 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of the complete series of B–N analogues of triptycene†
Ömer
Seven
,
Sebastian
Popp
,
Michael
Bolte
,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
Institut für Anorganische Chemie, J.W. Goethe-Universität Frankfurt, Max-von-Laue-Strasse 7, D-60438 Frankfurt (Main), Germany. E-mail: Matthias.Wagner@chemie.uni-frankfurt.de
First published on 27th February 2014
Abstract
The reaction between the bisborate Li2[o-C6H4(BH3)2] and 2 equivalents of an appropriate pyrazole derivative (HpzR) in the presence of Me3SiCl yields o-phenylene-bridged pyrazaboles HB(μ-pzR)2(μ-o-C6H4)BH (3a–3e; HpzR = 4-iodopyrazole (3a), 4-(trimethylsilyl)pyrazole (3b), 3,5-dimethylpyrazole (3c), 3,5-di(tert-butyl)pyrazole (3d), 3,5-bis(trifluoromethyl)pyrazole (3e)). The synthesis approach thus provides access to uncharged B–N triptycenes bearing (i) functionalisable groups, (ii) electron-donating or -withdrawing substituents and (iii) pyrazole rings of varying steric demand. Treatment of p-R*C6H4BBr2 with the potassium tris(pyrazol-1-yl)borates K[HBpz3] or K[p-R*C6H4Bpz3] yields cationic pyrazolyl-bridged pyrazaboles [p-BrC6H4B(μ-pz)3BH]Br ([4a]Br) and [p-R*C6H4B(μ-pz)3Bp-C6H4R*]Br (R* = Br ([4b]Br), I ([4c]Br), SiMe3 ([4d]Br)), which can be regarded as full B–N analogues of triptycene. The B–H bonds of 3b and [4a]Br are unreactive towards tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH even at temperatures of 80 °C, thereby indicating an appreciable thermal stability of the corresponding B–N cage bonds. Most of the cage compounds are sufficiently inert towards water to allow quick aqueous workup. However, NMR spectroscopy in CD3OD solution reveals degradation of 3b or [4a]Br to the corresponding pyrazoles and o-C6H4(B(OCD3)2)2 or p-BrC6H4B(OCD3)2/B(OCD3)3. The diphenylated species [4b]Br is significantly more stable under the same measurement conditions; even after 76 d, most of the material degrades only to the stage of the syn/anti-pyrazaboles p-BrC6H4(CD3O)B(μ-pz)2B(OCD3)p-C6H4Br (11a/11b). A derivatisation of [4c]Br with nBu3SnCCtBu through Stille-type coupling reactions furnishes the alkynyl derivative [p-tBuCCC6H4B(μ-pz)3Bp-C6H4CCtBu]Br ([4e]Br). Larger B–N aggregates are also accessible: treatment of the tetrakisborate Li4[1,2,4,5-C6H2(BH3)4] with 4 equivalents of HpzR in the presence of Me3SiCl leads to the corresponding B–N pentiptycenes 14a–14d (HpzR = 3,5-bis(trifluoromethyl)pyrazole (14a), 4-(trimethylsilyl)pyrazole (14b), 3,5-dimethylpyrazole (14c), 3,5-di(tert-butyl)pyrazole (14d)).
CH even at temperatures of 80 °C, thereby indicating an appreciable thermal stability of the corresponding B–N cage bonds. Most of the cage compounds are sufficiently inert towards water to allow quick aqueous workup. However, NMR spectroscopy in CD3OD solution reveals degradation of 3b or [4a]Br to the corresponding pyrazoles and o-C6H4(B(OCD3)2)2 or p-BrC6H4B(OCD3)2/B(OCD3)3. The diphenylated species [4b]Br is significantly more stable under the same measurement conditions; even after 76 d, most of the material degrades only to the stage of the syn/anti-pyrazaboles p-BrC6H4(CD3O)B(μ-pz)2B(OCD3)p-C6H4Br (11a/11b). A derivatisation of [4c]Br with nBu3SnCCtBu through Stille-type coupling reactions furnishes the alkynyl derivative [p-tBuCCC6H4B(μ-pz)3Bp-C6H4CCtBu]Br ([4e]Br). Larger B–N aggregates are also accessible: treatment of the tetrakisborate Li4[1,2,4,5-C6H2(BH3)4] with 4 equivalents of HpzR in the presence of Me3SiCl leads to the corresponding B–N pentiptycenes 14a–14d (HpzR = 3,5-bis(trifluoromethyl)pyrazole (14a), 4-(trimethylsilyl)pyrazole (14b), 3,5-dimethylpyrazole (14c), 3,5-di(tert-butyl)pyrazole (14d)).
Introduction
Triptycenes 1 (Fig. 1) are a class of organic compounds possessing a three-dimensional paddle-wheel structure. Due to their rigid bicyclo[2.2.2]octatriene core, angles close to 120° are maintained between the three o-phenylene ring panels, which leads to the formation of three equally sized compartments lined with π-electron clouds. On the one hand, this unique geometry often frustrates space-efficient packing and thereby leads to void spaces in a supramolecular assembly, called the “internal free volume” (IFV), which can be used to store small guest molecules.1 On the other hand, the three triptycene blades have also been used to create interlocking molecular structures acting as rotors, gearings or brakes in molecular machines.2,3 Triptycenes are therefore useful building blocks for the generation of sophisticated molecular architectures with well-defined geometries.4 During the last few decades, a wide spectrum of applications has opened up, ranging from materials5,6 and sensor science7 to host–guest5,8 and coordination chemistry.4,9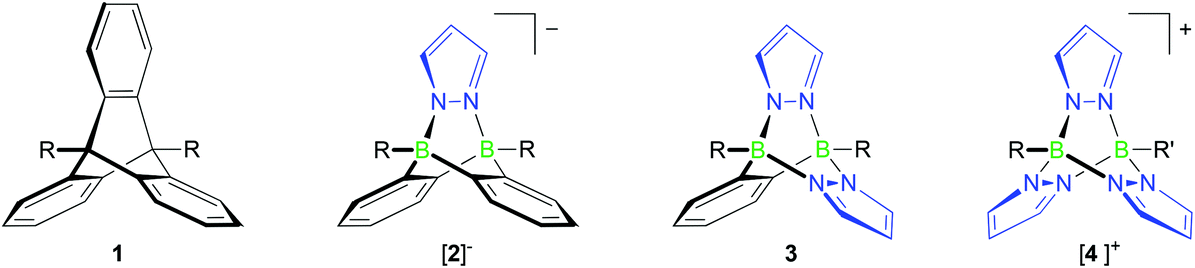 | ||
| Fig. 1 Triptycene (1) and its B–N analogues [2]−, 3 and [4]+. | ||
Further developments in these fields are critically dependent on the availability of broadly functionalised triptycene derivatives. To date, the Diels–Alder reactions between anthracenes and p-benzoquinones or benzynes have offered the most important methods for the assembly of triptycene scaffolds (Scheme 1). With respect to functional-group compatibility, it is therefore essential to be able to release benzyne intermediates in situ under mild conditions and with high efficiency; good results are usually obtained through aprotic diazotisation of anthranilic acids with amyl nitrite10 or upon treatment of (phenyl)[2-(trimethylsilyl)phenyl]iodonium triflate with an appropriate fluoride source.11 Even though various halogenated 1,2-bis(trimethylsilyl)benzenes have recently been made available,12 which are suitable starting materials for the synthesis of corresponding halogenated hypervalent iodine benzyne precursors, the crucial role of the benzyne intermediates in triptycene syntheses still constitutes a bottleneck on the way to novel triptycene-containing functional units.
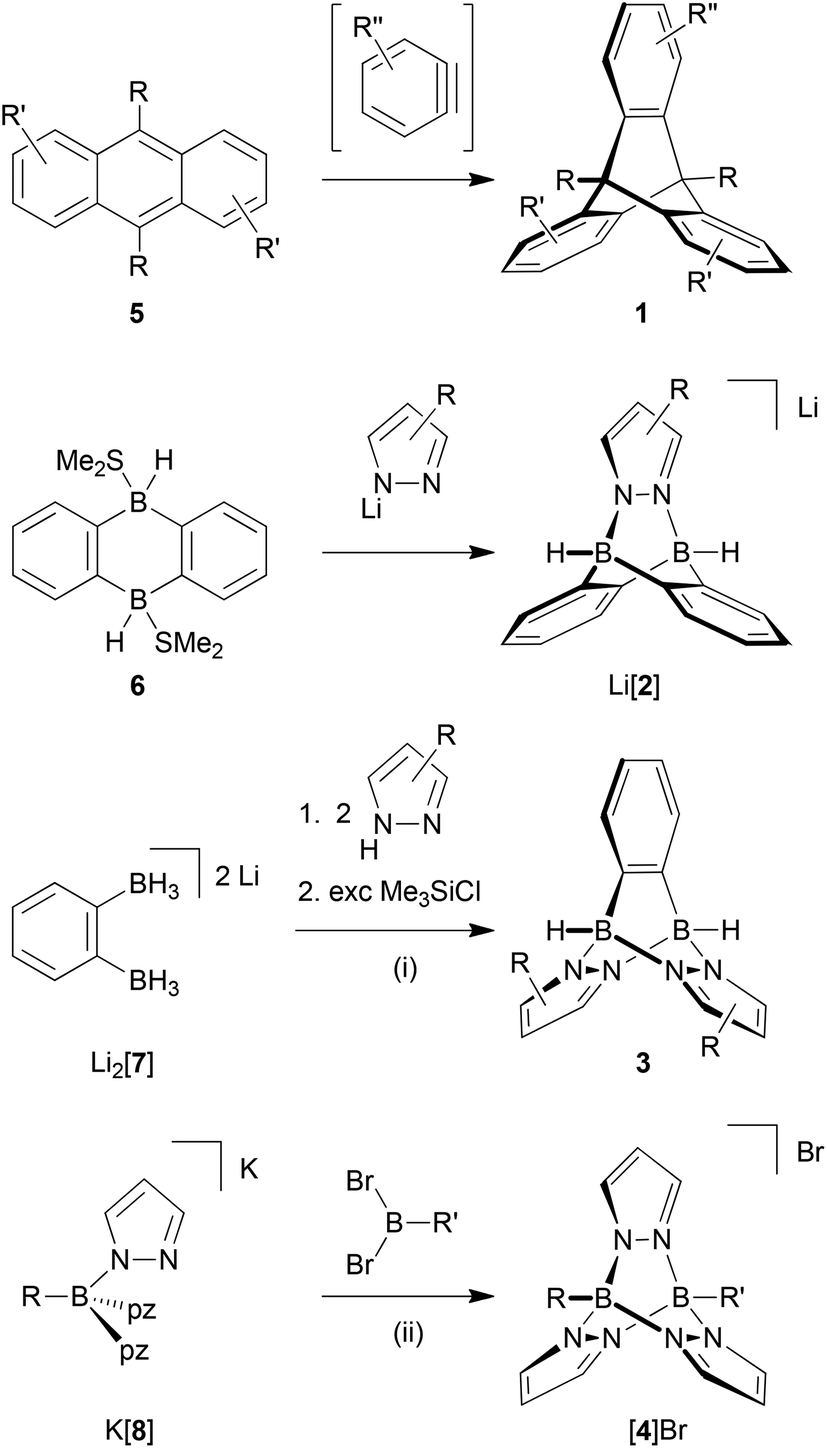 | ||
| Scheme 1 General synthesis methods of all-carbon triptycenes and of the complete series of B–N triptycenes. General reaction conditions: (i) Et2O, rt, between 12 h and 15 h. (ii) CH3CN-toluene or toluene, between rt and 120 °C, between 8 h and 24 h. | ||
Our group has a long-standing interest in exploiting the B–N/C–C isosterism for the facile synthesis of complex molecular frameworks.13–16 This approach takes advantage of the fact that B–N adduct bonds tend to form spontaneously in essentially quantitative yields whereas the isoelectronic C–C single bonds are usually much harder to establish.17,18 The general concept of B–N/C–C isosterism is very well applicable for the preparation of triptycene analogues (cf. compounds [2]−, 3 and [4]+; Fig. 1).
In a previous communication, we have already reported on the reaction between the Me2S adduct of 9,10-dihydro-9,10-diboraanthracene (6; Scheme 1) and pyridazine, which gives a soluble B–N congener of triptycene.19 The synthesis approach also works faithfully for phthalazine (benzo[d]pyridazine) or B-substituted 9,10-dihydro-9,10-diboraanthracenes as the starting materials. If lithium pyrazolides are used instead of pyridazine, ionic compounds of type Li[2] are obtained (Scheme 1; Fig. 1).19
While the third member of the family of B–N triptycenes, compound 3 (Fig. 1), has been unknown, scattered reports of cationic [4]+-type species can be found in the literature. Most of the times, the latter molecules have been observed as side products of the complexation of Lewis-acidic transition metal ions with tris(pyrazol-1-yl)borate ligands.20 Two early reports of targeted syntheses do, however, also exist: Trofimenko prepared [4]PF6 (R = R′ = Et) in yields of 22% through the reaction of pyrazolide ions with EtB(OTos)2 and subsequent salt metathesis (HOTos = toluenesulfonic acid); he also mentioned the relationship between [4]+ and triptycene.21 In a later report, Niedenzu et al. described the synthesis of derivatives of [4]+ carrying methyl groups at the pyrazolyl rings. The boron atoms were either equipped with one hydrogen atom each or with one hydrogen atom and one ethyl group.22 The synthesis approach relied on the reaction of tris(pyrazol-1-yl)borate anions (cf. [8]−; Scheme 1) with trigonal boranes containing two good leaving groups.
The purpose of this paper is to provide high-yield syntheses and full characterisations of selected examples of 3- and [4]+-type molecules together with compounds possessing extended B–N pentiptycene-type architectures. Special emphasis will be placed on the development of C-halogenated derivatives, which possess promising potential as future building blocks of (supra)molecular structures. Moreover, we will investigate and compare the stabilities of the bicyclo[2.2.2]octatriene cores of [2]−, 3 and [4]+ in order to explore the scope and limitations of these molecules as triptycene substitutes.
Results and discussion
Synthesis and NMR-spectroscopic characterisation of 3- and [4]+-type molecules
Neutral 3-type compounds were prepared from the o-phenylene-bridged bisborate Li2[7]23 and 2 equivalents of an appropriately substituted pyrazole (Scheme 1). The first two B–N bonds assemble spontaneously at room temperature through a Brønsted acid–base reaction with liberation of H2. Subsequent addition of excess Me3SiCl as a hydride scavenger creates free coordination sites at the boron centres; this leads to the formation of two intramolecular B–N adduct bonds and thereby closes the molecular cage. The synthesis protocol tolerates various degrees of steric bulk at the 3,5-positions of the pyrazolyl rings: for example, derivatives carrying hydrogen atoms (3a, 3b; Fig. 2), methyl groups (3c) or tert-butyl groups (3d) are all formed in good yields. It is also possible to employ both electron-richer (cf. the H3C-derivative 3c) and electron-poorer pyrazoles (cf. the CF3-derivative 3e) with similar success; 3e is, however, significantly more prone to hydrolysis than 3c. We also found that the o-phenylene ring may be replaced by other (metal)organic fragments, because the synthesis sequence can also be applied to the 1,1′-ferrocenylene-bridged bisborate Li2[1,1′-fc(BH3)2]24,25 to give compounds like 3f (Fig. 2). 3f can not only be viewed as an Fe-containing B–N triptycene, but also as an ansa-ferrocene with pyrazabole26 bridge.14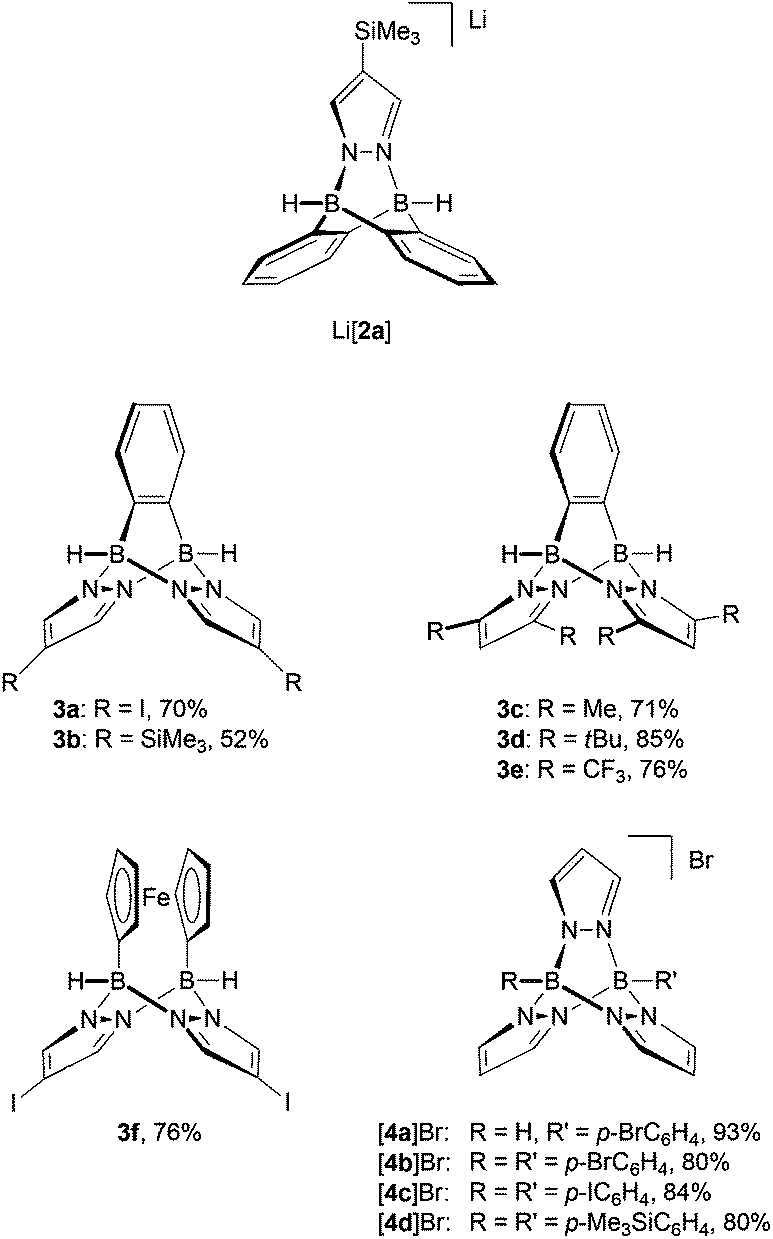 | ||
| Fig. 2 Compilation of the B–N triptycenes of types [2]−, 3 and [4]+ discussed in this paper. | ||
Cationic [4]+-type B–N triptycenes are accessible starting from readily available potassium tris(pyrazol-1-yl)borates27,28 (K[8]; cf. the ESI† for detailed information) and dibromo(organyl)boranes (Scheme 1). This synthesis approach provides access to unsymmetrically (R = H, R′ = p-BrC6H4 ([4a]Br); Fig. 2) as well as symmetrically B-functionalised compounds (R = R′ = p-BrC6H4 ([4b]Br), p-IC6H4 ([4c]Br) or p-Me3SiC6H4 ([4d]Br); Fig. 2). The combination of R = R′ = (substituted) phenyl with 3,5-dimethylpyrazole generally failed to give the desired B–N triptycenes, likely due to prohibitively high steric congestion. The use of 4-bromo- or 4-iodopyrazole led to very poorly soluble products, which were hard to purify and characterise and are consequently not considered further.
The 11B{1H} NMR shifts of all the B–N triptycenes under consideration here fall in the range between 0.2 ppm and −7.0 ppm, thereby testifying to the presence of tetracoordinated boron nuclei.29 Compound [4a]Br, which possesses two magnetically inequivalent boron centres, gives rise to two signals in the 11B{1H} NMR spectrum (−0.5 ppm (BC); −5.4 ppm (BH)); all other molecules show only one resonance each. B,H-coupling is resolved only in the cases of 3c (d, 1JB,H = 98 Hz) and [4a]Br (d, 1JB,H = 128 Hz). In the 1H NMR spectra, the boron-bonded hydrogen atoms give rise to very broad resonances in the interval between 5.5 ppm and 4.0 ppm; only for the signal of 3c the expected quartet multiplicity is visible.
All the proton spectra of 3a–3e contain two characteristic multiplets for the o-phenylene rings; the organometallic derivative 3f shows two virtual triplets at 4.12 ppm and 3.22 ppm for the 1,1′-ferrocenylene backbone. For each of the molecules, the overall integral ratio between the o-phenylene/1,1′-ferrocenylene resonances on the one hand and the pyrazolyl signals on the other confirms the proposed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of the two moieties in 3-type compounds. Moreover, the number of signals in the 1H and 13C{1H} NMR spectra is in line with the suggested symmetry of the molecular frameworks.
:2 ratio of the two moieties in 3-type compounds. Moreover, the number of signals in the 1H and 13C{1H} NMR spectra is in line with the suggested symmetry of the molecular frameworks.
In the 1H NMR spectrum of the unsymmetrically B-substituted cation [4a]+, the pyrazolyl rings lead to two doublets with characteristic low-field shifts and small 3JH,H coupling constants (8.24 ppm, 8.02 ppm; 2.5 Hz) and to one high-field shifted virtual triplet (6.51 ppm). The R substituent gives the two approximate doublet resonances that are expected for AA′BB′ spin systems. On going from [4a]Br to the symmetrically B-substituted species like [4b]Br, the two pyrazolyl doublets become one signal of double intensity and the pyrazolyl:phenylene integral ratios change from 9 H:4 H to 9 H:8 H.
X-ray crystal structure analyses of B–N triptycenes
Selected crystallographic data for the structure analyses discussed in the following paragraphs are compiled in Tables 1–3. (Note: In a crystallographic context, we are, for reasons of simplicity, treating compounds containing deuterium atoms as if they were containing exclusively hydrogen atoms.)| 3f*·3C6H6 | 3a | [4a]Br·CH2Cl2 | |
|---|---|---|---|
| Formula | C35H31B4Fe2I5N10·3 C6H6 | C12H10B2I2N4 | C15H14B2Br2N6·CH2Cl2 |
| M r | 1615.46 | 485.66 | 544.69 |
| Colour, shape | Orange, block | Colourless, block | Colourless, block |
| T [K] | 173(2) | 173(2) | 173(2) |
| Radiation, λ [Å] | MoKα, 0.71073 | MoKα, 0.71073 | MoKα, 0.71073 |
| Crystal system | Monoclinic | Trigonal | Orthorhombic |
| Space group | P21/c |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
Pnma |
| a [Å] | 21.7729(16) | 18.7020(7) | 12.0607(5) |
| b [Å] | 11.9678(12) | 18.7020(7) | 9.6544(6) |
| c [Å] | 24.4436(18) | 23.5495(9) | 18.1880(8) |
| α [°] | 90 | 90 | 90 |
| β [°] | 115.688(5) | 90 | 90 |
| γ [°] | 90 | 120 | 90 |
| V [Å3] | 5739.9(9) | 7133.3(6) | 2117.79(18) |
| Z | 4 | 18 | 4 |
| D calcd [g cm−3] | 1.869 | 2.035 | 1.708 |
| F(000) | 3096 | 4068 | 1072 |
| μ [mm−1] | 3.240 | 3.961 | 4.095 |
| Crystal size [mm3] | 0.10 × 0.10 × 0.10 | 0.20 × 0.20 × 0.10 | 0.20 × 0.10 × 0.05 |
| Rflns collected | 41538 |
35425 |
37320 |
| Independent rflns (Rint) | 10130 (0.1528) |
3057 (0.0689) | 2170 (0.1048) |
| Data/restraints/parameters | 10130/390/667 |
3057/0/190 | 2170/0/147 |
| GOF on F2 | 0.914 | 1.082 | 1.180 |
| R 1, wR2 [I > 2σ(I)] | 0.0678, 0.0973 | 0.0219, 0.0531 | 0.0353, 0.0780 |
| R 1, wR2 (all data) | 0.1459, 0.1167 | 0.0221, 0.0533 | 0.0383, 0.0794 |
| Largest diff. peak and hole [e Å−3] | 1.494, −0.848 | 1.246, −0.649 | 0.624, −0.367 |
| [4b]Br | 14a·3 C7H8 | 14b·2 CH2Cl2 | |
|---|---|---|---|
| Formula | C21H17B2Br3N6 | C26H10B4F24N8·3 C7H8 | C30H50B4N8Si4·2 CH2Cl2 |
| M r | 614.76 | 1210.06 | 848.23 |
| Colour, shape | Colourless, block | Light brown, block | Colourless, block |
| T [K] | 173(2) | 173(2) | 173(2) |
| Radiation, λ [Å] | MoKα, 0.71073 | MoKα, 0.71073 | MoKα, 0.71073 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a [Å] | 9.7625(8) | 8.8267(8) | 10.610(2) |
| b [Å] | 11.1593(8) | 11.3525(11) | 11.003(3) |
| c [Å] | 11.5455(9) | 14.2556(14) | 12.115(3) |
| α [°] | 90.305(6) | 99.583(8) | 63.855(17) |
| β [°] | 107.707(6) | 106.299(7) | 70.231(17) |
| γ [°] | 106.691(6) | 106.005(7) | 80.803(17) |
| V [Å3] | 1141.79(15) | 1271.1(2) | 1194.7(5) |
| Z | 2 | 1 | 1 |
| D calcd [g cm−3] | 1.788 | 1.581 | 1.179 |
| F(000) | 600 | 608 | 446 |
| μ [mm−1] | 5.322 | 0.156 | 0.380 |
| Crystal size [mm3] | 0.20 × 0.15 × 0.10 | 0.26 × 0.25 × 0.22 | 0.21 × 0.14 × 0.09 |
| Rflns collected | 10831 |
15695 |
10352 |
| Independent rflns (Rint) | 4185 (0.0570) | 4864 (0.0645) | 4202 (0.1236) |
| Data/restraints/parameters | 4185/0/289 | 4864/0/397 | 4202/0/243 |
| GOF on F2 | 1.015 | 1.046 | 0.856 |
| R 1, wR2 [I > 2σ(I)] | 0.0363, 0.0872 | 0.0451, 0.1127 | 0.0774, 0.1452 |
| R 1, wR2 (all data) | 0.0474, 0.0911 | 0.0580, 0.1189 | 0.1890, 0.1809 |
| Largest diff. peak and hole [e Å−3] | 0.596, −0.721 | 0.262, −0.265 | 0.681, −0.406 |
| 11a | 11b | |
|---|---|---|
| Formula | C20H20B2Br2N4O2 | C20H20B2Br2N4O2 |
| M r | 529.84 | 529.84 |
| Colour, shape | Colourless, block | Colourless, plate |
| T [K] | 173(2) | 173(2) |
| Radiation, λ [Å] | MoKα, 0.71073 | MoKα, 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n |
| a [Å] | 15.7852(7) | 7.6702(5) |
| b [Å] | 22.9613(10) | 12.4132(7) |
| c [Å] | 12.9046(5) | 11.2406(7) |
| α [°] | 90 | 90 |
| β [°] | 113.391(3) | 102.351(5) |
| γ [°] | 90 | 90 |
| V [Å3] | 4292.9(3) | 1045.47(11) |
| Z | 8 | 2 |
| D calcd [g cm−3] | 1.640 | 1.683 |
| F(000) | 2112 | 528 |
| μ [mm−1] | 3.801 | 3.902 |
| Crystal size [mm3] | 0.32 × 0.31 × 0.31 | 0.24 × 0.19 × 0.13 |
| Rflns collected | 92971 |
14759 |
| Independent rflns (Rint) | 9294 (0.0782) | 2137 (0.0915) |
| Data/restraints/parameters | 9294/0/541 | 2137/0/136 |
| GOF on F2 | 1.025 | 0.965 |
| R 1, wR2 [I > 2σ(I)] | 0.0598, 0.1343 | 0.0293, 0.0660 |
| R 1, wR2 (all data) | 0.1048, 0.1562 | 0.0415, 0.0696 |
| Largest diff. peak and hole [e Å−3] | 0.553, −1.016 | 0.347, −0.570 |
The reaction providing compound 3f is less selective than in the case of the o-phenylene congener 3a and the ferrocene derivative is more sensitive to air and moisture. Thus, even though we succeeded in an NMR spectroscopic and X-ray crystallographic characterisation of 3f, we have not been able to isolate the compound in an analytically pure form (cf. the ESI† for a plot of the molecular structure, which revealed no significant differences compared to other pyrazabole-bridged ansa-ferrocenes14). In two instances, single crystals of a side product/decomposition product of 3f grew upon prolonged storage of an NMR tube containing a solution of crude 3f in C6D6. X-ray crystallography revealed a neutral dinuclear ferrocene species (3f*; Fig. 3) containing 1 pyrazole ligand, 4 pyrazolyl moieties and 4 B–H bonds. As a remarkable feature of this side product, we note that the interannular bridge of each of the two ferrocene fragments consists of one pyrazolyl ring only. A third ring acts as a linker between the organometallic units, whereas the remaining two heterocyclic rings are terminal substituents. A proton residing on the nitrogen atom N(42) forms a hydrogen bond with the nitrogen atom N(2) to give a HB4N10 macrocycle.
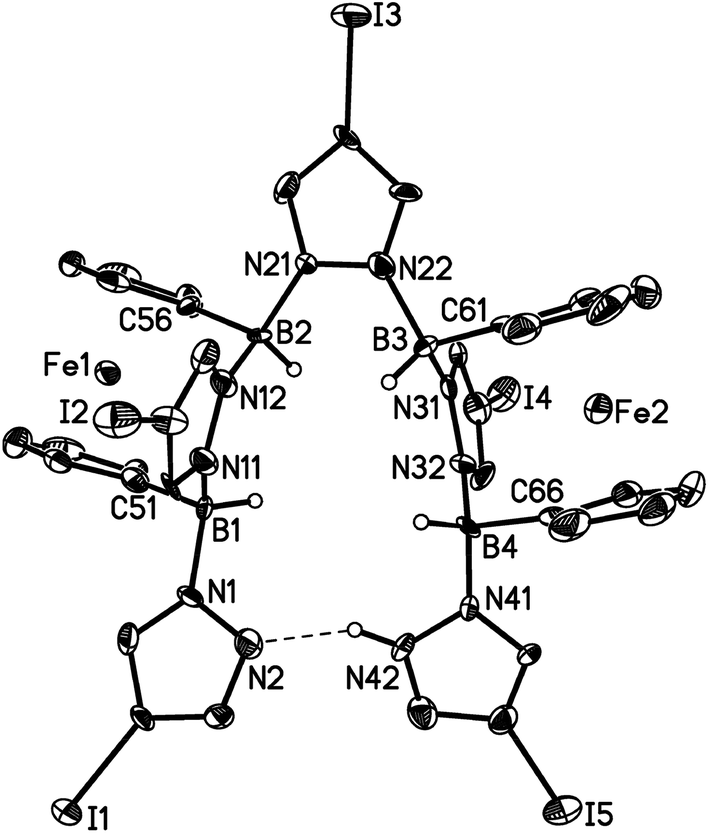 | ||
| Fig. 3 Molecular structure of 3f*·3C6H6 in the solid state. Hydrogen atoms (except on boron and N(42)) and the C6H6 molecules have been omitted for clarity; displacement ellipsoids are drawn at the 50% probability level. The bond lengths and bond angles are not given due to poor crystallographic data which lead to large error margins. The H atom at N(42) was found in the difference Fourier map, but it could not be isotropically refined. | ||
Compounds 3a, [4a]Br and [4b]Br have also been characterised by X-ray crystallography (Fig. 4 and 5). Compounds 3a and [4b]+ possess no symmetry element in the crystal lattice; the solid-state structure of the cation [4a]+ features a mirror plane containing the pyrazolyl ring pz(N(1)). All three molecules exhibit the aimed-for paddle-wheel structure with angles between the three core planes [B(1)N(1)N(2)B(2), B(1)N(11)N(12)B(2) and B(1)C(21)C(22)B(2) (3a) or B(1)N(11A)N(12A)B(2) ([4a]Br) or B(1)N(21)N(22)B(2) ([4b]Br)] close to 120°.30 The same is true for the angles between the corresponding planes in Li[2a] (Fig. 2).19 In the cases of the two cationic B–N triptycenes, we observe kinks along certain N–N bonds resulting in dihedral angles between the pyrazolyl blades that can be as small as pz(N(11))//pz(N(11A)) = 100.6(1)° ([4a]Br) or as large as pz(N(1))//pz(N(21)) = 138.7(2)° ([4b]Br).
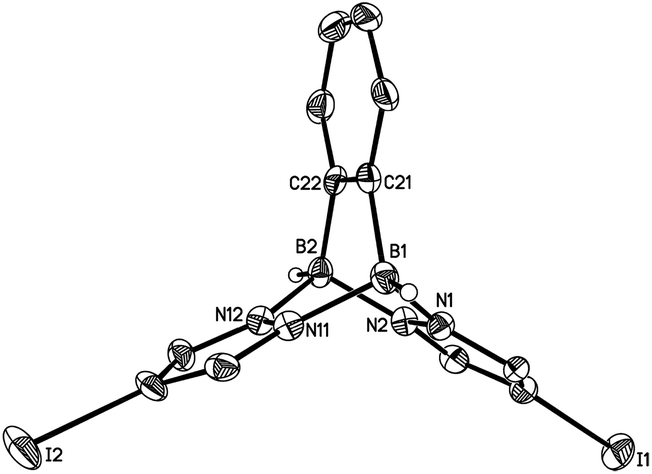 | ||
| Fig. 4 Molecular structure of 3a in the solid state. Hydrogen atoms except on boron have been omitted for clarity; displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å), atom⋯atom distance (Å), bond angles (°) and dihedral angles (°): B(1)–N(1) 1.575(4), B(1)–N(11) 1.588(4), B(1)–C(21) 1.611(5), B(2)–N(2) 1.574(4), B(2)–N(12) 1.579(4), B(2)–C(22) 1.604(5), B(1)⋯B(2) 2.707(5); N(1)–B(1)–N(11) 101.5(2), N(1)–B(1)–C(21) 105.7(3), N(11)–B(1)–C(21) 104.0(2), N(2)–B(2)–N(12) 100.9(2), N(2)–B(2)–C(22) 105.8(3), N(12)–B(2)–C(22) 104.4(3); N(1)B(1)N(11)//N(2)B(2)N(12) 84.1(3), N(1)B(1)N(11)//N(1)N(2)N(11)N(12) 47.7(2), N(2)B(2)N(12)//N(1)N(2)N(11)N(12) 48.2(3), B(1)N(1)N(2)B(2)//B(1)N(11)N(12)B(2) 117.3(1), B(1)N(1)N(2)B(2)//B(1)C(21)C(22)B(2) 122.4(2), B(1)N(11)N(12)B(2)//B(1)C(21)C(22)B(2) 120.3(2), pz(N(1))//pz(N(11)) 123.0(1), pz(N(1))//C6H4 122.5(1), pz(N(11))//C6H4 114.4(1); pz(N(X)) = pyrazolyl ring containing N(X). | ||
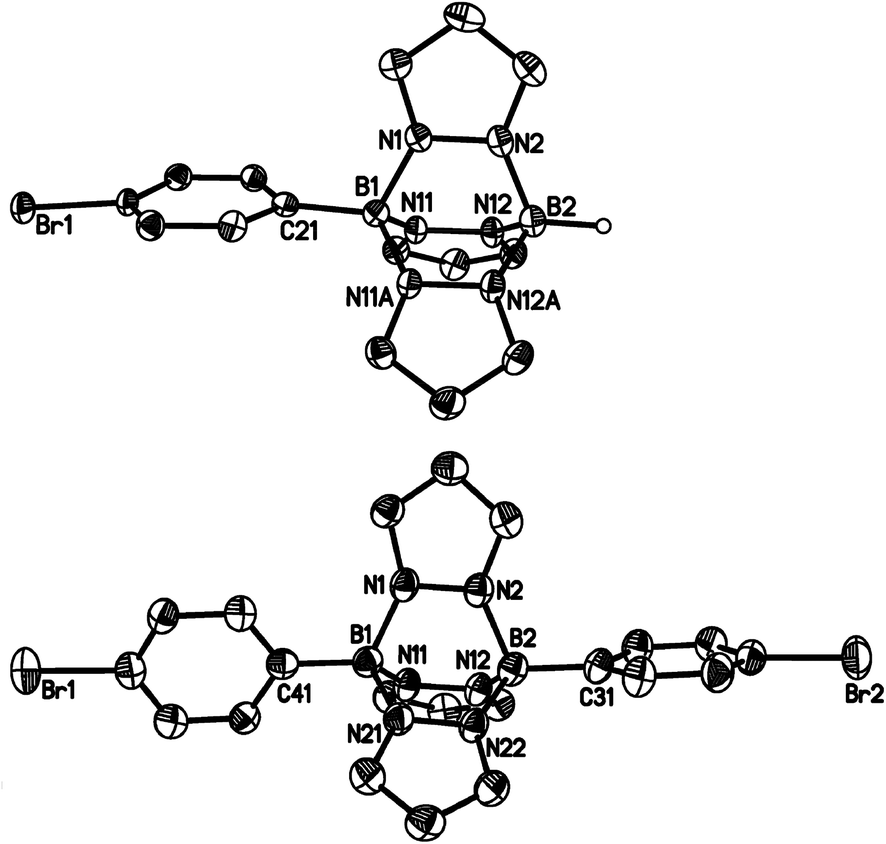 | ||
| Fig. 5 Molecular structures of [4a]Br·CH2Cl2 (top) and [4b]Br (bottom) in the solid state. Hydrogen atoms except on boron, the bromide counterions and the solvent molecule have been omitted for clarity; displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å), atom⋯atom distances (Å), bond angles (°) and dihedral angles (°): [4a]Br: B(1)–N(1) 1.561(5), B(1)–N(11) 1.574(3), B(1)–C(21) 1.582(5), B(2)–N(2) 1.541(6), B(2)–N(12) 1.549(3), B(1)⋯B(2) 2.676(6); N(1)–B(1)–N(11) 104.4(2), N(11)–B(1)–N(11A) 101.3(3), N(2)–B(2)–N(12) 104.8(2), N(12)–B(2)–N(12A) 102.7(3); B(1)N(1)N(2)B(2)//B(1)N(11)N(12)B(2) 121.3(1), B(1)N(11)N(12)B(2)//B(1)N(11A)N(12A)B(2) 117.5(1), pz(N(1))//pz(N(11)) 129.7(1), pz(N(11))//pz(N(11A)) 100.6(1); symmetry transformation used to generate equivalent atoms: A: x, −y + 3/2, z. [4b]Br: B(1)–N(1) 1.561(5), B(1)–N(11) 1.558(5), B(1)–N(21) 1.563(4), B(1)–C(41) 1.589(5), B(2)–N(2) 1.561(5), B(2)–N(12) 1.575(5), B(2)–N(22) 1.556(5), B(2)–C(31) 1.590(5), B(1)⋯B(2) 2.702(5); N(1)–B(1)–N(11) 102.8(3), N(1)–B(1)–N(21) 103.9(3), N(11)–B(1)–N(21) 103.2(3), N(2)–B(2)–N(12) 102.6(3), N(2)–B(2)–N(22) 104.5(3), N(12)–B(2)–N(22) 102.5(3); B(1)N(1)N(2)B(2)//B(1)N(11)N(12)B(2) 119.0(2), B(1)N(1)N(2)B(2)//B(1)N(21)N(22)B(2) 121.6(2), B(1)N(11)N(12)B(2)//B(1)N(21)N(22)B(2) 119.4(2), pz(N(1))//pz(N(11)) 105.6(1), pz(N(1))//pz(N(21)) 138.7(2), pz(N(11))//pz(N(21)) 115.5(1); pz(N(X)) = pyrazolyl ring containing N(X). | ||
The average B–N bond lengths decrease continuously upon going from Li[2a] (1.605 Å) to 3a (1.579 Å) to [4b]Br (1.562 Å). For [4a]Br, a comparison of the average B–N bond lengths about B(1) (1.570 Å) and B(2) (1.546 Å) indicates considerable steric repulsion between the p-BrC6H4 substituent and the bridging pyrazolide moieties. A related effect is obvious in the case of 9-phenyltriptycene, which shows longer endocyclic C–C bonds about the phenylated bridgehead carbon atom (average value = 1.554 Å) than about the unsubstituted bridgehead atom (average value = 1.513 Å).31
Similar to the B–N bonds, the intramolecular B⋯B distances contract along the series Li[2a] (2.734(6) Å) → 3a (2.707(5) Å) → [4a]Br (2.676(6) Å)/[4b]Br (2.702(5) Å). Even shorter through-space distances are found between the two bridgehead carbon atoms in 9-phenyltriptycene (2.630 Å)31 and in pristine triptycene (2.604 Å).32
Investigations into the relative cage stabilities of B–N triptycenes
Hydroboration reactions of alkenes and alkynes require three-coordinate hydroborane species.33 If borane adducts are employed (e.g., BH3·SMe2), the donor has to come off first in a preceding association–dissociation equilibrium step, before the B–H bond is able to add across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-double or a CC-triple bond. A comparison of the reactivities of Li[2a] (Fig. 2), 3b and [4a]Br toward the hydroboration of tBuCCH therefore provides useful first insight into the relative stabilities of the individual B–N cage bonds. Upon treatment with excess tBuCCH in THF at 60 °C, Li[2a] adds two equivalents of the alkyne to give Li[tBuC(H)(H)CB(μ-pzTMS)(μ-o-C6H4)2BC(H)(H)CtBu] (HpzTMS = 4-(trimethylsilyl)pyrazole).19 Under the same conditions, neither 3b nor [4a]Br undergoes a hydroboration reaction, but can be recovered in essentially quantitative yield; this result remains valid if the temperature is increased to 80 °C.
C-double or a CC-triple bond. A comparison of the reactivities of Li[2a] (Fig. 2), 3b and [4a]Br toward the hydroboration of tBuCCH therefore provides useful first insight into the relative stabilities of the individual B–N cage bonds. Upon treatment with excess tBuCCH in THF at 60 °C, Li[2a] adds two equivalents of the alkyne to give Li[tBuC(H)(H)CB(μ-pzTMS)(μ-o-C6H4)2BC(H)(H)CtBu] (HpzTMS = 4-(trimethylsilyl)pyrazole).19 Under the same conditions, neither 3b nor [4a]Br undergoes a hydroboration reaction, but can be recovered in essentially quantitative yield; this result remains valid if the temperature is increased to 80 °C.
In a second series of experiments, we investigated the sensitivity of B–N triptycenes toward air and moisture. While the B–H bonds of free 9,10-dihydro-9,10-diboraanthracene are extremely prone to hydrolysis,34 it takes several hours until the 1H NMR spectrum of compound Li[2a] in non-dried d8-THF shows significant signs of decomposition.19 In the cases of 3b, [4a]Br and [4b]Br, even quick aqueous workup is possible; 3b can also be purified by column chromatography under ambient conditions (silica gel, non-dried solvents). Further detailed protonolysis reactions were carried out in non-dried CD3OD at room temperature and were monitored by NMR spectroscopy (plots of the spectra are included in the ESI,† together with the spectra of selected authentic samples of proposed decomposition products). 1H NMR spectra recorded on solutions of compound 3b approximately 10 min after sample preparation already contained resonances assignable to degradation products. After 2 h, about 10% of the initial amount of 3b was left and after 2 d, the sample consisted exclusively of DpzTMS and o-C6H4(B(OCD3)2)2 (9) in a stoichiometric ratio of 2:1 (Scheme 2; for simplicity reasons, we have drawn the decomposition products as CD3OD esters, even though the corresponding boronic acids or mixed acid/ester species may well be present, too). Under the same measurement conditions, about 55% of [4a]Br was still present in the sample after 2 h. Two days later, however, the proton NMR spectrum showed only the resonances of free pyrazole, together with a broad multiplet at 7.48–7.52 ppm. The 11B{1H} NMR spectrum contained resonances at 28.3 ppm and 18.6 ppm with an integral ratio of 1:1.4. The first value is characteristic of boron nuclei surrounded by one carbon and two oxygen atoms (cf. C6H5B(OCH3)2: δ(11B) = 28.629), whereas the second signal corresponds to B(OCD3)3 (cf. B(OCH3)3: δ(11B) = 18.3; in CH3OH29,35). These data thus indicate that the cage of [4a]Br has completely been broken down to pyrazole, the p-bromophenylboronic acid methyl ester 10 (resulting from the substituted bridgehead boron atom) and B(OCD3)3 (resulting from the unsubstituted bridgehead boron atom; Scheme 2).
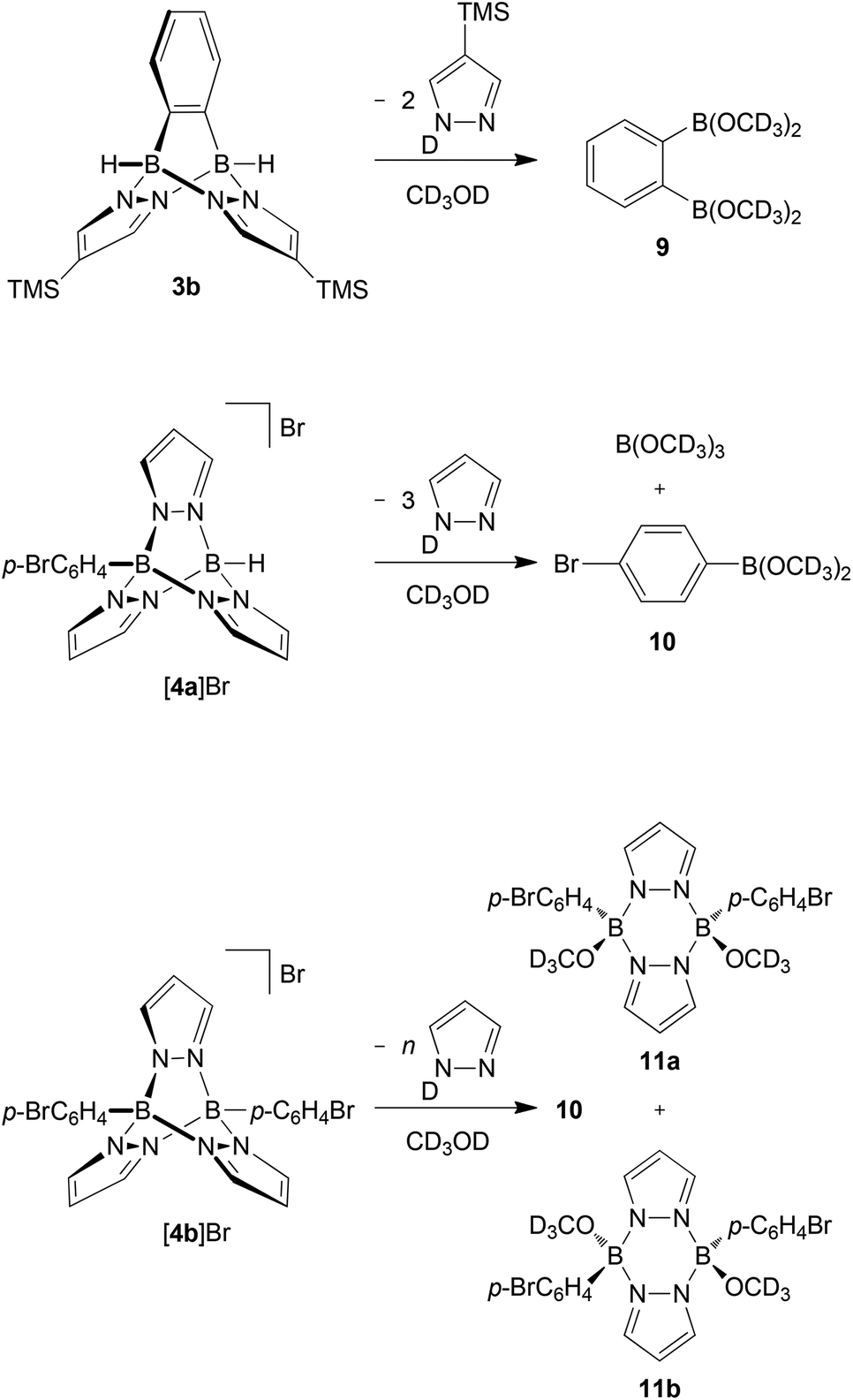 | ||
| Scheme 2 Degradation reactions of 3b, [4a]Br and [4b]Br in non-dried CD3OD. | ||
The diphenylated derivative [4b]Br turned out to be more stable towards CD3OD than 3b and [4a]Br. After 76 d, 1H NMR spectroscopy indicated the formation of four major products: (i) free pyrazole, (ii) 10 and (iii) two more species still consisting of both pyrazolide and p-BrC6H4 units. From the NMR tube, we were able to isolate the syn and anti pyrazaboles 11a36 and 11b in a single-crystal form (Fig. 6). Since the molecular structures of 11a and 11b are consistent with the proton resonance patterns of the two incompletely degraded species, we conclude that pyrazaboles are comparatively long-lived intermediates of the protonolysis pathway of [4b]Br.36 In line with that, the 11B NMR spectrum of [4b]Br in CD3OD after 76 d exhibited a resonance not only at 28.6 ppm (10), but also at 3.7 ppm (cf. (C6H5)(CH3O)B(μ-pz)2B(OCH3)(C6H5): δ(11B) = 3.5;37 in CDCl3).
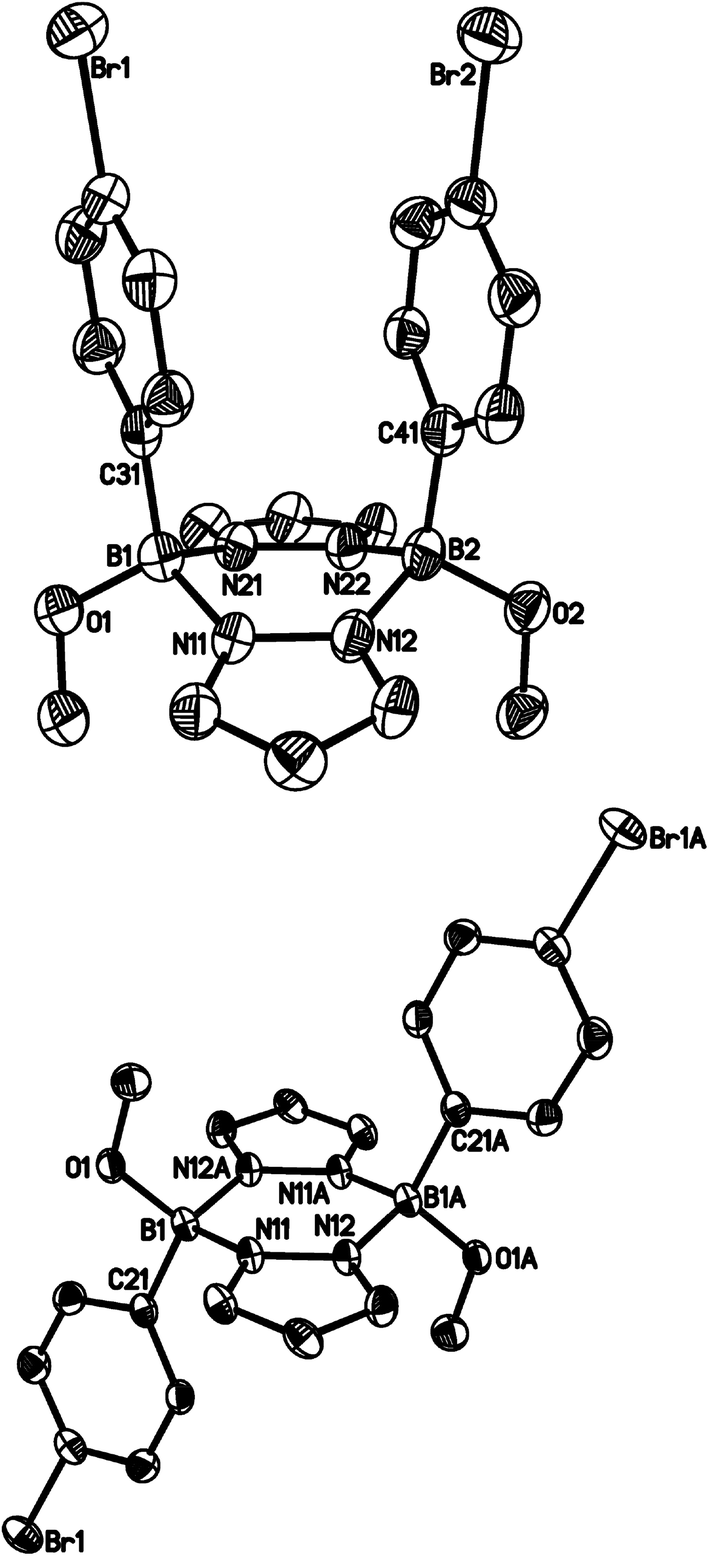 | ||
| Fig. 6 Molecular structures of 11aA (top) and 11b (bottom) in the solid state. Hydrogen atoms have been omitted for clarity; displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å), atom⋯atom distances (Å), bond angles (°) and dihedral angles (°): 11aA: B(1)–O(1) 1.415(6), B(1)–N(11) 1.577(6), B(1)–N(21) 1.587(6), B(1)–C(31) 1.602(7), B(2)–O(2) 1.442(6), B(2)–N(12) 1.600(6), B(2)–N(22) 1.578(6), B(2)–C(41) 1.589(7), B(1)⋯B(2) 3.115(7), COG(Ar(C(31)))⋯COG(Ar(C(41))) 3.808; N(11)–B(1)–N(21) 103.7(4), N(12)–B(2)–N(22) 103.0(3); N(11)B(1)N(21)//N(12)B(2)N(22) 54.6(3), N(11)B(1)N(21)//N(11)N(12)N(21)N(22) 27.5(3), N(12)B(2)N(22)//N(11)N(12)N(21)N(22) 27.1(3), B(1)N(11)N(12)B(2)//B(1)N(21)N(22)B(2) 140.2(2), pz(N(11))//pz(N(21)) 154.1(1), Ar(C(31))//Ar(C(41)) 9.3(1); Ar(C(X)) = aryl ring containing C(X); COG(Ar(C(X))) = centroid of the aryl ring containing C(X). 11b: B(1)–O(1) 1.428(3), B(1)–N(11) 1.568(3), B(1)–N(12A) 1.597(3), B(1)–C(21) 1.615(3), B(1)⋯B(1A) 3.266(5); N(11)–B(1)–N(12A) 104.9(2); N(11)B(1)N(12A)//N(11)N(12)N(11A)N(12A) 14.3(3), pz(N(11))//pz(N(11A)) 0; pz(N(X)) = pyrazolyl ring containing N(X). Symmetry transformation used to generate equivalent atoms: A: −x, −y + 1, −z + 1. | ||
Compound 11a crystallises from CD3OD with two crystallographically independent molecules in the asymmetric unit (11aA, 11aB). Since all key structural parameters of 11aA and 11aB are the same within the error margins, only the data for 11aA are compiled in Fig. 6. The pyrazabole core of 11aA adopts a boat conformation such that the two phenyl rings become almost coplanar. The two boron atoms are 3.115(7) Å apart from each other and the distance between the phenyl-ring centroids amounts to 3.808 Å. The Ci-symmetric molecule 11b features a central B2N4 ring in a shallow chair conformation and a crystallographically imposed dihedral angle between the two pyrazolide rings of 0°.
C–C coupling reactions on a B–N triptycene platform
Using compound [4c]Br, we compared the relative performance of Sonogashira-38versus Stille-type39,40 C–C-coupling protocols for the introduction of alkynyl substituents into B–N triptycene scaffolds (Scheme 3). The room-temperature reaction between [4c]Br and tBuCCH in dry NEt3 using PdCl2(dppf)·CH2Cl2/CuI as the catalyst system afforded [4e]Br in approximately 45% yield after 7 d. (Note: It took 7 d until the last traces of the starting material had vanished in the 1H NMR spectrum of the reaction mixture.) For reasons of simplicity, we still refer to the reaction product as [4e]Br (instead of [4e]X (X = Cl, Br or I)), even though some of the bromide counterions will likely be exchanged against chloride and/or iodide ions under the conditions applied. The ill-defined nature of the counterion in the reaction product is one shortcoming of the Sonogashira protocol, because it affects the calculation of precise stoichiometries as well as the determination of accurate yields. Another setback arises from the waste salt, [HNEt3]X, which is difficult to separate completely from the target compound. These problems did not occur upon application of the Stille protocol (nBu3SnCCtBu, CH3CN–toluene, 15 mol% Pd(PtBu3)2), which gave comparable yields of [4e]Br as the Sonogashira reaction and required a reaction time of only 24 h at room temperature.
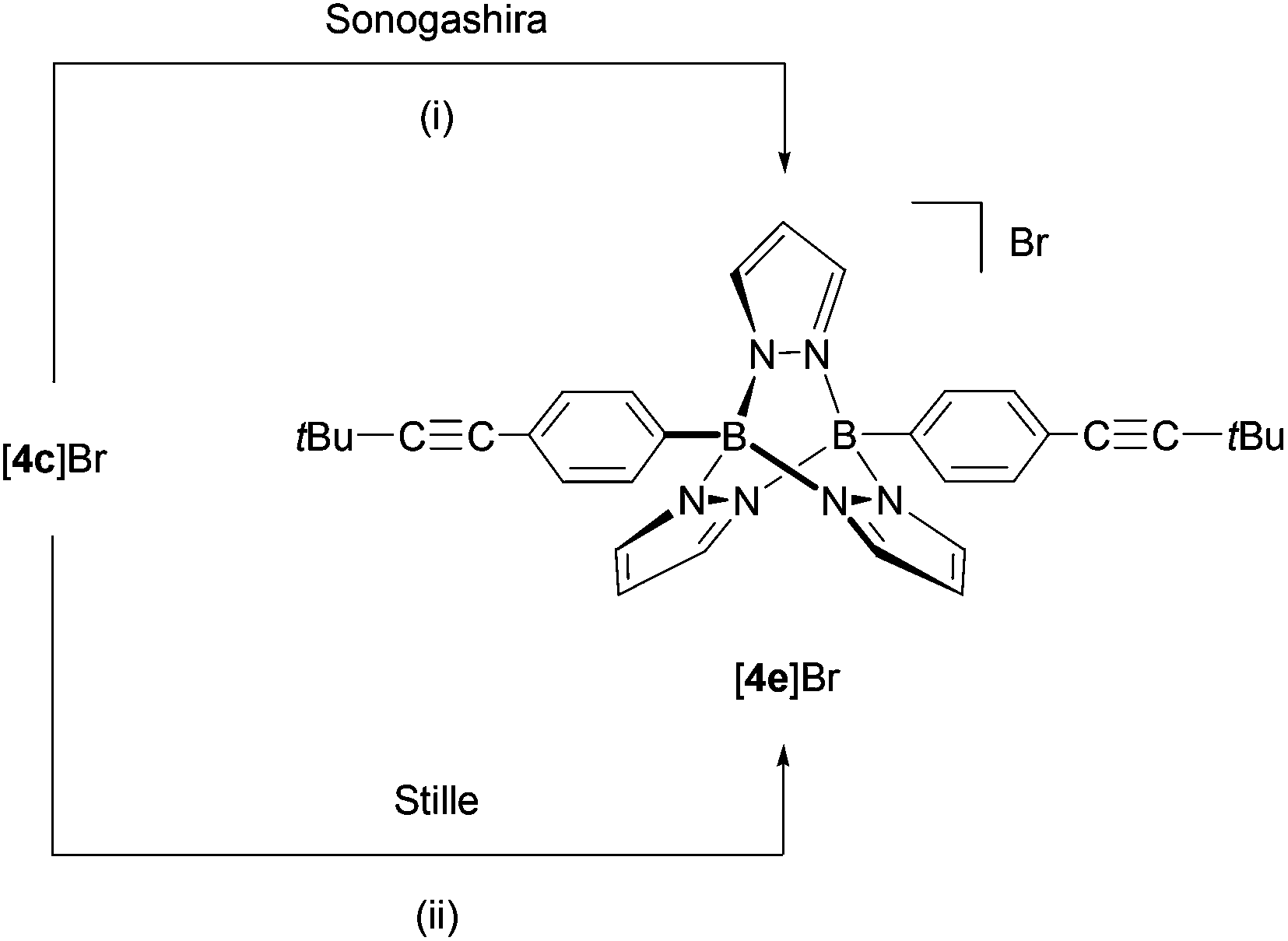 | ||
| Scheme 3 Derivatisation of [4c]Br through Sonogashira-type and Stille-type coupling reactions. Reaction conditions: (i) +2 tBuCCH, +PdCl2(dppf)·CH2Cl2/CuI (10 mol%), NEt3, rt, 7 d. (ii) +2 nBu3SnCCtBu, +Pd(PtBu3)2 (15 mol %), CH3CN–toluene, rt, 24 h. | ||
The B–N cage of [4e]Br revealed similar NMR characteristics to the other symmetrically substituted [4]+-type compounds. A singlet at 1.38 ppm in the 1H NMR spectrum is to be assigned to the tert-butyl substituents. The 13C NMR resonances of the alkynyl–carbon atoms appear at 101.7 ppm and 78.4 ppm.
Synthesis and characterisation of B–N-analogues of pentiptycenes
The key to success for the synthesis of 3-type B–N triptycenes was the availability of the lithium 1,2-bisborate Li2[7] (Scheme 1). Recently, we have also prepared the lithium 1,2,4,5-tetrakisborate Li4[13],41 which can consequently be employed for the synthesis of B–N pentiptycenes 14a–14d (Scheme 4). The general synthesis protocol for 14a–14d was essentially the same as that previously described in the cases of 3a–3e. We note, however, a markedly reduced solubility of some B–N pentiptycenes as compared to the B–N triptycenes (the same is true for the all-carbon frameworks).4 For example, the CF3 derivative 14a was obtained from toluene in an analytically pure, single-crystalline form; however, the crystals did not re-dissolve in any common inert solvent. Compound 14a was therefore characterised only by elemental analysis and X-ray crystallography (Fig. 7, top). The introduction of SiMe3 substituents in the 4-position of the pyrazolyl rings leads to good solubility of 14b in C6H6, toluene, CHCl3, CH2Cl2 or THF; NMR spectra were recorded in C6D6. The 1H NMR spectrum of 14b reveals a singlet at 8.66 ppm for the two protons on the central six-membered ring, another singlet at 7.35 ppm for the eight magnetically equivalent hydrogen atoms on the pyrazolyl fragments and one resonance at −0.08 ppm for the SiMe3 groups (36 H). The 11B{1H} NMR spectrum features one broad signal at −2.7 ppm (cf. 3b: −3.2 ppm). The molecular structure of 14b was finally confirmed also by X-ray crystallography (Fig. 7, bottom). The NMR spectra of the derivatives 14c and 14d (CDCl3) show no peculiarities compared to those of 14b; all corresponding data are compiled in the ESI.†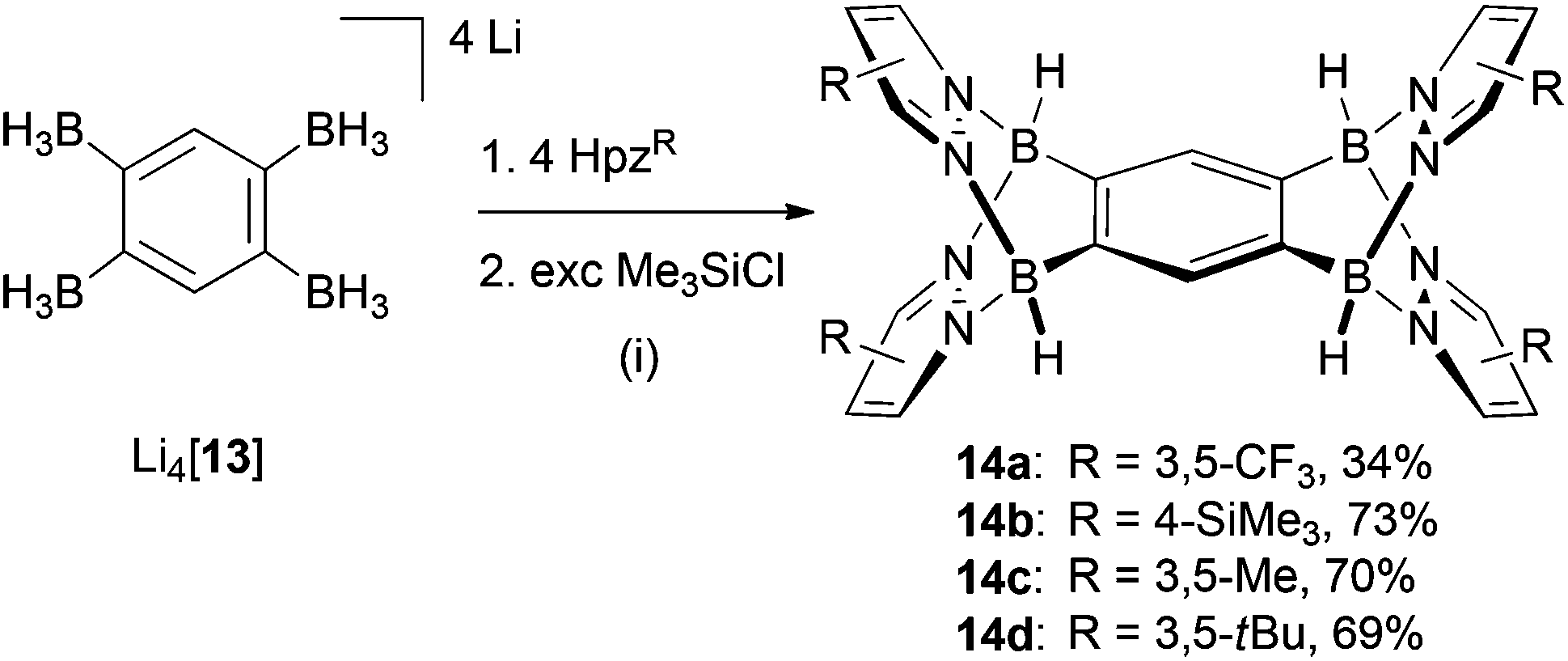 | ||
| Scheme 4 Synthesis of the B–N pentiptycenes 14a–14d. General reaction conditions: (i) THF or toluene, rt or 80 °C, between 1 d and 7 d. | ||
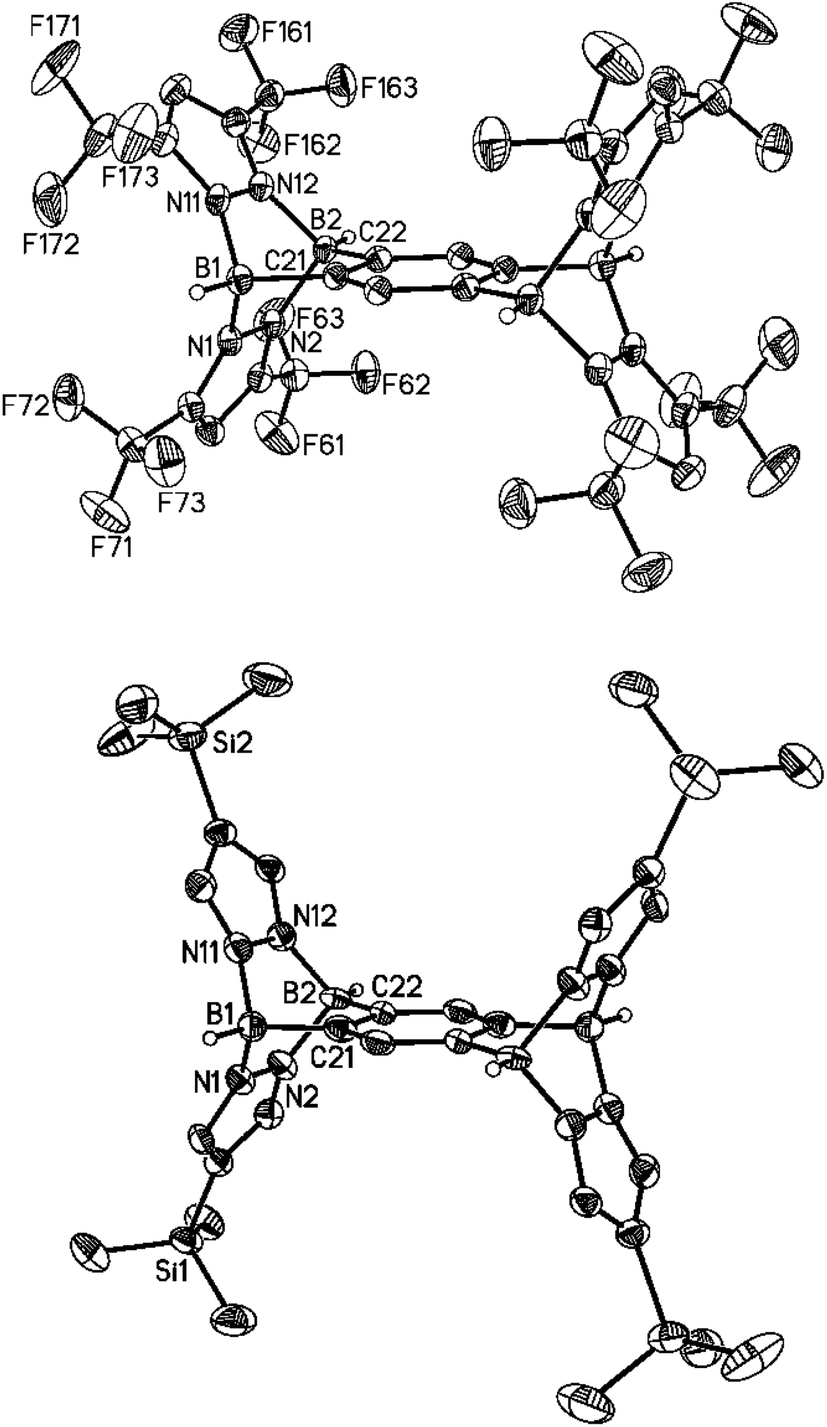 | ||
| Fig. 7 Molecular structures of 14a·3C7H8 (top) and 14b·2CH2Cl2 (bottom) in the solid state. Hydrogen atoms except on boron and the solvent molecules have been omitted for clarity; displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å), atom⋯atom distances (Å), bond angles (°) and dihedral angles (°): 14a: B(1)–N(1) 1.601(2), B(1)–N(11) 1.595(3), B(1)–C(21) 1.597(2), B(2)–N(2) 1.601(2), B(2)–N(12) 1.601(2), B(2)–C(22) 1.595(3), B(1)⋯B(2) 2.712(3); N(1)–B(1)–N(11) 99.7(1), N(1)–B(1)–C(21) 105.1(1), N(11)–B(1)–C(21) 106.1(1), N(2)–B(2)–N(12) 99.5(1), N(2)–B(2)–C(22) 105.5(1), N(12)–B(2)–C(22) 106.0(1); N(1)B(1)N(11)//N(2)B(2)N(12) 82.4(1), N(1)B(1)N(11)//N(1)N(2)N(11)N(12) 48.6(1), N(2)B(2)N(12)//N(1)N(2)N(11)N(12) 49.0(1), B(1)N(1)N(2)B(2)//B(1)N(11)N(12)B(2) 115.1(1), B(1)N(1)N(2)B(2)//B(1)C(21)C(22)B(2) 121.92(7), B(1)N(11)N(12)B(2)//B(1)C(21)C(22)B(2) 123.00(9), pz(N(1))//pz(N(11)) 117.77(8), pz(N(1))//C6H2 117.15(6), pz(N(11))//C6H2 125.07(8). 14b: B(1)–N(1) 1.574(9), B(1)–N(11) 1.590(8), B(1)–C(21) 1.597(9), B(2)–N(2) 1.593(7), B(2)–N(12) 1.578(8), B(2)–C(22) 1.597(9), B(1)⋯B(2) 2.701(1); N(1)–B(1)–N(11) 101.7(5), N(1)–B(1)–C(21) 105.9(5), N(11)–B(1)–C(21) 105.3(4), N(2)–B(2)–N(12) 100.9(5), N(2)–B(2)–C(22) 105.3(4), N(12)–B(2)–C(22) 104.1(5); N(1)B(1)N(11)//N(2)B(2)N(12) 84.0(6), N(1)B(1)N(11)//N(1)N(2)N(11)N(12) 48.7(5), N(2)B(2)N(12)//N(1)N(2)N(11)N(12) 47.3(4), B(1)N(1)N(2)B(2)//B(1)N(11)N(12)B(2) 117.3(3), B(1)N(1)N(2)B(2)//B(1)C(21)C(22)B(2) 122.0(3), B(1)N(11)N(12)B(2)//B(1)C(21)C(22)B(2) 120.7(3), pz(N(1))//pz(N(11)) 127.2(3), pz(N(1))//C6H2 120.4(3), pz(N(11))//C6H2 112.4(3); pz(N(X)) = pyrazolyl ring containing N(X). | ||
Both compounds, 14a and 14b, are Ci-symmetric in the solid state. All key metrical parameters of pentiptycene 14a are similar to those of its triptycene analogue 3e and the same applies to the bond lengths and angles of 14a compared to 14b. In the crystal lattice of 14a·3C7H8, the void spaces between the pyrazolyl rings pz(N(1)) and pz(N(11)) as well as pz(N(1)) and pz(N(11A)) are occupied by toluene molecules. In the case of 14b·2CH2Cl2, only the compartments above and below the central six-membered ring are hosting solvent molecules.
Conclusion
The concept of B–N/C–C isosterism was applied for the development of a complete series of B–N analogues of triptycene, i.e. [HB(μ-pzR)(μ-o-C6H4)2BH]− ([2]−), HB(μ-pzR)2(μ-o-C6H4)BH (3) and [RB(μ-pzR)3BR′]+ ([4]+). Instead of employing highly reactive benzyne intermediates, as they are required to prepare all-carbon triptycenes, the synthesis of the B–N congeners relies on the facile formation of B–N adduct bonds. We have also shown that the general design strategy can be extended to the preparation of B–N analogues of pentiptycene. Most of the compounds are sufficiently stable towards air and moisture to allow for a quick aqueous workup. As a proof-of-principle, we have also shown that the molecular framework of 4-type compounds is compatible with further derivatisation through Stille-type C–C-coupling protocols. Thus, B–N iptycenes are highly promising building blocks for the facile assembly of rigid supramolecular frameworks with selectable overall charges.Experimental methods
Unless otherwise specified, all reactions were carried out in carefully dried solvents under dry nitrogen or argon using Schlenk or glove-box techniques. n-Pentane, n-hexane, cyclohexane, C6H6, C6D6, toluene, THF, d8-THF and Et2O were dried over Na/benzophenone; NEt3, CH3CN, CD3CN, CHCl3, CDCl3, CH2Cl2 and Me3SiCl were dried over CaH2 and freshly distilled prior to use. Column chromatography was performed using silica gel 60 (Macherey-Nagel). NMR spectra were recorded at rt with Bruker AM 250, DPX 250, Avance II 300, Avance 400 and Avance III HD 500 spectrometers. Chemical shifts are referenced to (residual) solvent signals (1H/13C{1H}; C6D6: 7.16/128.06; d8-THF: 1.72/25.31; CD3CN: 1.94/1.32; CDCl3: 7.26/77.16; CD3OD: 3.31/49.00) or external BF3·OEt2 (11B,11B{1H}) and Si(CH3)4 (29Si INEPT). Abbreviations: s = singlet, d = doublet, tr = triplet, vtr = virtual triplet, q = quartet, m = multiplet, br = broad, n.o. = signal not observed; n.r. = multiplet expected in the 1H NMR spectrum but not resolved. Combustion analyses were performed by the Microanalytical Laboratory of the Goethe University Frankfurt; HRMS measurements were performed on a MALDI LTQ Orbitrap spectrometer (Thermo Scientific). The compounds Li2[7]·OEt2,23 Li2[1,1′-fc(BH3)2]·(OEt2)0.25,25 K[HBpz3],42 K[p-BrC6H4Bpz3],43p-BrC6H4BBr2,44 4-iodopyrazole,45nBu3SnCCtBu,46 4-(trimethylsilyl)pyrazole,47 and 3,5-di(tert-butyl)pyrazole48 are literature known; the amount of OEt2 present in the samples of Li2[1,1′-fc(BH3)2]·(OEt2)0.25 after prolonged storage in a screw-capped vial in the glove-box was determined by 1H NMR spectroscopy.
Synthesis of 3a
A Schlenk flask was charged with Li2[7]·OEt2 (50 mg, 0.26 mmol) and 4-iodopyrazole (101 mg, 0.521 mmol). Et2O (10 mL) was added at rt with stirring, whereupon vigorous gas evolution (H2) was observed, which lasted for approximately 2 min. After 30 min, neat Me3SiCl (0.1 mL, 0.08 g, 0.8 mmol) was added to the light yellow solution to give a white suspension. Stirring was continued for 12 h, the formed LiCl was removed by filtration and the filtrate was evaporated under vacuum to yield a colourless solid. Single crystals were grown by slow evaporation of a solution of 3a in Et2O. Yield: 89 mg, 70%. 1H NMR (300.0 MHz, C6D6): δ = 7.85–7.79 (2 H, m, H-3,6), 7.30–7.24 (2 H, m, H-4,5), 6.84 (4 H, s, pzH-3,5), 4.25* (2 H, br, BH); 13C{1H} NMR (75.4 MHz, C6D6): δ = 138.5 (pzC-3,5), 130.3 (C-3,6), 126.6 (C-4,5), 55.7 (pzC-4), n.o. (CB); 11B{1H} NMR (96.3 MHz, C6D6): δ = −3.7 (h1/2 = 240 Hz); HRMS (MALDI-TOF): m/z = 486.92435 ([M + H]+, calcd 486.92537). *This signal sharpens upon 11B decoupling.Synthesis of 3b
A Schlenk flask was charged with Li2[7]·OEt2 (0.1 g, 0.5 mmol) and 4-(trimethylsilyl)pyrazole (0.15 g, 1.1 mmol). Et2O (20 mL) was added at rt with stirring, whereupon vigorous gas evolution (H2) was observed, which lasted for approximately 2 min. After 30 min, neat Me3SiCl (0.20 mL, 0.17 g, 1.6 mmol) was added to the pale yellow solution to give a white suspension. Stirring was continued for 12 h, the formed LiCl was removed by filtration and the filtrate was evaporated under vacuum to yield a colourless solid. The solid was suspended in n-hexane (20 mL) and quenched with saturated brine (10 mL). The two liquid phases were separated from each other and the aqueous phase was extracted with n-hexane (20 mL). The combined organic phases were quickly extracted with H2O (20 mL), dried over anhydrous MgSO4, filtered, and the filtrate was evaporated to dryness under vacuum. The crude product was further purified by short column chromatography (silica gel, ethyl acetate, Rf = 0.86). Yield: 103 mg (52%). (Found: C, 57.22; H, 7.76; N, 14.52. Calc. for C18H28B2N4Si2 [378.24]: C, 57.16; H, 7.46; N, 14.81%.) 1H NMR (400.1 MHz, C6D6): δ = 8.04–8.00 (2 H, m, H-3,6), 7.35 (4 H, s, pzH-3,5), 7.32–7.28 (2 H, m, H-4,5), 4.84* (2 H, br, BH), −0.07 (18 H, s, SiMe3); 13C{1H} NMR (100.6 MHz, C6D6): δ = 138.2 (pzC-3,5), 130.3 (C-3,6), 126.4 (C-4,5), 114.5 (pz-C4), −0.6 (SiMe3), n.o. (CB); 11B{1H} NMR (128.4 MHz, C6D6): δ = −3.2 (h1/2 = 300 Hz); 29Si INEPT NMR (79.5 MHz, C6D6): δ = −10.6. *This signal sharpens upon 11B decoupling.Synthesis of 3f
A Schlenk flask was charged with Li2[1,1′-fc(BH3)2]·(OEt2)0.25 (50 mg, 0.20 mmol) and 4-iodopyrazole (81 mg, 0.42 mmol). Et2O (10 mL) was added at rt with stirring, whereupon vigorous gas evolution (H2) was observed, which lasted for approximately 2 min. After 30 min, neat Me3SiCl (0.1 mL, 0.08 g, 0.8 mmol) was added to the yellow solution to give a white suspension. Stirring was continued for 12 h, the formed LiCl was removed by filtration and the filtrate was evaporated under vacuum. The remaining orange solid was dissolved in Et2O and the solution was stored at −30 °C to grow single crystals of 3f·OEt2. Yield: 104 mg, 76%. 1H NMR (300.0 MHz, C6D6): δ = 7.03 (4 H, s, pzH-3,5), 4.12 (4 H, vtr, JH,H = 1.7 Hz, C5H4), 3.22 (4 H, vtr, JH,H = 1.7 Hz, C5H4), n.o. (BH); 13C{1H} NMR (75.4 MHz, C6D6): δ = 140.5 (pzC-3,5), 70.9 (C5H4), 70.3 (C5H4), 56.3 (pzC-4), n.o. (CB); 11B{1H} NMR (96.3 MHz, C6D6): δ = −3.5 (h1/2 = 240 Hz); MS (MALDI-TOF): m/z 593.8 ([M]+, 100%); HRMS (MALDI-TOF): m/z = 593.88323 ([M]+, calcd 593.88379).Synthesis of [4a]Br
A solution of p-BrC6H4BBr2 (194 mg, 0.594 mmol) in toluene (15 mL) was added dropwise with stirring at rt to a solution of K[HBpz3] (150 mg, 0.595 mmol) in CH3CN–toluene (1:1, 20 mL). The resulting mixture was stirred for 24 h at rt. All insolubles were collected on a frit and quickly washed with H2O (3 mL) to remove KBr. The crude product was immediately after that washed with n-pentane (2 × 15 mL) and dried under vacuum. Single crystals were grown by slow evaporation of a solution of [4a]Br in CH2Cl2. Yield: 255 mg, 93%. 1H NMR (500.2 MHz, CD3CN): δ = 8.24 (3 H, d, 3JH,H = 2.5 Hz, pzH-3 or pzH-5), 8.02 (3 H, d, 3JH,H = 2.5 Hz, pzH-3 or pzH-5), 7.91 (2 H, d, 3JH,H = 8.5 Hz, ArH), 7.84 (2 H, d, 3JH,H = 8.5 Hz, ArH), 6.51 (3 H, vtr, pzH-4), 5.35–4.30 (1 H, br m, BH). 13C{1H} NMR (125.8 MHz, CD3CN): 139.7 (pzC-3 or pzC-5), 139.2 (pzC-3 or pzC-5), 136.5 (ArC), 133.1 (ArC), 125.6 (CBr), 108.6 (pzC-4), n.o. (CB); 11B{1H} NMR (160.5 MHz, CD3CN): δ = −0.5 (h1/2 = 110 Hz, BC), −5.4 (h1/2 = 100 Hz, BH); 11B NMR (160.5 MHz, CD3CN): δ = −0.5 (s, BC), −5.4 (d, 1JB,H = 128 Hz, BH); MS (ESI+): m/z 379.2 ([M − Br]+, 100%); HRMS (MALDI-TOF): m/z = 379.06527 ([M − Br]+, calcd 379.06439).
Synthesis of [4b]Br
In a Schlenk vessel equipped with a Teflon Young's tab, p-BrC6H4BBr2 (40 mg, 0.12 mmol) was dissolved in toluene (15 mL). Neat solid K[p-BrC6H4Bpz3] (50 mg, 0.12 mmol) was added, the tab was closed and the vessel was heated with stirring for 8 h at 120 °C. After cooling to rt, all insolubles were collected on a frit and washed with n-pentane (6 × 5 mL). The solid material was suspended in CHCl3 (20 mL) and quenched with a saturated aqueous solution of NaHCO3 (10 mL). The two liquid phases were separated from each other and the aqueous phase was extracted with CHCl3 (20 mL). The combined organic phases were washed with H2O (20 mL), dried over anhydrous MgSO4, filtered and the filtrate was evaporated to dryness under vacuum. Single crystals were obtained as colourless blocks by slow evaporation of a solution of [4b]Br in CHCl3. Yield: 60 mg, 80%. (Found: C, 40.35; H, 2.87; N, 13.42. Calc. for C21H17B2Br3N6 [614.76]: C, 41.03; H, 2.79; N, 13.67%.) 1H NMR (500.2 MHz, CD3CN): δ = 8.08 (6 H, d, 3JH,H = 2.5 Hz, pzH-3,5), 7.94 (4 H, d, 3JH,H = 8.3 Hz, ArH), 7.87 (4 H, d, 3JH,H = 8.3 Hz, ArH), 6.52 (3 H, tr, 3JH,H = 2.5 Hz, pzH-4); 13C{1H} NMR (75.4 MHz, CD3CN): δ = 139.8 (pzC-3,5), 136.6 (ArC), 133.2 (ArC), 108.6 (pzC-4), n.o. (CBr), n.o. (CB); 11B{1H} NMR (96.3 MHz, CD3CN): δ = −0.5 (h1/2 = 160 Hz); HRMS (MALDI-TOF): m/z = 535.00411 ([M − Br]+, calcd 535.00416).Synthesis of [4c]Br
In a Schlenk vessel equipped with a Teflon Young's tab, p-IC6H4BBr2 (123 mg, 0.330 mmol) was dissolved in toluene (15 mL). Neat solid K[p-IC6H4Bpz3] (150 mg, 0.330 mmol) was added, the tab was closed and the vessel was heated with stirring for 6 h at 120 °C. After cooling to rt, all insolubles were collected on a frit, washed with H2O (20 mL), n-pentane (6 × 5 mL) and dried under vacuum. Yield: 196 mg, 84%. 1H NMR (300.0 MHz, CD3CN): δ = 8.11–8.04 (10 H, m, ArH, pzH-3,5), 7.79 (4 H, d, 3JH,H = 8.1 Hz, ArH), 6.52 (3 H, tr, 3JH,H = 2.5 Hz, pzH-4); 13C{1H} NMR (125.8 MHz, CD3CN): δ = 139.7 (pzC-3,5), 139.2 (ArC), 136.4 (ArC), 108.6 (pzC-4), 97.9* (CI), n.o. (CB); 11B NMR (96.3 MHz, CD3CN): δ = −0.9 (h1/2 = 230 Hz); HRMS (MALDI-TOF): m/z = 628.97883 ([M − Br]+, calcd 628.97847). *The position of this signal was confirmed by an HMBC experiment.Synthesis of [4d]Br
In a Schlenk vessel equipped with a Teflon Young's tab, p-Me3SiC6H4BBr2 (172 mg, 0.538 mmol) was dissolved in toluene (15 mL). Neat solid K[p-Me3SiC6H4Bpz3] (215 mg, 0.537 mmol) was added, the tab was closed and the vessel was heated with stirring for 6 h at 120 °C. After cooling to rt, all insolubles were collected on a frit, washed with n-pentane (6 × 5 mL), suspended in CHCl3 (20 mL) and quenched with a saturated aqueous solution of NaHCO3 (10 mL). The two liquid phases were separated from each other and the aqueous phase was extracted with CHCl3 (20 mL). The combined organic phases were washed with H2O (20 mL), dried over anhydrous MgSO4, filtered and the filtrate was evaporated to dryness under vacuum. Single crystals were obtained as colourless blocks by slow evaporation of a solution of [4d]Br in CHCl3. Yield: 259 mg, 80%. (Found: C, 54.29; H, 6.11; N, 13.86. Calc. for C27H35B2BrN6Si2 [601.30]: C, 53.93; H, 5.87; N, 13.98%.) 1H NMR (400.1 MHz, CDCl3): δ = 8.12 (6 H, d, 3JH,H = 2.5 Hz, pzH-3,5), 7.96 (4 H, d, 3JH,H = 8.0 Hz, ArH), 7.83 (4 H, d, 3JH,H = 8.0 Hz, ArH), 6.71 (3 H, tr, 3JH,H = 2.5 Hz, pzH-4), 0.36 (18 H, s, SiMe3); 13C{1H} NMR (100.6 MHz, CDCl3): δ = 144.4 (CSi), 138.8 (pzC-3,5), 134.6 (ArC), 132.5 (ArC), 108.9 (pzC-4), −1.1 (SiMe3), n.o. (CB); 11B NMR (128.4 MHz, CDCl3): δ = 0.2 (h1/2 = 400 Hz); 29Si INEPT NMR (99.4 MHz, CDCl3): δ = −3.3; MS (ESI+): m/z 521.8 ([M − Br]+, 100%).Synthesis of [4e]Br
Method A: [4c]Br (50 mg, 0.071 mmol), PdCl2(dppf)·CH2Cl2 (6 mg, 0.007 mmol) and CuI (2 mg, 0.01 mmol) were suspended in NEt3 (20 mL). tBuCCH (18 μL, 12 mg, 0.15 mmol) was added via a syringe and the solution was stirred for 7 d at rt. All volatiles were removed under reduced pressure. The remaining dark brown solid was washed with H2O (20 mL), THF (20 mL), C6H6 (20 mL) and n-pentane (30 mL) to obtain a colourless solid, which was dried in vacuo. Yield: 21 mg, 48% (note: the product was still contaminated with small quantities of [HNEt3]X (X = Cl, Br or I); NMR spectroscopic control).
Method B: [4c]Br (20 mg, 0.028 mmol) and Pd(PtBu3)2 (2 mg, 0.004 mmol) were dissolved in CH3CN (10 mL) and toluene (10 mL). Neat nBu3SnCCtBu (21 μL, 21 mg, 0.058 mmol) was added via a syringe and the clear yellow solution was stirred for 24 h at rt. All volatiles were removed under reduced pressure, the yellow solid residue was treated with C6H6 (10 mL) to give a yellow suspension, and the insoluble material was collected on a frit, washed with n-hexane (10 mL) and dried in vacuo. Yield: 8 mg, 46%.
1H NMR (500.2 MHz, CDCl3): δ = 8.11 (6 H, d, 3JH,H = 2.5 Hz, pzH-3,5), 7.95 (4 H, d, 3JH,H = 8.0 Hz, ArH), 7.72 (4 H, d, 3JH,H = 8.0 Hz, ArH), 6.66 (3 H, tr, 3JH,H = 2.5 Hz, pzH-4), 1.38 (18 H, s, CH3); 13C{1H} NMR (125.8 MHz, CDCl3): δ = 138.8 (pzC-3,5), 133.2 (ArC), 132.8 (ArC), 127.3* (CCC), 108.8 (pzC-4), 101.7* (tBuCC), 78.4* (ArCC), 31.1 (CH3), 28.3 (C(CH3)), n.o. (CB); 11B NMR (96.3 MHz, CD3CN): δ = 0.1 (h1/2 = 500 Hz); HRMS (MALDI-TOF): m/z = 537.31069 ([M − Br]+, calcd 537.31038). *The position of this signal was confirmed by an HMBC experiment.
Synthesis of 14a
A Schlenk flask was charged with Li4[13]·3thf (50 mg, 0.13 mmol) and 3,5-bis(trifluoromethyl)pyrazole (0.11 g, 0.54 mmol). Toluene (20 mL) was added at rt with stirring, whereupon a gas (H2) evolved. After 10 min, neat Me3SiCl (0.1 mL, 0.08 g, 0.8 mmol) was added to the colourless suspension and stirring was continued for 4 d. All insolubles were collected on a frit (77 mg; 100% LiCl = 22 mg) and the filtrate was stored at 4 °C for 14 d to obtain block-shaped crystals of 14a·3C7H8. Yield: 11 mg, 7%. The mother liquor was evaporated to dryness to give 14a as a colourless solid; an elemental analysis revealed that, in contrast to the single crystals, the microcrystalline product no longer contained toluene. Yield: 34 mg, 27%. (Found: C, 33.55; H, 1.24; N, 12.01. Calc. for C26H10B4F24N8 [933.62]: C, 33.45; H, 1.08; N, 12.00%.) Since 14a did not re-dissolve in any common inert solvent, NMR data cannot be provided.Synthesis of 14b
A Schlenk flask was charged with Li4[13]·3thf (50 mg, 0.13 mmol) and 4-(trimethylsilyl)pyrazole (75 mg, 0.53 mmol). THF (20 mL) was added at rt and the resulting solution was stirred for 15 min; the reaction proceeds with evolution of H2. Neat Me3SiCl (0.1 mL, 0.08 g, 0.8 mmol) was added to the pale yellow solution and stirring was continued for 24 h. All volatiles were removed under vacuum. The yellow solid residue was extracted with toluene (20 mL). The extract was evaporated to dryness under vacuum to obtain 14b as a colourless solid. Yield: 66 mg, 73%. Single crystals of 14b·2CH2Cl2 were obtained in the form of colourless blocks by slow evaporation of a solution of 14b in CH2Cl2.1H NMR (300.0 MHz, C6D6): δ = 8.66 (2 H, s, H-3,6), 7.35 (8 H, s, pzH-3,5), 4.96* (4 H, br, BH), −0.08 (36 H, s, SiMe3); 13C{1H} NMR (75.4 MHz, C6D6): δ = 137.8 (pzC-3,5), 132.6 (C-3,6), 113.8 (pz-C4), −0.6 (SiMe3), n.o. (CB); 11B{1H} NMR (96.3 MHz, C6D6): δ = −2.7 (h1/2 = 900 Hz); 29Si INEPT NMR (59.6 MHz, C6D6): δ = −10.8; HRMS (MALDI-TOF): m/z = 678.36164 ([M]+, calcd 678.36253 (C30H50B4N8Si4)). *This signal sharpens upon 11B decoupling.
X-ray crystal structure determinations
Data for 11a, 11b and 12a were collected on a STOE IPDS II two-circle diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) and corrected for absorption with an empirical absorption correction using the program PLATON.49 Data for 3a, 3c, 3d, 3e, 3f·OEt2, 3f*·3C6H6, [4a]Br·CH2Cl2, [4b]Br, 14a·3C7H8, and 14b·2CH2Cl2 were collected on a STOE IPDS II two-circle diffractometer with a Genix Microfocus tube with mirror optics using MoKα radiation (λ = 0.71073 Å) and were scaled using the frame scaling procedure in the X-AREA program system.50 The structures were solved by direct methods using the program SHELXS51 and refined against F2 with full-matrix least-squares techniques using the program SHELXL-97.51The H atoms bonded to B in 3a, 3c, 3e and [4a]Br·CH2Cl2 were isotropically refined. The coordinates of the H atoms bonded to B in 3d were refined. The crystal of 3a was merohedrally twinned (twin law: −1 −1 0/0 1 0/0 0 −1) with a fractional contribution of 0.1971(7) for the minor domain. The crystal of 3f·OEt2 was very weakly diffracting. The displacement parameters of all B, N and C atoms were restrained to an isotropic behaviour. In 3f*·3C6H6, the displacement parameters of B2, B4 and the benzene-C atoms C71 to C76 as well as C91 to C96 were restrained to an isotropic behaviour. Bond lengths and angles in the benzene solvent molecules were restrained to be equal. One toluene molecule in 14a·3C7H8 was disordered about a centre of inversion over two equally occupied positions; the H atoms bonded to B were isotropically refined. The crystal of 14b·2CH2Cl2 was just weakly diffracting; the H atoms bonded to B were isotropically refined.
CCDC reference numbers: 985503 (3a), 985504 (3c), 985505 (3d), 985506 (3e), 985507 (3f·OEt2), 985508 (3f*·3C6H6), 985509 ([4a]Br·CH2Cl2), 985510 ([4b]Br), 985511 (11a), 985512 (11b), 985513 (12a), 985514 (14a·3C7H8) and 985515 (14b·2CH2Cl2).
Acknowledgements
This work has been supported by the Beilstein-Institut, Frankfurt/Main, Germany, within the research collaboration NanoBiC through a Ph.D. grant for Ö.S.References
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS.
- H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175–182 CrossRef CAS.
- T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef CAS PubMed.
- C.-F. Chen and Y.-X. Ma, Iptycenes Chemistry: From Synthesis to Applications, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC.
- J. Liu, O. Shekhah, X. Stammer, H. K. Arslan, B. Liu, B. Schüpbach, A. Terfort and C. Wöll, Materials, 2012, 5, 1581–1592 CrossRef CAS.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed.
- C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC.
- A. Beyeler and P. Belser, Coord. Chem. Rev., 2002, 230, 29–39 CrossRef CAS.
- L. Friedman and F. M. Logullo, J. Am. Chem. Soc., 1963, 85, 1549–1549 CrossRef CAS.
- (a) T. Kitamura and M. Yamane, J. Chem. Soc., Chem. Commun., 1995, 983–984 RSC; (b) T. Kitamura, M. Yamane, K. Inoue, M. Todaka, N. Fukatsu, Z. Meng and Y. Fujiwara, J. Am. Chem. Soc., 1999, 121, 11674–11679 CrossRef CAS; (c) T. Kitamura, Z. Meng and Y. Fujiwara, Tetrahedron Lett., 2000, 41, 6611–6614 CrossRef CAS; (d) T. Kitamura, M. Todaka and Y. Fujiwara, Org. Synth., 2002, 78, 104–108 CrossRef CAS.
- (a) A. Lorbach, C. Reus, M. Bolte, H.-W. Lerner and M. Wagner, Adv. Synth. Catal., 2010, 352, 3443–3449 CrossRef CAS; (b) C. Reus, N.-W. Liu, M. Bolte, H.-W. Lerner and M. Wagner, J. Org. Chem., 2012, 77, 3518–3523 CrossRef CAS PubMed.
- K. Ma, M. Scheibitz, S. Scholz and M. Wagner, J. Organomet. Chem., 2002, 652, 11–19 CrossRef CAS.
- (a) F. Jäkle, T. Priermeier and M. Wagner, J. Chem. Soc., Chem. Commun., 1995, 1765–1766 RSC; (b) F. Jäkle, T. Priermeier and M. Wagner, Organometallics, 1996, 15, 2033–2040 CrossRef; (c) E. Herdtweck, F. Jäkle, G. Opromolla, M. Spiegler, M. Wagner and P. Zanello, Organometallics, 1996, 15, 5524–5535 CrossRef CAS; (d) E. Herdtweck, F. Jäkle and M. Wagner, Organometallics, 1997, 16, 4737–4745 CrossRef CAS.
- (a) F. Fabrizi de Biani, T. Gmeinwieser, E. Herdtweck, F. Jäkle, F. Laschi, M. Wagner and P. Zanello, Organometallics, 1997, 16, 4776–4787 CrossRef CAS; (b) L. Ding, K. Ma, M. Bolte, F. Fabrizi de Biani, P. Zanello and M. Wagner, J. Organomet. Chem., 2001, 637–639, 390–397 CrossRef CAS; (c) K. Ma, F. Fabrizi de Biani, M. Bolte, P. Zanello and M. Wagner, Organometallics, 2002, 21, 3979–3989 CrossRef CAS; (d) L. Ding, K. Ma, G. Dürner, M. Bolte, F. Fabrizi de Biani, P. Zanello and M. Wagner, J. Chem. Soc., Dalton Trans., 2002, 1566–1573 RSC; (e) C. Cui, J. Heilmann-Brohl, A. Sánchez Perucha, M. D. Thomson, H. G. Roskos, M. Wagner and F. Jäkle, Macromolecules, 2010, 43, 5256–5261 CrossRef CAS.
- (a) F. Jäkle, T. Priermeier and M. Wagner, Chem. Ber., 1995, 128, 1163–1169 CrossRef; (b) M. Fontani, F. Peters, W. Scherer, W. Wachter, M. Wagner and P. Zanello, Eur. J. Inorg. Chem., 1998, 1453–1465 CrossRef CAS; (c) M. Grosche, E. Herdtweck, F. Peters and M. Wagner, Organometallics, 1999, 18, 4669–4672 CrossRef CAS; (d) L. Ding, F. Fabrizi de Biani, M. Bolte, P. Zanello and M. Wagner, Organometallics, 2000, 19, 5763–5768 CrossRef CAS; (e) R. E. Dinnebier, M. Wagner, F. Peters, K. Shankland and W. I. F. David, Z. Anorg. Allg. Chem., 2000, 626, 1400–1405 CrossRef CAS; (f) K. Ma, H.-W. Lerner, S. Scholz, J. W. Bats, M. Bolte and M. Wagner, J. Organomet. Chem., 2002, 664, 94–105 CrossRef CAS.
- W. Luo, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 13006–13009 CrossRef CAS PubMed.
- For selected examples of B–N isosteres of aromatic molecules that give rise to new optoelectronic or biological properties, see: (a) A. J. Ashe III and X. Fang, Org. Lett., 2000, 2, 2089–2091 CrossRef; (b) A. J. Ashe III, X. Fang, X. Fang and J. W. Kampf, Organometallics, 2001, 20, 5413–5418 CrossRef; (c) C. A. Jaska, D. J. H. Emslie, M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, J. Am. Chem. Soc., 2006, 128, 10885–10896 CrossRef CAS PubMed; (d) M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, Angew. Chem., Int. Ed., 2007, 46, 4940–4943 CrossRef CAS PubMed; (e) A. J. V. Marwitz, E. R. Abbey, J. T. Jenkins, L. N. Zakharov and S.-Y. Liu, Org. Lett., 2007, 9, 4905–4908 CrossRef CAS PubMed; (f) Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed; (g) E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2008, 130, 7250–7252 CrossRef CAS PubMed; (h) A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS PubMed; (i) A. N. Lamm and S.-Y. Liu, Mol. BioSyst., 2009, 5, 1303–1305 RSC; (j) K. Raidongia, A. Nag, K. P. S. S. Hembram, U. V. Waghmare, R. Datta and C. N. R. Rao, Chem.–Eur. J., 2010, 16, 149–157 CrossRef CAS PubMed; (k) T. Taniguchi and S. Yamaguchi, Organometallics, 2010, 29, 5732–5735 CrossRef CAS; (l) T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed; (m) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (n) E. R. Abbey and S.-Y. Liu, Org. Biomol. Chem., 2013, 11, 2060–2069 RSC; (o) J.-S. Lu, S.-B. Ko, N. R. Walters, Y. Kang, F. Sauriol and S. Wang, Angew. Chem., Int. Ed., 2013, 52, 4544–4548 CrossRef CAS PubMed.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC.
- (a) D. N. Hendrickson, J. M. Hollander and W. L. Jolly, Inorg. Chem., 1970, 9, 612–615 CrossRef CAS; (b) E. M. Holt, S. L. Holt, K. J. Watson and B. Olsen, Cryst. Struct. Commun., 1978, 7, 613–616 CAS; (c) D. C. Bradley, M. B. Hursthouse, J. Newton and N. P. C. Walker, J. Chem. Soc., Chem. Commun., 1984, 188–190 RSC; (d) R. G. Ball, F. Edelmann, J. G. Matisons and J. Takats, Inorg. Chim. Acta, 1987, 132, 137–143 CrossRef CAS; (e) J. M. Boncella, M. L. Cajigal, A. S. Gamble and K. A. Abboud, Polyhedron, 1996, 15, 2071–2078 CrossRef CAS; (f) R. A. Kresiński, J. Chem. Soc., Dalton Trans., 1999, 401–405 RSC; (g) C.-L. Lee, Y.-Y. Wu, C.-P. Wu, J.-D. Chen, T.-C. Keng and J.-C. Wang, Inorg. Chim. Acta, 1999, 292, 182–188 CrossRef CAS; (h) J.-C. Hierso and M. Etienne, Eur. J. Inorg. Chem., 2000, 839–842 CrossRef CAS; (i) C.-L. Lee, P.-Y. Yang, C.-W. Su, J.-D. Chen, L.-S. Liou and J.-C. Wang, Inorg. Chim. Acta, 2001, 314, 147–153 CrossRef CAS; (j) A. E. Enriquez, B. L. Scott and M. P. Neu, Inorg. Chem., 2005, 44, 7403–7413 CrossRef CAS PubMed.
- S. Trofimenko, J. Am. Chem. Soc., 1969, 91, 5410–5411 CrossRef CAS.
- J. Bielawski and K. Niedenzu, Inorg. Chem., 1986, 25, 85–87 CrossRef CAS.
- Ö. Seven, Z.-W. Qu, H. Zhu, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Chem.–Eur. J., 2012, 18, 11284–11295 CrossRef PubMed.
- M. Scheibitz, J. W. Bats, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2004, 23, 940–942 CrossRef CAS.
- M. Scheibitz, H. Li, J. Schnorr, A. Sánchez Perucha, M. Bolte, H.-W. Lerner, F. Jäkle and M. Wagner, J. Am. Chem. Soc., 2009, 131, 16319–16329 CrossRef CAS PubMed.
- K. Niedenzu and H. Nöth, Chem. Ber., 1983, 116, 1132–1153 CrossRef CAS.
- S. Trofimenko, Scorpionates - The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed.
- C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed.
- H. Nöth and B. Wrackmeyer, in Nuclear Magnetic Resonance Spectroscopy of Boron Compounds. NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer, Berlin, 1978 Search PubMed.
- In most cases, we will give the smaller value for the angle between two planes as the interplanar/dihedral angle. The only exceptions are the angles between the blades of the B–N-triptycenes, for which we have chosen to consider the larger values, because throughout the literature angles close to 120° (instead of 60°) are given for triptycene.
- K. Nikitin, C. Fleming, H. Müller-Bunz, Y. Ortin and M. J. McGlinchey, Eur. J. Org. Chem., 2010, 5203–5216 CrossRef CAS.
- K. Anzenhofer and J. J. de Boer, Z. Kristallogr., 1970, 131, 103–113 CrossRef CAS.
- A. Pelter, K. Smith and H. C. Brown, Borane Reagents, Academic Press, London, 1988 Search PubMed.
- A. Lorbach, M. Bolte, H. Li, H.-W. Lerner, M. C. Holthausen, F. Jäkle and M. Wagner, Angew. Chem., Int. Ed., 2009, 48, 4584–4588 CrossRef CAS PubMed.
- H. Nöth and H. Vahrenkamp, Chem. Ber., 1966, 99, 1049–1067 CrossRef.
- Similar results as for [4b]Br were obtained upon targeted methanolysis of [4d]Br, which gave the corresponding syn pyrazabole (p-Me3SiC6H4)(CH3O)B(μ-pz)2B(OCH3)(p-C6H4SiMe3) (12a; cf. the ESI† for more information).
- M. K. Das, A. L. DeGraffenreid, K. D. Edwards, L. Komorowski, J. F. Mariategui, B. W. Miller, M. T. Mojesky and K. Niedenzu, Inorg. Chem., 1988, 27, 3085–3089 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS.
- Ö. Seven, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 1291–1299 CrossRef.
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170–3177 CrossRef CAS.
- J. Zagermann, M. C. Kuchta, K. Merz and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2009, 5407–5412 CrossRef CAS.
- S. Popp, K. Ruth, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o226–o230 CAS.
- M. I. Rodríguez-Franco, I. Dorronsoro, A. I. Hernández-Higueras and G. Antequera, Tetrahedron Lett., 2001, 42, 863–865 CrossRef.
- I. Meana, A. C. Albéniz and P. Espinet, Adv. Synth. Catal., 2010, 352, 2887–2891 CrossRef CAS.
- L. Birkofer and M. Franz, Chem. Ber., 1972, 105, 1759–1767 CrossRef CAS.
- C. Fernández-Castaño, C. Foces-Foces, N. Jagerovic and J. Elguero, J. Mol. Struct., 1995, 355, 265–271 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- Stoe & Cie, X-AREA. Diffractometer control program system, Stoe & Cie, Darmstadt, Germany, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including assigned NMR data of 3c, 3d, 3e, K[8] (R = p-Me3SiC6H4; p-IC6H4), 14c and 14d. Plots of the 13C{1H} NMR spectra of 3a, 3c, 3d, [4a]Br, [4c]Br, [4e]Br, 14b, 14c and 14d. Decomposition experiments of 3b, [4a]Br and [4b]Br in CD3OD and [4d]Br in CH3OH. X-ray crystal structure analyses of 3c, 3d, 3e, 3f·OEt2 and 12a. CCDC 985503–985515. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00442f |
| This journal is © The Royal Society of Chemistry 2014 |