DOI:
10.1039/C4DT00510D
(Paper)
Dalton Trans., 2014,
43, 8261-8272
Group 4 metal complexes with new chiral pincer NHC-ligands: synthesis, structure and catalytic activity†
Received
18th February 2014
, Accepted 6th March 2014
First published on 7th March 2014
Abstract
Chiral group 4 NHC–metal complexes were prepared in good yields by amine elimination from M(NR2)4 (M = Ti, Zr, Hf; R = Me, Et) and chiral pincer NHC-ligands, L4 (L4a and L4b), L5 and L6, which are derived from (S,S)-diphenyl-1,2-ethanediamine. Treatment of M(NR2)4 with 1 equiv. of L4 in THF gives, after recrystallization from a benzene solution, the chiral titanium amides (L4)Ti(NMe2)(Br)(THF) (7) and (L4)Ti(NMe2)(Cl)(THF) (11), zirconium amides (L4)Zr(NMe2)(Br)(THF) (8), (L4)Zr(NEt2)(Br)(THF) (10), (L4)Zr(NMe2)(Cl)(THF) (12) and (L4)Zr(NEt2)(Cl)(THF) (14), and hafnium amides (L4)Hf(NMe2)(Br)(THF) (9) and (L4)Hf(NMe2)(Cl)(THF) (13), respectively. Similarly, the reactions of L5 or L6 with 1 equiv. of M(NR2)4 yield the titanium amide (L6)Ti(NMe2)(Cl)(THF) (16), the zirconium amides (L5)Zr(NMe2)(Cl)(THF) (15), (L6)Zr(NMe2)(Cl)(THF) (17) and (L6)Zr(NEt2)(Cl)(THF) (19), and the hafnium amide (L6)Hf(NMe2)(Cl)(THF) (18), respectively. Complexes 7–19 were characterized by various spectroscopic techniques and elemental analyses. The molecular structures of 10 and 14–19 were also established by X-ray diffraction analyses, which represent the first example of the structurally characterized group 4 chiral NHC–metal complex. Furthermore, 7–19 are active catalysts for the polymerization of rac-lactide in the presence of isopropanol, leading to the heterotactic-rich polylactides.
Introduction
Biodegradable polymers derived from renewable resources such as polylactides have received much attention over the past decade because of their attractive physical and mechanical properties.1 In addition, the chain stereochemistry determines the polymer properties and the rate of degradation.2 For example, whereas the enantiopure polylactide melts at 180 °C, a much higher melting temperature (230 °C) is found for stereocomplex polymers formed by an equivalent mixture of poly(L-lactide) and poly(D-lactide).3 Therefore, the polymerization of rac-lactide with stereoselective catalysts remains a challenge and an opportunity for chemists. To date, numerous reviews have covered catalyst systems for the ring-opening polymerization (ROP) of cyclic esters based on metals such as magnesium, zinc, calcium, aluminum, lanthanides, tin, group 4 metals, germanium, indium and iron,1d,4 but among these, the chiral group 4 catalysts are especially promising.5 Unfortunately, compared to other metals, structurally well-characterized chiral group 4 complexes that initiate the controlled ring-opening polymerization of lactides are still scarce.5
In recent years, transition-metal complexes with chiral N-heterocyclic carbene (NHC) ligands have become increasingly popular because of their stability to air and moisture and their strong σ-donating, but poor π-accepting abilities.6 An additional driving force is the longstanding interest in catalysts for enantioselective reactions such as olefin metathesis,7 conjugate addition of enones,8 allylic alkylations,9 olefin hydrogenations10 and hydrosilylations.11 Encouraged by the attractive features of chiral NHC-ligands in general, we are now focusing on the preparation of group 4 metal complexes coordinated by chiral multi-dentate NHC-ligands, and to our knowledge no chiral group 4 metal NHC-catalyst has yet been structurally authenticated.6,12 More recently, we have designed and prepared a new series of tridentate chiral pincer NHC-ligands L4–L6 from (S,S)-diphenyl-1,2-ethanediamine, and found them to be useful ligands for group 4 metals, which are potential catalysts for the polymerization of lactides. Herein, we report on the synthesis of these chiral NHC-ligands, their use in group 4 chemistry, and the application of the resulting complexes as catalysts in the polymerization of rac-lactide (rac-LA).
Experimental
General methods
Group 4 complexes and catalytic reactions were performed under an atmosphere of dry dinitrogen with rigid exclusion of air and moisture using standard Schlenk or cannula techniques, or a glovebox. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. Racemic lactide (rac-LA) was recrystallized twice from dry toluene and then sublimed under vacuum prior to use. All chemicals were purchased from Aldrich Chemical Co. and Beijing Chemical Co. and used as received unless otherwise noted. Infrared spectra were obtained from KBr pellets on an Avatar 360 Fourier transform spectrometer. Molecular weights of the polymer were estimated by gel permeation chromatography (GPC) using a PL-GPC 50 apparatus. 1H and 13C NMR spectra were recorded on a Bruker AV 500 spectrometer at 500 and 125 MHz, respectively. All chemical shifts are reported in δ units with reference to the residual protons of the deuterated solvents for proton and carbon chemical shifts. Melting points were measured on X-6 melting point apparatus and were uncorrected. Elemental analyses were performed on a Vario EL elemental analyzer.
Syntheses
Preparation of 1.
Salicylaldehyde (1.22 g, 10.0 mmol) was mixed with (S,S)-diphenyl-1,2-ethanediamine (1.06 g, 5.0 mmol) in absolute ethanol (30 mL) and stirred for 4 h at room temperature. NaBH4 (2.00 g, 52.6 mmol) was added in small portions at 0 °C, and the solution was then warmed to 50 °C and kept at this temperature for 2 h. The solvent was removed, the residue was treated with H2O (20 mL), extracted with ethyl acetate (20 mL × 3) and washed with brine (20 mL). The combined organic layers were dried with anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was further purified by flash column chromatography (hexane–ethyl acetate = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 1 as a colorless oil. Yield: 2.01 g (95%) (Found: C, 79.12; H, 6.60; N, 6.64. C28H28N2O2 requires: C, 79.22; H, 6.65; N, 6.60%). 1H NMR (C6D6): δ 7.14–7.07 (m, 4H, aryl), 6.91 (m, 6H, aryl), 6.68 (m, 2H, aryl), 6.58 (m, 2H, aryl), 6.52 (m, 4H, aryl), 3.57 (s, 2H, CH), 3.51 (d, J = 13.8 Hz, 2H, CH2), 3.19 (d, J = 13.8 Hz, 2H, CH2); NH and OH protons were not observed. 13C NMR (C6D6): δ 158.9, 138.3, 129.4, 129.0, 128.5, 128.3, 128.2, 128.1, 127.9, 127.8, 66.7, 50.1. IR (KBr, cm−1):
:1) to give 1 as a colorless oil. Yield: 2.01 g (95%) (Found: C, 79.12; H, 6.60; N, 6.64. C28H28N2O2 requires: C, 79.22; H, 6.65; N, 6.60%). 1H NMR (C6D6): δ 7.14–7.07 (m, 4H, aryl), 6.91 (m, 6H, aryl), 6.68 (m, 2H, aryl), 6.58 (m, 2H, aryl), 6.52 (m, 4H, aryl), 3.57 (s, 2H, CH), 3.51 (d, J = 13.8 Hz, 2H, CH2), 3.19 (d, J = 13.8 Hz, 2H, CH2); NH and OH protons were not observed. 13C NMR (C6D6): δ 158.9, 138.3, 129.4, 129.0, 128.5, 128.3, 128.2, 128.1, 127.9, 127.8, 66.7, 50.1. IR (KBr, cm−1): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) 3427 (s), 2853 (w), 1616 (s), 1588 (s), 1489 (s), 1454 (s), 1251 (s), 755 (s).
3427 (s), 2853 (w), 1616 (s), 1588 (s), 1489 (s), 1454 (s), 1251 (s), 755 (s).
Preparation of 2.
This compound was prepared as colorless oil from the reaction of 3-tert-butylsalicylaldehyde (1.78 g, 10.0 mmol) with (S,S)-diphenyl-1,2-ethanediamine (1.06 g, 5.0 mmol) in absolute ethanol (30 mL) at room temperature, followed by reduction with NaBH4 (2.00 g, 52.6 mmol) in ethanol, and purification by flash column chromatography (hexane–ethyl acetate = 10:1) by a similar procedure as outlined in the synthesis of 1. Yield: 2.47 g (92%) (Found: C, 80.53; H, 8.30; N, 5.30. C36H44N2O2 requires: C, 80.56; H, 8.26; N, 5.22%). 1H NMR (C6D6): δ 10.84 (s, 2H, OH), 7.21–7.09 (m, 12H, aryl), 6.62 (m, 4H, aryl), 4.06 (s, 2H, CH), 3.78 (d, J = 13.4 Hz, 2H, CH2), 3.59 (d, J = 13.4 Hz, 2H, CH2), 3.33 (s, 2H, NH), 1.38 (s, 18H, C(CH3)3). 13C NMR (C6D6): δ 156.1, 137.1, 135.7, 127.1, 126.8, 126.7, 126.6, 126.4, 125.7, 125.2, 121.7, 117.6, 65.6, 49.4, 33.7, 28.6. IR (KBr, cm−1): 3427 (s), 2957 (s), 1590 (m), 1437 (s), 1389 (m), 1235 (s), 1087 (s), 749 (s).
Preparation of 3.
This compound was prepared as colorless oil from the reaction of 3,5-di-tert-butyl-salicylaldehyde (2.34 g, 10.0 mmol) with (S,S)-diphenyl-1,2-ethanediamine (1.06 g, 5.0 mmol) in absolute ethanol (30 mL) at room temperature, followed by reduction with NaBH4 (2.00 g, 52.6 mmol) in ethanol, and purification by flash column chromatography (hexane–ethyl acetate = 10:1) by a similar procedure as outlined in the synthesis of 1. Yield: 2.92 g (90%) (Found: C, 81.40; H, 9.40; N, 4.30. C44H60N2O2 requires: C, 81.43; H, 9.32; N, 4.32%). 1H NMR (C6D6): δ 7.08–7.01 (m, 10H, aryl), 6.97 (d, J = 2.1 Hz, 2H, aryl), 6.55 (d, J = 2.1 Hz, 2H, aryl), 3.84 (d, J = 5.2 Hz, 2H, CH2), 3.72 (s, 2H, CH), 3.42 (d, J = 5.2 Hz, 2H, CH2), 1.25 (s, 18H, C(CH3)3), 1.08 (s, 18H, C(CH3)3); NH and OH protons were not observed. 13C NMR (C6D6): δ 154.0, 149.4, 142.3, 142.2, 139.8, 137.5, 135.3, 127.3, 127.1, 127.0, 126.9, 126.8, 66.1, 50.3, 34.4, 33.3, 30.9, 29.1. IR (KBr, cm−1): 3428 (s), 2960 (s), 1604 (m), 1480 (s), 1233 (s), 875 (s).
Preparation of L4a.
Compound 1 (2.00 g, 4.71 mmol), NH4Br (0.69 g, 7.07 mmol) and triethyl orthoformate (15 mL) were heated at 120 °C for one day. After the reaction mixture was cooled to room temperature, diethyl ether (40 mL) was added to precipitate a colorless solid, which was filtered and washed with diethyl ether to give L4a as a colorless solid. Yield: 2.23 g (92%) (Found: C, 67.54; H, 5.30; N, 5.42. C29H27N2BrO2 requires: C, 67.58; H, 5.28; N, 5.43%). M.p.: 273–275 °C. 1H NMR (DMSO-d6): δ 10.17 (s, 2H, OH), 9.13 (s, 1H, NCHN), 7.40 (m, 6H, aryl), 7.17 (m, 6H, aryl), 6.95 (m, 4H, aryl), 6.75 (m, 2H, aryl), 4.83 (d, J = 14.3 Hz, 2H, CH2), 4.70 (s, 2H, CH), 4.10 (d, J = 14.3 Hz, 2H, CH2). 13C NMR (DMSO-d6): δ 158.3, 156.0, 135.8, 130.8, 130.3, 129.5, 129.4, 127.0, 119.1, 118.7, 115.6, 71.5, 45.9. IR (KBr, cm−1): 3435 (m), 3175 (s), 2927 (m), 1648 (s), 1599 (s), 1461 (s), 1373 (s), 1277 (s), 1106 (s), 757 (s).
Preparation of L4b.
This compound was prepared as a colorless solid from the reaction of 1 (2.00 g, 4.71 mmol), NH4Cl (0.38 g, 7.07 mmol) and triethyl orthoformate (15 mL) at 120 °C, followed by washing with diethyl ether by a similar procedure as described for the synthesis of L4a. Yield: 2.11 g (95%) (Found: C, 73.90; H, 5.78; N, 5.93. C29H27N2ClO2 requires: C, 73.95; H, 5.78; N, 5.95%). M.p.: 260–262 °C. 1H NMR (DMSO-d6): δ 10.48 (s, 2H, OH), 9.20 (s, 1H, NCHN), 7.38 (m, 6H, aryl), 7.17 (m, 6H, aryl), 7.07 (m, 2H, aryl), 6.93 (m, 2H, aryl), 6.72 (m, 2H, aryl), 4.85 (d, J = 14.3 Hz, 2H, CH2), 4.68 (s, 2H, CH), 4.09 (d, J = 14.3 Hz, 2H, CH2). 13C NMR (DMSO-d6): δ 158.3, 156.2, 135.9, 130.7, 130.1, 129.5, 129.4, 127.1, 119.0, 118.7, 115.8, 71.5, 46.0. IR (KBr, cm−1): 3424 (m), 3093 (s), 2954 (m), 1648 (s), 1600 (s), 1461 (s), 1374 (s), 1277 (s), 1197 (s), 1108 (s), 757 (s).
Preparation of L5.
This compound was prepared as a colorless solid from the reaction of 2 (2.00 g, 3.73 mmol), NH4Cl (0.30 g, 5.60 mmol) and triethyl orthoformate (15 mL) at 120 °C, followed by washing with diethyl ether by a similar procedure as described for the synthesis of L4a. Yield: 1.83 g (84%) (Found: C, 76.21; H, 7.43; N, 4.82. C37H43N2ClO2 requires: C, 76.20; H, 7.43; N, 4.80%). M.p.: 240–242 °C. 1H NMR (DMSO-d6): δ 9.25 (s, 1H, NCHN), 9.10 (s, 2H, OH), 7.41–7.32 (m, 6H, aryl), 7.21 (m, 2H, aryl), 7.10 (m, 4H, aryl), 6.70 (m, 4H, aryl), 5.33 (d, J = 14.6 Hz, 2H, CH2), 4.54 (s, 2H, CH), 4.17 (d, J = 14.6 Hz, 2H, CH2), 1.40 (s, 18H, C(CH3)3). 13C NMR (DMSO-d6): δ 166.0, 157.0, 154.3, 139.5, 136.0, 129.5, 129.3, 129.0, 127.4, 127.0, 121.9, 120.3, 71.2, 47.0, 34.5, 29.7. IR (KBr, cm−1): 3412 (m), 3090 (s), 2956 (s), 1638 (s), 1589 (m), 1438 (s), 1378 (s), 1208 (s), 1093 (s), 752 (s). Colorless crystals of 2(L5)·C6H6 suitable for X-ray structural analysis were grown from a benzene solution.
Preparation of L6.
This compound was prepared as a colorless solid from the reaction of 3 (2.00 g, 3.08 mmol), NH4Cl (0.25 g, 4.62 mmol) and triethyl orthoformate (15 mL) at 120 °C, followed by washing with diethyl ether by a similar procedure as in the synthesis of L4a. Yield: 1.84 g (86%) (Found: C, 77.69; H, 8.53; N, 4.05. C45H59N2ClO2 requires: C, 77.72; H, 8.55; N, 4.03%). M.p.: 235–237 °C. 1H NMR (DMSO-d6): δ 9.22 (s, 1H, NCHN), 8.84 (s, 2H, OH), 7.34 (m, 2H, aryl), 7.31 (m, 4H, aryl), 7.18 (d, J = 2.0 Hz, 2H, aryl), 7.04 (m, 4H, aryl), 6.61 (d, J = 2.0 Hz, 2H, aryl), 5.32 (d, J = 14.5 Hz, 2H, CH2), 4.44 (s, 2H, CH), 4.17 (d, J = 14.6 Hz, 2H, CH2), 1.38 (s, 18H, C(CH3)3), 1.14 (s, 18H, C(CH3)3). 13C NMR (DMSO-d6): δ 157.0, 151.8, 141.8, 138.5, 135.9, 129.5, 129.2, 127.2, 125.9, 123.9, 120.8, 71.1, 47.4, 34.7, 33.7, 31.2, 29.7. IR (KBr, cm−1): 3422 (w), 3085 (w), 2957 (s), 1637 (s), 1482 (s), 1362 (m), 1201 (s), 699 (s).
Preparation of (L4)Ti(NMe2)(Br)(THF) (7).
While stirring a THF solution (5 mL) of Ti(NMe2)4 (0.11 g, 0.50 mmol) was slowly added to a THF (15 mL) suspension of L4a (0.26 g, 0.50 mmol) at room temperature. After this mixture was stirred overnight at room temperature, the solution was filtered and the solvent was removed under reduced pressure. The resulting red solid was recrystallized from a benzene solution to give 7 as red microcrystals. Yield: 0.25 g (74%) (Found: C, 61.96; H, 5.62; N, 6.25. C35H38N3BrO3Ti requires: C, 62.14; H, 5.66; N, 6.21%). M.p.: 145–147 °C (dec.). 1H NMR (C6D6): δ 7.19 (m, 2H, aryl), 6.93 (m, 2H, aryl), 6.85 (m, 4H, aryl), 6.70 (m, 4H, aryl), 6.55 (m, 6H, aryl), 4.56 (br s, 1H, CH), 4.38 (m, 3H, CH and CH2), 3.74 (s, 6H, NCH3), 3.67 (m, 4H, THF), 3.49 (br s, 2H, CH2), 1.41 (m, 4H, THF). 13C NMR (C6D6): δ 208.4, 164.3, 163.8, 136.6, 129.4, 128.9, 128.6, 128.2, 127.8, 118.8, 118.1, 117.8, 75.1, 68.0, 53.1, 48.1, 25.0. IR (KBr, cm−1): 2962 (m), 1593 (s), 1482 (s), 1450 (s), 1260 (s), 1108 (s), 1034 (s), 886 (s), 799 (s).
Preparation of (L4)Zr(NMe2)(Br)(THF) (8).
This compound was prepared as pale yellow microcrystals from the reaction of L4a (0.26 g, 0.50 mmol) with Zr(NMe2)4 (0.14 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.24 g (68%) (Found: C, 58.27; H, 5.42; N, 5.80. C35H38N3BrO3Zr requires: C, 58.40; H, 5.32; N, 5.84%). M.p.: 177–179 °C (dec.). 1H NMR (C6D6): δ 7.20 (m, 2H, aryl), 6.90 (m, 8H, aryl), 6.68 (m, 8H, aryl), 4.54 (br s, 2H, CH2), 3.80 (m, 6H, CH2 and THF), 3.38 (m, 8H, CH and NCH3), 1.36 (m, 4H, THF). 13C NMR (C6D6): δ 209.0, 162.1, 138.1, 137.8, 137.2, 129.3, 129.2, 128.3, 125.7, 119.5, 119.2, 118.8, 117.9, 75.4, 69.4, 47.0, 45.5, 24.8. IR (KBr, cm−1): 2963 (m), 1595 (w), 1384 (m), 1260 (s), 1091 (s), 1018 (s), 798 (s).
Preparation of (L4)Hf(NMe2)(Br)(THF) (9).
This compound was prepared as colorless microcrystals from the reaction of L4a (0.26 g, 0.50 mmol) with Hf(NMe2)4 (0.18 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.29 g (72%) (Found: C, 52.15; H, 4.67; N, 5.26. C35H38N3BrO3Hf requires: C, 52.09; H, 4.75; N, 5.21%). M.p.: 230–232 °C (dec.). 1H NMR (C6D6): δ 7.24 (m, 2H, aryl), 6.90 (m, 8H, aryl), 6.70 (m, 8H, aryl), 4.53 (br s, 2H, CH2), 3.78 (m, 6H, CH and THF), 3.54 (s, 6H, NCH3), 3.30 (br s, 2H, CH2), 1.36 (m, 4H, THF). 13C NMR (C6D6): δ 213.6, 163.1, 138.7, 137.8, 130.1, 129.6, 128.8, 128.4, 127.6, 126.1, 120.5, 120.1, 118.0, 76.0, 69.5, 47.3, 45.2, 25.4. IR (KBr, cm−1): 2962 (s), 1595 (m), 1451 (m), 1384 (s), 1260 (s), 1090 (s), 1019 (s), 798 (s).
Preparation of (L4)Zr(NEt2)(Br)(THF) (10).
This compound was prepared as pale yellow crystals from the reaction of L4a (0.26 g, 0.50 mmol) with Zr(NEt2)4 (0.19 mg, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.27 g (73%) (Found: C, 59.50; H, 5.57; N, 5.64. C37H42N3BrO3Zr requires: C, 59.42; H, 5.66; N, 5.62%). M.p.: 120–122 °C (dec.). 1H NMR (C6D6): δ 7.18 (m, 4H, aryl), 6.94 (m, 6H, aryl), 6.81 (m, 2H, aryl), 6.66 (m, 6H, aryl), 5.33 (br s, 1H, CH), 4.56 (br s, 3H, CH and CH2), 4.26 (br s, 2H, CH2), 3.95 (m, 2H, THF), 3.83 (m, 2H, THF), 3.62 (br s, 2H, CH2), 3.35 (br s, 2H, CH2), 1.36 (m, 4H, THF), 1.25 (m, 6H, N(CH2CH3)2). 13C NMR (C6D6): δ 209.2, 162.1, 137.6, 137.4, 129.6, 129.3, 129.2, 128.7, 128.3, 125.6, 119.1, 118.2, 117.9, 75.6, 70.0, 47.3, 46.2, 24.7, 14.9. IR (KBr, cm−1): 2962 (s), 1595 (s), 1480 (s), 1453 (s), 1260 (s), 1090 (s), 1019 (s), 797 (s).
Preparation of (L4)Ti(NMe2)(Cl)(THF) (11).
This compound was prepared as red microcrystals from the reaction of L4b (0.24 g, 0.50 mmol) with Ti(NMe2)4 (0.11 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.22 g (70%) (Found: C, 66.58; H, 6.05; N, 6.60. C35H38N3ClO3Ti requires: C, 66.51; H, 6.06; N, 6.65%). M.p.: 154–156 °C (dec.). 1H NMR (C6D6): δ 7.20 (m, 2H, aryl), 6.95 (m, 4H, aryl), 6.86 (m, 6H, aryl), 6.67 (m, 6H, aryl), 4.49 (d, J = 10.1 Hz, 2H, CH2), 4.43 (d, J = 10.1 Hz, 2H, CH2), 3.86 (br s, 6H, CH and THF), 3.62 (s, 6H, NCH3), 1.41 (m, 4H, THF). 13C NMR (C6D6): δ 209.4, 166.1, 137.5, 129.4, 128.9, 128.2, 127.8, 126.0, 120.2, 119.5, 118.5, 118.3, 117.6, 75.4, 67.4, 52.9, 47.8, 25.0. IR (KBr, cm−1): 2962 (m), 1593 (m), 1480 (m), 1452 (m), 1260 (s), 1090 (s), 1018 (s), 798 (s).
Preparation of (L4)Zr(NMe2)(Cl)(THF) (12).
This compound was prepared as pale yellow crystals from the reaction of L4b (0.24 g, 0.50 mmol) with Zr(NMe2)4 (0.14 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.22 g (66%) (Found: C, 62.22; H, 5.66; N, 6.22. C35H38N3ClO3Zr requires: C, 62.24; H, 5.67; N, 6.22%). M.p.: 163–165 °C (dec.). 1H NMR (C6D6): δ 7.24 (m, 2H, aryl), 6.95 (m, 8H, aryl), 6.70 (m, 8H, aryl), 4.53 (m, 2H, CH2), 3.83 (s, 2H, CH), 3.63 (br s, 6H, CH2 and THF), 3.44 (br s, 3H, NCH3), 3.32 (br s, 3H, NCH3), 1.39 (m, 4H, THF). 13C NMR (C6D6): δ 210.1, 162.6, 138.5, 137.9, 137.3, 130.0, 129.6, 128.6, 128.3, 125.4, 119.3, 118.5, 118.0, 75.7, 68.1, 46.4, 45.5, 25.5. IR (KBr, cm−1): 2963 (m), 1593 (w), 1481 (m), 1384 (m), 1260 (s), 1091 (s), 1018 (s), 798 (s).
Preparation of (L4)Hf(NMe2)(Cl)(THF) (13).
This compound was prepared as colorless microcrystals from the reaction of L4b (0.24 g, 0.50 mmol) with Hf(NMe2)4 (0.18 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.27 g (70%) (Found: C, 55.11; H, 5.02; N, 5.56. C35H38N3ClHfO3 requires: C, 55.12; H, 5.02; N, 5.51%). M.p.: 214–216 °C (dec.). 1H NMR (C6D6): δ 7.23 (m, 2H, aryl), 6.95 (m, 8H, aryl), 6.65 (m, 8H, aryl), 4.54 (m, 2H, CH2), 3.86 (s, 2H, CH), 3.73–3.24 (m, 12H, CH2 and THF and NCH3), 1.39 (m, 4H, THF). 13C NMR (C6D6): δ 214.5, 164.3, 138.5, 137.4, 129.9, 129.6, 129.1, 128.5, 127.7, 125.4, 120.1, 119.2, 117.5, 76.1, 67.7, 47.8, 47.0, 25.5. IR (KBr, cm−1): 2962 (m), 1594 (w), 1450 (m), 1384 (m), 1260 (s), 1091 (s), 1019 (s), 798 (s).
Preparation of (L4)Zr(NEt2)(Cl)(THF) (14).
This compound was prepared as pale yellow crystals from the reaction of L4b (0.24 g, 0.50 mmol) with Zr(NEt2)4 (0.19 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.26 g (73%) (Found: C, 63.21; H, 6.06; N, 5.96. C37H42N3ClO3Zr requires: C, 63.18; H, 6.02; N, 5.97%). M.p.: 200–202 °C (dec.). 1H NMR (C6D6): δ 7.18 (m, 2H, aryl), 6.92 (m, 8H, aryl), 6.81 (m, 4H, aryl), 6.73 (m, 4H, aryl), 5.38 (br s, 1H, CH), 4.57 (br s, 4H, CH2), 4.27 (br s, 1H, CH), 3.81 (m, 2H, THF), 3.73 (m, 2H, THF), 3.58 (br s, 2H, CH2), 3.36 (br s, 2H, CH2), 1.37 (m, 4H, THF), 1.26 (m, 6H, N(CH2CH3)2). 13C NMR (C6D6): δ 209.3, 161.5, 136.8, 128.9, 128.5, 128.0, 127.5, 126.5, 125.0, 118.6, 117.4, 117.1, 74.8, 68.3, 46.6, 45.4, 24.1, 14.4. IR (KBr, cm−1): 2962 (m), 1594 (w), 1481 (m), 1384 (m), 1260 (s), 1089 (s), 1017 (s), 797 (s).
Preparation of (L5)Zr(NMe2)(Cl)(THF) (15).
This compound was prepared as pale yellow crystals from the reaction of L5 (0.29 g, 0.50 mmol) with Zr(NMe2)4 (0.14 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.28 g (70%) (Found: C, 65.55; H, 6.89; N, 5.34. C43H54N3ClO3Zr requires: C, 65.58; H, 6.91; N, 5.34%). M.p.: 156–158 °C (dec.). 1H NMR (C6D6): δ 7.44 (m, 2H, aryl), 6.93 (m, 8H, aryl), 6.69 (m, 6H, aryl), 4.75 (d, J = 14.1 Hz, 2H, CH2), 4.55 (s, 2H, CH), 3.64 (m, 4H, THF), 3.39 (d, J = 14.1 Hz, 2H, CH2), 3.29 (s, 6H, N(CH3)2), 1.76 (s, 9H, C(CH3)3), 1.39 (s, 9H, C(CH3)3), 1.37 (m, 4H, THF). 13C NMR (C6D6): δ 212.9, 162.7, 139.1, 138.6, 137.8, 137.3, 129.4, 129.2, 128.6, 127.8, 126.4, 125.6, 117.9, 78.4, 69.7, 50.1, 45.2, 35.7, 30.5, 25.6. IR (KBr, cm−1): 2961 (s), 1585 (s), 1416 (s), 1384 (m), 1257 (s), 1092 (s), 1020 (s), 870 (s), 810 (s).
Preparation of (L6)Ti(NMe2)(Cl)(THF) (16).
This compound was prepared as red crystals from the reaction of L6 (0.35 g, 0.50 mmol) with Ti(NMe2)4 (0.11 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.28 g (65%) (Found: C, 71.54; H, 8.23; N, 4.93. C51H70N3ClO3Ti requires: C, 71.52; H, 8.24; N, 4.91%). M.p.: 143–145 °C (dec.). 1H NMR (C6D6): δ 7.63 (d, J = 10.1 Hz, 2H, aryl), 6.97 (m, 4H, aryl), 6.86 (m, 6H, aryl), 6.40 (m, 2H, aryl), 4.58 (m, 3H, CH and CH2), 4.16 (br s, 1H, CH), 3.57 (m, 4H, THF), 3.53 (s, 6H, NCH3), 3.21 (br s, 2H, CH2), 1.98 (s, 9H, C(CH3)3), 1.92 (s, 9H, C(CH3)3), 1.38 (m, 4H, THF), 1.25 (s, 18H, C(CH3)3). 13C NMR (C6D6), δ 201.8, 157.3, 152.3, 140.9, 139.1, 136.5, 135.5, 128.2, 127.2, 126.8, 125.7, 124.3, 120.1, 70.4, 66.4, 48.5, 40.4, 34.3, 32.8, 30.2, 29.1, 25.8. IR (KBr, cm−1): 2962 (s), 1602 (w), 1443 (m), 1384 (m), 1260 (s), 1091 (s), 1018 (s), 799 (s).
Preparation of (L6)Zr(NMe2)(Cl)(THF) (17).
This compound was prepared as pale yellow crystals from the reaction of L6 (0.35 g, 0.50 mmol) with Zr(NMe2)4 (0.14 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.28 g (63%) (Found: C, 68.04; H, 7.85; N, 4.66. C51H70N3ClO3Zr requires C, 68.08; H, 7.84; N, 4.67%). M.p.: 158–160 °C (dec.). 1H NMR (C6D6): δ 7.57 (m, 4H, aryl), 7.09 (m, 4H, aryl), 7.02 (m, 2H, aryl), 6.92 (m, 4H, aryl), 4.40 (br s, 2H, CH2), 3.63 (br s, 8H, CH, CH2 and THF), 3.20 (s, 6H, NCH3), 1.91 (s, 9H, C(CH3)3), 1.89 (s, 9H, C(CH3)3), 1.22 (s, 13H, C(CH3)3 and THF), 1.20 (s, 9H, C(CH3)3). 13C NMR (C6D6): δ 206.2, 160.2, 139.3, 137.9, 137.6, 129.0, 128.9, 128.8, 128.7, 125.5, 125.4, 125.0, 124.1, 78.8, 68.1, 45.3, 39.1, 34.0, 33.9, 30.5, 30.4, 25.5. IR (KBr, cm−1): 2962 (s), 1384 (s), 1260 (s), 1092 (s), 1020 (s), 799 (s).
Preparation of (L6)Hf(NMe2)(Cl)(THF) (18)·2THF (18·2THF).
This compound was prepared as colorless crystals from the reaction of L6 (0.35 g, 0.50 mmol) with Hf(NMe2)4 (0.18 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.40 g (71%) (Found: C, 62.63; H, 7.67; N, 3.73. C59H86N3ClHfO5 requires: C, 62.64; H, 7.66; N, 3.71%). M.p.: 110–112 °C (dec.). 1H NMR (C6D6): δ 7.59 (s, 2H, aryl), 7.09 (m, 4H, aryl), 7.02 (m, 4H, aryl), 6.93 (d, J = 6.8 Hz, 2H, aryl), 6.11 (m, 2H, aryl), 5.15 (d, J = 13.0 Hz, 1H, CH), 4.64 (d, J = 11.1 Hz, 1H, CH), 4.40 (m, 2H, CH2), 3.72 (m, 12H, THF), 3.29 (s, 6H, NCH3), 3.15 (m, 2H, CH2), 1.93 (s, 9H, C(CH3)3), 1.88 (s, 9H, C(CH3)3), 1.22 (s, 18H, C(CH3)3), 1.21 (m, 12H, THF). 13C NMR (C6D6): δ 216.9, 160.7, 139.0, 138.7, 138.4, 138.2, 129.0, 128.9, 126.2, 125.6, 125.3, 124.8, 124.1, 78.9, 69.9, 50.5, 44.7, 35.8, 33.9, 31.7, 30.3, 25.4. IR (KBr, cm−1): 2962 (s), 1602 (w), 1442 (m), 1384 (m), 1260 (s), 1091 (s), 1018 (s), 799 (s).
Preparation of (L6)Zr(NEt2)(Cl)(THF) (19)·3C6H6 (19·3C6H6).
This compound was prepared as pale yellow crystals from the reaction of L6 (0.35 g, 0.50 mmol) with Zr(NEt2)4 (0.19 g, 0.50 mmol) in THF (20 mL) and recrystallization from a benzene solution by a similar procedure to the synthesis of 7. Yield: 0.42 g (73%) (Found: C, 73.40; H, 7.97; N, 3.64. C71H92N3ClO3Zr requires: C, 73.38; H, 7.98; N, 3.62%). M.p.: 102–104 °C (dec.). 1H NMR (C6D6): δ 7.57 (m, 2H, aryl), 7.12 (m, 24H, aryl), 7.08 (m, 2H, aryl), 6.90 (m, 2H, aryl), 6.12 (m, 2H, aryl), 5.48 (br s, 1H, CH), 4.76 (br s, 1H, CH), 4.46 (br s, 2H, CH2), 4.29 (br s, 2H, CH2), 3.75 (m, 4H, THF), 3.28 (m, 4H, CH2), 1.94 (s, 9H, C(CH3)3), 1.88(s, 9H, C(CH3)3), 1.21 (s, 18H, C(CH3)3), 1.14 (m, 10H, N(CH2CH3)2 and THF). 13C NMR (C6D6): δ 214.5, 160.2, 139.3, 138.4, 138.0, 137.9, 129.0, 128.0, 127.8, 127.5, 125.5, 125.1, 124.2, 79.0, 69.4, 51.0, 44.2, 35.8, 33.9, 31.7, 30.5, 25.4, 14.0. IR (KBr, cm−1): 2962 (s), 1603 (w), 1438 (m), 1384 (s), 1260 (s), 1092 (s), 1019 (s), 798 (s).
General procedure for polymerization of a rac-lactide
In a glovebox, rac-lactide (rac-LA) (0.360 g, 2.5 mmol), 2-propanol (0.01 mmol, in 0.5 mL of toluene or THF), complex (typically 0.01 mmol, in 0.5 mL of toluene or THF), and toluene or THF (4.0 mL) were added sequentially into a Schlenk flask with stirring. The flask containing the reaction mixture was subsequently placed in an oil bath and stirred for 0.5 h at 70 °C. The polymerization was quenched by the addition of cold acidified methanol. The precipitated polylactide was collected, washed with cold methanol several times, and dried in vacuum at 50 °C overnight.
X-ray crystallography
Single-crystal X-ray diffraction measurements were carried out on a Bruker SMART CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program.13 All structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELXL-97 program package.14 All the hydrogen atoms were geometrically fixed using the riding model. Disordered solvents in the voids of 15, 16 and 17 were modeled or removed using the SQUEEZE program.15 The crystal data and experimental data for L5, 10 and 14–19 are summarized in Tables 1 and 2. Selected bond lengths and angles are listed in Table 3.
Table 1 Crystal data and experimental parameters for compounds L5, 10 and 14–15
| Compound |
2(L5)·C6H6 |
10
|
14
|
15
|
| Formula |
C80H92N2Cl2O4 |
C37H42N3BrO3Zr |
C37H42N3ClO3Zr |
C43H54N3ClO3Zr |
| Formula weight |
1244.48 |
747.87 |
703.41 |
787.56 |
| Crystal system |
Monoclinic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
| Space group |
P21 |
P212121 |
P212121 |
C2221 |
|
a (Å) |
10.306(1) |
11.088(1) |
11.160(1) |
18.048(1) |
|
b (Å) |
31.646(4) |
14.680(1) |
14.834(2) |
24.570(1) |
|
c (Å) |
11.070(1) |
21.126(1) |
20.608(2) |
46.164(2) |
|
β (°) |
99.48(1) |
90 |
90 |
90 |
|
V (Å3) |
3561.2(7) |
3438.4(4) |
3411.5(7) |
20471.3(13) |
|
Z
|
2 |
4 |
4 |
16 |
|
D
calc (g cm−3) |
1.161 |
1.445 |
1.370 |
1.022 |
|
μ(Mo/Kα)calc (mm−1) |
0.143 |
1.521 |
0.440 |
0.300 |
| Size (mm) |
0.49 × 0.30 × 0.22 |
0.42 × 0.36 × 0.30 |
0.27 × 0.12 × 0.08 |
0.22 × 0.13 × 0.11 |
|
F(000) |
1332 |
1536 |
1464 |
6624 |
| 2θ range (°) |
4.00 to 50.50 |
3.86 to 55.24 |
4.16 to 55.10 |
3.76 to 50.50 |
| No. of reflns collected |
17778 |
20249 |
20265 |
18556 |
| No. of obsd reflns |
9062 |
7178 |
7149 |
15790 |
| Abs corr (Tmax, Tmin) |
0.97, 0.93 |
0.66, 0.57 |
0.97, 0.89 |
0.97, 0.94 |
|
R
|
0.055 |
0.029 |
0.032 |
0.051 |
|
R
w
|
0.135 |
0.060 |
0.063 |
0.121 |
| wR2 (all data) |
0.146 |
0.061 |
0.065 |
0.125 |
| Gof |
1.01 |
1.00 |
1.02 |
1.04 |
| CCDC |
975119
|
975120
|
975123
|
975124
|
Table 2 Crystal data and experimental parameters for compounds 16–19
| Compound |
16
|
17
|
18·2THF |
19·3C6H6 |
| Formula |
C51H70N3ClO3Ti |
C51H70N3ClO3Zr |
C59H86N3ClHfO5 |
C71H92N3ClO3Zr |
| Formula weight |
856.45 |
899.77 |
1131.25 |
1162.15 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
| Space group |
P212121 |
P212121 |
P212121 |
P212121 |
|
a (Å) |
12.392(1) |
12.729(2) |
12.781(1) |
15.211(1) |
|
b (Å) |
15.178(2) |
15.022(2) |
14.952(1) |
18.900(1) |
|
c (Å) |
29.978(3) |
30.104(4) |
30.090(2) |
22.591(2) |
|
V (Å3) |
5638.4(10) |
5756.1(12) |
5750.3(5) |
6494.5(8) |
|
Z
|
4 |
4 |
4 |
4 |
|
D
calc (g cm−3) |
1.009 |
1.038 |
1.307 |
1.189 |
|
μ(Mo/Kα)calc (mm−1) |
0.236 |
0.273 |
1.908 |
0.257 |
| Size (mm) |
0.45 × 0.22 × 0.18 |
0.60 × 0.18 × 0.13 |
0.45 × 0.40 × 0.26 |
0.45 × 0.42 × 0.39 |
|
F(000) |
1840 |
1912 |
2360 |
2480 |
| 2θ range (°) |
3.82 to 50.50 |
3.84 to 50.50 |
3.84 to 50.50 |
3.88 to 50.50 |
| No. of reflns collected |
10187 |
10419 |
28673 |
31878 |
| No. of obsd reflns |
9028 |
9645 |
9927 |
10522 |
| Abs corr (Tmax, Tmin) |
0.96, 0.90 |
0.97, 0.85 |
0.64, 0.48 |
0.91, 0.89 |
|
R
|
0.040 |
0.031 |
0.022 |
0.041 |
|
R
w
|
0.097 |
0.076 |
0.048 |
0.102 |
| wR2 (all data) |
0.101 |
0.077 |
0.048 |
0.108 |
| Gof |
1.04 |
1.04 |
1.01 |
1.04 |
| CCDC |
975125
|
975126
|
975127
|
975128
|
Table 3 Selected bond distances (Å) and bond angles (°) for compounds 10 and 14–19
| Compound |
M–O (av.) |
M–O(THF) |
M–X |
M–C |
M–N |
Sum angle of N(3) |
|
10 (Zr) |
2.136(2) |
2.401(2) |
Br: 2.661(1) |
2.416(2) |
2.046(2) |
359.3(2) |
|
14 (Zr) |
2.139(2) |
2.406(2) |
Cl: 2.498(1) |
2.419(2) |
2.046(2) |
359.5(2) |
|
15 (Zr) |
2.115(3) |
2.332(3) |
Cl: 2.499(1) |
2.397(4) |
2.046(4) |
359.7(4) |
|
16 (Ti) |
1.993(2) |
2.208(2) |
Cl: 2.410(1) |
2.252(2) |
1.901(2) |
359.9(2) |
|
17 (Zr) |
2.110(2) |
2.313(2) |
Cl: 2.501(1) |
2.401(2) |
2.044(2) |
359.6(2) |
|
18 (Hf) |
2.098(2) |
2.290(2) |
Cl: 2.478(1) |
2.370(3) |
2.041(3) |
359.7(3) |
|
19 (Zr) |
2.122(2) |
2.344(2) |
Cl: 2.515(1) |
2.410(4) |
2.052(3) |
360.0(3) |
Results and discussion
Synthesis and characterization of pro-ligands
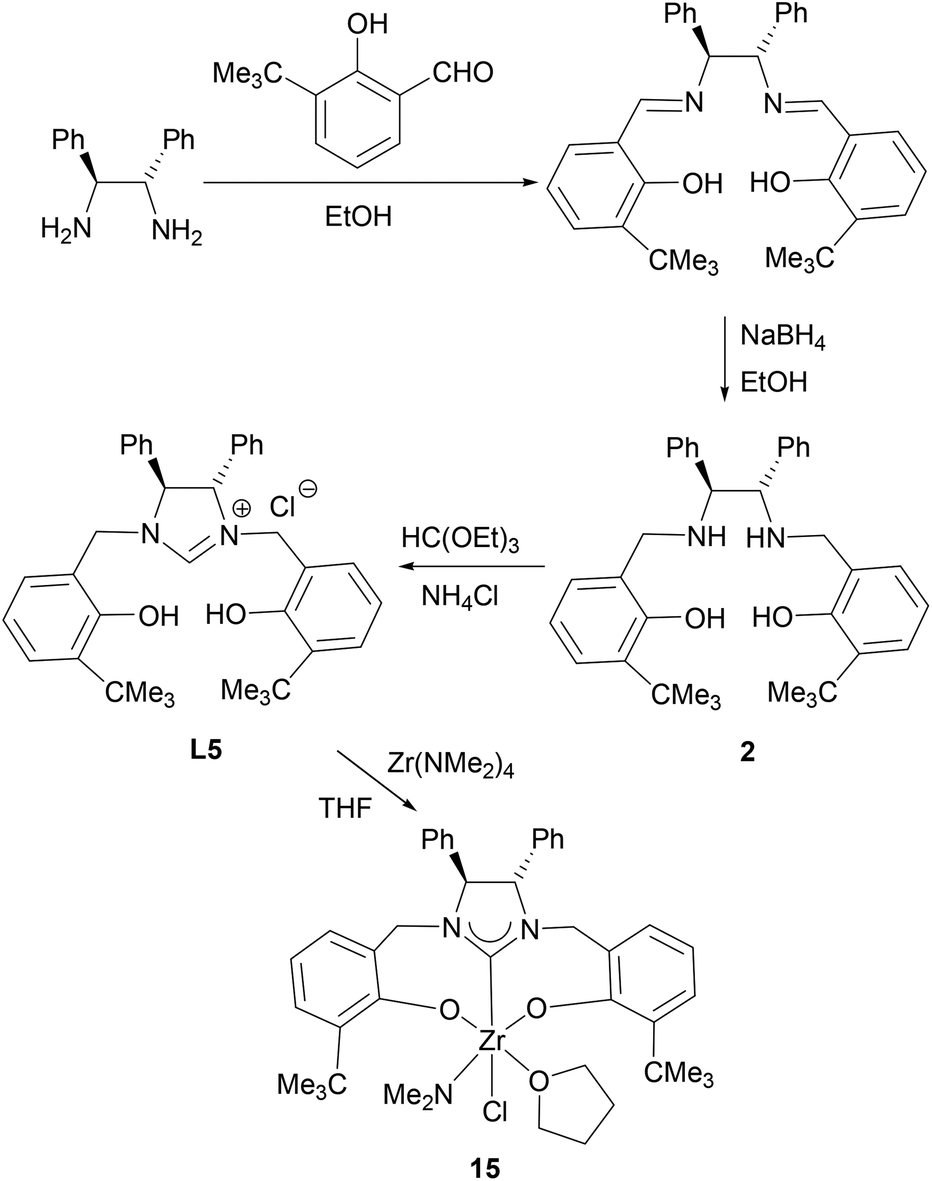
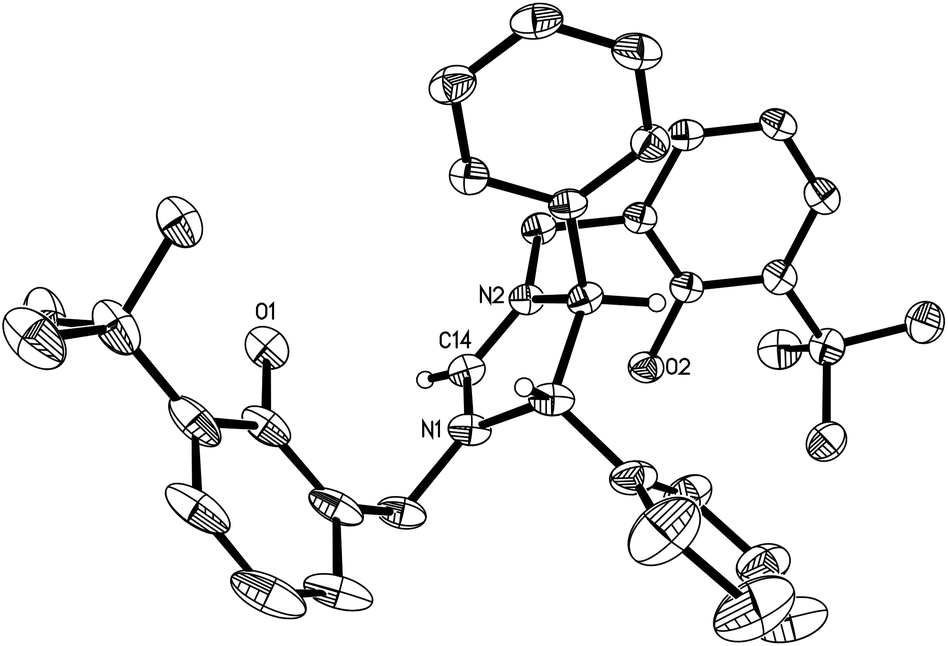
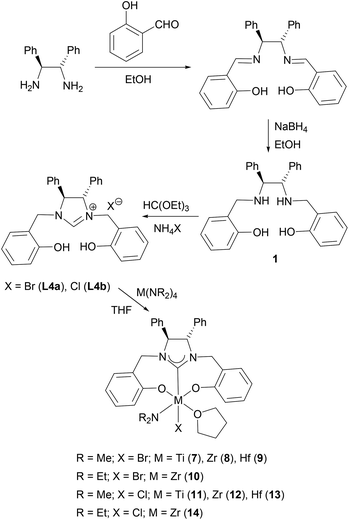
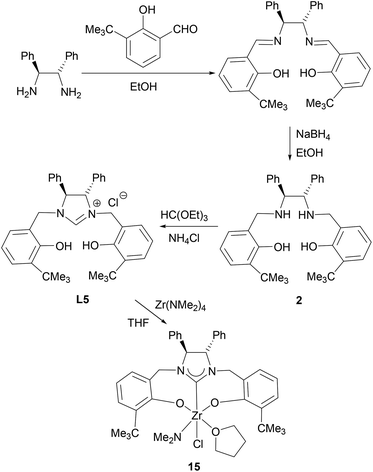
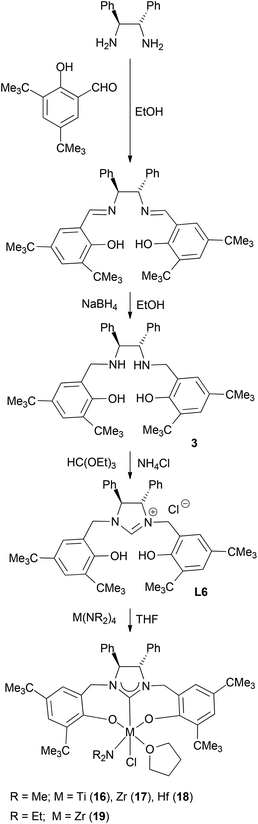
Condensation of (S,S)-diphenyl-1,2-ethanediamine with 1 equiv. of salicylaldehyde, 3-tert-butylsalicylaldehyde or 3,5-di-tert-butylsalicylaldehyde in absolute ethanol at ambient temperature, followed by reduction with an excess of NaBH4 in ethanol forms the chiral diamines 1–3 (Schemes 1–3). Subsequent cyclization of 1–3 with triethyl orthoformate in the presence of NH4Br or NH4Cl at 120 °C gives the imidazolium salts L4 (L4a and L4b), L5 and L6, respectively, in good yields (Schemes 1–3). All compounds are air-stable and have been characterized by various spectroscopic techniques and elemental analyses. The 1H and 13C NMR spectra are consistent with their C2-symmetric structure. In addition, besides aromatic stretches the IR spectra of L4–L6 also feature the characteristic O–H (at ca. 3420 cm−1) and strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretches (at ca. 1640 cm−1). The C2 symmetric structure of L5 was also confirmed by X-ray diffraction analysis (Fig. 1).
N stretches (at ca. 1640 cm−1). The C2 symmetric structure of L5 was also confirmed by X-ray diffraction analysis (Fig. 1).
 |
| | Scheme 1 Synthesis of complexes 7–14. | |
 |
| | Scheme 2 Synthesis of complex 15. | |
 |
| | Scheme 3 Synthesis of complexes 16–19. | |
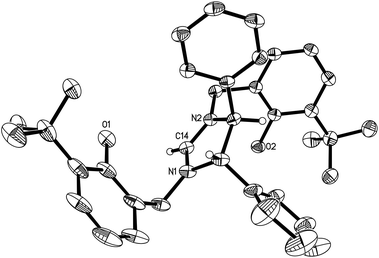 |
| | Fig. 1 Molecular structure of the cation in L5 (thermal ellipsoids drawn at the 35% probability level). | |
Synthesis and characterization of complexes
Amine elimination between M(NMe2)4 and protic reagents is a very efficient way for the synthesis of group 4 metal amide complexes.16 Hence, a similar reaction is expected for the acidic protons in the ligands L4 (L4a and L4b), L5 and L6 and metal amides. In fact, treatment of M(NR2)4 (M = Ti, Zr, Hf; R = Me, Et) with 1 equiv. of L4 in THF gives, after recrystallization from a benzene solution, the chiral titanium amides (L4)Ti(NMe2)(Br)(THF) (7) and (L4)Ti(NMe2)(Cl)(THF) (11), zirconium amides (L4)Zr(NMe2)(Br)(THF) (8), (L4)Zr(NEt2)(Br)(THF) (10), (L4)Zr(NMe2)(Cl)(THF) (12) and (L4)Zr(NEt2)(Cl)(THF) (14), and hafnium amides (L4)Hf(NMe2)(Br)(THF) (9) and (L4)Hf(NMe2)(Cl)(THF) (13), respectively, in good yields (Scheme 1). Similarly, the reactions of L5 or L6 with 1 equiv. of M(NR2)4 (M = Ti, Zr, Hf; R = Me, Et) also afford the chiral titanium amide (L6)Ti(NMe2)(Cl)(THF) (16), zirconium amides (L5)Zr(NMe2)(Cl)(THF) (15), (L6)Zr(NMe2)(Cl)(THF) (17) and (L6)Zr(NEt2)(Cl)(THF) (19), and the hafnium amide (L6)Hf(NMe2)(Cl)(THF) (18), respectively, in good yields (Schemes 2 and 3).
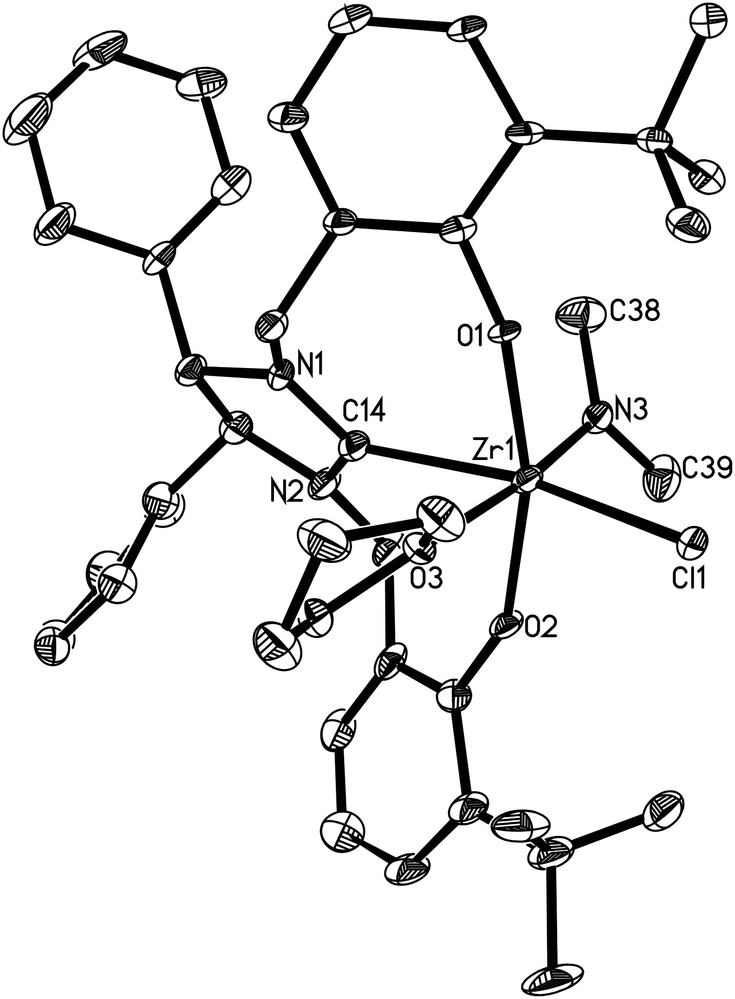
Complexes 7–19 are stable in a dry nitrogen atmosphere, while they are very sensitive to moisture. They are soluble in organic solvents such as THF, DME, pyridine, toluene, and benzene, and only sparingly soluble in aliphatic solvents such as n-hexane. They have been characterized by various spectroscopic techniques and elemental analyses. The 1:1:1 ratio between the NR2 (R = Me, Et) group, the coordinate THF and the ligand L4, L5 or L6 is established by 1H NMR spectroscopy. Furthermore, the characteristic O–H and CN stretches at ca. 3420 and 1640 cm−1 in L4–L6 disappear upon treatment with M(NR2)4, supporting the formation of complexes 7–19. The solid-state structures of 10 and 14–19 have further been confirmed by X-ray diffraction analyses.
Complexes 16 and 17 are isostructural. The M4+ ion features a distorted-octahedral ligand environment in (L4)Zr(NEt2)(Br)(THF) (10), (L4)Zr(NEt2)(Cl)(THF) (14), (L5)Zr(NMe2)(Cl)(THF) (15), (L6)Ti(NMe2)(Cl)(THF) (16), (L6)Zr(NMe2)(Cl)(THF) (17), (L6)Hf(NMe2)(Cl)(THF) (18) and (L6)Zr(NEt2)(Cl)(THF) (19) (Fig. 2–8). Complexes 10 and 14–19 represent, to our knowledge, the first example of the structurally characterized group 4 chiral NHC–metal complex. The average M–O distances are 1.993(2) Å for Ti, 2.110(2) Å to 2.139(2) Å for Zr, and 2.098(2) Å for Hf. The Zr–Br distance is 2.661(1) Å (for 10), and the M–Cl distance is 2.410(1) Å for Ti, 2.498(1) Å to 2.515(1) Å for Zr, and 2.478(1) Å for Hf. The M–C(carbene) distances of 2.252(2) Å for Ti, 2.397(4) Å to 2.419(2) Å for Zr, and 2.370(3) Å for Hf are in the typical range for a M–C σ-bond. These structural data can be compared with those found in [η3-O,C,O-{(3,5-(Me3C)2-C6H2O)2N2C3H4}]M(OiPr)(Cl)(THF) (M = Ti, Zr)17a,b and [η3-O,C,O-{(3,5-(Me3C)2-C6H2O)2N2C3H4}]MCl2(THF) (M = Ti, Zr).17c,d Furthermore, the M–NR2 (R = Me, Et) distances are short with 1.901(2) Å for Ti, 2.044(2) Å to 2.052(3) Å for Zr, and 2.041(3) Å for Hf. This in combination with the planar geometry around the nitrogen atom N(3) suggests that the sp2-hybridized N-atom engages in N(pπ)→M(dπ) interactions.16
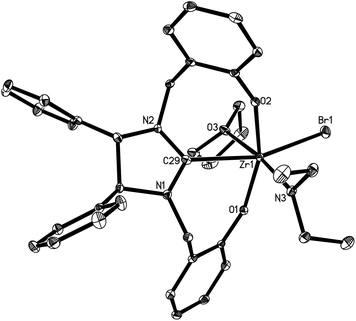 |
| | Fig. 2 Molecular structure of 10 (thermal ellipsoids drawn at the 35% probability level). | |
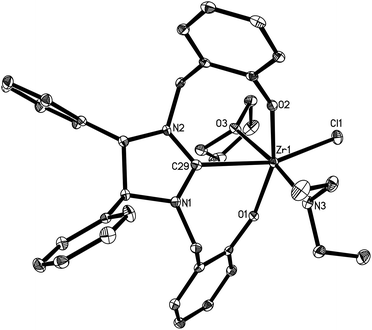 |
| | Fig. 3 Molecular structure of 14 (thermal ellipsoids drawn at the 35% probability level). | |
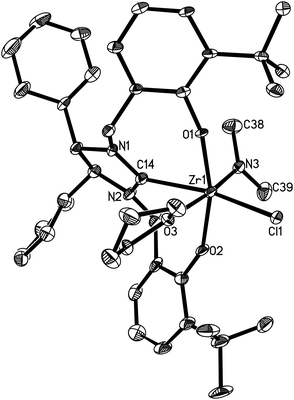 |
| | Fig. 4 Molecular structure of 15 (thermal ellipsoids drawn at the 35% probability level). | |
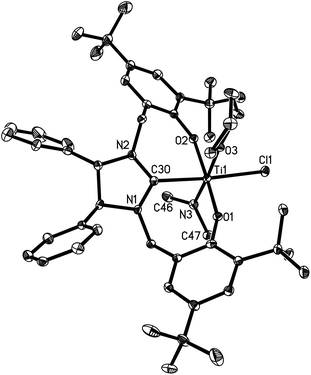 |
| | Fig. 5 Molecular structure of 16 (thermal ellipsoids drawn at the 35% probability level). | |
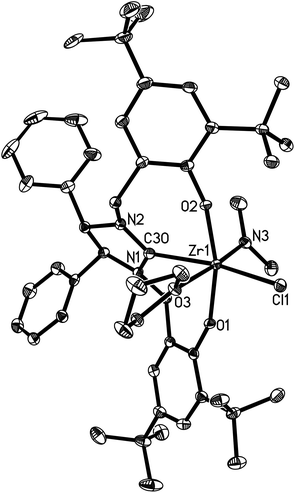 |
| | Fig. 6 Molecular structure of 17 (thermal ellipsoids drawn at the 35% probability level). | |
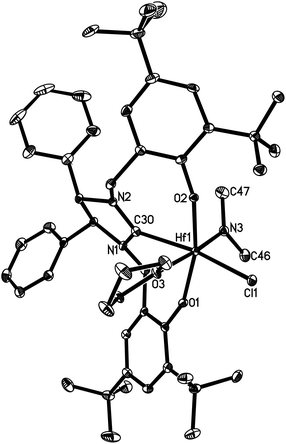 |
| | Fig. 7 Molecular structure of 18 (thermal ellipsoids drawn at the 35% probability level). | |
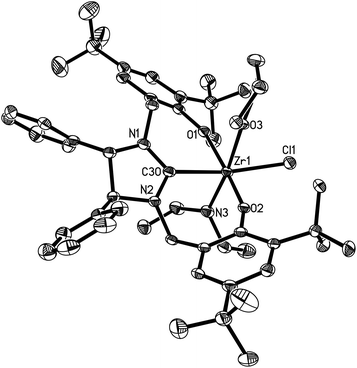 |
| | Fig. 8 Molecular structure of 19 (thermal ellipsoids drawn at the 35% probability level). | |
Polymerization of rac-lactide
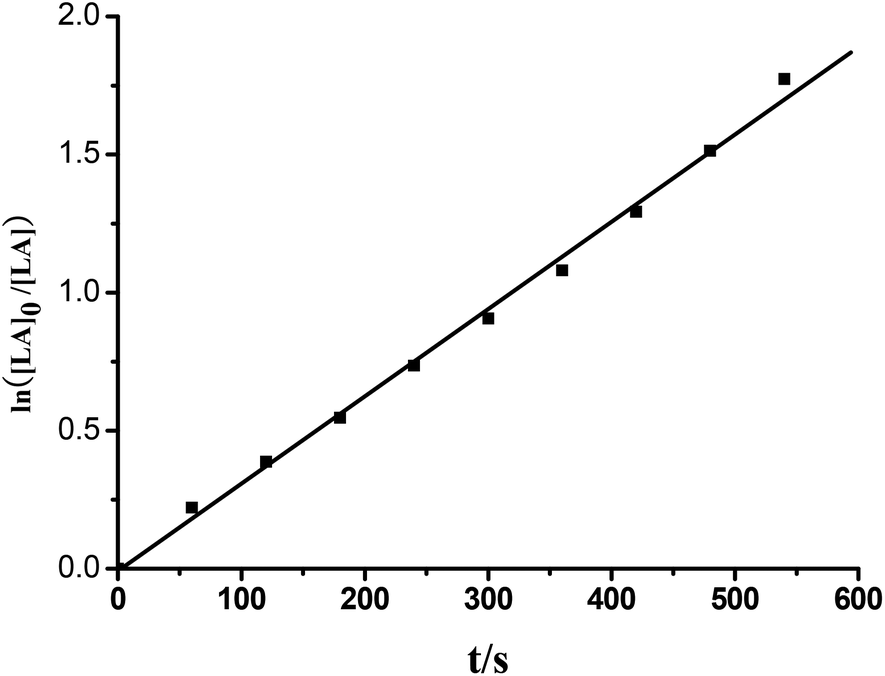
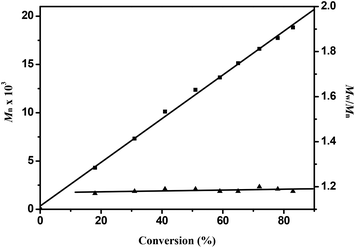
Efficient ring opening polymerization (ROP) of rac-lactide (rac-LA) is achieved by the chiral group 4 NHC–metal complexes 7–19 under the conditions listed in Table 4. With the zirconium and hafnium complexes 8–10 and 12–14 complete conversion of 250 equiv. of lactide is achieved within 0.5 h at 70 °C in toluene at [rac-LA] = 0.5 mol L−1 (Table 4, entries 2, 7, 8 and 10–12). A more detailed analysis was undertaken for 8 that acts as a single-site initiator for the controlled polymerization of rac- LA. The formed polylactides have experimental Mn values (Mn,exp) that are very close to the calculated Mn,calcd values and that the molar mass distributions are very narrow (Mw/Mn = 1.18–1.21; Table 4, entries 2–6). In addition, for complex 8 a first order kinetic dependence on the concentration of rac-LA and no induction period were observed (Fig. 9). The Mn,exp values increase linearly with the monomer conversion, whereas the Mw/Mn values remain in the narrow range of 1.17–1.22 (Fig. 10). However, when the more bulky ligands L5 and L6 are used, the polymerization with the zirconium and hafnium complexes 15 and 17–19 is slightly slower (Table 4, entries 13, and 15–17), presumably because of the increased steric hindrance at the metal centers. Although the zirconium and hafnium complexes are effective catalysts for the polymerization of rac-LA, the titanium complexes 7, 11 and 16 exhibit only poor catalytic activity (Table 4, entries 1, 9 and 14), consistent with the smaller ionic radius of Ti4+. These differences also prevail in THF solution (Table 4, entries 18–30), but the polymerization with these group 4 initiators/catalysts proceeds much more slowly in THF (Table 4, entries 18–30), most likely a consequence of competitive monomer–solvent coordination to the metal ion. A similar competition was observed for the organoyttrium and organoaluminum catalysts.16a,18 In the absence of isopropanol, no polymerization occurs in toluene or THF solution even when heated at 70 °C for 72 h. The polymer microstructure, as determined by homo-decoupled 1H NMR experiments,19 shows that the polylactides are heterotactic-rich polylactides under conditions examined. The catalytic activities of 7–19 resemble that of [η3-O,C,O-{(3,5-(Me3C)2-C6H2O)2N2C3H4}]M(OiPr)(Cl)(THF) (M = Ti, Zr),17a,b while the microstructures of the resulting polylactides are similar to those initiated by [(R)-(2-O-C6H4)CHNCH(Me)(C6H5)]2Zr(OiPr)2.5c
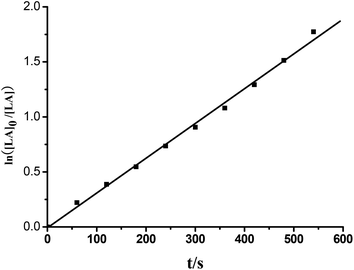 |
| | Fig. 9 Ln([LA]0/[LA]) vs. time plot for the ROP of rac-LA initiated by complex 8. Conditions: precat./isopropanol/LA (mol/mol/mol) = 1/1/150, [LA] = 0.5 mol L−1, solvent = toluene, T = 70 °C. kobs = 3.14 × 10−3 s−1. | |
 |
| | Fig. 10
M
n and Mw/Mnvs. conversion plots for the ROP of rac-LA initiated by complex 8. Conditons: precat./isopropanol/LA (mol/mol/mol) = 1/1/150, [LA] = 0.5 mol L−1, solvent = toluene, T = 70 °C. | |
Table 4 Polymerization of rac-lactide catalyzed by chiral group 4 NHC-complexes 7–19a
Conclusions
Chiral group 4 NHC–metal complexes were prepared and structurally characterized. These complexes represent the first example of the structurally characterized group 4 chiral NHC–metal complex, and they can initiate the ring-opening polymerization of rac-lactide in the presence of isopropanol, leading to the heterotactic-rich polylactides. Nevertheless, the reactivity is strongly influenced by the size of the metal ion and the solvents. For example, fast polymerization is observed in toluene, whereas the conversion is slow in THF because of competitive monomer–solvent coordination to the metal ions. The zirconium and hafnium complexes are efficient precatalysts for polymerization of rac-LA, while the titanium complexes exhibit only poor catalytic activity because of the smaller ionic radius of Ti4+. Further studies will focus on the application of these complexes towards other asymmetric reactions and the exploration of new group 4 metal complexes based on chiral ligands.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21172022, 21272026), the Specialized Research Fund for the Doctoral Program of Higher Education, the Fundamental Research Funds for the Central Universities, the Beijing Municipal Commission of Education, and the Deutsche Forschungsgemeinschaft (DFG) through the Emmy-Noether program (WA 2513/2-2).
Notes and references
- For selected reviews, see:
(a) E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305–313 CrossRef CAS PubMed;
(b) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS;
(c) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC;
(d) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS;
(e) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
-
(a) M. S. Reeve, S. P. McCarthy, M. J. Downey and R. A. Gross, Macromolecules, 1994, 27, 825–831 CrossRef CAS;
(b) J. R. Sarasua, R. E. Prud'homme, M. Wisniewski, A. LeBorgne and N. Spassky, Macromolecules, 1998, 31, 3895–3905 CrossRef CAS;
(c) N. Nomura, J. Hasegawa and R. Ishii, Macromolecules, 2009, 42, 4907–4909 CrossRef CAS.
-
(a) Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS;
(b) H. Tsuji and Y. Ikada, Polymer, 1999, 40, 6699–6708 CrossRef CAS.
- For selected recent reviews, see:
(a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC;
(b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed;
(c) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed;
(d) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC;
(e) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
-
(a) F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1993–1999 CrossRef CAS PubMed;
(b) R. H. Howard, C. Alonso-Moreno, L. M. Broomfield, D. L. Hughes, J. A. Wright and M. Bochmann, Dalton Trans., 2009, 8667–8682 RSC;
(c) A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437–1443 RSC;
(d) J. Lee, Y. Kim and Y. Do, Inorg. Chem., 2007, 46, 7701–7703 CrossRef CAS PubMed;
(e) M. Hu, M. Wang, P. Zhang, K. Jin, Y. Chen and L. Sun, Polym. Bull., 2012, 68, 1789–1799 CrossRef CAS.
- For selected reviews, see:
(a) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951–961 CrossRef CAS;
(b) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC;
(c) S. Roland and P. Mangeney, Top. Organomet. Chem., 2005, 15, 191–229 CrossRef CAS;
(d) R. E. Douthwaite, Coord. Chem. Rev., 2007, 251, 702–717 CrossRef CAS PubMed;
(e) L. H. Gade and S. Bellemin-Laponnaz, Coord. Chem. Rev., 2007, 251, 718–725 CrossRef CAS PubMed;
(f) D. R. Snead, H. Seo and S. Hong, Curr. Org. Chem., 2008, 12, 1370–1387 CrossRef CAS;
(g) L. N. Gu, G. B. Zhu, H. B. Song and G. F. Zi, Chin. J. Org. Chem., 2009, 29, 1499–1507 CAS.
- T. J. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225–3228 CrossRef CAS PubMed.
-
(a) D. Martin, S. Kehrli, M. d'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416–8417 CrossRef CAS PubMed;
(b) K. S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898–2900 CrossRef CAS PubMed.
- S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402–3415 CrossRef CAS PubMed.
- M. C. Perry, X. H. Cui, M. T. Powell, D. R. Hou, J. H. Reibenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113–123 CrossRef CAS PubMed.
-
(a) W. A. Herrmann, L. J. Goossen, C. Köcher and G. R. Artus, Angew. Chem., Int. Ed. Engl., 1996, 35, 2805–2807 CrossRef CAS PubMed;
(b) W.-L. Duan, M. Shi and G.-B. Rong, Chem. Commun., 2003, 2916–2917 RSC.
- B. Cardinal-David, D. E. A. Raup and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 5345–5347 CrossRef CAS PubMed.
-
G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
-
(a)
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structure from Diffraction Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- SQUEEZE: P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- For selected papers, see:
(a) G. Zi, Q. Wang, L. Xiang and H. Song, Dalton Trans., 2008, 5930–5944 RSC;
(b) L. Xiang, H. Song and G. Zi, Eur. J. Inorg. Chem., 2008, 1135–1140 CrossRef CAS PubMed;
(c) G. Zi, X. Liu, L. Xiang and H. Song, Organometallics, 2009, 28, 1127–1137 CrossRef CAS;
(d) G. Zi, F. Zhang, X. Liu, L. Ai and H. Song, J. Organomet. Chem., 2010, 695, 730–739 CrossRef CAS PubMed;
(e) G. Zi, F. Zhang, L. Xiang, Y. Chen, W. Fang and H. Song, Dalton Trans., 2010, 39, 4048–4061 RSC;
(f) Q. Wang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1583–1591 CrossRef CAS PubMed;
(g) G. Zi, J. Organomet. Chem., 2011, 696, 68–75 CrossRef CAS PubMed.
-
(a) C. Romain, B. Heinrich, S. Bellemin-Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC;
(b) C. Romain, L. Brelot, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2010, 29, 1191–1198 CrossRef CAS;
(c) H. Aihara, T. Matsuo and H. Kawaguchi, Chem. Commun., 2003, 2204–2205 RSC;
(d) D. Zhang, H. Aihara, T. Watanabe, T. Matsuo and H. Kawaguchi, J. Organomet. Chem., 2007, 692, 234–242 CrossRef CAS PubMed;
(e) S. Barroso, S. R. M. M. de Aguiar, R. F. Munhá and A. M. Martins, J. Organomet. Chem., 2013 DOI:10.1016/j.jorganchem.2013.11.041.
-
(a) Q. Wang, L. Xiang, H. Song and G. Zi, J. Organomet. Chem., 2009, 694, 691–696 CrossRef CAS PubMed;
(b) Q. Wang, F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2011, 696, 2186–2192 CrossRef CAS PubMed;
(c) A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS PubMed;
(d) N. Zhao, Q. Wang, G. Hou, H. Song and G. Zi, J. Organomet. Chem., 2014, 754, 51–58 CrossRef CAS PubMed;
(e) N. Zhao, Q. Wang, G. Hou, H. Song and G. Zi, Inorg. Chim. Acta, 2014, 413, 128–135 CrossRef CAS PubMed;
(f) W. Ren, L. Chen, N. Zhao, Q. Wang, G. Hou and G. Zi, J. Organomet. Chem., 2014, 758, 65–72 CrossRef CAS PubMed.
-
(a) J. E. Kasperczyk, Macromolecules, 1995, 28, 3937–3939 CrossRef CAS;
(b) K. A. M. Thakur, R. T. Kean, E. S. Hall, M. A. Doscotch, J. I. Siepmann and E. J. Munson, Macromolecules, 1997, 30, 2422–2428 CrossRef CAS;
(c) J. E. Kasperczyk, Polymer, 1999, 40, 5455–5458 CrossRef CAS;
(d) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed;
(e) M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 35, 7700–7707 CrossRef CAS;
(f) F. Drouin, P. O. Oguadinma, T. J. J. Whitehorne, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139–2147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of a representative polymer sample. CCDC 975119–975120 and 975123–975128. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00510d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a