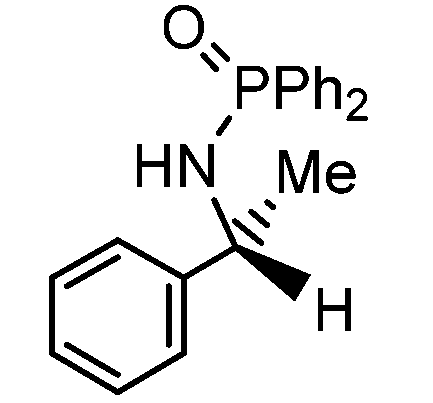
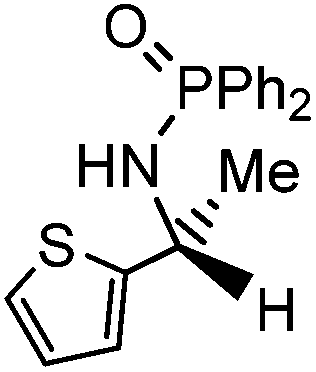
Initial attempts at reducing aryl- and alkyl-substituted ketimines using our catalysts were not successful. However in 2010 Beller and coworkers reported the ATH of activated imines using an iron carbonyl precursor and the tetradentate ligand employed in the first generation 6,5,6 systems.61 This work encouraged our group to try these substrates as well using our second generation 5,5,5 systems. Under similar conditions to analogous ketone reductions, a wide variety of imine substrates with an activating diphenylphosphoryl group were tested (see Table 3).62 The second generation catalysts were extremely active (TOF up to 150 h−1), and exceptionally selective (greater than 99% enantioselectivity).62 In addition, the iron catalysts were not poisoned by imines containing heteroaromatic groups, but were significantly slower for bulky or cyclic imines.62 Furthermore, the 5,5,5 systems were much less active for p-toluenesulfonyl activating groups on the imine substrates, and completely inactive for phenyl, benzyl and acetyl activating groups.62 Nevertheless, a wide variety of imines could be reduced and later hydrolyzed to give chiral amines.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (min) | Cat. [mol%] | Conv. [%] | ee [%] |
| 1 |
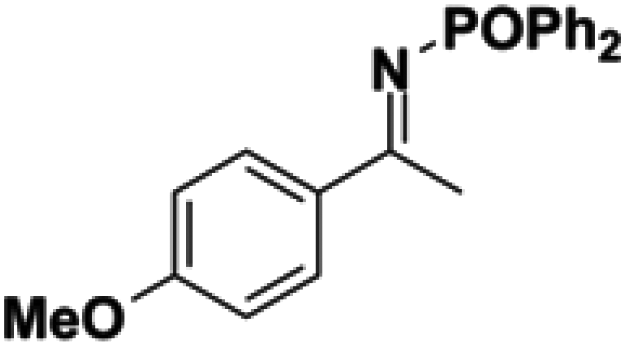
|
40 | 1 | 91 | 98 |
| 2 |
|
40 | 1 | 92 | 95 |
| 3 |
|
40 | 1 | 91 | >99 |
| 4 |
|
60 | 2 | 83 | 98 |
| 5 |
|
60 | 2 | 80 | 97 |
| 6 |
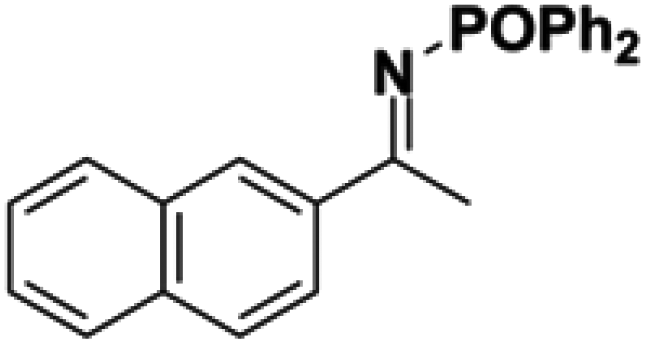
|
60 | 1 | 90 | 98 |
Development of the third generation amine–imine 5,5,5 system
Amine–imine ligand and iron precatalyst synthesis
Our DFT studies suggested that the rate-limiting step to form the active catalyst was the reduction of a single imine functionality of the 5,5,5-PNNP ligand by a molecule of isopropanol.59 This mechanism was supported by the isolation of a yellow solid, spectroscopically determined to be a partially reduced amine–imine diphosphine complex.56 When this complex was preactivated with base, we observed no induction period for ATH and a TOF of 55000 h−1, much higher than the second generation bis(imine)diphosphine iron carbonyl precatalyst.36 These results prompted the synthesis of an amine–imine ligand (P–N–NH–P), with the expectation that a more pure form of the amine–imine iron complex would be even more active.
As mentioned previously in the synthesis of the 5,5,5 precatalysts 8a and 9a (Fig. 5), we observed that a kinetic intermediate (2 or 3) with two tridentate P–N–NH2 ligands was formed initially before converting to the thermodynamic bis(acetonitrile)(PNNP) product.35,38 We therefore targeted the bis(tridentate) complexes as promising precursors for partially reduced tetradentate ligands. To begin, we treated the air- and water-stable phenyl (1a) or meta-xylyl (1g) phosphonium dimers with base in the presence of iron(II) and added an enantiopure diamine to produce the bis(tridentate ligand) complex containing two amine–imine–phosphine ligands as the chloride salts (26a or 26g, Fig. 23).63 This complex was then treated with lithium aluminum hydride to reduce the imine functionalities, and hydrolyzed to release the newly formed diamine–phosphine ligands in 90% purity (27a or 27b). Without further purification, the reduced tridentate ligand was mixed with half an equivalent of phosphonium dimer (1a, 1e or 1g) as well as base to produce tetradentate bis(acetonitrile)(P–N–NH–P)iron(II) compounds.63 These bis(acetonitrile) species were found to be inactive for ATH, and as such a ligand exchange was performed to generate the corresponding trans carbonyl-chloride complexes 28–30 (Fig. 23). This synthesis provides highly active precatalysts in acceptable yields.63 The single crystal X-ray diffraction structure of precatalyst 29 confirmed the presence of the amine and imine functionalities based on bond lengths, and also indicated that the amino proton and chloro ligand were located on opposite sides of the plane defined by the PNNHP ligand.63 Remarkably, 31P NMR indicated that only one diastereomer of the precatalyst was formed. It is important to note that this synthesis tolerated a large degree of variability, namely changes in the diamine backbone as well as both of the phosphine donor moieties (see the purple and green Ar groups in Fig. 23).
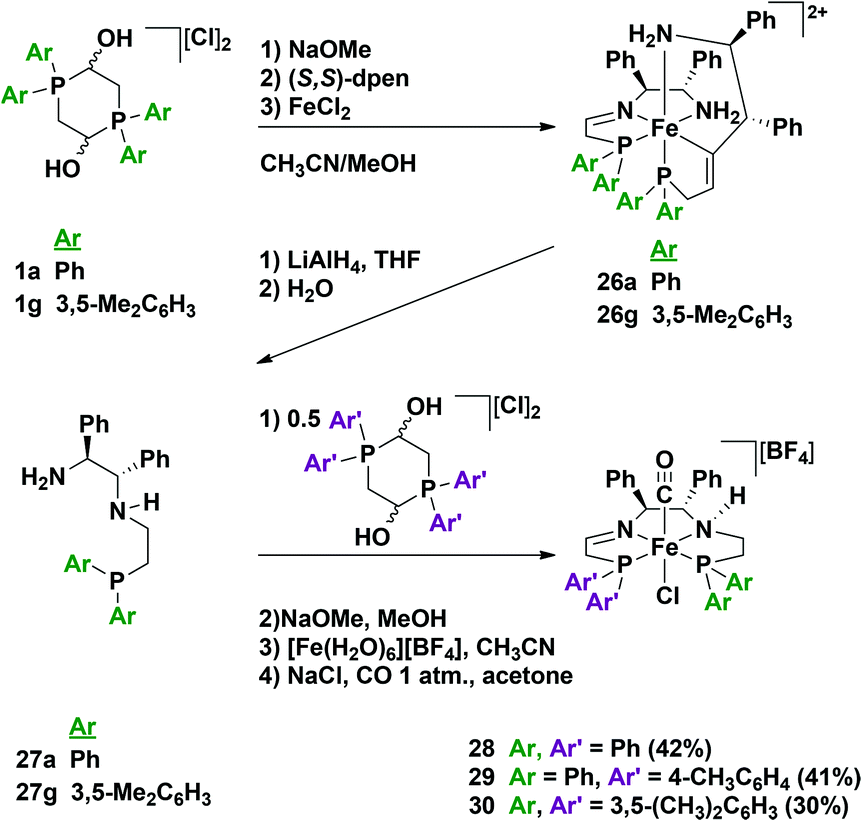 | ||
| Fig. 23 The reaction pathway for the formation of the amine–imine tetradentate ligands 27a and 27g as well as the third generation precatalysts 28–30. From ref. 63. Reprinted with permission from AAAS. | ||
Activity and scope
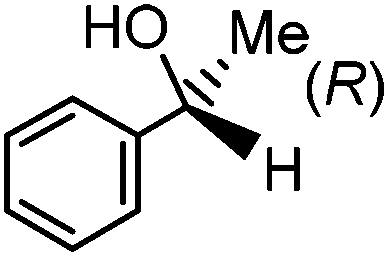
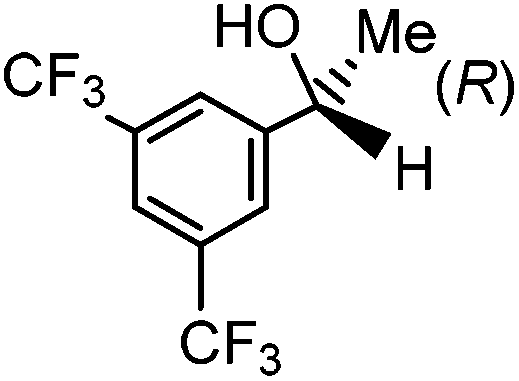
Having successfully synthesized the third generation precatalysts (28–30), we then investigated their activity towards a variety of ketone and imine substrates (Tables 4 and 5).63 The results indicated that the ATH of ketones gave moderate to high enantioselectivities (24–98%) with TOF as high as 242 s−1, while the ATH of imines yielded enantiopure amines (>99%), albeit with a lower TOF of 10 s−1. When the three isolated precatalysts were compared directly using the same conditions (acetophenone:KOtBu:precatalyst ratio = 6100:8:1), it was seen that the highest TOF of 152 s−1 at 28 °C was given by the para-tolyl complex 29, while complex 30 containing the xylyl group displayed the least drop in enantioselectivity over the course of the reaction.63 These highly active precatalysts surpassed the TOF demonstrated by rival ruthenium-based and osmium-based (R,S)-Josiphos systems, as well as the ruthenium-based P–NH–NH–P complex which obtained TOF of 89 s−1 and 92 s−1, respectively, at elevated temperatures of 60 °C.31,64 A substrate of particular interest is 3,5-bistrifluoromethylacetophenone, which was reduced with a TOF of 200 s−1 and an enantioselectivity of 98% when using precatalyst 30.63 The (R)-alcohol product is an intermediate for the synthesis of an efficient neurokinin antagonist used to combat nausea associated with cancer chemotherapy.65
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Product alcohols |
|
|
|
|
||||
|
a [Cat., 2b] = 6.73 × 10−5 M, [KOtBu] = 1.35 × 10−4 M, [substrate] = 0.412 M, [iPrOH] = 12.4 M, 28 °C.
b Ketone:cat. ratio = 2000:1 to prevent poisoning by the acidic alcohol product.
|
||||||||
| Precatalyst | 28 | 29 | 29 | 30 | 28 | 30 | 28 | 28 |
| Yield (%) | 82 | 83 | 83 | 83 | 99 | 99 | 98 | 100 |
| TON at equil. | 5000 | 5100 | 5100 | 5000 | 6060 | 2000 | 6000 | 6060 |
| TOF at 50% conv. | 119 s−1 | 152 s−1 | 12 s−1 | 70 s−1 | 147 s−1 | 200 s−1 | 100 s−1 | 242 s−1 |
| ee (S) at equil. | 78% | 70% | 80% | 90% | 90% | 98% | 24% | — |
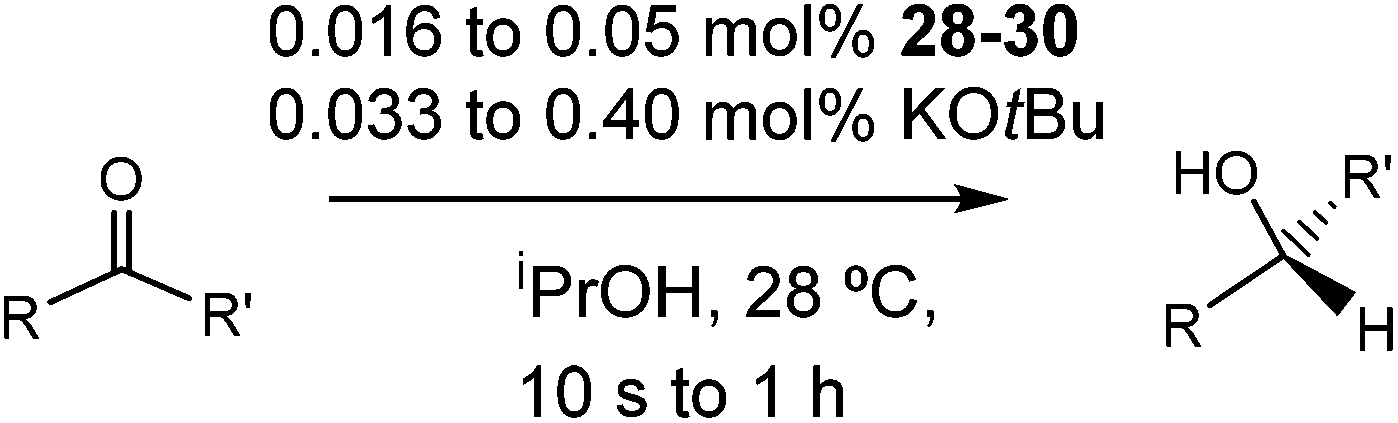
|
|
||
|---|---|---|
| Amine products |
|
|
| Yield: | 100% | 100% |
| TON at equil. | 100 | 100 |
| TOF at 50% conv. | 10 s−1 | 5 s−1 |
| ee (R) at equil. | >99% | >99% |
When the amount of base was reduced from eight to two equivalents (see 29a in Table 4), a dramatic decrease in TOF from 152 s−1 to 12 s−1 was observed. In comparison to previous generations, at least six equivalents of base were required to obtain maximum rates of catalysis for the 6,5,6 and 5,5,5 systems.23 Our group proposed that the additional base protected the amido and hydride reactants by decreasing the overall acidity of the alcohol medium.66,67
Verification of the proposed catalytic intermediates
Our DFT studies suggested that the bis(eneamido) complex 20 is partially reduced by a molecule of isopropanol to form the amido eneamido square-pyramidal iron(II) complex 22 (Fig. 21), featuring a carbonyl ligand in the apical position.59 Further mechanistic studies showed that 22 corresponds to complex 31 or 31′ which is generated quickly from the third generation precatalyst 28 by reaction with base (Fig. 24). Isopropanol can bind to this complex, triggering a proton transfer followed by a hydride transfer from the alkoxide anion in a stepwise fashion, to produce acetone and an amino hydride intermediate (32 in Fig. 24). In a similar manner, but in the reverse direction, acetophenone could be reduced to 1-phenethanol, while regenerating the amido eneamido iron(II) complex, thus completing the proposed catalytic cycle.63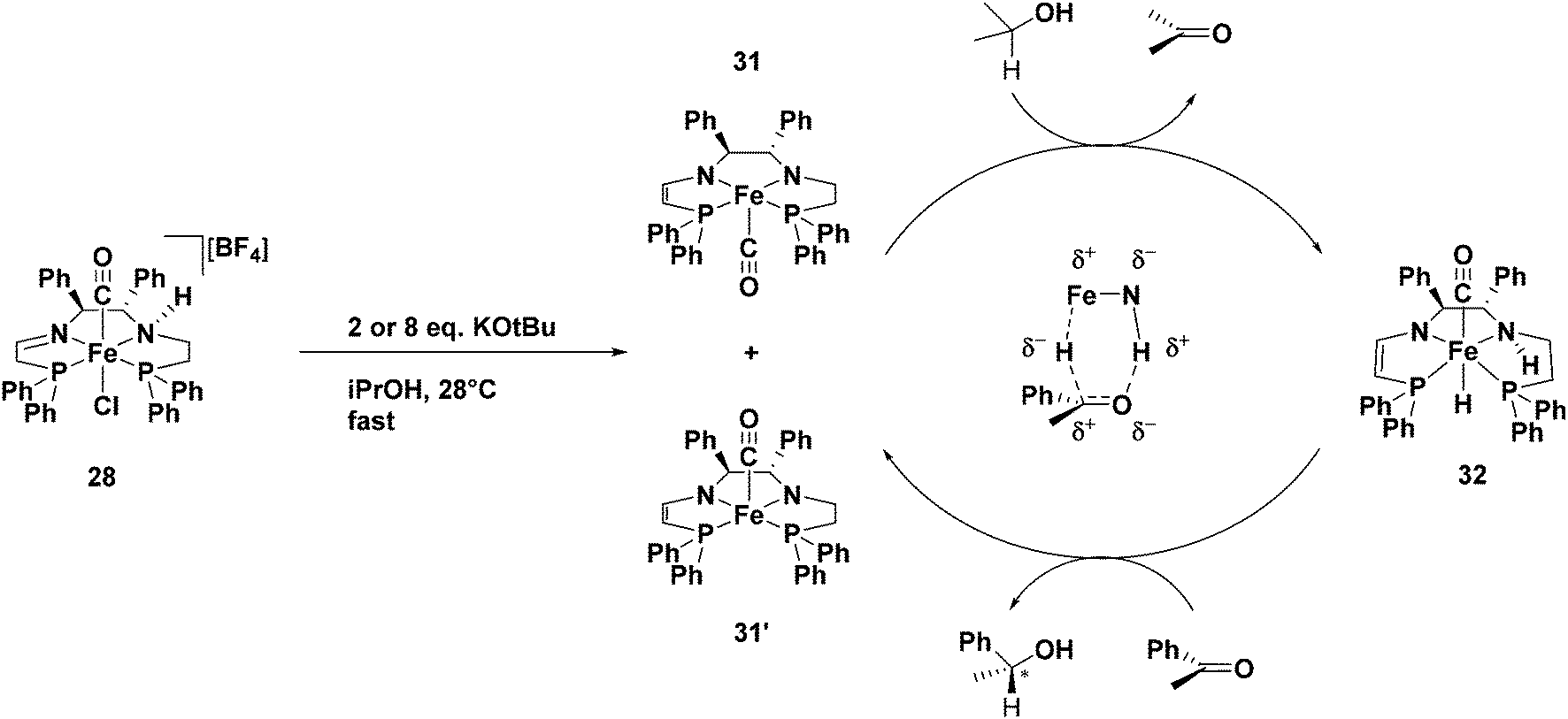 | ||
| Fig. 24 The proposed mechanism for the ATH of acetophenone using precatalyst 28. From ref. 63. Reprinted with permission from AAAS. | ||
To verify the formation of the amido enamido complex produced in the catalytic cycle, precatalyst 28 and two equivalents of KOtBu were stirred for several minutes in THF at room temperature prior to the evaporation of the solvent. The NMR spectra revealed the presence of a major and a minor diastereomer, consistent with either of the structures 31 or 31′ (Fig. 24). A mixture of these diastereomers were then dissolved in isopropanol and allowed to react without the addition of substrate. The depletion of complex 31′ with the rapid production of hydride complex 32, as well as the slightly delayed production of an isomer of complex 32, which was assumed to have the carbonyl ligand in the opposite apex of the octahedron, was seen (Fig. 24). It is important to note that the major isomer of 32 would have both the metal hydride and the amino proton below the plane formed by the tetradentate ligand in order to reduce the incoming ketone via proton and hydride transfer. As expected, when acetophenone was placed in a C6D6 solution containing 31, 31′, and 32 and the reaction was monitored by 1H NMR spectroscopy, the hydride signals of 32 disappeared alongside the appearance of the product (R)-1-phenylethanol. These results were fully consistent with the proposed mechanism in Fig. 24.63 The rigidity of the tetradentate PNNP ligands directly impacted the activity of the iron catalysts formed in situ by forcing the placement of the metal hydride trans to a carbonyl ligand, thus weakening the iron-hydride bond, and therefore activating the iron catalysts towards ketone hydrogenation.24,45,63
Conclusions and future outlook
In summary, we have developed three generations of iron catalysts bearing tetradentate PNNP ligands and explored their activity for the ATH of prochiral polar double bonds using mechanistic studies. The information gained from investigating each iteration of catalyst led to key insights that drove further rational catalyst design and, in the end, produced the most active system known to date. Although the development of the 5,5,5 ligand architecture was a large improvement upon the initial 6,5,6 iron catalysts, the most crucial breakthrough in our studies came with a better understanding of the catalyst activation process through experimental and computational methods. Partial reduction of the tetradentate ligand gave active catalytic species with amido and eneamido arms which could then reduce substrates through a bifunctional step-wise outer-sphere process. Independent syntheses of these partially reduced species yielded the third generation of iron catalysts which reached TOF of 242 s−1 and surpassed the activity of ruthenium-based ATH catalysts.1,63 Our iron-based catalysts, however, are significantly greener than their ruthenium counterparts and we hope that they will become commercially available in the near future. Our current focus now lies in adapting our iron systems for asymmetric hydrogenation reactions utilizing pressurized hydrogen gas, a process which is more amenable to large scale applications. With the knowledge gleaned from our ATH studies in hand, the development of a highly active and selective iron catalyst for the direct hydrogenation of prochiral substrates is an exciting possibility. Very recently reports in this area have appeared.68,69Acknowledgements
NSERC is thanked for a Discovery Grant to RHM. All of the members of the Morris group contributed to this work as cited.References
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703–1707 CrossRef CAS.
- K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- R. M. Bullock, Molybdenum and Tungsten Catalysts for Hydrogenation, Hydrosilylation and Hydrolysis, in Catalysis without Precious Metals, Wiley-VCH Verlag GmbH & Co., 2010, pp. 51–81 Search PubMed.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- R. Raja, J. M. Thomas, M. Xu, K. D. M. Harris, M. Greenhill-Hooper and K. Quill, Chem. Commun., 2006, 448–450 RSC.
- B. Pugin and H.-U. Blaser, Top. Catal., 2010, 53, 953–962 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS PubMed.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- Y.-Q. Wang, S.-M. Lu and Y.-G. Zhou, Org. Lett., 2005, 7, 3235–3238 CrossRef CAS PubMed.
- E. I. Solomon, A. Decker and N. Lehnert, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3589–3594 CrossRef CAS PubMed.
- B. Yu, W. C. Edstrom, J. Benach, Y. Hamuro, P. C. Weber, B. R. Gibney and J. F. Hunt, Nature, 2006, 439, 879–884 CrossRef CAS PubMed.
- Prices obtained from metalprices.com using Ruthenium Metal Powder and Scrap Iron from 2012.
- Data obtained from the U.S. Pharmacopeial Convention, “Elemental Impurities Limits and Elemental Impurities Procedures” published on February 1st, 2013. http://www.usp.org/usp-nf/official-text/revision-bulletins/elemental-impurities-limits-and-elemental-impurities-procedures-2010.
- R. R. Chowdhury, A. K. Crane, C. Fowler, P. Kwong and C. M. Kozak, Chem. Commun., 2008, 94–96 RSC.
- M. Haberberger, E. Irran and S. Enthaler, Eur. J. Inorg. Chem., 2011, 2011, 2797–2802 CrossRef CAS.
- A. Chanda, D.-L. Popescu, d. O. F. Tiago, E. L. Bominaar, A. D. Ryabov, E. Muenck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606–619 CrossRef CAS PubMed.
- C. Wang, D. Wang, F. Xu, B. Pan and B. Wan, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Stroemberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS.
- K. Abdur-Rashid, M. Faatz, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2001, 123, 7473–7474 CrossRef CAS.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- J.-X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087–1089 CrossRef CAS.
- K. Abdur-Rashid, A. J. Lough and R. H. Morris, Organometallics, 2000, 19, 2655–2657 CrossRef CAS.
- A. J. Lough, R. H. Morris, L. Ricciuto and T. Schleis, Inorg. Chim. Acta, 1998, 270, 238–246 CrossRef CAS.
- R. H. Morris, Can. J. Chem., 1996, 74, 1907–1915 CrossRef CAS.
- A. J. Lough, S. Park, R. Ramachandran and R. H. Morris, J. Am. Chem. Soc., 1994, 116, 8356–8357 CrossRef CAS.
- V. Rautenstrauch, X. Hoang-Cong, R. Churlaud, K. Abdur-Rashid and R. H. Morris, Chem. – Eur. J., 2003, 9, 4954–4967 CrossRef CAS PubMed.
- T. Li, R. Churlaud, A. J. Lough, K. Abdur-Rashid and R. H. Morris, Organometallics, 2004, 23, 6239–6247 CrossRef CAS.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940–943 CrossRef CAS PubMed.
- C. Sui-Seng, F. N. Haque, A. Hadzovic, A.-M. Putz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De Iuliis and R. H. Morris, Inorg. Chem., 2009, 48, 735–743 CrossRef CAS PubMed.
- J.-S. Chen, L.-L. Chen, Y. Xing, G. Chen, W.-Y. Shen, Z.-R. Dong, Y.-Y. Li and J.-X. Gao, Huaxue Xuebao, 2004, 62, 1745–1750 CAS.
- N. Meyer, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2009, 15, 5605–5610 CrossRef CAS PubMed.
- D. E. Prokopchuk, J. F. Sonnenberg, N. Meyer, M. Zimmer-De Iuliis, A. J. Lough and R. H. Morris, Organometallics, 2012, 31, 3056–3064 CrossRef CAS.
- R. H. Morris, Hydrogenases and model complexes, in Concepts and Models in Bioinorganic Chemistry, ed. H. B. Kraatz and N. Metzler-Nolte, Wiley-VCH Verlag GmbH & Co., 2006, pp. 331–362 Search PubMed.
- S. Dey, P. K. Das and A. Dey, Coord. Chem. Rev., 2013, 257, 42–63 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- J. F. Sonnenberg, N. Coombs, P. A. Dube and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 5893–5899 CrossRef CAS PubMed.
- D. Matt, R. Ziessel, A. De Cian and J. Fischer, New J. Chem., 1996, 20, 1257–1263 CAS.
- A. A. Mikhailine, E. Kim, C. Dingels, A. J. Lough and R. H. Morris, Inorg. Chem., 2008, 47, 6587–6589 CrossRef CAS PubMed.
- A. Mikhailine, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS PubMed.
- A. A. Mikhailine and R. H. Morris, Inorg. Chem., 2010, 49, 11039–11044 CrossRef CAS PubMed.
- P. O. Lagaditis, A. J. Lough and R. H. Morris, Inorg. Chem., 2010, 49, 10057–10066 CrossRef CAS PubMed.
- P. E. Sues, A. J. Lough and R. H. Morris, Organometallics, 2011, 30, 4418–4431 CrossRef CAS.
- A. A. Mikhailine, P. O. Lagaditis, P. E. Sues, A. J. Lough and R. H. Morris, J. Organomet. Chem., 2010, 695, 1824–1830 CrossRef CAS PubMed.
- P. O. Lagaditis, A. A. Mikhailine, A. J. Lough and R. H. Morris, Inorg. Chem., 2009, 49, 1094–1102 CrossRef PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- R. Guo, C. Elpelt, X. Chen, D. Song and R. H. Morris, Org. Lett., 2005, 7, 1757–1759 CrossRef CAS PubMed.
- C. A. Busacca, J. C. Lorenz, N. Grinberg, N. Haddad, M. Hrapchak, B. Latli, H. Lee, P. Sabila, A. Saha, M. Sarvestani, S. Shen, R. Varsolona, X. Wei and C. H. Senanayake, Org. Lett., 2005, 7, 4277–4280 CrossRef CAS PubMed.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed.
- P. O. Lagaditis, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2011, 133, 9662–9665 CrossRef CAS PubMed.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Ozkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS PubMed.
- D. E. Prokopchuk and R. H. Morris, Organometallics, 2012, 31, 7375–7385 CrossRef CAS.
- F. Hasanayn and R. H. Morris, Inorg. Chem., 2012, 51, 10808–10818 CrossRef CAS PubMed.
- S. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121–8125 CrossRef CAS PubMed.
- A. A. Mikhailine, M. I. Maishan and R. H. Morris, Org. Lett., 2012, 14, 4638–4641 CrossRef CAS PubMed.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed.
- W. Baratta, F. Benedetti, Z. A. Del, L. Fanfoni, F. Felluga, S. Magnolia, E. Putignano and P. Rigo, Organometallics, 2010, 29, 3563–3570 CrossRef CAS.
- K. M. J. Brands, J. F. Payack, J. D. Rosen, T. D. Nelson, A. Candelario, M. A. Huffman, M. M. Zhao, J. Li, B. Craig, Z. J. Song, D. M. Tschaen, K. Hansen, P. N. Devine, P. J. Pye, K. Rossen, P. G. Dormer, R. A. Reamer, C. J. Welch, D. J. Mathre, N. N. Tsou, J. M. McNamara and P. J. Reider, J. Am. Chem. Soc., 2003, 125, 2129–2135 CrossRef CAS PubMed.
- K. Abdur-Rashid, S. E. Clapham, A. Hadzovic, J. N. Harvey, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2002, 124, 15104–15118 CrossRef CAS PubMed.
- C. A. Sandoval, T. Ohkuma, K. Muniz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS PubMed.
- P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367–1380 CrossRef CAS PubMed.
- Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong and J. Gao, J. Am. Chem. Soc., 2014, 136, 4031–4039 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |