 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis and application of novel Ni(II) N-alkyl dipyridylaldiminato complexes as selective ethylene oligomerisation catalysts†
Andrew J.
Swarts
and
Selwyn F.
Mapolie
*
Stellenbosch University, Department of Chemistry and Polymer Science, Private Bag X1, Matieland, 7602, Stellenbosch, South Africa. E-mail: 15428958@sun.ac.za; smapolie@sun.ac.za; Fax: +27 (0)21 808 3849; Tel: +27 (0)21 808 2722
First published on 13th May 2014
Abstract
A series of N-alkyl 2,2′-dipyridylamine ligands of general formula (2-C5H3NR)2NR′, (a): R = H, R′ = Me; (b): R = H, R′ = benzyl; (c): R = H, R′ = methylcyclohexyl; (d): R = H, R′ = neopentyl; (e): R = Me, R′ = Me) were prepared by a modified method involving base-mediated N-alkylation with the respective alkyl halide. Reaction of the ligands, a–e, with NiCl2(DME) allowed for the isolation of μ-Cl Ni(II) complexes: [Ni(μ-Cl){a}Cl]2 (1a); [Ni(μ-Cl){b}Cl]2 (1b); [Ni(μ-Cl){c}Cl]2 (1c); [Ni(μ-Cl){d}Cl]2 (1d) and [Ni(μ-Cl){e}Cl]2 (1e). The complexes were characterised by FT-IR spectroscopy, magnetic susceptibility measurements, mass spectrometry, elemental analyses and in the case of 1a, SCD analysis. In the case of complex 1e, an acid-mediated hydrolysis process was identified. The product of hydrolysis, the protonated ligand and a tetrachloronickelate salt (1e-A), was characterised by SCD analysis. Activation of 1a–1e with alkyl aluminium reagents generated highly active catalysts for the oligomerisation of ethylene, with activities of up to 864 kgoligomers molNi−1 h−1 and high selectivity toward the formation of butenes. In general, trans 2-butene was observed as the major isomer, with the exception of 1e. In the case of 1e, the selectivity for 1-butene was 98%, thereby demonstrating the significant effect that the introduction of a low degree of steric pressure in the coordination sphere of the catalyst has on selectivity.
Introduction
The field of transition metal-mediated olefin oligomerisation and polymerisation has seen phenomenal growth over the past three decades. Initial reports detailed the application of early transition metal catalyst systems of Ti, Zr and Hf constrained geometry catalysts (CGC's) which displayed exceptional activity in ethylene polymerisation.1 Despite their high activity, these catalyst systems generally showed low levels of chain entrapment in ethylene homopolymerisation as well as low levels of co-monomer enchainment in ethylene/α-olefin co-polymerisation.2 In addition, the inherent oxophilicity of these catalysts necessitate the rigorous exclusion of oxygen and water and precluded the use of polar α-olefins in co-polymerisation reactions. Brookhart, Grubbs and Gibson paved the way for the development of late transition metal catalysts of Ni and Pd which were capable of ethylene polymerisation/co-polymerisation with non-polar and polar α-olefins.3 Of particular significance was the work done on Ni and Pd systems ligated by diimine ligands, which displayed both remarkable activity and selectivity when applied to olefin homo- and co-polymerisation or oligomerisation reactions, thereby circumventing the inherent limitations of early transition metal catalyst systems.4 Seminal experimental and computational contributions by Brookhart and others elucidated the key features in homo- and co-polymerisation reactions of this class of catalysts. In the case of palladium, low temperature spectroscopic and computational investigations identified the catalyst resting states to be Pd–alkyl π-olefin species, establishing zero-order kinetics in olefin; barriers to insertion were determined to be in the range 17–18 kcal mol−1; branching in homopolymerisation was established to proceed via the isomerisation of π-agostic Pd–alkyl species with isomerisation barriers of 8–9 kcal mol−1; chain transfer proceeded via associative exchange of free olefin and that the degree of branching in the obtained polymer was independent of ethylene pressure while the morphology of the polymer was pressure dependent.5 For Ni(II) analogues, the degree of branching within the polymer formed during Ni(II)–diimine catalysed polymerisation was found to differ significantly with varying reaction conditions and the structure of the catalytically active species. The degree of branching was shown to be negligible at low reaction temperatures and high ethylene pressures when employing pre-catalysts with decreased steric bulk. In contrast, employing sterically encumbered pre-catalysts at high temperatures and low ethylene pressure led to the formation of highly branched polyethylene.6 Ethylene insertion barriers were found to be 4–5 kcal mol−1 lower in energy in comparison to their Pd congeners,7 while the energy barriers for insertion from sterically encumbered analogues were slightly lower than the less sterically congested Ni–alkyl species. With key mechanistic insights established, numerous reports have since been published detailing the catalytic application of complexes bearing nitrogen donor ligands in olefin transformations.8Gambarotta and co-workers recently reported the synthesis and application of Cr(III) complexes bearing N-alkyl dipyridylaldimine ligands in selective ethylene tetramerisation.9 They found that these complexes, after activation with an aluminium alkyl co-catalyst, catalysed the formation 1-octene selectively as well as a significant amount of wax. During the course of their catalytic investigations they found that the introduction of steric bulk in the form of Me-groups ortho to the pyridine nitrogen atoms switched the product selectivity from 1-octene to 1-hexene (Scheme 1).
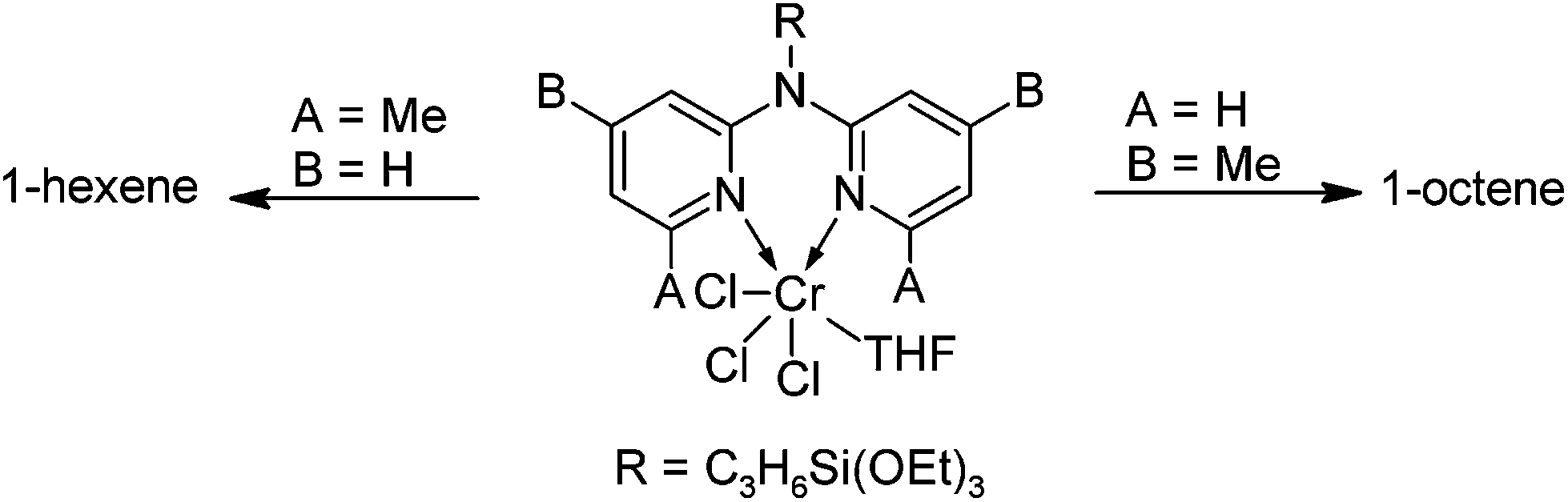 | ||
| Scheme 1 Switchable catalytic selectivity as a function of steric bulk in Cr(III)-catalysed ethylene tetramerisation. | ||
Intrigued by these results, we set out to evaluate the effect that increasing the electron-donating ability of the N-alkyl substituent and the introduction of steric bulk in the ortho position of the pyridyl ring would have on activity and selectivity in Ni(II)-catalysed ethylene oligo-/polymerisation. Herein we report our synthetic and catalytic application of a series of novel dinuclear Ni(II) N-alkyl dipyridylaldiminato complexes.
Results and discussion
Synthesis and characterisation
A series of known as well as novel N-alkyl-2,2′-dipyridylamine ligands of general formula (2-C5H3NR)2NR′, a: R = H, R′ = Me; b: R = H, R′ = benzyl; c: R = H, R′ = methylcyclohexyl; d: R = H, R′ = neopentyl; e: R = Me, R′ = Me) were prepared by a modified method involving base-mediated N-alkylation (Scheme 2). Ligands a,10b11 and d9 have been reported previously whereas c and e are novel. The ligands were isolated, after column chromatography, as yellow oils (a, c, d, e) or as a solid (b) in 60–90% yield. The ligands displayed solubility in most organic solvents and were characterised by FT-IR and 1H NMR spectroscopy (a, b, d) as well as 13C{1H} NMR spectroscopy, ESI-MS and elemental analysis (c, e).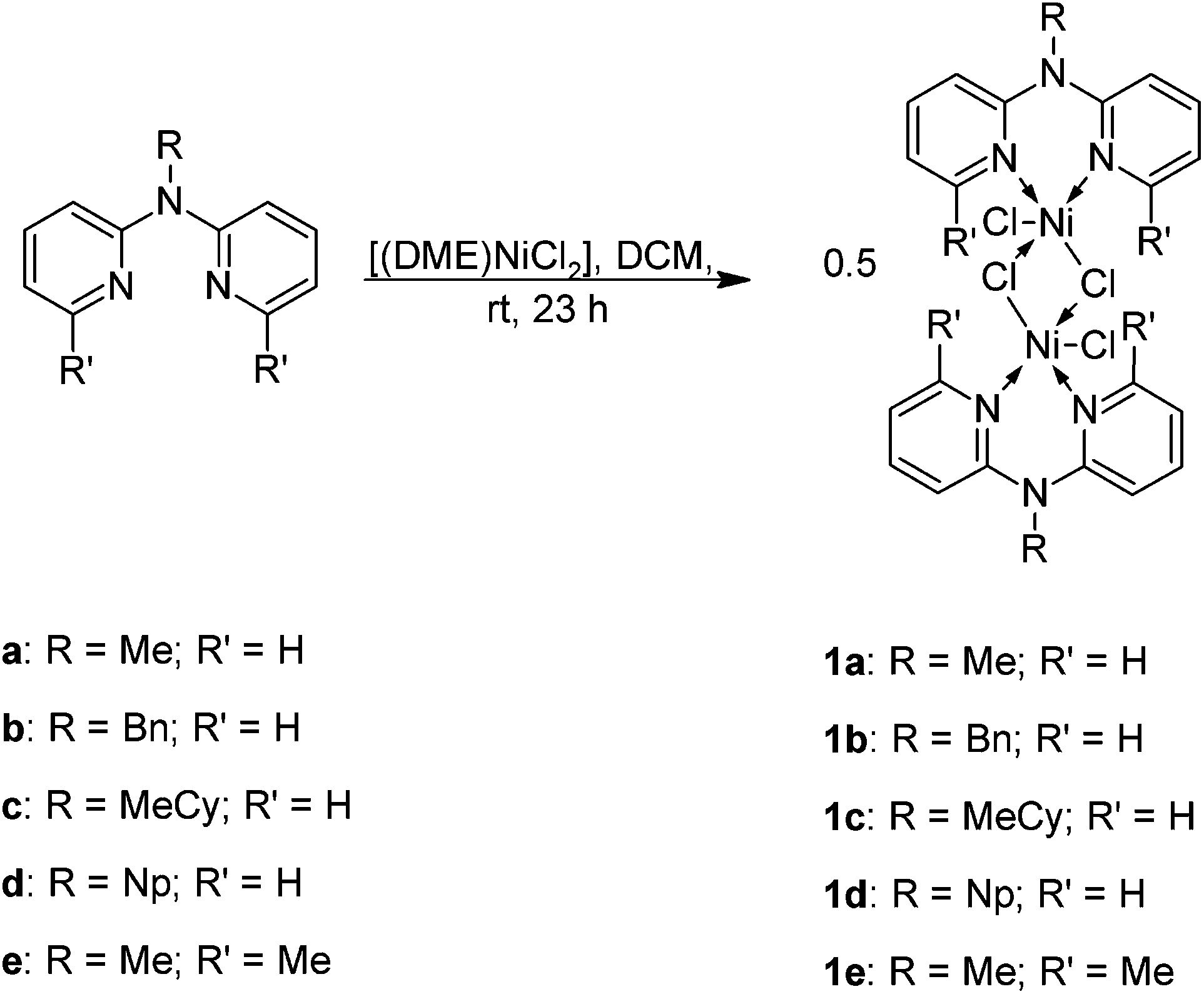 | ||
| Scheme 2 Synthesis of dinuclear Ni(II) N-alkyl 2,2′-dipyridylaldiminato complexes. | ||
The N-alkyl-2,2′-dipyridylamine ligands (a–e) were reacted with NiCl2(DME) to afford dimeric Ni(II) N-alkyl-2,2′-dipyridylaldiminato complexes, 1a–1e in high yields (Scheme 1). The complexes were isolated as blue-green (1a), green (1b and 1c) or yellow-green (1d) paramagnetic and hygroscopic solids which displayed solubility in polar coordinating solvents and alcohols. In the case of complex 1e, a pale purple-pink solid was isolated. The complexes were found to be insoluble in ethers, alkanes and chlorinated solvents. Complexes 1a–1e were characterised by a range of spectroscopic and analytical techniques.
Characterisation of the complexes by FT-IR spectroscopy showed a shift to higher wavenumbers of the pyridyl ring imine absorption bands, observed in the range 1597–1600 and 1565–1581 cm−1 for the complexes. Analogous shifts have been observed previously for related Ni(II) complexes, which is indicative of ligand coordination to nickel.12 Characterisation by ESI-MS spectrometry of complexes 1a–1e revealed interesting solvent-dependent fragmentation behaviour. When acetonitrile–0.1% formic acid solution was employed as the solvent during the ESI-MS experiment, mass fragments corresponding to mononuclear, dinuclear and trinuclear species were observed (Fig. S1†). Species aggregation is a common phenomenon during the ESI-MS experiment and has been reported previously for palladium complexes.13 In contrast to what was observed when employing acetonitrile–formic acid as solvent, the ESI-MS spectra obtained when employing 100% MeOH as dissolution and introduction solvent show the absence of dinuclear and trinucleas species. Instead, in all cases isotope clusters were observed which correspond to a doubly-charged species with the formulation [M + Na + MeOH]2+ where M = [Ni(μ-Cl){L}Cl]2 (Fig. S2†). The experimental solid state magnetic susceptibility values for complexes 1a–1e are in the range μeff 3.89–4.47μB, higher than the spin-only value of 2.83μB expected for two unpaired electrons.14 Despite this, experimentally determined values are within the range observed reported for high-spin dimeric Ni(II) complexes with S = 1.15
The structure of complex 1a was unambiguously determined by SCD analysis on single crystals grown by vapor diffusion of diethyl ether into a methanol solution of the complex. The molecular structure consists of a MeOH-solvated μ-Cl Ni(II)2 dimer, 1a·2MeOH (Fig. 1), residing on a crystallographic inversion center.
 | ||
| Fig. 1 Molecular structure of 1a·2MeOH drawn at 50% probability ellipsoids. Hydrogen atoms omitted for clarity. | ||
Metric parameters and crystallographic data are tabulated in Tables 1 and 2 respectively. The coordination sphere of the Ni(II) centre is distorted octahedral with the equatorial plane occupied by two bridging Cl atoms and the N atoms of the chelate ligand while the axial positions are occupied by a terminal Cl atom and a bound MeOH molecule.
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| Ni1–N1 | 2.036(2) | N1–Ni1–N2 | 85.76(6) |
| Ni1–N2 | 2.044(1) | N1–Ni1–Cl1 | 92.51(4) |
| Ni1–O1 | 2.165(1) | N1–Ni1–Cl2 | 175.19(4) |
| Ni1–Cl1 | 2.394(6) | N1–Ni1–O1 | 93.90(5) |
| Ni1–Cl2 | 2.415(5) | N2–Ni1–Cl1 | 94.00(4) |
| N2–Ni1–Cl2 | 173.61(4) | ||
| 1a·2MeOH | |
|---|---|
| Empirical formula | C24H30Cl4N6Ni2O2 |
| Temperature | 100(2) |
| Crystal system | Monoclinic |
| Space group | P 2/n |
| a/Å | 8.4860(14) |
| b/Å | 14.430(2) |
| c/Å | 11.5135(19) |
| α (°) | 90.00 |
| β (°) | 95.387(2) |
| γ (°) | 90.00 |
| V/Å3 | 1403.64 |
| Z | 1 |
| F(000) | 712 |
| D c (g cm−3) | 1.6411 |
| μ/mm−1 | 1.757 |
| Reflections [Fo > 4(Fo)] | 3237 |
| Parameters | 178 |
| GOF | 1.045 |
| R 1 (I > 2δ) | 0.0208 |
| wR2 | 0.0263 |
The Ni1–Cl1 and Ni1–Cl2 bond lengths of 2.394(6) and 2.415(5) Å respectively fall within the expected range reported for other μ-Cl Ni(II) dimeric structures.8i,16 In addition the N1–O1 bond length of 2.165(1) Å falls within the range observed for MeOH solvated-Ni(II) complexes.17 The deviation from planarity in the octahedral geometry is as a result of chelation of the ligand to the metal centre, with a N1–Ni1–N2 angle of 85.77°. The bound MeOH molecule is also slightly tilted within the crystal structure as observed by the O1–Ni1–Cl2 angle of 81.37°. All other bond lengths and angles fall within the expected range for this class of complexes.
Complex 1e displayed interesting solution behaviour during attempts at recrystallisation. The complex was isolated as a pale purple-pink solid, the colour of which suggested a square planar geometry around the metal centre, alluding to the metal centre being diamagnetic (Fig. S3†).
When 1e was dissolved in dichloromethane it formed a pink-red coloured solution. Previous literature reports have described square planar Ni(II) complexes as red or pink coloured solids.18 However, analytical data, specifically magnetic susceptibility data determined in the solid state, was consistent with a paramagnetic dinuclear chloro-bridged Ni(II) complex (μeff: 4.47μB). Analysis of the complex by 1H NMR spectroscopy in CD2Cl2 also showed broad, poorly resolved resonances, consistent with a paramagnetic transition metal complex (Fig. S4†). Prolonged storage of a solution of 1e in dichloromethane resulted in the formation of a pale-yellow precipitate, together with blue crystals (Fig. S3†). Removal of the supernatant and analysis of the pale-yellow precipitate by FT-IR spectroscopy showed absorption bands identical to commercially available NiCl2 (Fig. S5†). This initial result suggested that the complex dissociates in solution to generate the uncoordinated dipyridylamine ligand and nickel chloride. Layering of the pink-red solution with pentane and storage at 5 °C generated blue crystals which were analysed crystallographically (1e-A). The asymmetric unit consists of a tetrachloronickelate anion which is charge-balanced by two N-protonated 6,6′-dimethyl-2,2′-dipyridylamine ligands, which is effectively a tetrachloronickelate salt (Fig. 2). Selected bond lengths and angles (Table 3) as well as crystallographic parameters (Table 4), are tabulated. The cationic and anionic portions are effectively dissociated, as evidenced by Ni–N1 and Ni–N1′ bond distances of 5.540 and 5.768 Å. The Ni- and O-atoms are located on an inversion centre and display H⋯Cl interactions related by symmetry of 2.198 Å. In addition, H⋯Cl interactions are observed between the N-Me and the Cl atoms bound to nickel with a distance of 3.619 Å (Fig. 3). The molecules are packed in rows parallel to the c axis, with the rows stabilised by π–π stacking interactions of 3.645 Å and 3.490 Å. Consecutive rows along the a axis are linked via H-bonding to each other.
 | ||
| Fig. 2 Molecular structure of the crystals isolated as 1e-A, drawn at 50% probability. Ni and O atoms located on a centre of inversion. Selected hydrogen atoms omitted for clarity. | ||
 | ||
| Fig. 3 Crystal packing for 1e-A along the b-axis, showing the rhombus-shaped packing around the Ni-centre, with interleaving layers annotated as a and b. | ||
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| Ni1–Cl1 | 2.275(6) | Cl1–Ni1–Cl2 | 116.44(2) |
| Ni1–Cl2 | 2.258(7) | Cl1–Ni1–Cl1 | 100.12(2) |
| Cl1⋯H1 | 2.321 | ||
| Cl2⋯H7A | 3.620 | ||
| 1e-A | |
|---|---|
| Empirical formula | C26H32Cl4N6NiO |
| Temperature | 100(2) |
| Crystal system | Monoclinic |
| Space group | C 2/c |
| a/Å | 21.751(4) |
| b/Å | 11.949(2) |
| c/Å | 13.415(2) |
| α (°) | 90.00 |
| β (°) | 123.276(2) |
| γ (°) | 90.00 |
| V/Å3 | 2915.0(8) |
| Z | 4 |
| F(000) | 1336 |
| D c (g cm−3) | 1.6411 |
| μ/mm−1 | 1.064 |
| Reflections [Fo > 4(Fo)] | 3380 |
| Parameters | 180 |
| GOF | 1.058 |
| R 1 (I > 2δ) | 0.0326 |
| wR2 | 0.0749 |
Interleaving layers of metal anion, water and ligand pack infinitely along the b axis, in an a/b motif. A number of reports in literature detail the crystallographic characterisation of transition metal-/pyridinium ion-pairs,19 with only five reports corresponding to the preparation of tetrachloronickelate species.20
In all cases, the synthetic procedure for the isolation of the ion-pairs requires the presence of hydrochloric acid to protonate the pyridine ligand and generate the tetrachloronickelate anion. We thus propose that the formation of species 1e-A is mediated by hydrochloric acid, known to be present in dichloromethane in trace quantities.
Reactivity of complexes 1a–1e toward ethylene
We evaluated the reactivity of complexes 1a–1e, in the presence of alkylaluminium reagents as co-catalysts, in ethylene oligo-/polymerisation. Our initial catalysis optimisation runs were conducted with complex 1a as pre-catalyst (Table 5). We found that pre-catalyst/co-catalyst interactions had an effect on the generation of an active catalyst system. Initial catalyst screening with complex 1a/MAO at Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni ratio of 500:1 did not generate an active catalyst system (Table 5, entry 1), as evidenced by the absence of both oligomers and polymers in the reaction mixture. Previous reports of Ni(II)-catalysed ethylene oligomerisation demonstrated an increase in catalytic activity when employing chlorobenzene as solvent.21
:Ni ratios employing complex 1a, comparative evaluation of complexes 1b–1e and evaluation of reaction parameters on activitya
:Ni ratio of 500:1 did not generate an active catalyst system (Table 5, entry 1), as evidenced by the absence of both oligomers and polymers in the reaction mixture. Previous reports of Ni(II)-catalysed ethylene oligomerisation demonstrated an increase in catalytic activity when employing chlorobenzene as solvent.21
:Ni ratios employing complex 1a, comparative evaluation of complexes 1b–1e and evaluation of reaction parameters on activitya
| Entry | Complex | Co-catalyst | Al:Ni |
Activity (kgoligomers molNi−1 h−1) | TOF (×103 h−1) | % C4 | % C6e | % C8e | 1-C4:trans 2-C4:cis 2-C4 |
|---|---|---|---|---|---|---|---|---|---|
| a n(pre-catalyst): 10 μmol. Solvent (V): PhMe, 50 ml. Ethylene P: 5 bar. Reaction T: 30 °C. Reaction time: 30 min. TOF: n(ethylene consumed)/n(nickel)·h. MAO: methylaluminoxane. MMAO: modified methylaluminoxane. DEAC: diethyl aluminium chloride. b Chlorobenzene as solvent. c Ethylene P: 10 bar. d Reaction time: 90 min. e In all cases, the percentage of 1-hexene and 1-octene within the C6- and C8-fraction is less than 1%. | |||||||||
| 1 | 1a | MAO | 500:1 |
— | — | — | — | — | — |
| 2b | 1a | MAO | 500:1 |
— | |||||
| 3 | 1a | MAO | 1000:1 |
123 | 4 | 98 | 2 | 0 | 47:42:21 |
| 4 | 1a | MMAO | 500:1 |
— | — | — | — | — | |
| 5 | 1a | MMAO | 1000:1 |
— | — | — | — | — | |
| 6 | 1a | DEAC | 125:1 |
421 | 10 | 97 | 2.5 | 0.5 | 33:39:28 |
| 7 | 1a | DEAC | 250:1 |
589 | 21 | 99 | 1 | 0 | 26:46:28 |
| 8 | 1b | DEAC | 250:1 |
662 | 24 | >99 | 0 | 0 | 36:38:26 |
| 9 | 1c | DEAC | 250:1 |
527 | 19 | >99 | 0 | 0 | 22:48:30 |
| 10 | 1d | DEAC | 250:1 |
600 | 21 | >99 | 0 | 0 | 34:40:26 |
| 11 | 1e | DEAC | 250:1 |
499 | 18 | >99 | 0 | 0 | 98:1.2:0.8 |
| 12c | 1a | DEAC | 250:1 |
864 | 31 | >99 | 1 | 0 | 27:44:29 |
| 13d | 1a | DEAC | 250:1 |
249 | 9 | 98 | 2 | 0 | 20:50:30 |
When conducting our reactions in chlorobenzene as solvent, no ethylene oligomerisation or polymerisation was observed (Table 5, entry 2). We found that activation of complex 1a with MAO at Al:Ni ratios of ≥1000:1 in toluene generated a catalyst system capable of oligomerising ethylene with activities of 123 kgoligomers molNi−1 h−1 and turn-over frequencies of 4 × 103 h−1 (Table 5, entry 3). The formation of long-chain oligomers or polymer was not observed. The catalyst system displayed high selectivity toward the formation of 1- and 2-butenes (98%), with up to 47% 1-butene formed. The formation of small amounts of internal C6-isomers and C8-isomers was also observed. However, less than 1% of the higher olefin fraction corresponds to 1-hexene and 1-octene. Contrary to what has been reported previously for Ni(II) phosphinito-imine complexes,18 activation of complex 1a with MMAO with Al:Ni ratios of 500:1 and 1000:1 did not generate an active catalyst system (Table 5, entries 4 and 5).
Activation of complex 1a with DEAC at an Al:Ni ratio 125:1 generated highly active species, capable of oligomerising ethylene with high activities and at low co-catalyst loading (Table 5, entry 6). Increasing the amount of DEAC to an Al:Ni ratio of 250:1 resulted in the formation of an active species capable of dimerising ethylene to butenes with an activity of 589 kgoligomers molNi−1 h−1 and a turn-over frequency of 21 × 103 h−1 (Table 5, entry 7). A selectivity of 99% to butenes was observed, with the selectivity toward 1-butene found to be 26%. No significant differences were observed in the selectivity towards butenes and specifically 1-butene, when employing activated complex 4a with either MAO or DEAC as co-catalyst. The significant difference in catalytic activity as a function of the co-catalyst employed may be as a result of the different active species which is formed during activation employing MAO or DEAC. Brookhart and co-workers demonstrated experimentally that ethylene insertion to produce oligomers and polymers proceed from the catalyst resting state, the Ni–ethyl π-ethylene species.22 Assuming that an analogous Ni–ethyl π-ethylene species is the catalyst resting state for our catalyst system, alkylation and alkyl-abstraction by DEAC, in the presence of ethylene would generate the catalyst resting state directly. In contrast, alkylation and alkyl-abstraction by MAO under identical conditions would generate the Ni-methyl π-ethylene species, which would need to undergo a number of chain propagation and termination steps to generate the catalyst resting state. It is this reason which we believe account for the increased catalytic activity for ethylene oligomerisation observed when employing DEAC as co-catalyst. Next, we evaluated complexes 1a–1e comparatively in ethylene oligomerisation, employing DEAC as co-catalyst, with an Al:Ni ratio of 250:1. Under the evaluated reaction conditions, complexes 1a–1e were highly active catalysts for the oligomerisation of ethylene, forming butenes as major products (Table 5, entries 7–11). The observed activity varied between 499–662 kgoligomers molNi−1 h−1, while the observed turn-over frequencies varied between 18–24 × 103 h−1, in the range observed for previously reported Ni(II) phenyl-ether pyrazole complexes.8j
The observed activity of complexes 1a–1e is slightly lower than that reported previously for Ni(II) complexes in ethylene oligomerisation, while the selectivity for 1-butene displayed by 1e is higher than most reported in literature. Cámpora and co-workers reported the application of Ni(II) phosphinito-imine complexes as catalysts in ethylene oligomerisation.18 When activated with MMAO as co-catalyst, activities of up to 20.1 × 10−3 h−1 and selectivities of up to 100% for the formation of 1-butene was observed. Employing DEAC as co-catalyst generated even more active catalysts, consistent with our observation, with activities of up to 1685.3 × 10−3 h−1, while the maximum selectivity for 1-butene was found to be 50%. Braunstein, Giambastiani and co-workers reported the catalytic application of Ni(II) phosphinito-oxazoline complexes in ethylene oligomerisation.23 Activation with ethyl aluminium dichloride (EADC) generated catalysts with activities of up to 79 × 103 h−1 and selectivities for 1-butene of up to 22%. In contrast, activation with MAO produced catalysts with significantly higher activities of up to 230 × 103 h−1, while the selectivity for 1-butene was observed to decrease to 16%. Recently, Piers and co-workers demonstrated the ethylene oligomerisation behaviour of Ni(II) phosphine-borate complexes in the absence of a co-catalyst. These catalysts displayed TON's of up to 2000 in one hour. Selectivities for 1-butene was reported up to 70% with the remainder of the olefin products corresponding to hexenes.
When considering the activation of complexes 1a–1d with DEAC as co-catalyst, no clear correlation between the observed reactivity and the bridgehead N-alkyl substituents could be established. Thus the effect of the N-alkyl substituents on the observed catalytic activity is negligible. On the contrary, comparing the activity of 1a and 1e, the introduction of steric bulk in the ortho positions of the pyridylamine ligand result in a decrease in activity, from 589 kgoligomers molNi−1 h−1 to 499 kgoligomers molNi−1 h−1. The observed decrease is as a result of increased barriers to associative exchange and β-hydrogen transfer with increased steric bulk, leading to lower observed catalytic activity, as well as a less electrophilic metal centre which decreases the rate of ethylene insertion.22,24 When considering the selectivity toward 1-butene for complexes 1a–1d, no clear trend emerges (Table 5, entries 7–10). This again highlights the negligible effect the bridgehead N-alkyl substituents has on the observed selectivity. In general, for complexes 1a–1d, the major isomer formed during catalysis is the thermodynamically favoured trans 2-butene, in the range 38–46% (Table 5, entries 7–10, Fig. S6†). The percentages of 1-butene and cis 2-butene formed was found to vary as well, in the range 22–36% and 26–30% respectively. The varying selectivity for 1- and 2-butenes may be attributed to secondary isomerisation reactions which are difficult to control. These processes have been identified employing low temperature spectroscopic techniques,22 and have been observed previously in Ni(II)-catalysed ethylene oligomerisation.18 In contrast, when employing complex 1e as catalyst, the observed selectivity for 1-butene is 98% (Table 5, entry 11, Fig. S7†). This marked increase in 1-butene selectivity is attributed to the presence of the o-Me substituents which retards the isomerisation of 1-butene, by increasing steric pressure within the nickel coordination sphere. This is as a result of the re-inserted olefin being oriented within the plane of the metal centre prior to elimination, which destabilises the active species and decreases the propensity toward isomerisation. In addition, it has been established by both experiment and theory, that the olefin isomerisation barriers are much higher for sterically bulky Ni–alkyl olefin species, in comparison to their less sterically bulky analogues.6,7,24c It is these factors, a combination of steric and electronic effects, which we believe account for the observed difference in butene selectivity for complexes 1a and 1e.
Finally, we evaluated the effect of varying reaction parameters on activity employing complex 1a and DEAC as co-catalyst. It should be noted that we did not evaluate the effect of temperature due to the highly exothermic nature of the oligomerisation reaction. We found that increasing ethylene pressure from 5 bar to 10 bar, resulted in a significant increase in catalytic activity, which was found to be 864 kgoligomers molNi−1 h−1, corresponding to a TOF of 31 × 103 h−1 (Table 5, entry 12). This increase is attributable to an increased equilibrium concentration of monomer present in solution at higher ethylene pressures and has been observed previously for Ni(II) catalyst systems capable of oligomerising and polymerising ethylene.17c,18,25 Increasing the reaction time, from 30 minutes to 90 minutes resulted in a dramatic decrease in catalytic activity, from 589 kgoligomers molNi−1 h−1 to 249 kgoligomers molNi−1 h−1 (Table 5, entry 13). This decrease is consistent with decomposition of the catalytically active species and is generally observed for nickel- and palladium-catalysed olefin oligo- or polymerisation reactions.3a Our results have demonstrated the significant impact that tuning the coordination sphere of the catalyst can have on the observed product selectivity. In this example, the introduction of methyl groups in the ortho position of the dipyridylamine ligands increases olefin isomerisation barriers, thereby leading to the preferential formation of 1-butene.
Conclusions
A series of chloro-bridged dinuclear Ni(II) complexes, 1a–1e, ligated by N-alkyl dipyridylaldimine ligands were prepared. Characterisation by various spectroscopic and analytical techniques identified the complexes as paramagnetic Ni(II) complexes. During ESI-MS analysis of the complexes, interesting solvent-dependent fragmentation and aggregation processes were identified. In the case of complex 1e a unique acid-mediated hydrolysis process was identified, the product of which was characterised crystallographically. Following activation with alkylaluminium reagents, complexes 1a–1e generated species capable of oligomerising ethylene with high activity, up to 864 kgoligomers molNi−1 h−1 and high selectivity toward 1-butene, 98% in the case of 1e. Consistent with previous literature reports on Ni(II)-catalysed ethylene oligomerisation, an increase in ethylene pressure was found to increase the observed catalytic activity, while an increase in reaction time resulted in a decrease in the observed catalytic activity.Experimental section
General considerations
All transformations were performed using standard Schlenk techniques under a nitrogen atmosphere. Solvents were dried by refluxing over the appropriate drying agents followed by distillation prior to use and all other reagents were employed as obtained. ESI-MS (positive ion mode) analyses were performed on Waters API Quattro Micro and Waters API Q-TOF Ultima instruments by direct injection of sample. FT-IR analysis was performed on a Thermo Nicolet AVATAR 330 instrument, and was recorded as neat spectra (ATR) unless otherwise specified. Melting point determinations were performed on a Stuart Scientific SMP3 melting point apparatus and are reported as uncorrected. Magnetic susceptibility measurements were recorded on a Sherwood Scientific MK1 magnetic susceptibility balance. Samples were weighed and sealed in a glovebox prior to analysis. Measurements were conducted in duplicate. MAO (1.0 M in toluene, Sigma-Aldrich), MMAO (7 wt% in heptanes, Azko-Nobel) and DEAC (1.8 M in toluene, Sigma-Aldrich) was purchased from commercial sources and used as received. NiCl2(DME) was prepared according to a modified literature procedure, where the NiCl2·2H2O in the literature procedure was replaced with NiCl2·6H2O.26 Satisfactory micro-analysis could not be obtained for all the prepared complexes and in some cases (1a, 1b, 1c and 1d) the data was fitted to species which showed trace solvent inclusion.Synthesis of N-alkyl-2,2′-dipyridylamine ligands, 1a–1e
:1) as eluent. The pure product was obtained as a yellow oil. Yield: 582 mg, 89%. FT-IR (ATR, neat, ν): 1580 and 1557 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 1H NMR (CDCl3, 300 MHz): δ 8.33–8.36 (dd, 2H, 3JH–H 7.92 Hz, H1); δ 7.51–7.57 (m, 2H, H3); δ 7.15–7.19 (dt, 2H, 3JH–H 8.51 Hz, H4); δ 6.84–6.88 (m, 2H, H2); δ 3.63 (s, 3H, H6).
N). 1H NMR (CDCl3, 300 MHz): δ 8.31–8.33 (dd, 2H, 3JH–H 7.63 Hz, H1); δ 7.48–7.54 (m, 2H, H3); δ 7.33–7.36 (br. d, 2H, 3JH–H 7.34 Hz, H4); δ 7.25–7.28 (m, 2H, H9,10); δ 7.15–7.20 (m, 3H, H8,8′,9); δ 6.82–6.87 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 5.51 (s, 2H, H6).
:4). Yield: 823 mg, 88%. FT-IR (ATR, neat, ν): 1580 and 1558 cm−1 (CN). 1H NMR (CDCl3, 300 MHz): δ 8.32–8.34 (dd, 2H, 3JH–H 7.78 Hz, H1); δ 7.48–7.54 (m, 2H, H3); δ 7.06–7.10 (dt, 2H, 3JH–H 8.51 Hz, H4); δ 6.82–6.86 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 4.06 (d, 2H, 3JH–H 7.34 Hz, H6); δ 1.79–1.87 (m, 1H, H7); δ 1.59–1.74 (m, 5H, H8,9,10); δ 1.13–1.22 (m, 3H, H9′,10′); δ 0.92–1.03 (m, 2H, H8′). 13C{1H} (CDCl3, 75.38 MHz,): δ 158.05 (C5); δ 148.17 (C1); 136.95 (C3); δ 116.73 (C2); δ 114.86 (C4); δ 54.18 (C6); δ 37.24 (C7); δ 30.98 (C8,8′); δ 26.54 (C10); δ 26.00 (C9,9′). ESI-MS: m/z 268.2 [M + H]+. % Found (% calc.) for C17H21N3: C: 76.01 (76.37); H: 7.80 (7.92); N: 15.38 (15.72).
:4). Yield: 823 mg, 88%. FT-IR (ATR, neat, ν): 1581 and 1559 cm−1. 1H NMR (CDCl3, 300 MHz): δ 8.31–8.34 (dd, 2H, 3JH–H 7.78 Hz, H1); δ 7.47–7.53 (m, 2H, H3); δ 7.03–7.06 (dt, 2H, 3JH–H 8.36 Hz, H4); δ 6.82–6.87 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 4.20 (s, 2H, H6); δ 0.88 (s, 9H, H7).
N). 1H NMR (CDCl3, 300 MHz): δ 8.33–8.36 (dd, 2H, 3JH–H 7.92 Hz, H1); δ 7.51–7.57 (m, 2H, H3); δ 7.15–7.19 (dt, 2H, 3JH–H 8.51 Hz, H4); δ 6.84–6.88 (m, 2H, H2); δ 3.63 (s, 3H, H6).
N). 1H NMR (CDCl3, 300 MHz): δ 8.31–8.33 (dd, 2H, 3JH–H 7.63 Hz, H1); δ 7.48–7.54 (m, 2H, H3); δ 7.33–7.36 (br. d, 2H, 3JH–H 7.34 Hz, H4); δ 7.25–7.28 (m, 2H, H9,10); δ 7.15–7.20 (m, 3H, H8,8′,9); δ 6.82–6.87 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 5.51 (s, 2H, H6).
:4). Yield: 823 mg, 88%. FT-IR (ATR, neat, ν): 1580 and 1558 cm−1 (CN). 1H NMR (CDCl3, 300 MHz): δ 8.32–8.34 (dd, 2H, 3JH–H 7.78 Hz, H1); δ 7.48–7.54 (m, 2H, H3); δ 7.06–7.10 (dt, 2H, 3JH–H 8.51 Hz, H4); δ 6.82–6.86 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 4.06 (d, 2H, 3JH–H 7.34 Hz, H6); δ 1.79–1.87 (m, 1H, H7); δ 1.59–1.74 (m, 5H, H8,9,10); δ 1.13–1.22 (m, 3H, H9′,10′); δ 0.92–1.03 (m, 2H, H8′). 13C{1H} (CDCl3, 75.38 MHz,): δ 158.05 (C5); δ 148.17 (C1); 136.95 (C3); δ 116.73 (C2); δ 114.86 (C4); δ 54.18 (C6); δ 37.24 (C7); δ 30.98 (C8,8′); δ 26.54 (C10); δ 26.00 (C9,9′). ESI-MS: m/z 268.2 [M + H]+. % Found (% calc.) for C17H21N3: C: 76.01 (76.37); H: 7.80 (7.92); N: 15.38 (15.72).
:4). Yield: 823 mg, 88%. FT-IR (ATR, neat, ν): 1581 and 1559 cm−1. 1H NMR (CDCl3, 300 MHz): δ 8.31–8.34 (dd, 2H, 3JH–H 7.78 Hz, H1); δ 7.47–7.53 (m, 2H, H3); δ 7.03–7.06 (dt, 2H, 3JH–H 8.36 Hz, H4); δ 6.82–6.87 (dt, 2H, 3JH–H 7.19 Hz, H2); δ 4.20 (s, 2H, H6); δ 0.88 (s, 9H, H7).
Synthesis of μ-Cl Ni(II) N-alkyl dipyridylaldiminato complexes, 1a–1e
N). ESI-MS m/z 342 [M + Na + MeOH]2+. μeff: 4.15μB. % Found (Calc.) for [C22H22Cl4N6Ni2·0.5 CH2Cl2]: C: 40.23 (40.21); H: 3.41 (3.45); N: 12.42 (12.50).
N). ESI-MS m/z 418 [M + Na + MeOH]2+. μeff: 3.89μB. % Found (Calc.) for [C34H30Cl4N6Ni2·0.2 CH2Cl2]: C: 51.82 (51.85); H: 4.33 (4.39); N: 10.75 (10.78).
N). ESI-MS m/z 424 [M + Na + MeOH]2+. μeff: 4.01μB. % Found (Calc.) for [C34H42Cl4N6Ni2·0.6 CH2Cl2]: C: 48.45 (48.95); H: 4.83 (5.14); N: 10.11 (9.88).
N). ESI-MS m/z 398 [M + Na + MeOH]2+. μeff: 4.32μB. % Found (Calc.) for [C34H42Cl4N6Ni2·0.3 CH2Cl2]: C: 45.71 (45.86); H: 4.71 (4.71); N: 11.24 (11.32).
651 cm−1 (CN). ESI-MS m/z 370 [M + Na + MeOH]2+. μeff: 4.47μB. % Found (Calc.) for C26H30Cl4Ni6Ni2: C: 45.49 (45.54); H: 3.98 (4.41); N: 12.06 (12.06).
Isolation of [6-methylpyridinium-6′-methyl-N-methylpyridin-2-amine]2+[Ni(Cl)4]2−
A solution of complex 1e (30 mg, 0.043 mmol) was dissolved in dichloromethane (0.5 ml). The pink-red solution was layered with pentane and stored at 5 °C. The formation of a yellow precipitate was observed, with concomitant formation of blue crystals. The yellow precipitate was analysed by FT-IR, spectral features was consistent with NiCl2. The blue crystals were isolated and characterised by X-ray crystallography.X-Ray crystal structure determination
Single crystals of complexes 1a·2MeOH and 1e-A were mounted on a nylon loop and centred in a stream of cold nitrogen at 100(2) K. Crystal evaluation and data collection were performed on a Bruker-Nonius SMART Apex II CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å). Data collection, reduction and refinement were performed using SAINT27 and SADABS,28 which forms part of the APEX II software package. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using SHELX-9729 within the X-Seed graphic user interface.30 All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were placed using calculated positions and riding models.Procedure for preparative-scale ethylene oligomerisation
In a glovebox under a nitrogen atmosphere, a 250 ml Parr high-pressure autoclave was charged with the required amount of solvent and co-catalyst, and was sealed prior to being attached to the ethylene feed. The reactor was brought to temperature at which point a dispersion of the pre-catalyst in toluene (total volume: 50 ml) was added via syringe under positive ethylene pressure. The ethylene feed was maintained at the required pressure throughout the catalytic run, with the volume of ethylene consumed monitored throughout the catalytic run. After the allotted time, the reactor was cooled to −78 °C and the reaction mixture quenched with MeOH (5 ml). A liquid sample was filtered through a syringe filter and analyzed by GC-FID employing p-xylene as internal standard, taking care to maintain the temperature below −20 °C to minimize the loss of volatile reaction products. The observed oligomers were quantified against calibrated standards. Quenching the reaction mixture did not precipitate any polymers and no long-chain oligomers were observed after work-up.References
- (a) D. D. Devore, F. J. Timmers, D. L. Hasha, R. K. Rosen, T. J. Marks, P. A. Deck and C. L. Stern, Organometallics, 1995, 14, 3132–3134 CrossRef CAS; (b) H. G. Alt, K. Föttinger and W. Milius, J. Organomet. Chem., 1999, 572, 21–30 CrossRef CAS; (c) H. Sinn and W. Kaminsky, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1980, vol. 18, pp. 99–149 Search PubMed.
- (a) K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault, P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics, 2011, 30, 3318–3329 CrossRef CAS; (b) L. Li, M. V. Metz, H. Li, M.-C. Chen, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12725–12741 CrossRef CAS PubMed; (c) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- (a) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed; (b) C. Wang, S. Friedrich, T. R. Younkin, R. T. Li, R. H. Grubbs, D. A. Bansleben and M. W. Day, Organometallics, 1998, 17, 3149–3151 CrossRef CAS; (c) T. R. Younkin, E. F. Connor, I. H. Jason, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS; (d) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2002, 103, 283–316 CrossRef PubMed.
- (a) S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, Organometallics, 2009, 28, 2127 CrossRef CAS; (b) W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235–244 CrossRef.
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS; (b) F. C. Rix and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 1137–1138 CrossRef CAS; (c) F. C. Rix, M. Brookhart and P. S. White, J. Am. Chem. Soc., 1996, 118, 4746–4764 CrossRef CAS; (d) D. J. Tempel and M. Brookhart, Organometallics, 1998, 17, 2290–2296 CrossRef CAS; (e) S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888–899 CrossRef CAS; (f) D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS; (g) L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS PubMed; (h) T. K. Woo, P. M. Margl, P. E. Blöchl and T. Ziegler, J. Phys. Chem. B, 1997, 101, 7877–7880 CrossRef CAS; (i) A. Michalak and T. Ziegler, J. Am. Chem. Soc., 2001, 123, 12266–12278 CrossRef CAS PubMed; (j) M. J. Szabo, N. M. Galea, A. Michalak, S.-Y. Yang, L. F. Groux, W. E. Piers and T. Ziegler, J. Am. Chem. Soc., 2005, 127, 14692–14703 CrossRef CAS PubMed; (k) S.-Y. Yang, M. J. Szabo, A. Michalak, T. Weiss, W. E. Piers, R. F. Jordan and T. Ziegler, Organometallics, 2005, 24, 1242–1251 CrossRef CAS; (l) A. Haras, A. Michalak, B. Rieger and T. Ziegler, Organometallics, 2006, 25, 946–953 CrossRef CAS.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- (a) S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634–10635 CrossRef CAS; (b) J. C. Jenkins and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 5827–5842 CrossRef CAS PubMed; (c) L. Deng, P. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 1094–1100 CrossRef CAS; (d) D. G. Musaev, R. D. J. Froese, M. Svensson and K. Morokuma, J. Am. Chem. Soc., 1997, 119, 367–374 CrossRef CAS.
- (a) B. M. Boardman and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1597–1606 CrossRef CAS PubMed; (b) J. Flapper, H. Kooijman, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen, C. J. Elsevier and P. C. J. Kamer, Organometallics, 2009, 28, 3272–3281 CrossRef CAS; (c) S. Abraham, C.-S. Ha and I. Kim, Macromol. Rapid Commun., 2006, 27, 1386–1392 CrossRef CAS; (d) B. Blom, M. J. Overett, R. Meijboom and J. R. Moss, Inorg. Chim. Acta, 2005, 358, 3491–3496 CrossRef CAS PubMed; (e) R. D. Broene, M. Broookhart, W. M. Lamanna and A. F. Volpe Jr., J. Am. Chem. Soc., 2005, 127, 17194–17195 CrossRef CAS PubMed; (f) M. P. Conley, C. T. Burns and R. F. Jordan, Organometallics, 2007, 26, 6750–6759 CrossRef CAS; (g) M. D. Doherty, S. Trudeau, P. S. White, J. P. Morken and M. Brookhart, Organometallics, 2007, 26, 1261–1269 CrossRef CAS; (h) S. P. Meneghetti, P. J. Lutz and J. Kress, Organometallics, 1999, 18, 2734–2737 CrossRef CAS; (i) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Inorg. Chem., 2004, 43, 1649–1658 CrossRef CAS PubMed; (j) A. H. D. P. S. Ulbrich, R. R. Campedelli, J. L. S. Milani, J. H. Z. d. Santos and O. d. L. Casagrande Jr., Appl. Catal., A, 2013, 453, 280–286 CrossRef CAS PubMed; (k) Y.-y. Wang, S.-a. Lin, F.-m. Zhu, H.-y. Gao and Q. Wu, Inorg. Chim. Acta, 2009, 362, 166–172 CrossRef CAS PubMed.
- S. Licciulli, I. Thapa, K. Albahily, I. Korobkov, S. Gambarotta, R. Duchateau, R. Chevalier and K. Schuhen, Angew. Chem., Int. Ed., 2010, 49, 9225–9228 CrossRef CAS PubMed.
- M. J. Rauterkus, S. Fakih, C. Mock, I. Puscasu and B. Krebs, Inorg. Chim. Acta, 2003, 350, 355–365 CrossRef CAS.
- S. Fakih, W. C. Tung, D. Eierhoff, C. Mock and B. Krebs, Z. Anorg. Allg. Chem., 2005, 631, 1397–1402 CrossRef CAS.
- (a) R. Malgas-Enus, S. F. Mapolie and G. S. Smith, J. Organomet. Chem., 2008, 693, 2279–2286 CrossRef CAS PubMed; (b) R. Malgas-Enus, PhD thesis, Stellenbosch University, 2010.
- (a) R. Ares, M. López-Torres, A. Fernández, D. Vázquez-García, M. T. Pereira, J. M. Vila, L. Naya and J. J. Fernández, J. Organomet. Chem., 2007, 692, 4197–4208 CrossRef CAS PubMed; (b) J.-P. Taquet, O. Siri, P. Braunstein and R. Welter, Inorg. Chem., 2006, 45, 4668–4676 CrossRef CAS PubMed.
- G. L. Miessler and D. A. Tarr, Inorganic Chemistry, Pearson Prentice Hall, London, United Kingdom, 3rd edn, 2004 Search PubMed.
- M. Schär, D. Saurenz, F. Zimmer, I. Schädlich, G. Wolmershäuser, S. Demeshko, F. Meyer, H. Sitzmann, O. M. Heigl and F. H. Köhler, Organometallics, 2013, 32, 6298–6305 CrossRef.
- (a) J. Peng and Y. Kishi, Org. Lett., 2011, 14, 86–89 CrossRef PubMed; (b) J. Yu, X. Hu, Y. Zeng, L. Zhang, C. Ni, X. Hao and W.-H. Sun, New J. Chem., 2011, 35, 178–183 RSC; (c) T. V. Laine, M. Klinga and M. Leskelä, Eur. J. Inorg. Chem., 1999, 1999, 959–964 CrossRef.
- (a) W.-H. Sun, S. Song, B. Li, C. Redshaw, X. Hao, Y.-S. Li and F. Wang, Dalton Trans., 2012, 41, 11999–12010 RSC; (b) L. Zhao, V. Niel, L. K. Thompson, Z. Xu, V. A. Milway, R. G. Harvey, D. O. Miller, C. Wilson, M. Leech, J. A. K. Howard and S. L. Heath, Dalton Trans., 2004, 1446–1455 RSC; (c) R. Gao, M. Zhang, T. Liang, F. Wang and W.-H. Sun, Organometallics, 2008, 27, 5641–5648 CrossRef CAS.
- L. Ortiz de la Tabla, I. Matas, P. Palma, E. Álvarez and J. Cámpora, Organometallics, 2012, 31, 1006–1016 CrossRef CAS.
- (a) C. Tamuly, N. Barooah, A. S. Batsanov, R. Kataky and J. B. Baruah, Inorg. Chem. Commun., 2005, 8, 689–691 CrossRef CAS PubMed; (b) X.-F. Shi, Z.-Y. Xing, L. Wu and W.-Q. Zhang, Inorg. Chim. Acta, 2006, 359, 603–608 CrossRef CAS PubMed; (c) L. Li, M. M. Turnbull, C. P. Landee, J. Jornet, M. Deumal, J. J. Novoa and J. L. Wikaira, Inorg. Chem., 2007, 46, 11254–11265 CrossRef CAS PubMed; (d) J. Kuhnert, I. Cisarova, M. Lamac and P. Stepnicka, Dalton Trans., 2008, 2454–2464 RSC; (e) G. D. Potter, M. C. Baird and S. P. C. Cole, J. Organomet. Chem., 2007, 692, 3508–3518 CrossRef CAS PubMed.
- (a) M. R. Bond and R. D. Willett, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 861–865 CrossRef; (b) L. Cao, U. Englert and Q. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m377 CAS; (c) Z.-X. Du and J.-P. Qu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1656 CAS.
- C. Obuah, B. Omondi, K. Nozaki and J. Darkwa, J. Mol. Catal. A: Chem., 2014, 382, 31–40 CrossRef CAS PubMed.
- M. D. Leatherman, S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 2003, 125, 3068–3081 CrossRef CAS PubMed.
- P. Chavez, I. G. Rios, A. Kermagoret, R. Pattacini, A. Meli, C. Bianchini, G. Giambastiani and P. Braunstein, Organometallics, 2009, 28, 1776–1784 CrossRef CAS.
- (a) C. Zhang, W.-H. Sun and Z.-X. Wang, Eur. J. Inorg. Chem., 2006, 4895 CrossRef CAS; (b) M. Brookhart, F. C. Rix, J. M. DeSimone and J. C. Barborak, J. Am. Chem. Soc., 1992, 114, 5894–5895 CrossRef CAS; (c) L. Deng, T. K. Woo, L. Cavallo, P. M. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 6177–6186 CrossRef CAS.
- (a) B. A. Rodriguez, M. Delferro and T. J. Marks, Organometallics, 2008, 27, 2166–2168 CrossRef CAS; (b) H.-L. Mu, W.-P. Ye, D.-P. Song and Y.-S. Li, Organometallics, 2010, 29, 6282–6290 CrossRef CAS; (c) D.-P. Song, J.-Q. Wu, W.-P. Ye, H.-L. Mu and Y.-S. Li, Organometallics, 2010, 29, 2306–2314 CrossRef CAS.
- L. G. L. Ward, Inorg. Synth., 1971, 13, 154–164 CrossRef CAS.
- Bruker AXS Inc., Madison, WI, Version 6.45, 2003.
- R. H. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS; (b) J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Contains additional experimental data, Fig S1–S7: includes ESI-MS spectra, the solid state and solution appearance of complex 1e, 1H NMR and FT-IR spectra of 1e and the additional species formed in solution as well as GC-FID spectra showing the formation of 1- and 2-butenes during catalysis. CCDC 981834 and 981835 for 1a and 1e-A. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00537f |
| This journal is © The Royal Society of Chemistry 2014 |