 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmericium(III) and europium(III) complex formation with lactate at elevated temperatures studied by spectroscopy and quantum chemical calculations†
Astrid
Barkleit
*ab,
Jerome
Kretzschmar
a,
Satoru
Tsushima
a and
Margret
Acker
c
aInstitute of Resource Ecology, Helmholtz-Zentrum Dresden – Rossendorf, P.O. Box 510119, 01314 Dresden, Germany. E-mail: a.barkleit@hzdr.de
bRadiochemistry, Department of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
cCentral Radionuclide Laboratory, Technische Universität Dresden, 01062 Dresden, Germany
First published on 6th May 2014
Abstract
Thermodynamic parameters for the complex formation of Am(III) and Eu(III) with lactate were determined with UV-vis and time-resolved laser-induced fluorescence spectroscopy (TRLFS) in a temperature range between 25 and 70 °C. The reaction enthalpy decreased with increasing ionic strength. ATR FT-IR and NMR spectroscopy in combination with density functional theory (DFT) calculations revealed structural details of the Eu(III) lactate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex: a chelating coordination mode of the lactate with a monodentate binding carboxylate group and the hydroxyl group being deprotonated.
:1 complex: a chelating coordination mode of the lactate with a monodentate binding carboxylate group and the hydroxyl group being deprotonated.
Introduction
Small organic molecules, which can bind heavy metal ions, are ubiquitous in nature. They can be found in nearly all biological systems like microorganisms, plants and animals as a product of various biochemical processes. They are also part of organic matter of argillaceous rocks, which are potential host rocks for nuclear waste repositories.The understanding of the complex formation behavior of radionuclides with such small organic molecules and the thermodynamic quantification of the interaction is of great importance to simulate and predict their migration behavior in the environment. Especially data at elevated temperatures are crucial, because not only in various organisms but particularly in the near field of nuclear waste disposals higher temperatures are prevailing.1
We investigated the complex formation and thermodynamic data of Am(III) and its non-radioactive analogue lanthanide Eu(III) with lactate. Lactate was selected as a representative ubiquitous small organic molecule that exists as metabolite in all organisms and also in significant amounts in clay rock formations.2 Experiments were performed at ambient and elevated temperatures with time-resolved laser-induced fluorescence spectroscopy (TRLFS), and for Am(III) additionally with UV-vis spectrometry. Furthermore, spectroscopic investigations concerning structural features have been carried out for Eu(III) lactate with attenuated total reflection Fourier transform infrared (ATR FT-IR) spectroscopy and nuclear magnetic resonance (NMR) spectroscopy, supported by calculations with density functional theory (DFT).
For the Eu(III) as well as the Am(III) lactate system, several investigations have been published (Eu(III),3–12 Am(III)3,10,13–15). But studies about the complex formation behavior at trace metal concentration, lower ionic strength and higher temperatures, which are important parameters influencing the migration behavior of radionuclides in the environment, are still missing. The proposed combination of methods is highly suitable to fill this gap. TRLFS as a sensitive and selective technique has been extensively used to analyze actinide and lanthanide complex formation with inorganic and organic ligands at trace metal concentrations.16,17 The application of TRLFS onto Am(III) complexation was up to now limited because of its much lower luminescence intensity and much shorter lifetime in comparison to Cm(III) or Eu(III). Some publications about TRLFS with Am(III) at ambient18–26 or low temperatures22 exist, but no studies at elevated temperatures have been published until now.
Some structural suggestions for the Eu(III) lactate, which do exist are only assumptions from indirect methods.4,5,12 In this work, we want to provide direct structural information. ATR FT-IR spectroscopy combined with calculations of structure and spectroscopic data using DFT gives useful information about structural features as it has been shown previously for the Eu(III) complexes with pyromellitic and citric acid.27,28 Lanthanide induced shifts in NMR spectroscopy as caused by the interaction of nuclear spins with electronic unpaired spins can be used as a helpful tool for signal separation, probing the potential binding sites and structure including geometries and distances.29
The combination of all these methods should offer new insights concerning the structure of the Eu(III) lactate complex thereby resolving contradicting suggestions in the previous works whether the hydroxyl group is protonated or not.4,5,12,30
Experimental
Solutions and reagents
Na-L-lactate (Fluka) and EuCl3·6H2O (Aldrich) were used without further purification. Am-243 was supplied by the Oak Rich National Laboratory as AmO2. The oxide was dissolved in HNO3 and then aliquoted for experiments. The purity of the isotope was verified by α- and γ-spectrometry. Only the daughter nuclide of α-decay, Np-239 (half-life: 2.355 d), in equilibrium was determined (about 0.0001% of Am). For experiments except NMR, solutions were prepared with deionized water, the ionic strength was adjusted with NaClO4 or NaCl (Merck) to 0.1 M. Necessary pH adjustments were done with NaOH and HClO4 or HCl with an accuracy of 0.05 units. For NMR experiments, deuterated chemicals (all purchased from Sigma-Aldrich) were used. D2O (99.9 atom% D) was used as solvent and pD was adjusted by diluted solutions of NaOD (40 wt% in D2O, 99.5 atom% D) and DCl (35 wt% in D2O, 99 atom% D) in D2O.UV-vis measurements
For spectrophotometric UV-vis titration, 2.5 mL of 5 × 10−6 M Am3+ at pH 3.0 or 6.0 and 0.1 M NaClO4 were titrated with aliquots (5, 10 or 20 μL) of 5 × 10−3 M, 1 × 10−2 M or 0.1 M lactate solution (pH 3.0 or 6.0, 0.1 M NaClO4). 21 to 26 titration steps up to a lactate concentration of 0.1 M were performed; every mixture was allowed to equilibrate for at least 15 min. The measurements were carried out in a quartz cuvette with screw cap (1 cm path length). UV-vis absorption spectra were recorded in the wavelength range between 400 and 700 nm using a CARY G5 UV-vis-NIR spectrophotometer (Varian Inc., USA).TRLFS measurements
For spectrophotometric TRLFS titration 2.5 mL of 5 × 10−6 M Am3+ or Eu3+ at pH 3.0, 5.0 or 6.0 and 0.1 M NaClO4 were titrated with aliquots (5, 10 or 20 μL) of 5 × 10−3 M, 1 × 10−2 M or 0.1 M lactate solution (pH 3.0, 5.0 or 6.0, 0.1 M NaClO4). 26 to 31 titration steps up to a lactate concentration of 0.1 M were performed; every mixture was allowed to equilibrate for at least 15 min. The temperature was adjusted using a stirred temperature-controlled cuvette holder (Flash 300™, Quantum Northwest, USA).The TRLFS measurements for Am(III) were carried out with a pulsed Nd:YAG-MOPO laser system from Spectra Physics (Mountain View, USA), combined with a Spectrograph M270 and an ICCD camera system Spectrum One from Horiba-Jobin Yvon. The time difference between the trigger of the laser system and the start of the camera was adjusted by a delay generator from Spectrum One. The excitation wavelength of the laser source was varied between 503 and 508 nm with pulse energies of 10 mJ. Emission spectra were recorded between 625 and 773 nm, averaging 10 spectra with accumulating 80 laser pulses for each spectrum. The gate width of the camera was set to be 1 μs. The step width between two spectra in time-resolved mode was 2 ns, 50 to 60 delay steps (up to 120 ns) were measured for every sample. The spectrograph and the camera system were controlled by Spectramax from Horiba-Jobin Yvon.
The TRLFS measurements for Eu(III) were carried out with a pulsed flash lamp pumped Nd:YAG-OPO laser system from Continuum as described31 at an excitation wavelength of 394 nm and a gate width of 1 ms for all measurements. Static and time-resolved luminescence spectra of Eu(III) were recorded in the range of 565–650 nm (1200 lines mm−1 grating, 0.2 nm resolution, 2000 accumulations) and 440–780 nm (300 lines mm−1 grating, 0.7 nm resolution, 200 accumulations), respectively. For time-resolved measurements, 41 spectra were recorded with 20–50 μs separation.
ATR FT-IR measurements
ATR FT-IR measurements were carried out with 1 × 10−2 M lactate and Eu3+ each at pH 5.0. The infrared spectra were recorded at room temperature with an FT-IR spectrometer (Bruker VERTEX 80/v) equipped with a diamond ATR cell (crystal diameter: 4 mm, 9 reflections; Smiths Inc.) and a mercury cadmium telluride detector. Details on the experimental setup are given elsewhere.32,33 For the complex species, difference spectra were calculated from single-beam spectra consecutively recorded from solutions containing the Eu(III) lactate complexes and pure lactate with the same concentration and pH. For the experiments, an ATR flow cell (volume: 200 μL) with a constant flow rate of 200 μL min−1 was used. Each single-beam spectrum was co-added from 128 scans at a spectral resolution of 4 cm−1.DFT calculations
DFT calculations were performed using Gaussian 09.34 Geometries were optimized in the aqueous phase at the B3LYP level35,36 using the PCM solvation model implemented in Gaussian program. The large core effective core potential as well as the corresponding basis set suggested by Dolg37 was used on Eu as in the previous studies.27,28 For C, O, and H, all-electron valence triple-zeta basis set plus double polar and diffuse functions have been used.38 The final geometries were confirmed to be the energy minimum through vibrational frequency analysis where no imaginary frequency was found to be present. The spectra were fitted with the half-width of 8 cm−1 at half-height using the calculated harmonic frequencies and IR intensities.The spin-orbit effect and multiconfigurational character of the system were neglected. The first coordination sphere around Eu was saturated with water molecules fixing the coordination number to 8 or 9. The rest of the solvation shells were also considered through the use of the PCM model.
NMR measurements
NMR samples with 0.1 M in lactate and Eu3+ concentrations ranging from 0 to 0.15 M were prepared by the admixture of lactate and Eu3+ stock solutions in D2O, adjusting the pD to 5.4, corresponding to pH 5.0, by addition of appropriate amounts of diluted DCl or NaOD solutions in D2O. The sample containing La(III) instead of Eu(III) was prepared similarly. All measurements were performed on a Varian Unity Inova 400 spectrometer with a field strength of 9.4 T and resonance frequencies of 400 and 100 MHz for 1H and 13C, respectively, using an ATB 1H/19F/{15N-31P} PFG 5 mm broadband probe. For both nuclei the number of accumulations was increased, whereas the acquisition time and the relaxation delay were decreased with increasing Eu(III) concentration.Data analysis
The infrared, absorption and luminescence spectra were analyzed using OriginPro 7.5G (OriginLab, Northhampton, MA, USA) to obtain peak positions and lifetimes (TRLFS).Static luminescence spectra of Eu(III) have been normalized to the peak area of the 5D0→7F1 transition, which is a magnetic dipole and therefore not influenced by complexation.
The fluorescence decay lifetimes were calculated by fitting the integrated luminescence signal to a sum of exponential decay functions:
 | (1) |
E(t) is the total luminescence intensity at time t, Ei0 the luminescence intensity of the species i at the time t = 0, and τi the corresponding decay lifetime.
The number of water molecules in the first coordination shell was determined from the luminescence lifetimes τ (in ms). For Am(III), the empirical formula from Kimura and Kato19 (eqn (2)), and for Eu(III), the linear relationship developed by Horrocks and Sudnick39 and the resultant empirical formula from Kimura40 (eqn (3)), were used:
| n(H2O) ± 0.5 = 2.56 × 10−4 × τ−1 − 1.43 for Am(III) | (2) |
| n(H2O) ± 0.5 = 1.07 × τ−1 − 0.62 for Eu(III) | (3) |
The complex stability constants were determined from the absorption or luminescence spectra by using the factor analysis program specfit.41 Input parameters for the data fitting were the total concentrations of the metal ion and the ligand, the pH, and the pKa of lactate from literature (pKa1 = 3.69,42 pKa2 = 11.20,30 recalculated to I = 0.1 M). A brief description of the operation mode of this program43 and of the fitting procedure is given elsewhere.44
Thermodynamic data were calculated with the modified linear form of the van't Hoff equation:
 | (4) |
The extrapolation of the constants to infinite dilution, I = 0, was done applying the Specific Interaction Theory (SIT) using the IUPAC software for Ionic Strength Corrections.45 The ion interaction parameters ε were taken from ref. 46 (based on ref. 47,48) for Eu3+,ClO4−, from ref. 48 for Am3+,ClO4− and from ref. 48,49 for Na+,ClO4−, and Na+,CH3COO− (acetate as analog for lactate as it is proposed in ref. 50), whereas that for Eu3+,Lac− and Am3+,Lac− were calculated using the guidelines given in ref. 50. The temperature dependencies of ε and the Debye–Hückel parameter B can be neglected.48,50 Values for the Debye–Hückel parameter A as function of temperature have been calculated from literature data51 within the program.45
Results and discussion
Am(III) UV-vis spectroscopy
The absorption maximum of Am(III), usually equivalent to the excitation wavelength for fluorescence spectroscopy, namely the 7F0→5L6 transition, strongly depends on the chemical surrounding of the ion. The shift of the absorption maximum of Am(III) with lactate as ligand is depicted in Fig. 1A. For the aqueous Am(III), the absorption maximum is at 503 nm. At the highest lactate concentration of 0.1 M we observe a red-shift of about 3 nm to 506 nm. | ||
| Fig. 1 Absorption spectra of 5 μM Am(III) in dependence of the lactate concentration (50 μM to 0.1 M), pH = 6.0, I = 0.1 M, T = 25 °C (left), deconvoluted spectra of the single species (right). | ||
The quantitative analysis of the spectra clearly shows the formation of three Am(III) lactate complexes with a lactate concentration up to 0.1 M, Am(Lac)2+, Am(Lac)2+ and Am(Lac)3, as it was expected from literature.3,13Fig. 1B shows the deconvoluted single spectra of each individual species. They are very similar to those recently determined in trifluoromethansulfonate media.13 The simultaneous determination of all three complex formation constants results in relatively large uncertainties. Only for the 1:1 complex, the quality could be increased by using only the spectra up to a lactate concentration of 0.01 M. In this concentration range the 1:1 complex should be the dominating species. The complex formation constant was determined to be log β11 = 2.22 ± 0.11, providing that only the carboxylic group of lactate is deprotonated. This is in accordance with literature3,10,13,15 (see Table 1). The quality of the formation constants of the 1:2 and 1:3 complexes, respectively, could not be increased properly; possibly the spectral changes are too small to get more precise results than log β12 = 4.5 ± 0.3 for Am(Lac)2+, and log β13 = 6.3 ± 0.3 for Am(Lac)3. Nevertheless, these values are in the range of published data3,10,13,15 (see Table 1).
| T/°C | I/M (NaClO4) | Am(Lac)2+ | Am(Lac)2+ | Am(Lac)3 | Ref. |
|---|---|---|---|---|---|
| log β11(1) | log β12(2) | log β13(3) | Method | ||
| log βML(H) are the stability constants with protonated hydroxyl group(s), not considering the pKa2 of lactic acid.a Electrophoresis.b Solvent extraction.c UV-vis.d TRLFS. NaTf = Na-trifluoromethansulfonate; p.w. = present work. | |||||
| 10 | 1.5 | 2.57 | 4.21 | 10 | |
| 25 | 2 | 2.52 ± 0.04 | 4.77 ± 0.05 | 5.98 ± 0.08 | 15 |
| 1 | 2.43 ± 0.09 | 4.23 ± 0.27 | 5.65 ± 0.15 | 3 | |
| 1 (NaTf) | 2.60 ± 0.06 | 4.7 ± 0.1 | 6.2 ± 0.2 | 13 | |
| 0.1 | 2.27 ± 0.05 | 4.5 ± 0.3 | 6.3 ± 0.3 | p.w.c | |
| 0.1 | 2.22 ± 0.11 | p.w.d | |||
| 0 | 2.87 ± 0.26 | 5.5 ± 0.4 | 7.5 ± 0.4 | p.w. | |
| 45 | 0.1 | 2.17 ± 0.19 | p.w.d | ||
| 0 | 2.82 ± 0.31 | p.w. | |||
| 65 | 0.1 | 2.35 ± 0.31 | p.w.d | ||
| 0 | 3.03 ± 0.39 | p.w. | |||
Am(III) time-resolved laser-induced fluorescence spectroscopy
The best excitation wavelength for luminescence emission corresponds to the absorption maximum of the luminescent species. The highest luminescence intensity for the Am(III) aqua ion can be observed at 503–504 nm excitation (Fig. 2A), whereas the best excitation in the presence of 0.1 M lactate is at 506–507 nm (Fig. 2B). For the determination of the complex stability constants, an excitation wavelength of 504 nm was used, where the luminescence intensity of both the uncomplexed and complexed Am(III) species is satisfactory for quantitative determinations. | ||
| Fig. 2 Emission spectra of (A) 5 μM Am(III) and (B) 5 μM Am(III) + 0.1 M lactate in dependence of the excitation wavelength. | ||
The Am(III) aqua ion shows at pH 6.0 and different temperatures a luminescence lifetime of 23.8 ± 2.4 ns (25 °C), 22.8 ± 0.9 ns (40 °C) or 23.3 ± 1.5 ns (65 °C), corresponding to approximately 9 coordinating water molecules. This agrees with previous measurements.18,25 Complex formation with lactate causes a strong increase of the luminescence intensity and a red shift of the luminescence maximum of about 5 nm (Fig. 3, left). The luminescence decay is always mono-exponential, irrespective of the number of expected different Am(III) species (see Fig. S1 and S2, ESI†). This is caused by an exchange of the Am(III) coordination environments, which is faster than the luminescence decay rate of the excited state and results in a concentration-weighted average number of water molecules of all Am(III) species.52 The luminescence lifetime is prolonged up to 38.3 ± 0.4 ns (25 °C, 0.1 M lactate, pH 6.0). This value corresponds to 5 remaining water molecules, indicating an exchange of 4 water molecules with ligand molecules’ coordination sites. It implies the formation of not only a 1:1 complex but also a certain amount of complexes with higher stoichiometry like 1:2 or 1:3 complexes.
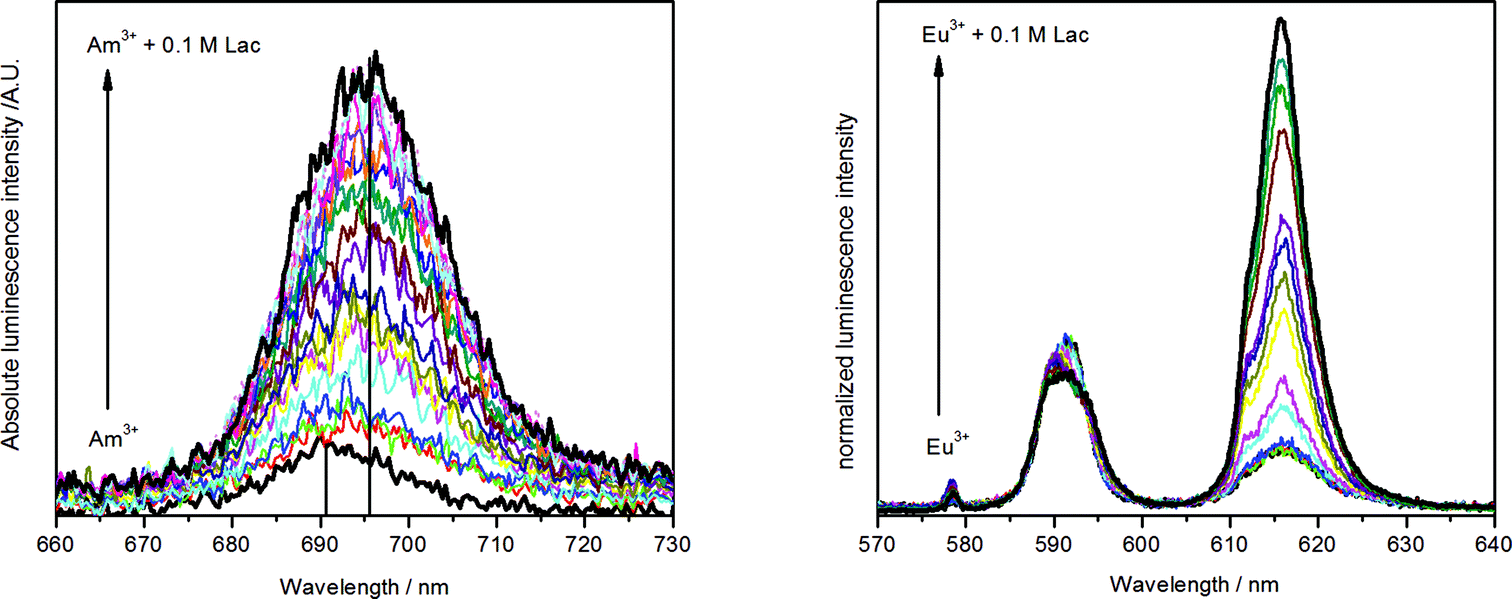 | ||
| Fig. 3 Emission spectra of 5 μM Am(III) (left) and 10 μM Eu(III) (right) in dependence of lactate concentration (10 μM to 0.1 M each, pH = 6.0, I = 0.1 M, T = 25 °C). | ||
The quantitative deconvolution of the luminescence spectra in order to determine complex stability constants for all three complexes failed. Possibly the spectral changes are too small to discriminate all three complexes. A reasonable stability constant could only be determined for the 1:1 complex. For determination of the formation constant of the 1:1 complex, the spectra with a lactate concentration up to 0.01 M were considered in analogy to the UV-vis measurements, yielding a log β11 = 2.27 ± 0.05 (25 °C). This is in very good accordance to the value from UV-vis spectroscopy and to literature values (see Table 1).
TRLFS measurements were also done at elevated temperatures (45 °C and 65 °C). The stability constant of the 1:1 complex shows no relevant tendency with rising temperature, indicating that the complex formation reaction causes only a very small enthalpy change. The van't Hoff plot (see Fig. 4) results in an enthalpy change closed to zero within the error bars (see Table 3). Other studies calculated a negative reaction enthalpy, corresponding to an exothermic reaction3,13 (Table 3). This discrepancy is possibly due to different ionic strengths and ionic media. A detailed discussion to this effect is provided in the next section.
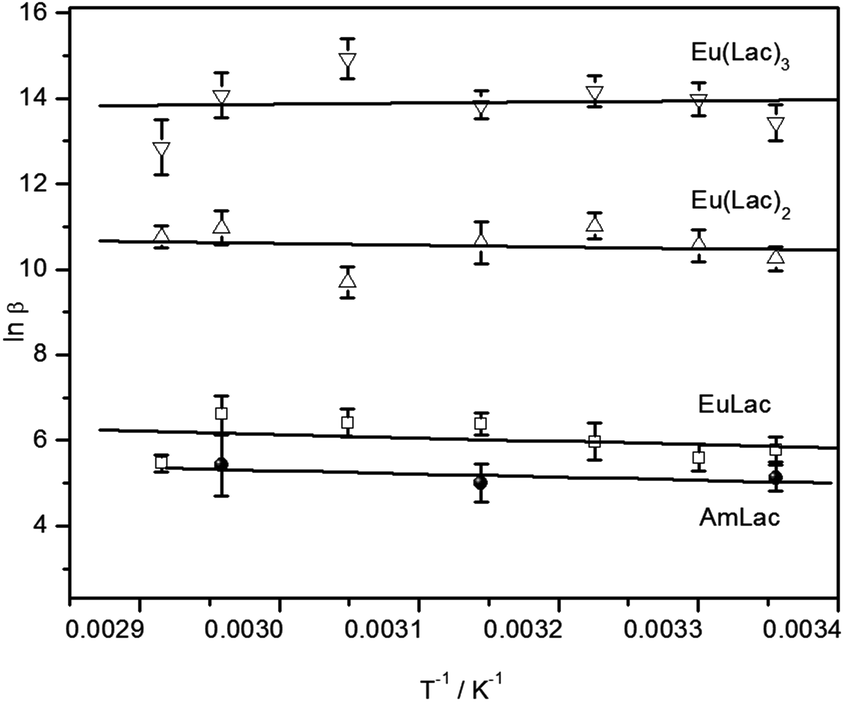 | ||
| Fig. 4 Van't Hoff plots of the Am(III) lactate and Eu(III) lactate complex formation. | ||
Eu(III) time-resolved laser-induced fluorescence spectroscopy
The complex formation of Eu(III) with lactate was studied with TRLFS at near neutral pH (3.0–6.0) and ambient to higher temperature (25–70 °C). Because of the high importance of lactate in metabolism the biological relevant temperature of 37 °C was included. The luminescence spectra in dependence of the lactate concentration are depicted in Fig. 3 (right). During ligand addition, the characteristic changes in Eu(III) luminescence spectrum caused by complex formation can be observed: a strong increase of the hypersensitive 5D0→7F2 transition at about 615 nm and the appearance of the symmetry forbidden 5D0→7F0 transition at around 578 nm (see Fig. 3, right). The luminescence lifetime is prolonged from 111.5 ± 2.2 μs (25 °C) for the Eu(III) aqua ion up to 223.9 ± 1.3 μs (25 °C, 0.1 M lactate, pH 6.0). The former corresponds to 9 water molecules in the first hydration shell of the Eu(III) aqua ion and is in accordance with literature.53,54 As it was observed for Am(III), also the Eu(III) lactate system shows always mono-exponential decay due to a fast ligand exchange in the first coordination sphere (see Fig. S3 and S4, ESI†). The resultant concentration-weighted average lifetime consists of certain contributions of the expected species. Upon complex formation with lactate, the average number of water molecules is reduced to 4.2 ± 0.5 at the highest lactate concentration (0.1 M). This indicates the formation of complexes with different metal-to-ligand ratios, possibly 1:1, 1:2 and 1:3 complexes as it was suggested for the lactate complex formation with Am(III) and already described in previous studies.4,6,7,9
Complex stability constants were determined for all three complexes and various temperatures between 25 °C and 70 °C, provided that only the carboxylic group of lactate is deprotonated (see Table 2). The stability constants show no significant trend with rising temperature, however, the van't Hoff plot (Fig. 4) results in a small positive reaction enthalpy change for the 1:1 and 1:2 complexes and a very small negative reaction enthalpy for the 1:3 complex (see Table 3), equivalent to an endothermic reaction for the former and an exothermic reaction for the latter complex. Previous investigations from Tian4 gave small negative reaction enthalpy changes for all three complexes. However, these measurements were done at an ionic strength of 1 M. The reaction enthalpies determined from Aziz7 and Choppin6 at an ionic strength of 2 M are even more negative (see Table 3). This gives the following ionic strength dependency of the reaction enthalpy: the higher the ionic strength the lower the reaction enthalpy. A comparison of literature data in data collections has shown a decrease of reaction enthalpy with increasing ionic strength for several metal–ligand systems; even changes from positive to negative enthalpy can be observed.55 This is caused by the ionic strength dependent variation in the activity coefficients of the reagents.56 In general, two contrary effects contribute to the reaction enthalpy: (1) the partially dehydration of the reactants which is usually endothermic, and (2) the complex formation which is expected to be exothermic.57 At low ionic strength the solvation spheres are tightly bound, equivalent to lowering the activity coefficients of the ions. This results in a larger endothermic dehydration enthalpy and the effect (1) mainly contributes to the reaction enthalpy. With higher ionic strength the solvation spheres loosen up because of a higher amount of competition ions. This causes increased activity coefficients of the ions and a smaller endothermic dehydration enthalpy term. In consequence, the contribution of the effect (2) might become dominant and even cause a change of sign of the reaction enthalpy.58
| T/°C | I/M (NaClO4) | Eu(Lac)2+ | Eu(Lac)2+ | Eu(Lac)3 | Ref. |
|---|---|---|---|---|---|
| log β11(1) | log β12(2) | log β13(3) | Method | ||
| log βML(H) are the stability constants with protonated hydroxyl group(s), not considering the pKa2 of lactic acid.a Electrophoresis.b Potentiometry.c TRLFS.d Solvent extraction. p.w. = present work. | |||||
| 10 | 1.5 | 2.62 | 4.22 | 10 | |
| 1.0 | 2.90 ± 0.36 | 4.90 ± 0.37 | 6.24 ± 0.30 | 4 | |
| 1.0 | 2.91 ± 0.24 | 5.02 ± 0.22 | 6.03 ± 0.28 | 4 | |
| 25 | 2.0 | 2.53 | 4.60 | 5.88 | 9 |
| 1.0 | 2.80 ± 0.02 | 4.76 ± 0.02 | 6.33 ± 0.02 | 4 | |
| 1.0 | 2.99 ± 0.17 | 5.09 ± 0.23 | 6.09 ± 0.27 | 4 | |
| 1.0 | 2.46 ± 0.09 | 4.28 ± 0.25 | 5.87 ± 0.10 | 3 | |
| 1.0 (NaCl) | 2.95 | 4.40 | 5.47 | 11 | |
| 0.2 | 2.55 ± 0.05 | 4.67 ± 0.06 | 5.55 ± 0.18 | 8 | |
| 0.1 | 2.51 ± 0.13 | 4.45 ± 0.12 | 5.83 ± 0.18 | p.w.c | |
| 0 | 3.14 ± 0.28 | 5.49 ± 0.28 | 7.07 ± 0.31 | p.w. | |
| 30 | 0.1 | 2.43 ± 0.14 | 4.58 ± 0.16 | 6.07 ± 0.17 | p.w.c |
| 0 | 3.06 ± 0.29 | 5.63 ± 0.30 | 7.32 ± 0.30 | p.w. | |
| 37 | 0.1 | 2.59 ± 0.19 | 4.78 ± 0.13 | 6.15 ± 0.16 | p.w.c |
| 0 | 3.24 ± 0.31 | 5.85 ± 0.28 | 7.42 ± 0.30 | p.w. | |
| 40 | 1.0 | 2.78 ± 0.15 | 4.57 ± 0.11 | 6.25 ± 0.21 | 4 |
| 1.0 | 2.91 ± 0.17 | 5.04 ± 0.27 | 6.17 ± 0.25 | 4 | |
| 45 | 0.1 | 2.77 ± 0.11 | 4.61 ± 0.21 | 6.01 ± 0.14 | p.w.c |
| 0 | 3.42 ± 0.27 | 5.69 ± 0.33 | 7.30 ± 0.29 | p.w. | |
| 55 | 1.0 | 2.70 ± 0.11 | 4.43 ± 0.15 | 6.28 ± 0.11 | 4 |
| 1.0 | 3.04 ± 0.24 | 5.00 ± 0.27 | 5.95 ± 0.26 | 4 | |
| 0.1 | 2.78 ± 0.14 | 4.21 ± 0.16 | 6.48 ± 0.20 | p.w.c | |
| 0 | 3.45 ± 0.29 | 5.32 ± 0.30 | 7.79 ± 0.32 | p.w. | |
| 65 | 0.1 | 2.87 ± 0.19 | 4.76 ± 0.17 | 6.11 ± 0.23 | p.w.c |
| 0 | 3.55 ± 0.31 | 5.89 ± 0.30 | 7.46 ± 0.34 | p.w. | |
| 70 | 1.0 | 2.81 ± 0.19 | 4.49 ± 0.21 | 6.33 ± 0.20 | 4 |
| 1.0 | 2.99 ± 0.23 | 4.88 ± 0.25 | 5.98 ± 0.26 | 4 | |
| 0.1 | 2.37 ± 0.09 | 4.67 ± 0.11 | 5.58 ± 0.28 | p.w.c | |
| 0 | 3.06 ± 0.27 | 5.82 ± 0.27 | 6.94 ± 0.38 | p.w. | |
| I/M (NaClO4) | Am(Lac)2+ | Eu(Lac)2+ | Eu(Lac)2+ | Eu(Lac)3 | |
|---|---|---|---|---|---|
| a Solvent extraction. b Calorimetry. c TRLFS. d Potentiometry. NaTf = Na-trifluoromethansulfonate; p.w. = present work. | |||||
| ΔrH*/kJ mol−1 | 2.0 | −4.3 ± 0.87a |
−8.6 ± 1.67a |
−23 ± 47a |
|
| 2.0 | −8.17 ± 1.056b |
−4.8 ± 2.56b |
−23.3 ± 5.96b |
||
| 1.0 | −163a |
−2.14 ± 0.774a |
−4.31 ± 0.424a |
−12.37 ± 0.674a |
|
| 1.0 (NaTf) | −5.38 ± 0.0713a |
||||
| 0.1 (p.w.) | 3.7 ± 4.6c |
6.7 ± 6.2c |
3.3 ± 6.6c |
−2.1± 6.9c |
|
| ΔrH0/kJ mol−1 | 0 (p.w.) | 6.2 ± 4.6 | 9.3 ± 6.2 | 8.0 ± 6.6 | 3.1 ± 6.9 |
| ΔrS*/J mol−1 K−1 | 2.0 | 33 ± 37a |
58 ± 67a |
34 ± 127a |
|
| 2.0 | 21 ± 36b |
72 ± 96b |
36 ± 206b |
||
| 1.0 | −63a |
46 ± 34a |
76 ± 24a |
78 ± 294a |
|
| 1.0 (NaTf) | 32 ± 213a |
||||
| 0.1 (p.w.) | 55 ± 15c |
71 ± 29c |
100 ± 31c |
109 ± 34c |
|
| ΔrS0/J mol−1 K−1 | 0 (p.w.) | 75 ± 15 | 92 ± 29 | 133 ± 31 | 150 ± 34 |
| ΔrG*/kJ mol−1 (25 °C) | 2.0 | −14.15 ± 0.127a |
−26.0 ± 0.27a |
−33.2 ± 0.47a |
|
| 2.0 | −14.57 ± 0.049d |
−26.23 ± 0.129d |
−34.06 ± 0.209d |
||
| 1.0 (NaTf) | −14.813a |
||||
| 0.1 (p.w.) | −12.5 ± 4.6c |
−1.6 ± 6.2c |
−29.2 ± 6.6c |
−53.36 ± 6.9c |
|
| ΔrG0/kJ mol−1 | 0 (p.w.) | −16.2 ± 4.6 | −18.2 ± 6.2 | −31.7 ± 6.6 | −41.62 ± 6.9 |
It is worth mentioning that the entropy changes are quite high (see Table 3), so we can assume that the complex formation reaction is predominantly entropy driven. The number of water molecules in the first coordination shell of Eu(III) (deduced from the luminescence lifetimes) helps to explain this. As written earlier, up to 5 water molecules are replaced by 3 ligand molecules which increases the entropy in the system.
The challenge is to receive information about the coordination type of Eu(III) lactate. Possible structures for the 1:1 complex are depicted in Fig. 5. Is it monodentate coordination of the carboxylate group with a high sterical requirement of the lactate (A) or bidentate coordination with carboxylate and hydroxyl group, protonated (B) or deprotonated (C) or bidentate coordination of the carboxylate group (D)? Spectroscopic (FT-IR, NMR) and computational (DFT) techniques were carried out to get an idea about the coordination behavior of the Eu(III) lactate 1:1 complex.
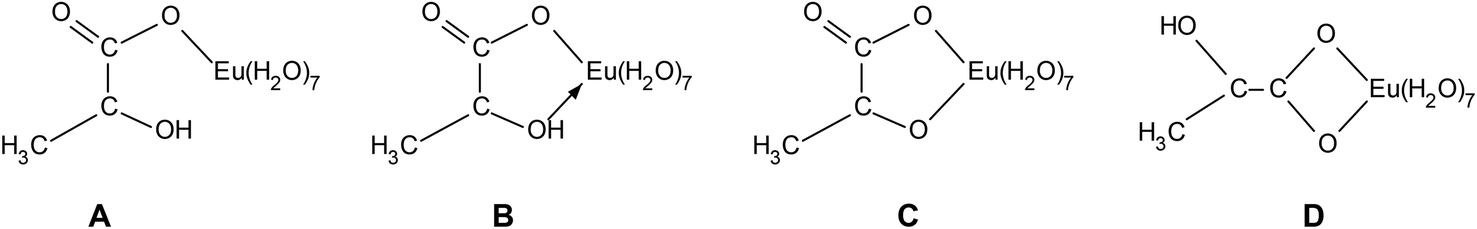 | ||
| Fig. 5 Possible structures of the Eu(III) lactate 1:1 complex: (A) monodentate, (B) bidentate with coordinating hydroxyl group, (C) bidentate with deprotonated hydroxyl group, (D) bidentate binding of the carboxylate group. | ||
Vibrational spectroscopy and DFT analysis
 | ||
| Fig. 6 Experimental infrared spectra of lactate at pH 5 (a), Eu(III) lactate at pH 5 (b), and the calculated difference spectrum Eu(III) lactate minus lactate (c). | ||
:1 complex (Fig. 6b) shows in principle the same vibration modes as the pure ligand (Fig. 6a). The difference spectrum (Fig. 6c) shows the shifts of the modes more clearly; hereby positive peaks represent vibrational modes of the Eu(III) lactate complex and negative peaks those of the pure ligand. Upon complex formation, the asymmetric and symmetric stretching vibrations νas and νs of the carboxylate group are shifted to higher (νas, 1588 cm−1) and lower (νs, 1388 cm−1) frequencies, respectively. By the degree of spectral splitting of the νs/as(COO) (Δν) the type of coordination of the carboxylate group can be derived. The Δν value of the uncomplexed ligand serves as reference; in our case it is 159 cm−1. In general, bidentate coordination is expected to show a significantly lower spectral splitting, whereas monodentate coordination usually exhibits a larger splitting compared to the uncomplexed ligand.59,60 The Eu(III) lactate 1:1 complex shows a Δν of 200 cm−1, which is a strong indication for monodentate carboxylate coordination. So we can rule out structure D (Fig. 5) with bidentate coordination mode of the carboxylate group.
The difference spectrum (Fig. 6c) shows additionally significant changes of the spectral modes at around 1120 cm−1 and 1040 cm−1 (strong negative bands in the difference spectrum). According to DFT calculations (see Fig. 7 and Fig. S7, ESI†), these modes can be assigned to the C–O stretching vibration of the hydroxyl group and the subsequent C–C stretching vibration of the C–CH3 unit, respectively.
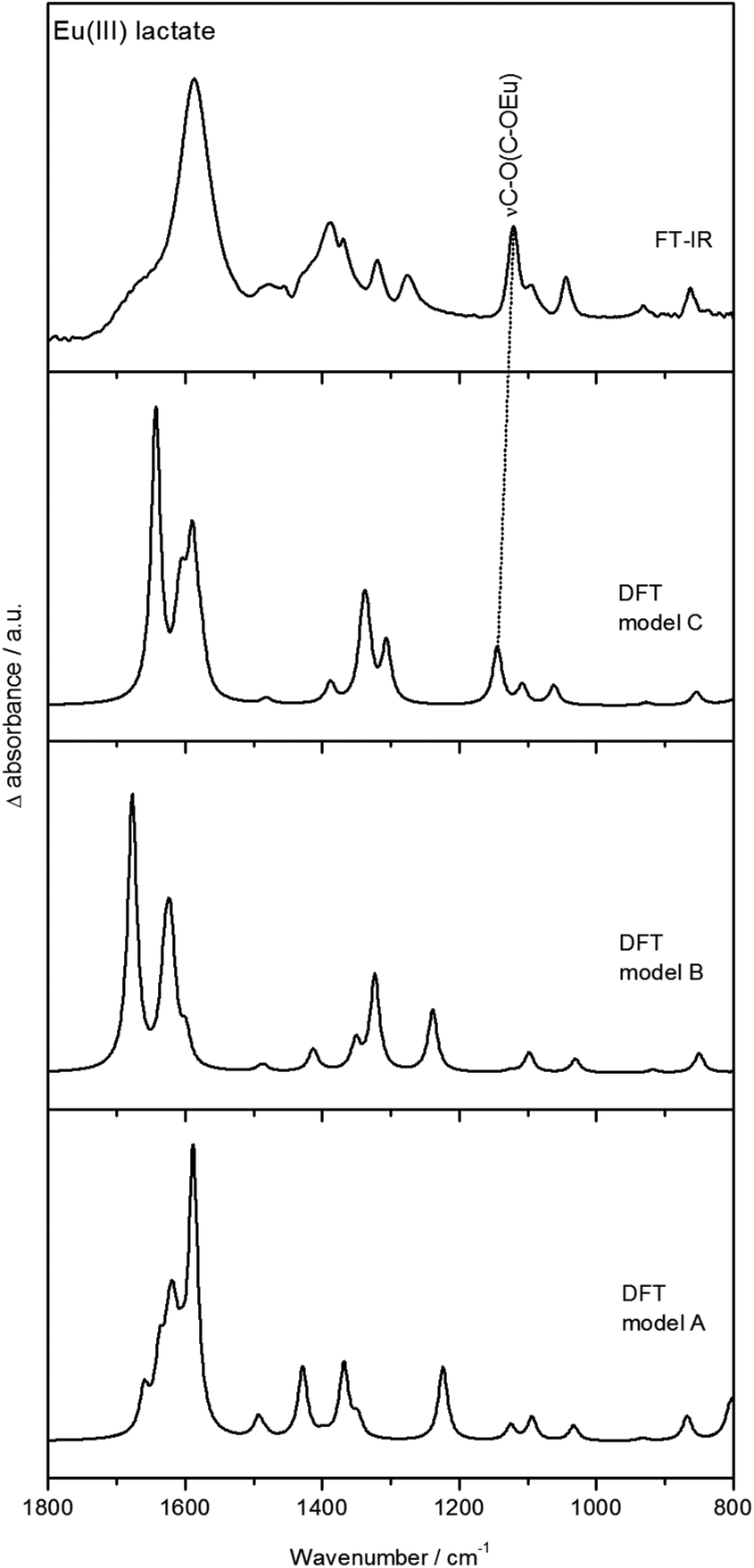 | ||
| Fig. 7 Experimental (ATR FT-IR) and calculated (DFT) spectra of Eu(III) lactate. Calculations were done for structures A, B, and C (from Fig. 5). | ||
Fig. 7 shows the DFT calculated vibrational spectra for the models A, B, and C from Fig. 5 in comparison with the measured ATR FT-IR spectrum of Eu(III) lactate. The best accordance to the measured spectrum is given by model C. Especially the peak at around 1120 cm−1 (measured spectrum) finds its equivalent only in the calculated IR spectrum of model C (1145 cm−1). In the calculated IR spectra of models A and B, this peak is missing or shifted strongly to 1224 cm−1 (model A) and 1238 cm−1 (model B), respectively. This mode is caused by the C–O stretching vibration of the hydroxo group which is protonated in models A and B. In model C this functionality is deprotonated resulting in a covalent binding to the Eu(III) cation. Due to the position of this stretching vibration mode in the measured spectrum compared to the calculated spectra it is assumed that model C reflects best the binding behavior of the Eu(III) lactate complex. The coordinating hydroxyl group seems to become deprotonated under complex formation with Eu(III).
NMR spectroscopy
 | ||
| Fig. 8 1H-NMR spectra of 0.1 M lactate with varying Eu(III) concentration (from bottom to top: 0, 1, 5, 10, 50, 100 mM). The inset shows expansions of lactate samples without Eu (A) and with equimolar concentration of Eu (B). | ||
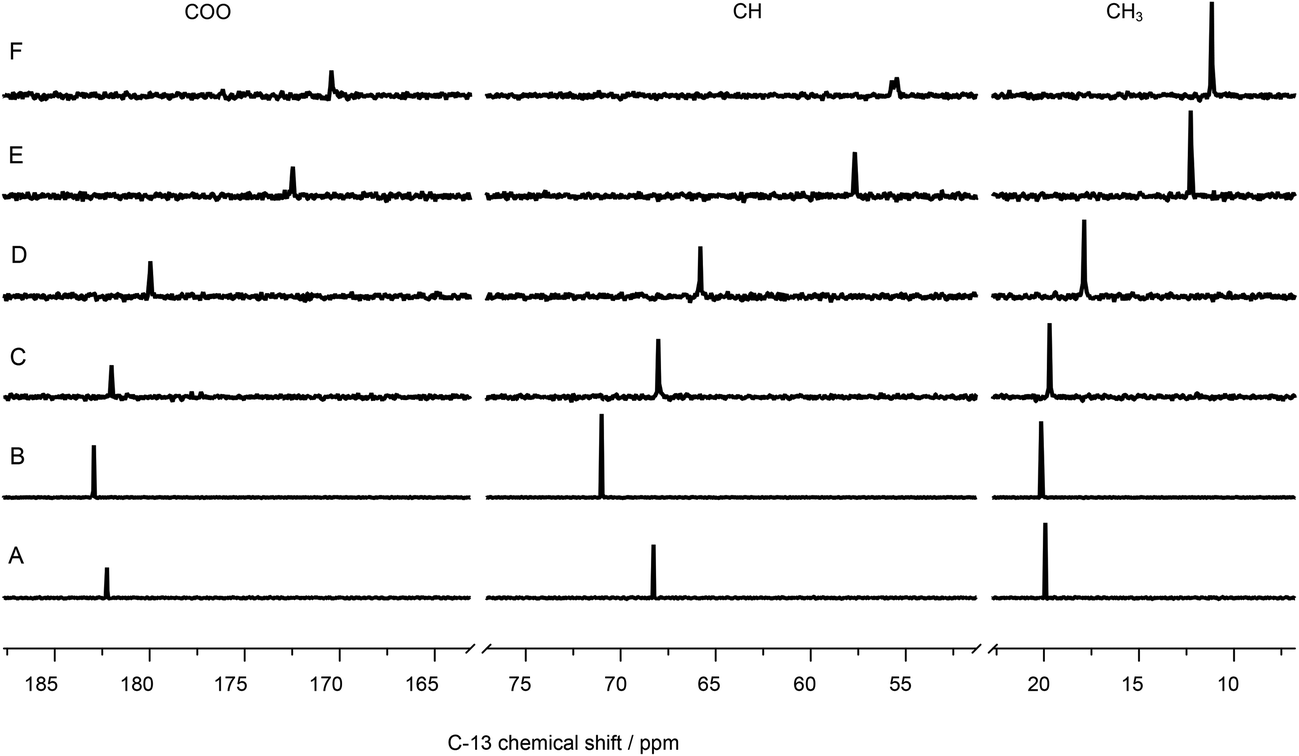 | ||
| Fig. 9 13C-NMR spectra of 100 mM lactate, (A) without metal, (B) containing 120 mM La(III), and (C) 5, (D) 10, (E) 50, (F) 100 mM Eu(III). | ||
 | ||
| Fig. 10 Plot of 13C chemical shift differences vs. Eu(III) concentration. ■CH, ▲COO, ●CH3, lines drawn for better visualization. | ||
Using La(III) (4f0 configuration, closed shell) as a diamagnetic analogue of Eu(III), the chemical shift changes induced by interaction of the unpaired f-electrons (LIS) can be separated from the pure charge and complex formation induced shifts. As expected, the positive charge of the trivalent metal ion causes a de-shielding, i.e. reduction of electron density of the nuclei at or near the binding site, cf.Fig. 9B. Therefore, the carbon signals are shifted to higher chemical shift values. Interestingly, also in this case the CH carbon is affected most, pointing towards a strong participation of the hydroxyl oxygen in the complex formation. In the case of Eu(III), magnitude and direction of the shift are related to the distribution of f-electron-density at the nuclei of interest, overcompensating the pure charge induced effects.
The LIS has two contributions: (1) contact term, i.e., interaction via bonds and (2) pseudo-contact term, i.e., through space. The contact term depends on type and number of bonds between the (open shell) metal center and the atom of interest. The pseudo-contact term is mediated through dipolar interaction and strongly distance dependent. Both terms can contribute to different extent, depending on, e.g., the electronic configuration and the energy of the ground state or the ligand field splitting.61 Neither for 1H nor for 13C do the shifts of signals of adjacent atoms show alternating signs, indicating that the contact contribution can be neglected.62 Thus, the observed differences in LIS can be fully attributed to spatial europium distances.
Interestingly, the CH carbon atom shows the strongest LIS, indicating that this carbon is affected mostly by the europium's unpaired electrons. This can be explained only by participation of the hydroxyl oxygen in Eu(III) coordination. Therefore, model A (Fig. 5) can be excluded.
Model B (Fig. 5) contains the coordination by the hydroxyl oxygen, but being protonated. Due to this hydrogen, the distance between Eu(III) and this particular oxygen as well as the adjacent carbon is bigger than for both the carboxylic oxygen and carbon (distances calculated with DFT, see Fig. S7, ESI†). This is in contradiction to the 13C-NMR results.
Model C (Fig. 5), however, reflects perfectly the NMR findings: same distance between the carbons of interest and Eu(III) (3.24 Å, calculated with DFT, cf. Fig. S7, ESI†), resulting in similar magnitude of LIS. The small discrepancy in the chemical shift differences of these two particular carbons (cf.Fig. 10) is probably related to the angle between the crystal field axis of the complex and the radius vector from Eu(III) to the respective carbon. The NMR findings strongly support the results obtained from ATR FT-IR measurements in combination with DFT calculations.
With respect to this new findings, the complex formation constants of the Eu(III) lactate 1:1 complex have to be recalculated considering the pKa of the hydroxyl group.30 The resultant constants and thermodynamic data are listed in Table 4. Interestingly, the recalculation process of the complex formation constants shows that only the 1:1 complex seems to exist with deprotonated hydroxyl group. The calculation of stability constants with further fully deprotonated lactate ligands failed. At pH 3, the hydroxyl group even of the first lactate remains protonated.
| T/°C | log β*110 | log β°110a |
|---|---|---|
| a Uncertainty ± 0.25. | ||
| 25 | 7.52 ± 0.07 | 8.80 |
| 30 | 7.28 ± 0.07 | 8.57 |
| 37 | 7.33 ± 0.06 | 8.64 |
| 45 | 7.66 ± 0.04 | 8.99 |
| 55 | 7.73 ± 0.05 | 9.08 |
| 65 | 7.53 ± 0.05 | 8.92 |
| 70 | 7.23 ± 0.12 | 8.63 |
| ΔrH/kJ mol−1 | 7.7 ± 3.0 | 12.9 ± 3.0 |
| ΔrS/J mol−1 K−1 | 169 ± 10 | 210 ± 10 |
| ΔrG/kJ mol−1 (25 °C) | −42.7 ± 3.0 | −49.7 ± 3.0 |
Conclusion
The combination of spectroscopic and theoretical methods for the investigation of the Am(III)/Eu(III) lactate system provides a broad set of information. From UV-vis and TRLFS measurements thermodynamic data were achieved. A negative correlation between ionic strength and reaction enthalpy has been noticed. With higher ionic strength the enthalpies become smaller and the reactions change from endothermic to exothermic. This indicates that with higher temperature the complexes become more stable at lower ionic strengths and weaker at higher ionic strength. On the other hand, at ambient temperature the complexes seem to become stronger with higher ionic strength. Systematic studies to confirm and quantify this assumption are in progress.The results from ATR FT-IR and NMR measurements combined with DFT calculations provided detailed structural information for the Eu(III) lactate 1:1 complex. The finding that the hydroxyl group seems to be deprotonated under complex formation (model C, Fig. 5) contradicts former structure suggestions, which suppose a coordination of the trivalent metal ion with the protonated hydroxyl group (model B, Fig. 5).4,5,12,63 Both experimental methods, ATR FT-IR and NMR, as well as the DFT calculations yielded an impressively homogeneous structural explanation of the investigated Eu(III) lactate 1:1 species.
The thermodynamic results indicate that the complex formation of trivalent actinides and lanthanides with organic matter is strongly influenced by different parameters like temperature and ionic strength. This makes it difficult to simulate and predict the migration behavior of the metal ions in the environment. Insights in the structural behavior of the complexes in aqueous solution (like it is provided with this study) improve understanding and may result in a more reliable prediction of such migration processes.
Acknowledgements
This study was partly funded by the German Federal Ministry of Economy (BMWi) under Contract No. 02E11021 and 02E10417 and the Deutsche Forschungsgemeinschaft (DFG) under Contract No. BE 2234/10-1/2. The authors are grateful for providing of Am-243 from the transplutonium element production facilities at Oak Ridge National Laboratory within collaboration between the Helmholtz-Zentrum Dresden – Rossendorf and Prof. Nitsche from the Lawrence Berkeley National Laboratory. All the quantum chemical calculations were performed using PC–Farm Atlas at the Zentrum für Informationsdienste und Hochleistungsrechnen (ZIH) at the Dresden University of Technology, Germany. The authors thank K. Heim for the ATR FT-IR measurements and M. Zegke for assistance with practical work in the laboratory.References
- L. F. Rao, Chem. Soc. Rev., 2007, 36, 881–892 RSC.
- A. Courdouan, I. Christl, S. Meylan, P. Wersin and R. Kretzschmar, Appl. Geochem., 2007, 22, 2926–2939 CrossRef CAS PubMed.
- R. Lundqvist, J. F. Lu and I. Svantesson, Acta Chem., Scand. Ser. A, 1984, 38, 501–512 CrossRef PubMed.
- G. X. Tian, L. R. Martin and L. F. Rao, Inorg. Chem., 2010, 49, 10598–10605 CrossRef CAS PubMed.
- R. S. Dickins, S. Aime, A. S. Batsanov, A. Beeby, M. Botta, J. Bruce, J. A. K. Howard, C. S. Love, D. Parker, R. D. Peacock and H. Puschmann, J. Am. Chem. Soc., 2002, 124, 12697–12705 CrossRef CAS PubMed.
- G. R. Choppin and H. G. Friedman, Inorg. Chem., 1966, 5, 1599–1603 CrossRef CAS.
- A. Aziz, S. J. Lyle and J. E. Newbery, J. Inorg. Nucl. Chem., 1971, 33, 1757–1765 CrossRef CAS.
- H. Deelstra and F. Verbeek, Anal. Chim. Acta, 1964, 31, 251–257 CrossRef CAS.
- G. R. Choppin and J. A. Chopoorian, J. Inorg. Nucl. Chem., 1961, 22, 97–113 CrossRef CAS.
- M. Sakanoue and M. Nakatani, Bull. Chem. Soc. Jpn., 1972, 45, 3429–3433 CrossRef CAS.
- P. R. Danesi, C. Cianetti and E. P. Horwitz, Sep. Sci. Technol., 1982, 17, 507–519 CrossRef CAS.
- A. P. G. Kieboom, C. A. M. Vijverberg, J. A. Peters and H. Vanbekkum, Recl. Trav. Chim. Pays-Bas-J. Roy. Neth. Chem. Soc., 1977, 96, 315–316 CrossRef CAS.
- P. R. Zalupski, K. L. Nash and L. R. Martin, J. Solution Chem., 2010, 39, 1213–1229 CrossRef CAS.
- P. R. Zalupski, K. L. Nash, Y. Nakamura, M. Yamamoto and L. R. Martin, in Actinides 2009. IOP Conference Series-Materials Science and Engineering, IOP Publishing Ltd, Bristol, England, 2010, vol. 9 Search PubMed.
- A. Aziz and S. J. Lyle, J. Inorg. Nucl. Chem., 1971, 33, 3407–3408 CrossRef CAS.
- G. Geipel, Coord. Chem. Rev., 2006, 250, 844–854 CrossRef CAS PubMed.
- C. Moulin, Radiochim. Acta, 2003, 91, 651–657 CrossRef CAS.
- J. V. Beitz, J. Alloys Compd., 1994, 207, 41–50 CrossRef.
- T. Kimura and Y. Kato, J. Alloys Compd., 1998, 271, 867–871 CrossRef.
- T. Kimura, R. Nagaishi, Y. Kato and Z. Yoshida, Radiochim. Acta, 2001, 89, 125–130 CrossRef CAS.
- W. Runde, C. Van Pelt and P. G. Allen, J. Alloys Compd., 2000, 303–304, 182–190 CrossRef CAS.
- T. Stumpf, M. M. Fernandes, C. Walther, K. Dardenne and T. Fanghänel, J. Colloid Interface Sci., 2006, 302, 240–245 CrossRef CAS PubMed.
- P. Thouvenot, S. Hubert, C. Moulin, P. Decambox and P. Mauchien, Radiochim. Acta, 1993, 61, 15–21 CAS.
- A. B. Yusov, J. Radioanal. Nucl. Chem., 1990, 143, 287–294 CrossRef CAS.
- A. Barkleit, G. Geipel, M. Acker, S. Taut and G. Bernhard, Spectrochim. Acta, Part A, 2011, 78, 549–552 CrossRef PubMed.
- B. Raditzky, S. Sachs, K. Schmeide, A. Barkleit, G. Geipel and G. Bernhard, Polyhedron, 2013, 65, 244–251 CrossRef CAS PubMed.
- A. Barkleit, S. Tsushima, O. Savchuk, J. Philipp, K. Heim, M. Acker, S. Taut and K. Fahmy, Inorg. Chem., 2011, 50, 5451–5459 CrossRef CAS PubMed.
- A. Heller, A. Barkleit, H. Foerstendorf, S. Tsushima, K. Heim and G. Bernhard, Dalton Trans., 2012, 41, 13969–13983 RSC.
- B. C. Mayo, Chem. Soc. Rev., 1973, 2, 49–74 RSC.
- A. K. Patel and J. D. Joshi, J. Indian Chem. Soc., 1996, 73, 71–73 CAS.
- H. Moll, A. Johnsson, M. Schäfer, K. Pedersen, H. Budzikiewicz and G. Bernhard, BioMetals, 2008, 21, 219–228 CrossRef CAS PubMed.
- K. Müller, H. Foerstendorf, S. Tsushima, V. Brendler and G. Bernhard, J. Phys. Chem. A, 2009, 113, 6626–6632 CrossRef PubMed.
- B. Li, J. Raff, A. Barkleit, G. Bernhard and H. Foerstendorf, J. Inorg. Biochem., 2010, 104, 718–725 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS.
- T. Kimura, G. R. Choppin, Y. Kato and Z. Yoshida, Radiochim. Acta, 1996, 72, 61–64 CrossRef CAS.
- R. A. Binstead, A. D. Zuberbühler and B. Jung, SPECFIT Global Analysis System, Version 3.0.37, Spectrum Software Associates, Marlborough, MA, USA, 2005 Search PubMed.
- K. Gajda-Schrantz, L. Nagy, T. Fiore, L. Pellerito and T. Gajda, J. Chem. Soc., Dalton Trans., 2002, 152–158 RSC.
- A. Günther, R. Steudtner, K. Schmeide and G. Bernhard, Radiochim. Acta, 2011, 99, 535–541 CrossRef.
- A. Barkleit, M. Acker and G. Bernhard, Inorg. Chim. Acta, 2013, 394, 535–541 CrossRef CAS PubMed.
- L. D. Pettit, I. Puigdomenech and H. Wanner, Ionic Strength Corrections for Stability Constants using Specific Interaction Theory (SIT), Academic Software, York, UK, 2004 Search PubMed.
- T. Vercouter, B. Amekraz, C. Moulin, E. Giffaut and P. Vitorge, Inorg. Chem., 2005, 44, 7570–7581 CrossRef CAS PubMed.
- K. Spahiu, Ph. D. Thesis, The Royal Institute of Technology Stockholm, Sweden, 1983.
- I. Grenthe and H. Wanner, in Chemical Thermodynamics of Uranium., Elsevier, Amsterdam, 1992, Appendix B: Ionic strength corrections Search PubMed.
- L. Ciavatta, Anal. Chim., 1980, 70, 551–567 CAS.
- L. Ciavatta, Anal. Chim., 1990, 80, 255–263 CAS.
- H. C. Helgeson, D. H. Kirkham and G. C. Flowers, Am. J. Sci., 1981, 281, 1249–1516 CrossRef CAS.
- R. M. Supkowski and W. D. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- J. I. Kim, R. Klenze, H. Wimmer, W. Runde and W. Hauser, J. Alloys Compd., 1994, 213/214, 333–340 CrossRef CAS.
- G. Plancque, V. Moulin, P. Toulhoat and C. Moulin, Anal. Chim. Acta, 2003, 478, 11–22 CrossRef CAS.
- K. J. Powell and L. D. Pettit, The IUPAC Stability Constants Database, Academic software, York, UK, 2005 Search PubMed.
- G. A. Gamov, K. V. Grazhdan, M. A. Gavrilova, S. V. Dushina, V. A. Sharnin and A. Baranski, Russ. J. Phys. Chem. A, 2013, 87, 967–972 CrossRef CAS.
- R. E. Mesmer, W. L. Marshall, D. A. Palmer, J. M. Simonson and H. F. Holmes, J. Solution Chem., 1988, 17, 699–718 CrossRef CAS.
- Z. F. Gesse, G. I. Repkin, V. A. Isaeva and V. A. Sharnin, J. Therm. Anal. Calorim., 2012, 110, 1457–1462 CrossRef CAS.
- M. Kakihana, T. Nagumo, M. Okamoto and H. Kakihana, J. Phys. Chem., 1987, 91, 6128–6136 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- I. Bertini, C. Luchinat and G. Parigi, in Current methods in inorganic chemistry, Elsevier, Amsterdam, 2001, vol. 2 Search PubMed.
- E. W. Stout and H. S. Gutowsky, J. Magn. Reson., 1976, 24, 389–398 CAS.
- X. Y. Chen, G. S. Goff, W. C. Ewing, B. L. Scott and W. Runde, Inorg. Chem., 2012, 51, 13254–13263 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00440j |
| This journal is © The Royal Society of Chemistry 2014 |