 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile coordination modes of bis[5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl]alkanes in Cu(II) complexes†
Alexey N.
Gusev
*a,
Ivan
Nemec
b,
Radovan
Herchel
b,
Eziz
Bayjyyev
a,
Galyna A.
Nyshchimenko
a,
Grigory G.
Alexandrov
c,
Igor L.
Eremenko
c,
Zdeněk
Trávníček
b,
Miki
Hasegawa
d and
Wolfgang
Linert
*e
aGeneral Chemistry Department, Taurida National V.I. Vernadsky University, Simferopol, Ukraine. E-mail: galex0330@gmail.com
bDepartment of Inorganic Chemistry, Faculty of Science, Palacky University, Olomouc, Czech Republic
cKurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
dDepartment of Chemistry and Biological Science, College of Science and Engineering, Aoyama Gakuin University, Kanagawa 252-5258, Japan
eInstitute for Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163-AC, A-1060 Vienna, Austria. E-mail: wolfgang.linert@tuwien.ac.at
First published on 27th February 2014
Abstract
Nine new mononuclear and polynuclear Cu(II) complexes [Cu(H2L2)Cl]Cl·3H2O (1), [Cu(H2L3)Cl]Cl·H2O (2), [Cu(H2L4)Cl]Cl·2.5H2O (3), [Cu3(μ3-L1)2(H2O)3](ClO4)2·H2O (4), [Cu4(μ-HL1)4](ClO4)8·CH3OH·5H2O (5), [Cu2(HL3)2](ClO4)2·2H2O (6a), [Cu2(μ-HL3)2](ClO4)2·H2O (6b), [Cu2(μ-HL3)(L3)Cu(teta)](ClO4)3·2H2O (7) and [Cu2(H2L3)2(ox)](ClO4)2·2H2O·2MeOH (8) containing [5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl]alkanes (H2Ln, n = 1–4) in combination with other ligands, such as chlorido, aqua, triethylenetetramine (teta) and/or oxalato (ox2−), were synthesized and characterized by various techniques such as elemental analysis, FTIR, NMR and UV-Vis spectroscopy. X-ray structures of H2L3 and H2L4 as well as complexes 1–8 were determined. The X-ray structures revealed that relatively small composition and structural changes in the H2Ln ligands have a substantial impact on the coordination geometries of the complexes themselves as well as on their resulting magnetic properties. It has been found that the geometries of the complexes vary from square-pyramidal to trigonal-bipyramidal (with τ ranging from 0.00 to 0.96) and, moreover, that the trigonal bipyramidal geometry becomes more preferable with the increase in the length of the polymethylene chain within the corresponding H2Ln ligand. The magnetic properties of the polynuclear compounds 4, 5, 6, 7 and 8 were analysed using the spin Hamiltonian formalism, which revealed the presence of antiferromagnetic exchange in the polynuclear systems mediated by the title ligands. The significant effect of the geometric parameters on the Cu⋯Cu exchange interactions in the polynuclear complexes is discussed.
Introduction
The coordination chemistry of flexible ditopic ligands has attracted increasing interest due to their demonstrated versatility in the formation of exciting new polynuclear coordination complexes. The interest in these systems can be justified by the numerous applications arising from the unusual properties they may exhibit, such as new electronic, optical, magnetic and catalytic properties.1 Ditopic ligands, in which chelating units are joined by a flexible moiety, can lead to the formation of a large family of coordination cages showing remarkable variety and complexity.2,3 In recent years, a great deal of attention has been devoted to the synthesis of a new type of ligand containing five-membered polyazole rings.4 Among them, pyrazole and its derivatives have drawn widespread attention, due to their practical applications (as new optic and magnetic materials) and the surprising structural variability of the resulting complexes. The group of Prof. M. Ward has demonstrated numerous examples of polyhedral cages based on relatively simple bis(pyrazolyl-pyridine) bridging ligands and transition metal cations. A great number of polyhedral shapes of polynuclear complexes, e.g. M4L6 tetrahedra, M6L9 trigonal prisms, M8L12 cubes and ‘cuneane’, M12L18 truncated tetrahedra, and M16L24 tetra-capped truncated tetrahedra, have been described.5In our previous work we described a new type of bis-bidentate bridging ligand, namely bis[5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl]propane, which is structurally related to the above-mentioned pyrazolyl ligands.6 We demonstrated that the ligand can act as a tetradentate chelate towards a single Ln(III) atom. However, considering the conformational flexibility of such ligands and the large number of donor atoms, other coordination modes resulting from different preparation procedures can be expected.
In the present paper we report on the Cu(II) coordination chemistry of four different bis[5-(2-pyridyl)-1,2,4-triazole-3-yl]alkanes – new tetradentate ligands in which two bidentate chelating pyridyl-triazole fragments are linked by a flexible polymethylene chain (Scheme 1): H2L1 (linked via methylene), H2L2 (ethylene), H2L3 (propylene) and H2L4 (butylene). The target of this work was to study how: (a) the presence of three nitrogen atoms (pyridyl-triazole fragments) potentially capable of coordination to metal ions gives new coordination possibilities not accessible using pyrazolyl-pyridine chelating sites; (b) different lengths of the aliphatic linkers imply different overall molecular shapes and nuclearities; (c) the presence of the acidic hydrogen atom allows varying the composition and structure of the target complexes by changing the acidity of the medium. In this work we report on the synthesis, crystal structures and magnetic properties of a series of new Cu(II) complexes with various nuclearities: [Cu(H2L2)Cl]Cl·3H2O (1), [Cu(H2L3)Cl]Cl·H2O (2), [Cu(H2L4)Cl]Cl·2.5H2O (3), [Cu3(L1)2(H2O)3](ClO4)2·H2O (4), [Cu4(HL1)4](ClO4)8·CH3OH·5H2O (5), [Cu2(HL3)2](ClO4)2·2H2O (6a), [Cu2(HL3)2](ClO4)2·H2O (6b), [Cu2(HL3)(L3)Cu(teta)](ClO4)3·2H2O (7, teta = triethylenetetramine) and [Cu2(H2L3)2(ox](ClO4)2·2H2O·2MeOH (8, ox = oxalate anion).
 | ||
| Scheme 1 Schematic representation of H2Ln. | ||
Experimental
General details
The reagents and solvents employed were commercially available and used as received without further purification. The C, H, and N microanalyses were carried out with a Perkin-Elmer 240 elemental analyser. The IR spectra were recorded on a Nicolet Magna FT-IR 750 spectrometer using KBr pellets in the range of 4000–400 cm−1. Thermogravimetric analysis (TGA) data were collected with a Paulik–Paulik–Erdey Q-derivatograph in air at a heating rate of 10 K min−1. UV-Vis spectra were obtained on a Perkin-Elmer Lambda 900 spectrometer using the diffuse reflectance technique, with MgO as a reference. 1H NMR spectra were recorded on a Bruker VXR-400 spectrometer. The samples for NMR spectral measurements were dissolved in DMSO-d6. Magnetic measurements were carried out on a Quantum Design PPMS-9SQUID magnetometer under an external magnetic field of 5000 G in the temperature range of 2–300 K. The diamagnetic contributions of the samples were estimated from Pascal's constants.The commercially available CuCl2·2H2O and Cu(ClO4)2·6H2O were used as reactants. The syntheses of bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes were described previously,6 but here they were prepared by a modified method described below.
Synthesis of bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes (a general method)
Sodium (0.8 g) was added carefully to 35 cm3 of methanol followed by addition of 2-pyridinecarbonitrile (10.9 g, 104 mmol). The solution was left undisturbed for 30 min. Dihydrazide of the related dicarboxylic acid (malonic, succinic, glutaric and adipic) (45 mmol) was added to the solution followed by addition of 1 cm3 of acetic acid. The reaction mixture was stirred and refluxed for 5 hours. A light yellow solid formed during the reaction was filtered off upon cooling and air dried for one day. The product was heated in a vacuum at 200–210 °C for 30 min. The resulting triazole was recrystallized from the MeOH–water solution.Preparation of complexes 1–3 (a general method)
A mixture of the appropriate H2L2–4 ligand (1 mmol), and CuCl2·2H2O (0.171 g, 1 mmol) in a water–MeOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture (10 cm3) afforded a clear blue-green solution after stirring for a few minutes. The solution was allowed to stand for two weeks and yielded greenish-blue X-ray quality crystals.
:1 v/v) mixture (10 cm3) afforded a clear blue-green solution after stirring for a few minutes. The solution was allowed to stand for two weeks and yielded greenish-blue X-ray quality crystals.
[Cu(H2L2)Cl]Cl·3H2O (1) Anal. calc. for C16H20Cl2CuN8O3. Calcd C 37.92; H 3.98; N 22.11; found: C 37.79; H 3.82; N 22.18.
[Cu(H2L3)Cl]Cl·H2O (2) Anal. calc. for C17H18Cl2CuN8O. Calcd C 42.11; H 3.74; N 23.11; found: C 42.26; H 3.97; N 23.24.
[Cu(H2L4)Cl]Cl·2.5H2O (3) Anal. calc. for C18H23Cl2CuN8O2.5. Calcd C 41.11; H 4.41; N 21.31; found: C 41.18; H 4.68; N 21.21.
Preparation of complex 4
H2L1 (0.302 g, 1 mmol) was suspended in a water–MeOH (1:1 v/v) mixture (10 cm3), and a solution of Cu(ClO4)2·6H2O (0.371 g, 1 mmol) in MeOH (10 cm3) was added. Then, the solution of 80 mg of NaOH in 5 cm3 of water was added. The reaction mixture was stirred upon heating for 3 hours and complex 4 precipitated as a blue solid; it was filtered off, washed with water and dried in air. Single crystals suitable for X-ray analysis were obtained upon recrystallization from water.
[Cu3(L1)2(H2O)3](ClO4)2·H2O (4) Anal. calc. for C30H28Cl2Cu3N16O12: C 33.79; H 2.64; N 21.02; found: C 33.60; H 3.11; N 20.78.
Preparation of complex 5
H2L1 (0.302 1 mmol) was suspended in a water–MeOH (1:1 v/v) mixture (10 cm3), and a solution of Cu(ClO4)2·6H2O (0.371 g, 1 mmol) in MeOH (10 cm3) was added. The reaction solution was stirred at room temperature for 1 hour. The solution was allowed to stand for two days and yielded blue crystals as blue needles.
[Cu4(HL1)4](ClO4)8·CH3OH·5H2O (5) Anal. calc. for C61H62Cl8Cu4N32O38: C 30.67; H 2.61; N 18.76; found: C 30.91; H 2.27; N 18.58.
Preparation of complex 6a
1,3-Bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane 0.365 g (1.1 mmol) was suspended in 15 cm3 of a water–MeOH (1:1 v/v) mixture. Then, 0.371 g (1 mmol) of Cu(ClO4)2·6H2O was added to the suspension of the ligand. The mixture was stirred with heating for one hour to obtain a clear blue solution. Then, 92 mg (0.5 mmol) of potassium oxalate monohydrate was added to the solution. A greenish-blue precipitate formed immediately, and it was filtered off and dried to give the crude product. X-ray quality crystals were grown by slow evaporation of methanol solution prepared from the crude material.
[Cu2(HL3)2](ClO4)2·2H2O (6a) Anal. calc. for C34H34Cl2Cu2N16O10: C 39.85; H 3.34; N 21.87; found: C 39.89; H 3.26; N 21.82.
Preparation of complex 6b
1,3-Bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane 0.332 g (1 mmol) was suspended in 10 cm3 of an acetone–water solution (1:1 v/v). Then, 0.371 g (1 mmol) of Cu(ClO4)2·6H2O was added to the suspension of the ligand. The mixture was stirred with heating for one hour to obtain a clear blue solution and then 112 mg (2 mmol) of potassium hydroxide was added to the solution. The colour of the solution changed from blue to greenish-blue. The resulting solution was allowed to stand overnight, yielding pine colour crystals.
[Cu2(HL3)2](ClO4)2·H2O (6b) Anal. calc. for C34H32Cl2Cu2N16O9. Calcd C 40.56; H 3.20; N 22.26; found: C 40.71; H 3.41; N 22.05.
Preparation of complex 7
1,3-Bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane 0.365 g (1.1 mmol) was dissolved in 15 cm3 of methanol. A mixture of triethylenetetramine (teta) hydrate (0.330 g, 2 mmol) and Cu(ClO4)2·6H2O (0.745 g, 2 mmol) in 15 ml MeOH was added to the solution of the ligand. The deep blue solution was stirred for 1 hour, filtrated and left to stand for three days to obtain big greenish-blue crystals.[Cu2(HL3)(L3)Cu(teta)](ClO4)3·2H2O (7) Anal. calc. for C40H50Cl3Cu3N20O14. Calcd C 36.07; H 3.78; N 21.03; found: C 35.88; H 4.07; N 20.92.
Preparation of complex 8
1,3-Bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane 0.365 g (1.1 mmol) was suspended in 15 cm3 of acetone–water solution (1:1 v/v). 0.371 g (1 mmol) of Cu(ClO4)2·6H2O was added to the suspension of the ligand. The mixture was stirred with heating for one hour to obtain a clear blue solution and then 63 mg (0.5 mmol) of the oxalic acid dihydrate was added to the solution. After 10 minutes a blue solid precipitated. The mixture was left undisturbed overnight and the target complex was filtered off and washed twice with cold water and methanol, and dried carefully in vacuo. Single crystals suitable for X-ray analysis were grown from the mother liquid.
[Cu2(H2L3)2(μ-ox](ClO4)2·2H2O·2MeOH (8) Anal. calc. for C38H44Cl2Cu2N16O16: C 38.71; H 3.76; N 19.01; found: C 39.02; H 4.02; N 18.80.
X-ray crystallography
Single crystal X-ray diffraction data for H2L4 were collected using a Rigaku VariMax diffractometer with a Saturn CCD detector equipped with a monochromatic radiation source (MoKα radiation, λ = 0.71073 Å). Data for H2L3, H2L4 and Cu(II) complexes (2–8) were collected using a Bruker SMART APEX II diffractometer with a CCD detector and a monochromatic radiation source (MoKα radiation, λ = 0.71073 Å), and an Oxford diffraction Xcalibur2 CCD diffractometer with a Sapphire CCD detector (MoKα radiation, λ = 0.71073 Å) and equipped with an Oxford Cryosystems nitrogen gas-flow apparatus.The structure of H2L4 was solved by a direct method and refined in the full-matrix anisotropic approximation for all non-hydrogen atoms. All calculations were performed using the CrystalStructure software package (RIGAKU).7a
The structures of H2L3 and complexes 2–8 were solved by direct methods and refined in the full-matrix anisotropic approximation for all non-hydrogen atoms. All hydrogen atoms were found in differential Fourier maps and their parameters were refined using the riding model, with Uiso(H) = 1.2 or 1.5Ueq.. All the calculations were performed by direct methods and using the SHELX-97 program package.7b–d Some crystals exhibit a low percentage of observed reflections due to the presence of solvation water molecules and disordered perchlorate anions, which leads in some cases to a large R-factor.
The crystallographic parameters and refinements are given in Table 1. More details can be found in ESI† and in CCDC (956318–956326 and 956570).
| Parameter | H2L3 | H2L4 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| Formula | C17H16N8 | C18H18N8 | C17H17.25Cl2CuN8O0.75 | C36H46Cl4Cu2N16O5 | C30H30Cl2Cu3N16O13 |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21212 | P21/c | P | Cc | P21/m |
| Temperature | 150 | 293 | 296 | 173 | 150 |
| a (Å) | 12.162(3) | 9.543(6) | 9.6933 (8) | 20.907 (2) | 7.5521(13) |
| b (Å) | 15.550 (4) | 10.017(5) | 21.0519 (18) | 8.7394 (8) | 18.127 (3) |
| c (Å) | 4.2446 (11) | 9.650(5) | 22.3646 (19) | 24.795 (2) | 14.725 (3) |
| α (°) | 90.0 | 90.0 | 70.4200 (10) | 90.0 | 90.0 |
| β (°) | 90.0 | 117.012(13) | 88.867(2) | 95.685 (2) | 99.004(3) |
| γ (°) | 90.0 | 90.0 | 89.447(2) | 90.0 | 90.0 |
| V (Å3) | 802.7 (4) | 821.8(8) | 4299.0(6) | 4508.2 (7) | 1991.1(6) |
| Z | 2 | 2 | 8 | 4 | 2 |
| μ Mo. (mm−1) | 0.09 | 0.091 | 1.29 | 1.24 | 1.808 |
| Parameters | 117 | 118 | 1071 | 605 | 320 |
| No. unique | 1481 | 1621 | 15216 |
11846 |
4757 |
| No. I > 2σ(I) | 1028 | 1588 | 5404 | 10601 |
2884 |
| R int | 0.072 | 0.0487 | 0.099 | 0.023 | 0.096 |
| R(I > 2σ(I))a | 0.059 | 0.0364 | 0.098 | 0.033 | 0.079 |
| wR2 (all data)b | 0.125 | 0.0950 | 0.184 | 0.081 | 0.232 |
| Parameter | 5 | 6a | 6b | 7 | 8 |
|---|---|---|---|---|---|
| a R 1 = ∑(|Fo| − |Fc|)/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |||||
| Formula | C61H62Cl8Cu4N32O38 | C34H34Cl2Cu2N16O10 | C34H32Cl2Cu2N16O9 | C40H51Cl3Cu3N20O14 | C40H48Cl2Cu2N16O16 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/n | C2/c | P | P21/c | P21/c |
| Temperature | 120 | 296 | 150 | 120 | 293 |
| a (Å) | 17.7867(13) | 22.2091 (17) | 11.1178(3) | 15.662(3) | 12.411(3) |
| b (Å) | 26.4574(18) | 20.9808 (17) | 13.3569(3) | 22.852(4) | 15.274(3) |
| c (Å) | 19.7560(13) | 19.0829 (16) | 16.0478(3) | 15.481(3) | 16.177(3) |
| α (°) | 90.0 | 90.0 | 76.917(2) | 90.0 | 90.0 |
| β (°) | 98.5580(10) | 104.2400 (10)° | 75.935(2) | 107.791(4) | 128.84(2) |
| γ (°) | 90.0 | 90.0 | 85.931(2) | 90.0 | 90.0 |
| V (Å3) | 9193.5(11) | 8618.7 (12) | 2251.32(9) | 5275.6(16) | 2388.5(12) |
| Z | 4 | 8 | 2 | 4 | 2 |
| μ Mo. (mm−1) | 1.25 | 1.19 | 1.131 | 1.43 | 1.089 |
| Parameters | 1169 | 588 | 568 | 721 | 347 |
| No. unique | 21065 |
9909 | 7755 | 11099 |
4082 |
| No. I > 2σ(I) | 12420 |
5504 | 5598 | 3900 | 3894 |
| R int | 0.080 | 0.049 | 0.0261 | 0.237 | 0.078 |
| R(I > 2σ(I)a | 0.079 | 0.074 | 0.043 | 0.077 | 0.0579 |
| wR2 (all data)b | 0.241 | 0.220 | 0.125 | 0.223 | 0.1515 |
Results and discussion
Ligands
A series of new ligands H2Ln was prepared by a two-step reaction of 2-cyanopyridine and dihydrazide of malonic, succinic, glutaric or adipic acid as shown in Scheme 2.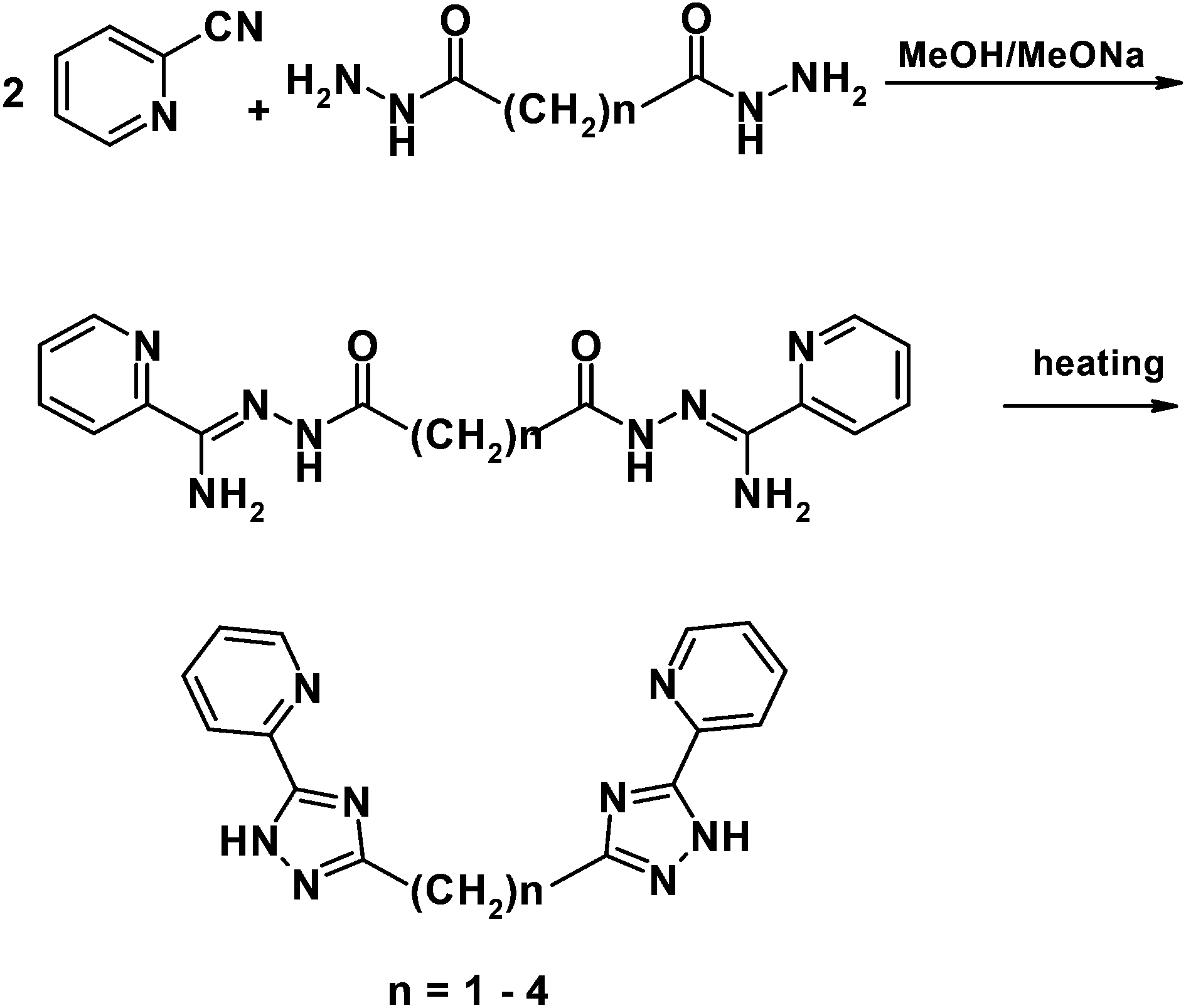 | ||
| Scheme 2 General pathway showing the preparation of bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes. | ||
It should be noted that synthesis of H2Ln (n = 1, 3 and 4) was described previously but the moderate yield forced us to optimize this method.6 We carried out cyclization by heating of the intermediate amidrazones in a vacuum that led to the target triazoles with a high yield and good purity.
The bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes have been analysed by UV-Vis, FTIR, 1H NMR spectroscopy and elemental analysis. The NMR spectra of title triazoles were relatively simple, with slightly broadened signals, which could be easily assigned. Broadening of the signals is observed due to the existence of several conformational forms because of polymethylene bridge mobility with a lifetime smaller than the characteristic NMR time. The solid state-electronic absorption spectrum was measured by using diffuse reflectance spectroscopy. The electronic bands were observed at 240 and 280–282 nm (the corresponding spectra for H2L1 are depicted in Fig. S1†). Both signals are attributed to the π–π* transition of the triazole and pyridine rings, respectively.
X-ray quality crystals of 1,3-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane and 1,4-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)butane were obtained by recrystallization from MeOH and the molecular structures are shown in Fig. 1.
 | ||
| Fig. 1 Molecular structures of H2L3 (above) and H2L4 (below) with thermal ellipsoids at the 50% probability level. | ||
The H2L3 ligand crystallizes in the orthorhombic space group P212121. The molecule has crystallographic symmetry C2 – the twofold axis passes through atom C(9) and divides the angle HC (9)H in half. The molecule adopts a cisoid geometry with the arms on opposite sides of the propane linker. The pyridine-triazole rings within the ‘arms’ of the ligand are slightly removed from being co-planar with the angle of 17.81(2)° and 18.30(2)° between the mean planes.
The H2L4 ligand crystallizes in the monoclinic space group P21/c. The chelating arms of the H2L4 ligand are arranged in a transoid manner around the tetramethylene spacer. The central C13–C13′ bond lies on the c2 axis, and therefore the triazolyl-pyridine arms are symmetrically equivalent. The pyridyl and triazole rings located in the very same arm of the H2L4 molecule are almost co-planar, with the dihedral angle between their least-square planes being 3.21(2)°.
Coordination chemistry of bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes
A series of the space-armed pyridyl-triazole ligands H2Ln was left to react with copper(II) salts yielding several coordination compounds. The complexes that were obtained vary in their nuclearity: from discrete mononuclear species to tetranuclear clusters (Scheme 3). The reaction of the ligands with di-, tri- and tetramethylene chains with equimolar quantity of copper(II) chloride led to the formation of mononuclear cationic complexes 1–3. It should be noted that the variation in the ratio of the reagents does not change the stoichiometric composition of the resulting products. Elemental analysis suggested a general formula [Cu(H2Ln)Cl]Cl·xH2O (n = 2, 3, 4) for 1–3, which was confirmed by X-ray crystallographic methods for compounds 2 and 3. Complex 2 crystallizes in the triclinic symmetry with the space group of P and contains four crystallographically independent [Cu(H2L3)Cl]Cl molecules within the unit cell with different Bond's parameters. The molecular structure of complex 2 is displayed in Fig. 2. | ||
| Scheme 3 Coordination mode of bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes. | ||
 | ||
| Fig. 2 Molecular structure of 2 (above, one of four symmetrically independent complex cations is shown as a representative) and 3 (below). Hydrogen atoms, counter-anions and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (in Å) and angles (in °): 2, Cu1–N2 = 1.937(10), Cu1–N6 = 1.969(8), Cu1–N1 2.040(8), Cu1–N5 = 2.202(8), Cu1–Cl1 = 2.305(3), Cu2–N6A 1.949(9), Cu2–N2A = 2.005(8), Cu2–N1A = 2.029(9), Cu2–N5A = 2.200(9), Cu2–Cl2 = 2.319(3), Cu3–N2B = 1.975(9), Cu3–N5B = 2.009(8), Cu3–N6B = 2.018(8), Cu3–N1B = 2.183(9), Cu3–Cl3 = 2.332(3), Cu4–N2C = 1.941(8), Cu4–N6C = 1.997(9), Cu4–N5C = 2.020(8), Cu4–N1C = 2.182(8), Cu4–Cl4 = 2.335(3), N5–Cu1–N2 = 125.3(4), N1–Cu1–N6 = 174.5(4), N6C–Cu4–N1C = 131.6(4), N5C–Cu4–N2C = 177.0(4), N1A–Cu2–N6A = 173.7(4), N2A–Cu2–N5A = 129.8(4), N5B–Cu3–N2B = 175.7(4), N6B–Cu3–N1B = 131.6(3); 3, Cu1–N5 = 1.979(2), Cu1–N1 = 2.011(2), Cu1–N2 = 2.084(2), Cu1–N8 = 2.164(2), Cu1–Cl1 = 2.3178(7), N1–Cu1–N5 = 169.82(8), N2–Cu1–N8 = 120.80(8), N13–Cu2–N9 = 169.71(8), N10–Cu2–N16 = 112.40(8). | ||
In all four symmetrically independent [Cu(H2L3)Cl]+ molecules the {CuN4Cl} chromophore adopts a distorted trigonal-bipyramidal coordination geometry (τ = 0.73–0.82, Table 2) where the Cu(II) atom is coordinated by one chloride-ligand and four nitrogen atoms from H2L3 ligand (N2, N5 in the basal plane and N1 and N6 in the apical positions). The H2L3 ligand is coordinated to the Cu atom as a tetradentate chelate, which leads to closure of the eight-membered rings.
| Complex | 1 | 2 | 3 | 4 | 5 | 6a | 6b | 7 |
|---|---|---|---|---|---|---|---|---|
| Copper numbering – τ parameter | Cu1–0.819 | Cu1–0.034 | Cu1–0.479 | |||||
| Cu1–0 | Cu2–0.744 | Cu1–0.820 | Cu1–0.0 | Cu2–0.022 | Cu1–0.454 | Cu1–0.388 | Cu2–0.656 | |
| Cu2–0.101 | Cu3–0.738 | Cu2–0.956 | Cu2–0.010 | Cu3–0.064 | Cu2–0.474 | Cu2–0.496 | Cu3–0.083 | |
| Cu4–0.734 | Cu4–0.011 |
Bond's parameters of four molecules from the independent cell appear to be markedly asymmetrical. The Cu–N bond lengths vary from 1.93 to 2.20 Å where the shortest Cu–N bond lengths belong to the bonds between the Cu and nitrogen atoms of the triazole rings (Ntz), whereas the longest bonds are found for bonds with nitrogen atoms from pyridyl moieties (Npy) (Table S1†). The Cu–Cl bond lengths are in the narrow range from 2.30 to 2.33 Å. One half of the non-coordinated chloride anions are disordered over two positions with the occupation factor of 0.5. The four crystallographically independent molecules form two pairs of complexes oriented in an edge-to-face (d(Cu2⋯Cu3) = 6.846(2) Å) and a face-to-face (d(Cu1⋯Cu4) 5.437(2) Å) manner, respectively.
Increased length of the spacer alkyl chain in 3 in comparison to 2 does not change the nuclearity of the product of the reaction between CuCl2 and H2L4. The structure of the [Cu(H2L4)Cl]+ cation is depicted in Fig. 2. Complex 3 crystallizes in monoclinic symmetry, the Cc space group, with two [Cu(H2L4)Cl]+ cations in the asymmetric unit. The pentacoordinate copper atom is in a distorted trigonal-bipyramidal surrounding (τ = 0.820 and 0.956, Table 2). Three equatorial donor atoms are Cl1, N2 and N8; the two axial ones are N5 and N1. Despite a rather long spacer involved in H2L4 the coordination of this ligand to the copper atom leads to the closure of the nine-membered metallocycle with a boat-like conformation (Fig. 2).
The complex cation [Cu(H2L2)Cl]+ in 1 has same basic structural features seen for the other members of this series (Fig. S2†). Crystals have triclinic symmetry with the cell parameters: a = 7.2244(1) Å, b = 16.1267(3) Å, c = 18.0322(3) Å, and α = 101.193(1)°, β = 93.725(1)°, γ = 101.572(1)°. The coordination polyhedron is close to square pyramidal (Table 2, Fig. S2†). Full refinement of the crystal structure failed due to the poor diffraction quality of the single-crystals. In summary, it should be noted that the complexes 1–3 involving the H2L2–4 ligand have very similar structures in comparison with complexes involving the 1,3-bis[3-(2-pyridyl)pyrazol-1-yl]propane ligand prepared and characterized by the group of Prof. M. Ward.5a,b
In contrast to the herein reported complexations of the ligands H2L2–4, the reaction of copper(II) perchlorate with H2L1 in the presence of a base leads to the formation of a new trinuclear complex with formula [Cu3(μ3-L1)2(H2O)3](ClO4)·H2O (4). The monomethylene group is not sufficiently flexible to allow two bidentate arms to chelate one metal ion, so a difference in the coordination mode is expectable. Single-crystal X-ray structural analysis revealed that complex 4 crystallizes in the monoclinic space group P21/m with an asymmetric unit consisting of one half of the trinuclear complex with the central copper atom at the special position and two halves of the ClO4 anions and disordered lattice water molecules. Three Cu(II) atoms are arranged in a bent linear formation with a Cu2–Cu1–Cu2′ angle of 139.32(1)° (Fig. 3).
 | ||
| Fig. 3 Molecular structure of 4. Hydrogen atoms, counter-anions and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (in Å) and angles (in °): Cu1–N7 = 1.993(5), Cu1–N7′ = 1.993(5), Cu1–N3 = 2.015(6), Cu1–N3′ = 2.015(6), Cu1–O2W = 2.253(9), Cu2–N2 = 1.945(6), Cu2–N6 = 1.950(5), Cu2–N5 = 2.061(6), Cu2–N1 = 2.122(6), Cu2–O1W = 2.198(5), N5–Cu2–N2 = 167.4(2), N6–Cu2–N1 = 161.4(2), N3–Cu1–N7′ = 174.5(2). | ||
The distance between neighbouring copper atoms is 4.011 (3) Å, which is typical of the μ-triazolyl bridging mode8 and 7.521(2) Å between terminal Cu2 and Cu2′. The molecular structure of 4 shows that each (L1)2− ligand acts as a bridge between the three copper(II) atoms, with the two ligands in a ‘face-to-face’ arrangement (Fig. 3). Basic conditions during the synthesis of 4 lead to the loss of the acidic hydrogen atoms from the H2L1 ligands. The copper atoms are coordinated in two types of coordination geometry. The Cu2/2′ centres are pentacoordinate with distorted square pyramidal geometry (τ = 0.010, Table 2) surrounded by four cis-related nitrogen atoms from two pairs of chelated (L1)2− ligands in the base plane and the oxygen atoms from one monodentate water molecule. The central copper atom Cu1 of the trimer is five-coordinated with a square pyramidal geometry (τ = 0.0, Table 2) formed by four equivalent nitrogen atoms from four triazolyl moieties and the oxygen atom from the coordinated water molecule. The copper atom is out of the basal plane by 0.097(2) Å. Perchlorate anions are involved in several non-covalent contacts with coordinated and non-coordinated water molecules.
The reaction of equimolar amounts of copper(II) perchlorate and H2L1 without a base leads to the formation of the homoleptic tetranuclear complex 5. The crystal structure determination revealed that complex 5 (Fig. 4) consists of the discrete complex cations [Cu4(HL1)4]4+ and eight perchlorate anions. The complex is a homoleptic [2 × 2] grid involving four copper(II) centres bridged by the μ-triazolyl units. All the copper atoms are pentacoordinated with N5 donor set. Each copper(II) atom displays a slightly distorted square-pyramidal geometry with the nitrogen atom from the pyridine ring in the axial position (τ = 0.01–0.07, Table 2). Four copper(II) centres are arranged in an unusual Cu4N8 U-like core. The distances between the bridged ions in the complex (3.93–3.98 Å) are similar to each other and have the values typical for the μ-triazolyl coordination mode.8 The ligand strands are oriented in a “head-to-tail” arrangement at the Cu(II) sites; the “head” and “tail” terms refer to the tridentate and bidentate donor pockets, respectively.
 | ||
| Fig. 4 Molecular structure of 5 with highlighted chromophores of the Cu4 core (above). Counter-anions and solvent molecules are omitted for clarity. Detailed view on the chromophores of the Cu4 core with atom labelling (below). Selected bond lengths (in Å) and angles (in °): Cu1–N31 = 1.916(7), Cu1–N14 = 1.980(7), Cu1–N27 = 2.007(7), Cu1–N32 = 2.129(7), Cu1–N16 = 2.284(7), Cu2–N12 = 1.920(7), Cu2–N22 = 1.985(7), Cu2–N13 = 1.996(7), Cu2–N9 = 2.110(8), Cu2–N24 = 2.292(8), Cu3–N20 = 1.914(7), Cu3–N2 = 1.993(7), Cu3–N21 = 2.006(8), Cu3–N17 = 2.141(8), Cu3–N1 = 2.317(8), Cu4–N7 = 1.923(7), Cu4–N26 = 1.989(7), Cu4–N3 = 2.027(7), Cu4–N8 = 2.122(7), Cu4–N25 = 2.263(7), N8–Cu4–N3 = 164.4(3), N26–Cu4–N7 = 163.7(3), N17–Cu3–N21 = 164.8(3), N20–Cu3–N2 = 161.0(3), N22–Cu2–N12 = 166.2(3), N9–Cu2–N13 = 164.8(3), N14–Cu1–N31 = 166.6(3), N32–Cu1–N27 = 164.5(3). | ||
The complexes 1–5 with bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkanes have the coordination polyhedra geometry intermediate between trigonal bipyramidal and rectangular pyramidal. This is consistent with their electronic spectra which showed the relatively low-energy and high intensity d–d transition at 711–755 nm in addition to strong ligand-centered transitions in the UV region.9 The degree of distortion of the coordination polyhedron was evaluated by the Addison parameter τ.10 The values of parameter τ increase with increasing length of the spacer (see Table 2). So the extent of distortion of these pentacoordinate structures towards a specific geometry is clearly limited by the length of the polymethylene chain.
In order to prepare binuclear complexes with the trimethylene spacer the reaction with the molar ratio of Cu(ClO4)2:H2L2:K2(ox) = 2:2:1 was done (ox2− = oxalate anion). It is well known that the oxalate anion can act as a rigid bridging ligand that binds two copper atoms and prevents the H2L3 ligand from closure to metallocycle. Similar MOFs on triazole-oxalato bridged ligands were described earlier.11 It was found that oxalate anions do not bind a copper ion upon reaction. This can be explained on the basis that potassium oxalate acts as a base deprotonating the ligand and the resulting formula of the complex prepared in this way is [Cu2(μ-HL3)2](ClO4)2·2H2O (6a) and it crystallizes in the monoclinic space group C2/c (Fig. 5). The asymmetric unit of 6a consists of one complex dication, two disordered perchlorate anions and two lattice water molecules. The triazolate groups bridge the Cu1 and Cu2 atoms to form a rigid, non-planar 6-membered {Cu1–N6–N7–Cu2–N12–N11} ring with a boat conformation. The metallocycle is quite distorted with the copper–triazolate angles having 137.68(2)° (Cu1–N11–N12) and 119.07(2)° (Cu1–N6–N7) (Fig. 5). The distance between the copper ions within the dimer is 3.814(2) Å. Each Cu(II) atom is pentacoordinate having square pyramidal geometry of the chromophore (τ = 0.45–0.47), provided by three nitrogen atoms of one pyridyl-triazole arm of the first and by two nitrogen atoms from the second HL3− ligand. Pyridyl-triazole arms of each HL3− ligand are coordinated to the Cu(II) ion in an asymmetric manner and bent with respect to each other by the angle of 68.71(3)°. From an inter-molecular perspective, complexes are associated via hydrogen bonding between the triazole nitrogen atoms, lattice water molecules and oxygen atoms from the perchlorate anions. The solid state sample shows a weak band at 690 nm in the UV-Vis spectrum which can be assigned to a d–d transition.
 | ||
| Fig. 5 Molecular structure of 6a. Hydrogen atoms, counter-anions and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (in Å) and angles (in °): Cu1–N11 = 1.952(5), Cu1–N2 = 1.990(5), Cu1–N6 = 1.999(5), Cu1–N9 = 2.109(6), Cu1–N1 = 2.190(5), Cu2–N7 = 1.950(4), Cu2–N13 = 1.989(5), Cu2–N12 = 2.021(4), Cu2–N8 = 2.112(5), Cu2–N15 = 2.189(5), N11–Cu1–N2 = 169.3(2), N9–Cu1–N6 = 142.0(2), N13–Cu2–N7 = 170.9(2), N12–Cu2–N8 = 142.4(2). | ||
In order to obtain a coordination compound with fully deprotonated H2L3 the reaction between Cu(ClO4)2 and H2L3 was carried out in the presence of two equivalents of a base. Nevertheless, binuclear complex 6b, very similar to 6a, was found to be the main product of the reaction. The coordination mode of the (HL3)− ligand is the same as in the case of 6a and the bond lengths in 6b within its chromophore are very similar to those in 6a (Fig. 5 and Table S2†). The main difference between these two compounds is in the absence of the lattice water molecules in 6b and the space group this compound crystallize in (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). It can be concluded that in a basic medium, the dominant reaction products are dinuclear complexes with the monodeprotonated (HL3)− form of the ligand. The resulting dimers have potential to be used as precursors for the preparation of polynuclear complexes due to non-coordinated triazole nitrogen atoms available for further coordination. This hypothesis was verified by the reaction of two equivalents of [Cu(teta)](ClO4)2 (teta = triethylenetetraamine) and one equivalent of H2L3 which resulted in isolation of the trinuclear complex 7 (Fig. 6). Compound 7 crystallizes in the monoclinic space group P21/c and it is composed of the discrete trinuclear complexes [Cu2(μ-HL3)(L3)Cu(teta)](ClO4)3·2H2O. In 7 the [Cu2(μ-HL3)2]+ fragment coordinates the [Cu(teta)]2+ moiety by the nitrogen atom of the triazole ring (Fig. 6).
). It can be concluded that in a basic medium, the dominant reaction products are dinuclear complexes with the monodeprotonated (HL3)− form of the ligand. The resulting dimers have potential to be used as precursors for the preparation of polynuclear complexes due to non-coordinated triazole nitrogen atoms available for further coordination. This hypothesis was verified by the reaction of two equivalents of [Cu(teta)](ClO4)2 (teta = triethylenetetraamine) and one equivalent of H2L3 which resulted in isolation of the trinuclear complex 7 (Fig. 6). Compound 7 crystallizes in the monoclinic space group P21/c and it is composed of the discrete trinuclear complexes [Cu2(μ-HL3)(L3)Cu(teta)](ClO4)3·2H2O. In 7 the [Cu2(μ-HL3)2]+ fragment coordinates the [Cu(teta)]2+ moiety by the nitrogen atom of the triazole ring (Fig. 6).
 | ||
| Fig. 6 Molecular structure of 7. Hydrogen atoms, counter-anions and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (in Å) and angles (in °): Cu1–N11 = 1.962(8), Cu1–N2 = 1.977(8), Cu1–N6 = 2.011(8), Cu1–N9 = 2.102(8), Cu1–N1 = 2.150(9), Cu2–N7 = 1.961(7), Cu2–N13 = 1.965(8), Cu2–N12 = 1.996(8), Cu2–N8 = 2.138(8), Cu2–N16 = 2.158(8), Cu3–N18 = 2.010(9), Cu3–N20 = 2.019(9), Cu3–N19 = 2.027(9), Cu3–N17 = 2.041(8), Cu3–N14 = 2.188(8), N2–Cu1–N11 = 168.8(3), N6–Cu1–N9 = 140.2(3), N13–Cu2–N7 = 169.4(3), N8–Cu2–N12 = 130.1(3), N20–Cu3–N18 = 158.9(4), N17–Cu3–N19 = 154.0(4). | ||
Bond lengths and angles within the chromophore of the binuclear [Cu2(μ-HL3)(L3)]+ fragment are comparable to those found in 6a and 6b. The trinuclear complex has an asymmetric angular structure with a Cu1–Cu2–Cu3 angle of 130.99(4)°. The nitrogen atoms of triethylenetetramine together with the axial triazole core nitrogen atom form a very slightly distorted square pyramidal CuN5 coordination environment (τ = 0.083). Cu3 is located above the basal plane, defined by the teta nitrogen atoms by 0.384(2) Å. The apical Cu3–N14 bond length is 2.188(8) Å and it is significantly longer than the remaining bonds of the coordination polyhedron.
As was mentioned above, attempts to obtain binuclear complexes with the oxalate bridging ligand failed when potassium oxalate was used in the synthesis and this acted only as a base not coordinating the metal ions. Therefore, the preparation method was modified by involvement of the oxalic acid in order to prevent deprotonation of the H2L3 ligand. Then, the binuclear complex [Cu2(μ-H2L3)2(μ-ox)](ClO4)2·2H2O·2MeOH (8) was obtained. The molecular structure of 8 is illustrated in Fig. 7.
 | ||
| Fig. 7 Molecular structure of 8. Hydrogen atoms, counter-anions and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (in Å): Cu1–O1 = 2.091(5), Cu1–O2 = 2.188(5), Cu1–N9 = 2.069(5), Cu1–N1 = 2.073(4), Cu1–N2 = 2.128(7), Cu1–N8 = 2.130(6). | ||
The crystal structure of 8 consists of the centrosymmetric dinuclear copper(II) complex cation [Cu2(μ-H2L3)2(μ-ox)]2+ (Fig. 7) and two ClO4− anions together with methanol as crystallization solvent molecules. The coordination environment of two centrosymmetrically related Cu1 and Cu1′ atoms adopts an elongated tetragonal bipyramid coordination geometry defined by four nitrogen atoms from two triazolyl-pyridine moieties and two carboxylate oxygen atoms from the oxalate anion. Each H2L3 ligand coordinates the Cu1 atom in a bis(bidentate) N,N′-bridging coordination mode while the ditopic ox2− ligand coordinates the Cu1 atom in a η2-tetradentate bridging mode to form a dimeric structure. These coordination modes led to the formation of two metallo-macrocycles separated by a bridging oxalate anion. The Cu–O bond lengths are non-equivalent (d(Cu–O1) = 2.091(5) Å and d(Cu–O2) = 2.188(5) Å) as a result of Jahn–Teller distortion.12 The UV-vis spectrum is consistent with the structural observations because of a broad d–d band centred at 680 nm, which is typical for the octahedral copper(II) complexes with tetragonal distortion. The intramolecular Cu⋯Cu distance is 5.414(4) Å. The two bidentate triazolyl-pyridine arms are bent each to the other (Fig. 7); the angle between the two CuN2 planes involving each bidentate fragment is 87.37(3)°.
Magnetic properties
Mononuclear copper(II) complexes 1–3 show typical Curie-like variable temperature magnetic properties consistent with well isolated metal centres, with the effective magnetic moment values of 1.75–1.79μB which are close to the spin-only value expected for Cu(II) (S = 1/2). The magnetic properties of the polynuclear complexes 4–7 were investigated in more detail to elucidate magnetic interactions among paramagnetic centres. The evolution of the molar magnetization and the effective magnetic moment with the temperature for complex 4 is shown in Fig. 8. The room temperature value of the effective magnetic moment is 2.76μB and it decreases gradually upon cooling to 1.82μB at 5 K indicating antiferromagnetic exchange. The magnetic properties were treated with the spin Hamiltonian for a linear trimer formulated as13 | (1) |
 | ||
| Fig. 8 The magnetic data for 4–8. Left: the temperature dependence of the effective magnetic moment. Right: the molar magnetization measured at B = 0.5 T. Empty circles – experimental data; full lines – calculated data. | ||
The magnetic properties of tetranuclear complex 5 are displayed in Fig. 8. For four non-interacting spins (Si = 1/2) we would expect a theoretical value of μeff/μB which should be equal to 3.46 and indeed the effective magnetic moment is progressively decreasing from 3.25μB at 300 K to 0.21μB at 3 K. Moreover, the maximum is observed on the curve Mmolvs. T at 75 K. All these facts suggest the occurrence of antiferromagnetic interactions within the tetrameric unit, which results in the diamagnetic ground state S = 0. However, the non-zero effective magnetic moment at low temperature is assigned to the presence of a small amount of monomeric paramagnetic impurity (PI). As the bond distances of Cu1–Cu2, Cu2–Cu3, Cu3–Cu4 and Cu4–Cu1 in 5 are almost the same as evidenced from the structural data, the exchange interactions between the triazolate-bridged Cu(II) ions are considered to be identical. The exchange interactions between non-bridging copper atoms were not considered. Thus, the Heisenberg spin Hamiltonian was applied in the form13
 | (2) |
The experimental data can be fitted with J = −70 cm−1, g = 1.96, xPI = 0.53%, where xPI is the molar fraction of monomeric paramagnetic impurities and the final magnetization was calculated as Mmol = (1 − xPI)Mtetramer + 4xPIMPI.
Magnetic behaviour of binuclear complexes 6a and 8 shown in Fig. 8 is characteristic of the antiferromagnetic interaction between copper centres, but with different intensities, which can be supported by comparing the room temperature and the lowest temperature effective magnetic moment values for 6a (2.37μB → 0.0μB) and for 8 (2.59μB → 2.22μB). The magnetism of these compounds was analysed with the following spin Hamiltonian:
 | (3) |
The parameters obtained from the best fits are: J = −93 cm−1 and g = 2.06 for 6a and J = −1.6 cm−1 and g = 2.10 for 8, indicating a moderate antiferromagnetic coupling for 6a and a very weak one for 8.
The magnetic data for trinuclear complex 7 are typical for an antiferromagnetically coupled system (Fig. 8) with the effective magnetic moment decreasing from 3.01μB at 298 K to 1.83μB at 2.6 K. Three copper(II) centres are arranged at the corner of an irregular triangle as observed from the crystal structure of 7. The experimental magnetic data were fitted using the exchange-Hamiltonian for a non-equivalent trinuclear core13
 | (4) |
The obtained data on the magnetic behaviour of the title complexes along with the X-ray diffraction data provide the ability to search structural correlations. For complexes 4 and 5 a superexchange interaction is realized through the bridging triazolyl moiety. As was shown earlier for related pyrazole bridged complexes, the deviations from the co-planarity of the copper–pyrazolate planes (dihedral angle α – the angle between the least-square planes formed by the copper atom and two coordinated nitrogen atoms from bridging pyrazolate/triazolate planes) and the Ntr–Cu–Ntr angle were identified as crucial factors that determine the strength of the antiferromagnetic coupling.14 The correlation data for the triazole moiety indicate that the coupling via the triazole bridge is usually less efficient than that via a pyrazolate bridge.15 It was shown that a more symmetric bridging mode leads to stronger antiferromagnetic exchange.15,16 However, further complex analysis is difficult due to the fact that most of the binuclear complexes on the triazole bridging ligand basis have the geometry of the metallocycle as planar or nearly planar. Therefore, the question on the effects of planarity on the singlet–triplet splitting is still open.
As follows from the structural data, the bridging fragments Cu–(N–N)2–Cu for complexes 4 and 6a are far from being planar. This is further illustrated by Table 3 which summarizes several results.
| Complex | d(Cu⋯Cu)/Å | −J/cm−1 | Ntz–Cu–Ntz° | α/° | Cu–Ntz–Ntz/° |
|---|---|---|---|---|---|
| a The data given in ref. 16a,b were recalculated for the spin Hamiltonian used in this article. | |||||
| 4 | 4.011(3) | 138 | 91.5(2) | 28.50(2) | 127.8(4) |
| 93.4(2) | 136.1(4) | ||||
| 6a | 3.814(2) | 93 | 92.8(2) | 52.43(2) | 137.2(2) |
| 92.6(2) | 119.1(2) | ||||
| 7 | 3.749(2) | 93(fixed) | 91.8(3) | 58.19(3) | 136.1(6) |
| 94.0(3) | 117.9(5) | ||||
| Ref. 16b | 4.085(1) | 118 | 90.2(1) | Near planar | 135.2(2) |
| 90.1(1) | 134.9(2) | ||||
| Ref. 16c | 3.854(6) | 36 | 102.8(5) | Near planar | 128.0(8) |
| 129.2(8) | |||||
| Ref. 16a | 4.0265(8) | 49 | 95.0(2) | Near planar | 124.4(3) |
| 94.3(1) | 139.8(3) | ||||
It is observed that the value of the Ntz–Cu–Ntz angle does not affect the value of the exchange parameter noticeably in the case of compounds 4 and 6a with α ≠ 0. The deviation of the Ntz–Cu–Ntz angle from its ideal value of 90° was identified as a crucial factor that determines the strength of the antiferromagnetic coupling, which is consistent with previous work with compounds having α close to 0.16c
The planarity of the six-membered metallocycle seems to play a leading role in the exchange mechanism in 4 and 6a. The compound 4 (J = −138 cm−1, α = 28.50(2)°) has significantly stronger antiferromagnetic exchange interaction than the less planar compound 6 (J = −93 cm−1, α = 52.43(2)°), while the mean values of the Ntz–Cu–Ntz angles are approximately the same (92.5 in 4vs. 92.7 in 6).
A search for magnetostructural correlations for the tetranuclear complex is even more difficult because there are practically no literature data for such grids on the pyridyl-triazole basis17 in contrast to the fact that the antiferromagnetic coupling mediated by the pyrazolate bridge itself is well understood.18 Using the results of the study made on tetranuclear complexes based on pyrazole derivatives it is known that: (i) shorter Cu⋯Cu separations give larger exchange parameter values, (ii) deviation from orthogonality of the copper coordination planes leads to an increase in antiferromagnetic coupling, (iii) the most effective exchange interaction is realized when the angle N–Cu–N approaches 130.5°. Although many of the previously described systems satisfy the above mentioned conditions, relatively small J values are usually observed. The authors explain this fact as a result of the mutual orthogonality of the d-orbitals of neighbouring Cu atoms for S4 symmetry of the tetranuclear core. Lowering the symmetry to S2 and C1 contributes to more efficient overlap of the magnetic orbitals and leads to greater J-parameter.19 The tetranuclear core of 5 has symmetry close to C1 which together with the other geometrical parameters gives a high value of the exchange parameter: −70 cm−1.
Complex 7 has two different pairs of copper ions which are involved in the exchange interactions. In the first pair (Cu1–Cu2), quite a strong interaction through the N–N triazole moiety was found, while in the second pair (Cu2–Cu3), only a weak interaction was observed mediated by the triazole ring which acts in this case as an imidazole analogue. It is well known that all imidazole-bridged Cu(II) dimers exhibit antiferromagnetic interaction with J varying from 0 to −88 cm−1 depending on geometric factors.20 Another fact that supports very weak exchange interaction in a Cu2–Cu3 pair is that the pentacoordinated Cu3 ion displays typical Jahn–Teller elongation with the bond lengths Cu–Neq. of 2.01–2.04 Å and Cu–Nax of 2.19 Å, thus suggesting that the magnetic dx2–y2 orbital of Cu3 occupies the equatorial plane and is not capable of efficiently overlapping with the magnetic orbitals from Cu2.
The value of the exchange interaction parameter calculated for complex 8 was unexpectedly small, because most of the oxalato-bridged Cu(II) complexes show strong antiferromagnetic coupling with J up to −400 cm−1.21 However, structural analysis can explain the apparent contradiction. Examination of the structure of complex 8 reveals lengthening of the Cu–O2 (2.188(5) Å) and Cu–N2 (2.128(7) Å) bonds, which leads to the situation in which one oxalate oxygen atom is in the basal plane while the other is axial; the magnetic orbital is situated in the plane perpendicular to the oxalate σ-orbitals and has a poor overlap with them. It should be noted that such an interpretation was proposed earlier for some oxalate-bridged copper(II) dimers.22
Conclusions
Nine new crystal structures of the copper(II) coordination compounds involving flexible bis-pyridyltriazole ligands H2L1–4 with different lengths of the spacer group are reported. The different coordination possibilities of these ligands were demonstrated by varying the length of the spacer, the pH and other conditions. The resulting compounds vary also in their nuclearity: from mononuclear compounds (1–3), to dinuclear (6a, 6b and 8), trinuclear (4 and 7) and tetranuclear (5). Furthermore, it has been shown that complexes 6a/b can be used as precursors for the preparation of polynuclear complexes such as 7.The analysis of the magnetic properties of these complexes was performed using the spin-Hamiltonian approach and it revealed the presence of antiferromagnetic exchange interactions in the binuclear μ-triazole bridged Cu dimers (4, J = −138 cm−1; 6a and 7, J = −93 cm−1) and the possible dependence of the exchange parameter J on the planarity of the dimeric metallocycle was proposed. Furthermore, compound 7 contains besides the [Cu2(μ-HL3)(L3)]2+ cation also the [Cu(teta)]+ moiety bonded by the peripheral nitrogen atom of the dimeric entity. This exchange pathway was found to be small in comparison with the dimeric pathway (J = −2.7 cm−1). The tetranuclear compound (5) exhibits a relatively strong antiferromagnetic coupling within the Cu4 core (J = −70 cm−1). The oxalato bridged Cu2 dimer (8) exhibits unexpectedly a small antiferromagnetic coupling (J = −1.6 cm−1), which can be explained on the basis of the metal–ligand orbital orthogonality.
Acknowledgements
I.N., R.H. and Z.T. would like to acknowledge financial support from the Operational Program Research and Development for Innovations – the European Regional Development Fund (CZ.1.05/2.1.00/03.0058), the Operational Program Education for Competitiveness European Social Fund (CZ.1.07/2.3.00/20.0017) of the Ministry of Education, Youth and Sports of the Czech Republic.Notes and references
- (a) A. Klug, Angew. Chem., Int. Ed., 1983, 22, 565 CrossRef; (b) V. Soghomonian, Q. Chen, R. C. Haushalter, J. Zubieta and C. J. O'Connor, Science, 1993, 259, 1596 CAS; (c) A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. Schroder and M. A. Withersby, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS; (d) D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375 CrossRef CAS PubMed.
- (a) J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed; (b) W. L. Leong and J. J. Vitta, Chem. Rev., 2011, 111, 688 CrossRef CAS PubMed.
- (a) A.-M. Stadler and J. Harrowfield, Inorg. Chim. Acta, 2009, 362, 4298 CrossRef CAS; (b) G. M. Larin and V. F. Shul'gin, Russ. J. Inorg. Chem, 2006, 51(Suppl. 1), S28 CrossRef.
- (a) G. Aromi, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485 CrossRef CAS; (b) F. H. Allen, Acta. Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef; (c) Y. P. Prananto, D. R. Turner, J. Lu and S. R. Batten, Aust. J. Chem., 2009, 62, 108 CrossRef CAS.
- (a) K. L. V. Mann, J. C. Jeffery, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 3029 RSC; (b) K. L. V. Mann, J. C. Jeffery, J. A. McCleverty, P. Thornton and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 89 RSC; (c) J. S. Fleming, K. L. V. Mann, S. M. Couchman, J. C. Jeffery, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 2047 RSC; (d) R. L. Paul, Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4883 CrossRef CAS PubMed; (e) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (f) A. M. Najar, I. S. Tidmarsh, H. Adams and M. D. Ward, Inorg. Chem., 2009, 48, 11871 CrossRef CAS PubMed.
- A. N. Gusev, V. F. Shul'gin, S. B. Meshkova, P. G. Doga, M. Hasegawa, G. G. Aleksandrov, I. L. Eremenko and W. Linert, Inorg. Chim. Acta, 2012, 387, 321 CrossRef CAS.
- (a) CrystalStructure 3.8: Crystal Structure Analysis Package, Rigaku and Rigaku Americas (2000–2007). 9009 New Trails Dr, The Woodlands TX 77381 USA; (b) G. M. Sheldrick, SADABS. Program for Scanning and Correction of Area Detector Data, Güttingen Univ., Güttingen, 1997 Search PubMed; (c) G. M. Sheldrick, SHELX97. Program for the Solution of Crystal Structures, Güttingen Univ., Güttingen, 1997 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- J. G. Haasnoot, Coord. Chem. Rev., 2000, 200–202, 131 CrossRef CAS.
- N. Wei, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1994, 33, 6093 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) U. García-Couceiro, O. Castillo, A. Luque, J. P. Garcia-Teran, G. Beobide and P. Roman, Eur. J. Inorg. Chem., 2005, 4280 CrossRef; (b) U. Garcia-Couceiro, O. Castillo, J. Cepeda, A. Luque, S. Perez–Yanez and P. Roman, Inorg. Chim. Acta, 2009, 362, 4212 CrossRef CAS; (c) A. L. Spek, P. J. van Koningsbruggen and J. G. Haasnoot, private communication, 2004.
- (a) W. Fitzgerald, J. Foley, D. McSweeney, N. Ray, D. Sheahan and S. Tyagi, J. Chem. Soc., Dalton Trans., 1982, 1117 RSC; (b) H. Oshio and U. Nagashima, Inorg. Chem., 1992, 3295 CrossRef CAS.
- (a) R. Boča, A Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed; (b) O. Kahn, Molecular Magnetism, VCH Publishers, 1993 Search PubMed.
- (a) H. Matsushima, H. Hamada, K. Watanabe, M. Koikawa and T. Tokii, J. Chem. Soc., Dalton Trans., 1999, 971 RSC; (b) H. Matsushima, H. Hamada, K. Watanabe, M. Koikawa and T. Tokii, J. Chem. Soc., Dalton Trans., 1999, 971 RSC; (c) S. Tanase, I. A. Koval, E. Bouwman, R. De Gelder and J. Reedijk, Inorg. Chem., 2005, 44, 7860 CrossRef CAS PubMed.
- (a) V. P. Hanot, T. D. Robert, J. Kolnaar, J. G. Haasnoot, J. Reedijk, H. Kooijiman and A. L. Spek, J. Chem. Soc., Dalton Trans., 1996, 4275 RSC; (b) S. Ferrer, P. J. van Koningsbruggen, J. G. Haasnoot, J. Reedijk, H. Kooijiman, A. L. Spek, L. Lezama, A. M. Arif and J. S. Miller, J. Chem. Soc., Dalton Trans., 1999, 4269 RSC.
- (a) P. M. Slangen, P. J. van Koningsbruggen, K. Goubitz, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1994, 33, 112 CrossRef; (b) R. Prins, P. J. M. W. L. Birker, J. G. Haasnoot, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1985, 24, 4128 CrossRef CAS; (c) S. Ferrer, P. J. van Koningsbruggen, J. G. Haasnoot, J. Reedijk, H. Kooijman, A. L. Spek, L. Lezama, A. M. Arif and J. S. Miller, J. Chem. Soc., Dalton Trans., 1999, 4269 RSC.
- R. Prins, R. A. G. de Graaff, J. G. Haasnoot, C. Vader and J. Reedijk, J. Chem. Soc., Chem. Commun., 1986, 1430 RSC.
- (a) D. S. Cati, J. Ribas, J. Ribas-Arino and H. Stoeckli-Evans, Inorg. Chem., 2004, 43, 1021 CrossRef CAS PubMed; (b) J. I. van der Vlugt, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2008, 47, 1576 CrossRef CAS PubMed; (c) J. Klingele, A. I. Prikhod'ko, G. Leibeling, S. Demeshko, S. Dechert and F. Meyer, Dalton Trans., 2007, 2003 RSC; (d) Y. Chen, L. Zheng, S. She, Z. Chen, B. Hu and Y. Li, Dalton Trans., 2011, 40, 4970 RSC; (e) X. Feng, L.-Ya Wang, J.-S. Zhao, B. Liu, J.-G. Wang and X.-G. Shi, Inorg. Chim. Acta, 2009, 362, 5127 CrossRef CAS.
- K. L. V. Mann, E. Psillakis, J. C. JeVery, L. H. Rees, N. M. Harden, J. A. McCleverty, M. D. Ward, D. Gatteschi, F. Totti, F. E. Mabbs, E. J. L. McInnes, P. C. Riedi and G. M. Smith, J. Chem. Soc., Dalton Trans., 1999, 339 RSC.
- (a) Z.-W. Mao, Q.-W. Hang and W.-X. Tang, Polyhedron, 1996, 15, 321 CrossRef CAS; (b) T. Higa, M. Moriya, Y. Shimazaki, T. Yajima, F. Tani, S. Karasawa, M. Nakano, Y. Naruta and O. Yamauchi, Inorg. Chim. Acta, 2007, 360, 3304 CrossRef CAS; (c) N. Matsumoto, T. Nozaki, H. Ushio, K. Motoda, M. Ohba and G. Mago, J. Chem. Soc., Dalton Trans., 1993, 2157 RSC.
- M. Julve, M. Verdaguer, O. Kahn, A. Gleizes and M. Philoche Levisalles, Inorg. Chem., 1983, 22, 368 CrossRef CAS.
- J. Glerup, P. A. Goodson, D. J. Hodgson and K. Michelsen, Inorg. Chem., 1995, 34, 6255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 956318-956326 and 956570. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00462k |
| This journal is © The Royal Society of Chemistry 2014 |