 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiplying the electron storage capacity of a bis-tetrazine pincer ligand†
Christopher R.
Benson
a,
Alice K.
Hui
a,
Kumar
Parimal
a,
Brian J.
Cook
a,
Chun-Hsing
Chen
a,
Richard L.
Lord
*b,
Amar H.
Flood
*a and
Kenneth G.
Caulton
*a
aIndiana University, Department of Chemistry, 800 East Kirkwood Avenue, Bloomington, IN, USA. E-mail: caulton@indiana.edu; Fax: +1 812-855-8300; Tel: +1 812-855-4798
bGrand Valley State University, Department of Chemistry, Allendale, MI, USA
First published on 13th February 2014
Abstract
An unexpected doubling in redox storage emerging from a new pincer ligand upon bis-ligation of iron(II) is described. When tetrazine arms are present at the two ortho positions of pyridine, the resulting bis-tetrazinyl pyridine (btzp) pincer ligand displays a single one-electron reduction at ca. −0.85 V vs. Ag/AgCl. Complexation to iron, giving the cation Fe(btzp)22+, shows no oxidation but four reduction waves in cyclic voltammetry instead of the two expected for the two constituent ligands. Mossbauer, X-ray diffraction and NMR studies show the iron species to contain low spin Fe(II), but with evidence of back donation from iron to the pincer ligands. CV and UV-Vis spectroelectrochemistry, as well as titration studies as monitored by CV, electronic spectra and EPR reveal the chemical reversibility of forming the reduced species. DFT and EPR studies show varying degrees of delocalization of unpaired spin in different species, including that of a btzp−1 radical anion, partnered with various cations.
Introduction
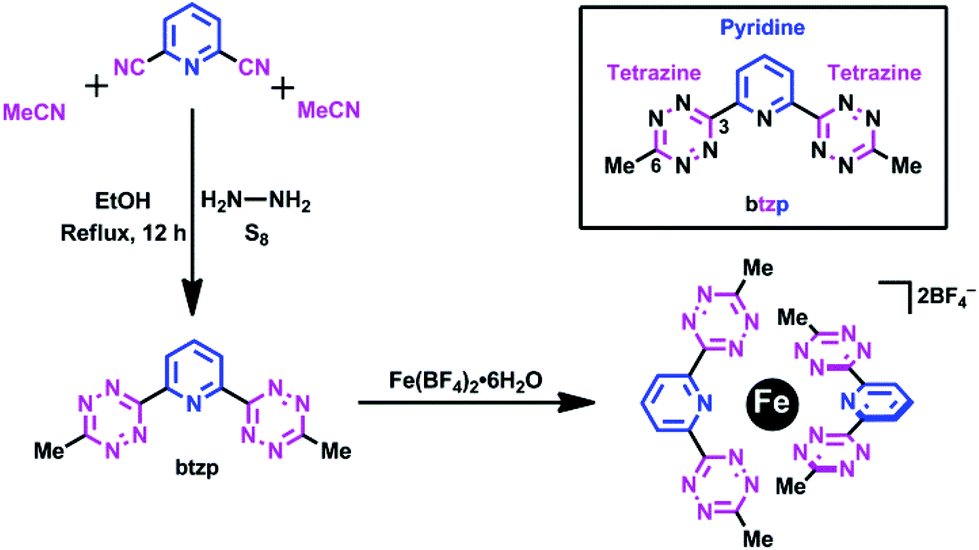
Redox equivalents delivered into the manifold of orbitals defined within transition metal complexes have been utilized in myriad strategies for chemical transformations.1–4 Recent attention has turned from metal-centered redox processes to those centered on the attendant ligands (Fig. 1). This interest emerged out of studies seeking to clarify the ambiguity surrounding the site of reduction or oxidation, in complexes characterized by “redox non-innocent” chemistry that could be ascribed to either the metal or the ligand, particularly when using ligands derived from quinones.5–7 As a sign of the times, this question has evolved to the point where the unusual quality of a ligand's non-innocence is now replaced by the deliberate design8,9 of ligands that will predictably display multiple redox states. The design of molecular switches10–12 or electrocatalytic processes13–15 involving ligand-derived processes benefits from this evolution in our understanding. We have also reviewed recently the exceptional potential of polyazine aromatics to accept electrons and hence to serve as components of a new class of redox active ligands.16 Thus, opportunities exist to explore novel variations on known, and unknown, motifs that could serve as the origin of a ligand's redox activity. To this end, we present the synthesis, electronic properties and representative metal complexation character of a C2-symmetric, pincer-type terdentate ligand where a central pyridine is flanked by two redox-active and highly π-acidic tetrazines.17 Unexpectedly, from one ligand with one reduction, we add a coordinating metal ion and multiply the redox activity twofold. This type of behavior, where the ligand's ability to accept electron doubles, is unprecedented in the behavior of complexes of polyazine ligands. | ||
| Fig. 1 Some redox-active ligands: (a) benzene dithiolate,6 (b) pyridine bisdiimine,1 (c) bispyridyltetrazine,17 and (d) diazinyl pyridine.8 | ||
In coordination chemistry, Kaim17 has explored the potential of 1,2,4,5 tetrazines (Fig. 1c) when located between two pyridines to define a redox-active ligand that can couple two metal centers together via its accessible lowest unoccupied molecular orbital (LUMO) giving rise to Class II through to Class III mixed-valent character.18 Recently, some of us have employed this ligand's redox activity to drive molecular switching in pseudorotaxanes.10–12 The LUMO which gives rise to these properties is localized on the nitrogens of the central tetrazine ring with negligible orbital amplitude at the bridgehead carbons of the π* orbitals of the two pyridines. Thus, we wondered if this orbital situation could be engineered in reverse such that the pyridine serves as the bridge between two tetrazines (btzp, Scheme 1), which also offers the possibility of redox states changing by two electrons. The symmetrical terdentate ligand we designed represents the most expedient formulation of this idea.
 | ||
| Scheme 1 Synthetic approach. | ||
Tetrazine rings are synthesized from organonitriles (R–CN) in reaction with hydrazine, followed by oxidation, to form the 1,2,4,5-tetrazine core disubstituted at the 3 and 6 positions. With few exceptions, these tetrazines are prepared as coordinating ligands in a symmetrical manner. The recent emergence of metal-free click chemistry has seen an exploration19 of non-symmetrical tetrazines, which is the class of substitution needed for btzp. With the exception of one multistep synthesis reported in the patent literature,20 these tetrazine-forming reactions are statistical in nature. We reasoned that our desired non-symmetrically substituted tetrazine could subsequently be separated from symmetrical co-products given the large differences in the structure between btzp and the other two by-products: dimethyl tetrazine and a pyridyl-tetrazine polymer.
For comparison, a study of terpy analogs (Fig. 1d) of M(pincer)22+ complexes (Fig. 1, M = Fe and Co), where the outer arms contained 2 or 3 nitrogens, has been reported.8 This revealed low spin behavior for iron, and Fe–N bond lengths were interpreted as showing weaker metal binding to polyazine rings than to pyridine. In marked contrast to our results with btpz, the reported iron complexes all showed one oxidation somewhere between +1.5 and +2.0 V vs. SCE. Multiple reductions were observed for the iron complexes between −0.5 and −1.5 V vs. SCE, and attributed to the usual stabilizations of ligand-centered radical anions that occur upon coordination to cationic metals, but these reduction products were not further characterized.
Herein we report the successful synthesis and iron(II) coordination chemistry of the redox-active bis-tetrazinyl pyridine ligand, “btzp” (Scheme 1). We verified that the highly π-acidic character of the ligand originates from the low-lying LUMO delocalized across the tetrazines and the redox properties of the ligand are explored. Complexation with iron(II) leads to multiple redox states becoming accessible with the iron(II) oxidation state being highly stabilized in this environment; the iron's oxidation was shifted by more than ∼900 mV to beyond +2.2 V vs. Ag/AgCl compared to an analogous bis-terpyridine complex. This work summarizes our results to establish the veracity of the novel doubling in redox storage displayed by a bis-tetrazine pincer ligand that shows a single electron process as a free ligand, and then gains access to a second reduction upon complexation.
Results
Synthesis of the bis-tetrazine ligand
Generation of this pincer proceeds (Scheme 1) from the addition of hydrazine to 2,6-dicyano pyridine, which can couple with acetonitrile in the presence of sulfur.19 Under the conditions of the reaction, we were able to minimize the generation of pyridyl-tetrazine polymers through the addition of a large excess of acetonitrile. Residual side products were removed by column chromatography with pyridyl-tetrazine polymer binding to the column gel, and the highly volatile dimethyl tetrazine subliming during the evaporation of solvent under vacuum or during chromatography. This molecule has a lilac or bright fuchsia color typical of tetrazines, due to an n→π* transition, and showing the small HOMO–LUMO gap in the molecule (contrast colorless 2,2′-terpyridyl). The molecule shows a 1H NMR singlet for the ring methyls, together with an AX2 pattern in the aromatic region for the symmetrically substituted pyridyl.X-ray diffraction from a single crystal of the free ligand provided confirmation of the structure (Fig. SI-1†) in the solid state. The three rings are co-planar with deviations out of plane attributed to crystal packing forces. The solid-state packing shows the formation of anti-parallel rows of the btzp ligands with close intermolecular contacts made by virtue of multiple CH⋯N hydrogen bonds. Each btzp ligand is also engaged in π stacking and while the ligands are offset in a slip-stack manner, they are located approximately above and below each other consistent with stacking typical of the largely π-deficient character of the tetrazine rings.21
Synthesis and characterization of Fe(btzp)22+
Addition of [Fe(H2O)6](BF4)2 to btzp at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratio, each in acetonitrile, occurs with color change to dark blue within minutes at 25 °C to form a dark solid. This solid was purified by re-precipitation from acetonitrile with ether or benzene and was established to be [Fe(btzp)2](BF4)2 by a variety of characterization methods. Its 1H NMR spectrum shows a doublet (intensity 2) and a triplet (intensity 1) for the pyridyl hydrogens, as well as an intensity 6 signal for equivalent tetrazine methyls, all in a chemical shift range consistent with diamagnetism and proving twofold symmetry of two equivalent pincer ligands. Addition of free btzp to a solution of Fe(btzp)22+ shows separate 1H NMR signals for free and coordinated ligand protons, showing integrity, both thermodynamic and kinetic, of the complex. The 19F NMR spectrum shows only a singlet for free BF4−1, resolved into 1:4 intensities due to 10,11B isotope effect on chemical shift. The positive ion mass spectrum of this compound in MeCN shows Fe(btzp)22+ (m/z = 295) and also the ion pair Fe(btzp)2F+ (609, from F− capture from BF4−), but also the monocation Fe(btzp)2+ (590), a product of reduction during electrospray, which speaks for the ready reduction of the pincer ligands in this dication. The dication is unaffected by dissolved oxygen, even to the point of no broadening of the 1H NMR spectrum, and no EPR signal.
:2 mole ratio, each in acetonitrile, occurs with color change to dark blue within minutes at 25 °C to form a dark solid. This solid was purified by re-precipitation from acetonitrile with ether or benzene and was established to be [Fe(btzp)2](BF4)2 by a variety of characterization methods. Its 1H NMR spectrum shows a doublet (intensity 2) and a triplet (intensity 1) for the pyridyl hydrogens, as well as an intensity 6 signal for equivalent tetrazine methyls, all in a chemical shift range consistent with diamagnetism and proving twofold symmetry of two equivalent pincer ligands. Addition of free btzp to a solution of Fe(btzp)22+ shows separate 1H NMR signals for free and coordinated ligand protons, showing integrity, both thermodynamic and kinetic, of the complex. The 19F NMR spectrum shows only a singlet for free BF4−1, resolved into 1:4 intensities due to 10,11B isotope effect on chemical shift. The positive ion mass spectrum of this compound in MeCN shows Fe(btzp)22+ (m/z = 295) and also the ion pair Fe(btzp)2F+ (609, from F− capture from BF4−), but also the monocation Fe(btzp)2+ (590), a product of reduction during electrospray, which speaks for the ready reduction of the pincer ligands in this dication. The dication is unaffected by dissolved oxygen, even to the point of no broadening of the 1H NMR spectrum, and no EPR signal.
Crystals grown from MeCN–hexane were shown (Fig. 2) by single crystal X-ray diffraction to contain two pincer ligands per iron, with high crystallographic symmetry of the cation so that only one octant of the species is unique (i.e., Fe, one tetrazine and half of one pyridyl). Bond lengths of iron to N are longer by 0.04 Å to the tetrazine than to pyridyl, but all are short, consistent with the low spin state and hence empty σ*FeN orbitals of eg symmetry on an octahedron.
 | ||
| Fig. 2 ORTEP drawing (50% probabilities) of the non-hydrogen atoms of [Fe(btzp)2]2+ as its BF4− salt, showing selected atom labeling. Unlabeled atoms are carbon or (shaded) nitrogen. The dication has an S4 axis along the N1/Fe1 line. Selected structural parameters: Fe1–N1, 1.900(11) Å; Fe1–N4, 1.947(7); N4–Fe1–N4*, 91.38(6)°; N4–Fe1–N4′, 162.2(4); N4–Fe1–N1′, 98.9(2); N4–Fe1–N1, 81.1(2). | ||
The oxidation state and the spin state of the iron center within the solid state structure of the [Fe(btzp)2](BF4)2·MeCN complex were determined using Mössbauer spectroscopy22 (Fig. SI-3†). A reproducible Mössbauer spectrum of the bis-ligand complex with an isomer shift of δ = 0.18 mm s−1 and quadrupole splitting of ΔEQ = 1.32 (mm s−1) are typical of low spin iron(II) complexes.23–25
The electronic structure of the ligand and its complex was further investigated using UV-Vis spectroscopy (Fig. 3). The free ligand shows a visible absorption at 536 nm consistent with the characteristic pink color of tetrazine-containing compounds with similarly modest absorptivity, ε = 700 M−1 cm−1. Addition of up to 0.5 equivalent of iron(II) salt in MeCN led to the appearance (Fig. 3) of two new absorptions at 584 nm (7000 M−1 cm−1) and 419 nm (4700 M−1 cm−1) that are tentatively assigned to metal-to-ligand charge-transfer (MLCT) transitions largely in line with other Fe(II) complexes of polypyridyl-derived ligands.26,27 The lowest energy electronic transition corresponds to a band gap of 2.1 eV. Assuming a formal MLCT state with modest mixing between the metal-centered and the ligand-centered HOMO and LUMO respectively, this optical energy gap provides a reasonable estimate of the voltage difference between the first reduction and first oxidation of the complex in solution.
 | ||
| Fig. 3 UV-Vis titration to form Fe(btzp)22+ in solution (purple trace) from the free ligand (pink trace), demonstrating the emergence of both n→π* and d→π* transitions in the complex. | ||
Solution characterization of the ligand and its iron(II) complex
To better understand the mechanism of complex formation, an 1H NMR titration (Fig. 4) was carried out in which aliquots of iron(II) tetrafluoroborate hexahydrate were added to a 3 mM solution of btzp in CD3CN. Titration with aliquots of iron(II) salt resulted in the complete formation of the Fe(btzp)22+ complex after addition of 0.5 equivalent with no residual peaks associated with the remnant ligand, matching a 2:1 ligand:metal ratio. The shifts in the pyridyl peaks by 0.59 and 0.45 ppm for H4 and H3,5 respectively, are consistent with prior studies on Fe(II) coordination of terpyridine and its analog,28,29 and indicative of proton deshielding on account of the strong σ-donor properties of the pyridine. Shifts of protons in similar structures have been seen with methylene protons in the terdentate ligand on 2,6-bis(1,2,3-triazol-4-yl)pyridine28 as well as on the terminal methyl protons of the Cu(I) complex of 3,6-bis-pyridyl tetrazine.11
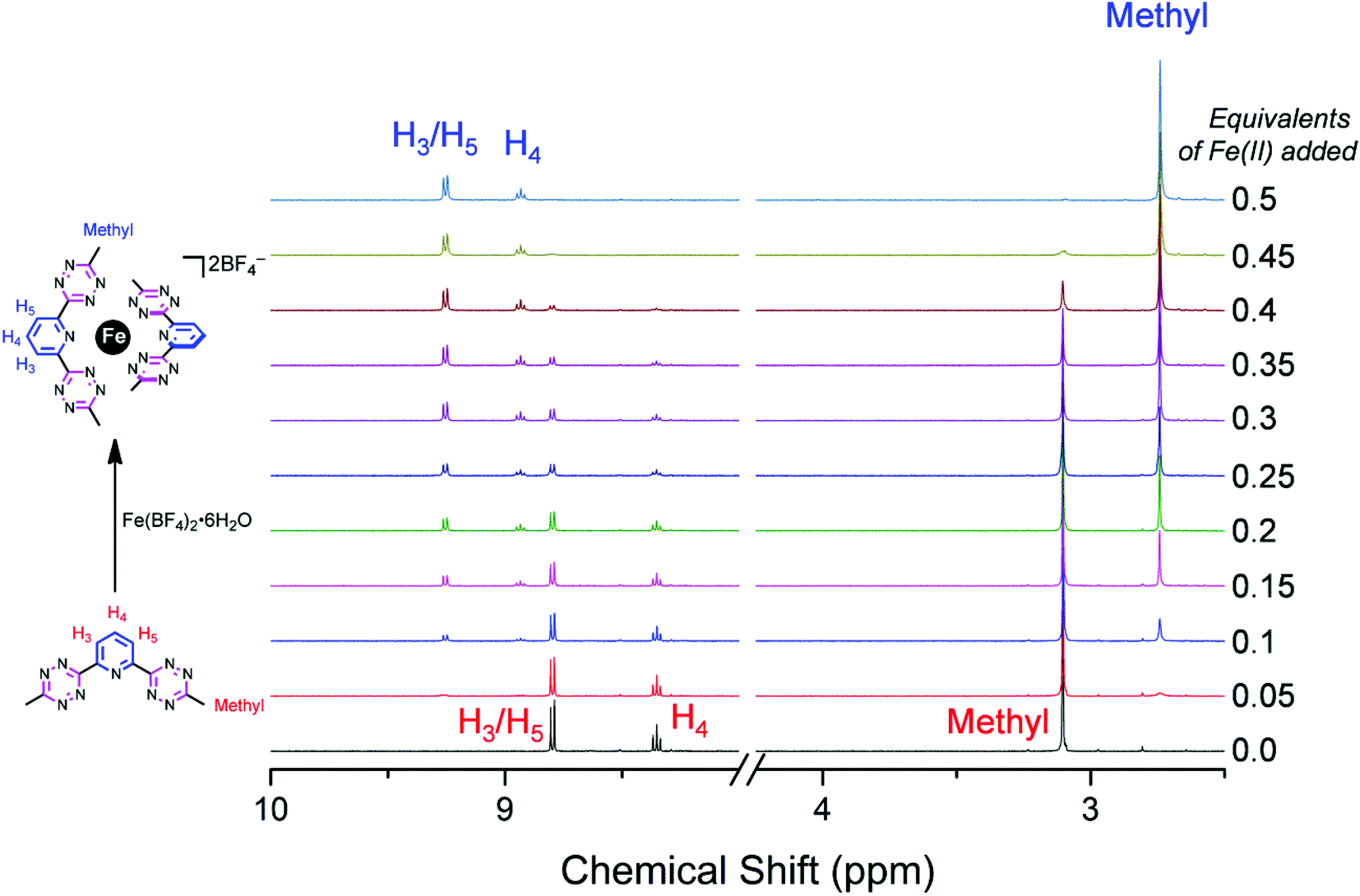 | ||
| Fig. 4 1H NMR titration to form Fe(btzp)22+ in solution beginning from a solution of btzp. Simultaneous appearance of product peaks with disappearance of ligand peaks is indicative of tight binding conditions with slow chemical exchange on the time scale of the experiment. | ||
The simultaneous decrease in intensity of peaks for the free ligand and growth of peaks associated with the complex indicates tight binding conditions, resulting in a slow exchange process in which the intermediary solutions are composed of clearly defined species (either free ligands or ML2 complexes) with no detectable population of intermediates (e.g., complexes with metals on the exo-directed nitrogen atoms on the tetrazine moieties). Consequently, the structure observed in the solid state exists as the dominant species in CD3CN solution under these conditions. This case is further bolstered by a CV taken of the titration solution at the end of the NMR experiment (Fig. 5) which shows an identical number and relative intensity of peaks as does the CV of the synthesized Fe(btzp)22+ complex (vide infra).
 | ||
| Fig. 5 CV of the 0.5 equivalent NMR end point solution (blue) showing identical features to the voltammetry profile of synthesized Fe(btzp)22+ (inset, purple). Upper trace involves a different iron concentration. | ||
Electron transfer reactivity of btzp and its Fe(II) complex
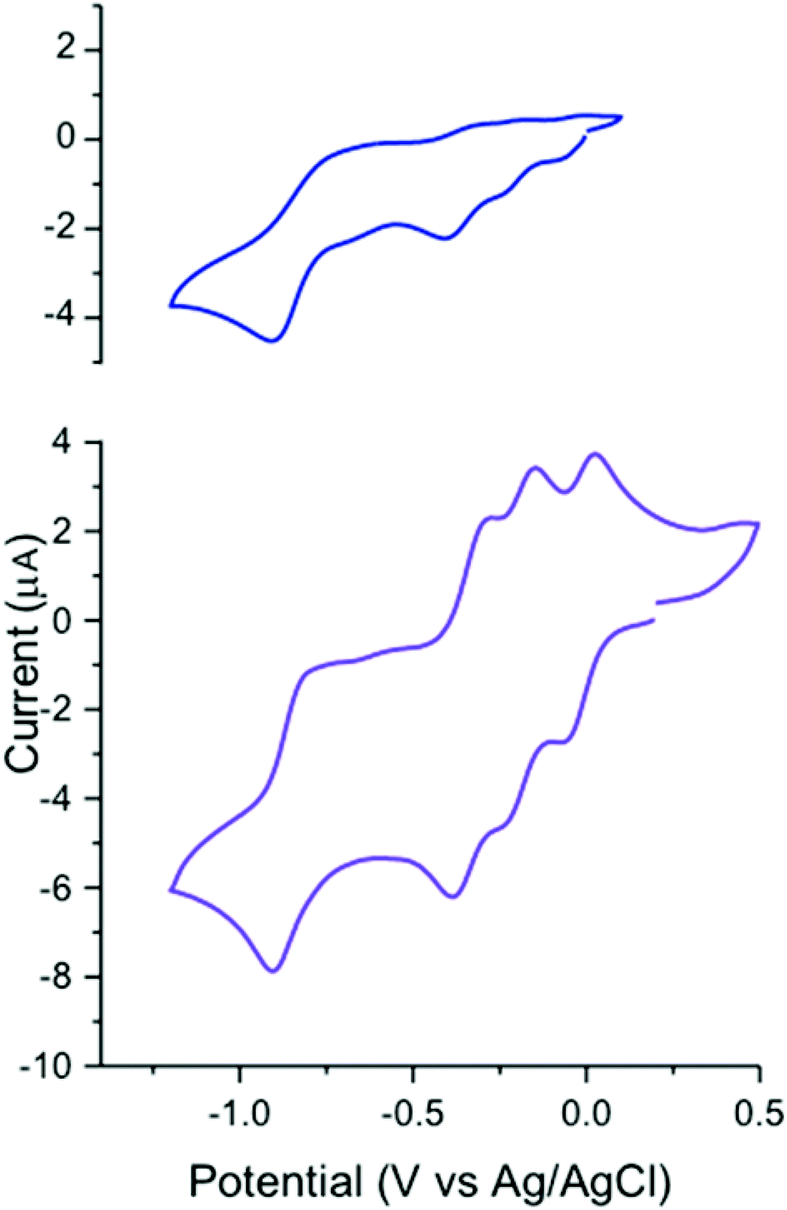
Free btzp (Fig. 6) exhibits a single sharply-defined redox process with E1/2 = –850 mV vs. Ag/AgCl in MeCN, a value consistent with the redox chemistry of uncoordinated monomeric tetrazines.17 Peak-to-peak potential separation (ΔE) was found to be 108 mV demonstrating reversibility, and with comparable ipc and ipa. Although there are two tetrazine moieties per btzp ligand that could take up one electron each, coulometry (see ESI†) shows that the reduction at −850 mV represents only a single electron process. No oxidation peaks were observed in the solvent window. | ||
| Fig. 6 Cyclic voltammetry on (a) free btzp ligand and (b) Fe(btzp)22+ complex, showing near-ideal reversibility. Black arrow indicates the direction of sweep. | ||
Our previous work with tetrazine ligands10–12 would predict that addition of an electropositive transition metal center would simply cause anodic shifts of the free ligand redox potential. Surprisingly, Fe(btzp)22+ shows (Fig. 6) four reversible redox couples with half-wave potentials at E1/2 = −25 mV, −190 mV, and −335 mV and −850 mV with no other processes, oxidative or reductive, in the solvent window. This latter observation is consistent with the optical band gap of 2.1 eV, i.e., that given the position of the first reduction of the complex near 0 V, the Fe(II/III) oxidation process would be anticipated to be more positive than 2.1 V. Recent work8 on similar disubstituted pyridine ligands also bolsters this claim, finding that increased nitrogen character on the flanking azine heterocycles drives the Fe(II)/Fe(III) couple towards increasingly anodic potentials. In contrast, Fe(terpy)22+ shows the oxidation to the ferric ion at ∼+1.3 V, consistent with the significantly poorer σ donor/strong π acceptor properties of tetrazines and illustrating their inability to stabilize the Fe(III) oxidation state. The open circuit potential measured for a sample of Fe(btzp)22+ from isolated solid was +0.004 V, an observation consistent with all peaks observed during the cathodic sweep being reductions from species Fe(btzp)22+.
The complexity of the CV of Fe(btzp)22+ was unexpected, motivating additional experiments. To discount the possibility that the observed results were the consequence of non-redox equilibration processes (e.g., ligand substitution by solvent) intrinsic to Fe(btzp)22+ in MeCN, variable scan rates between 5 V s−1 and 50 mV s−1 were recorded, revealing no unexpected change in relative peak heights or peak positions.
To estimate the number of electrons transferred with each redox process, a CV was recorded of a solution containing an equimolar mixture of Fe(btzp)22+ and its dimethyl-terpyridine homolog, Fe(Me2terpy)22+ (Fig. S8†). These structurally similar complexes should have similar diffusion coefficients, thereby allowing us to directly compare the peak current for the characteristic one electron Fe(II)/Fe(III) couple in Fe(terpy)22+ to the observed processes within the tetrazine complex. All peak currents appeared to be roughly equal, suggesting that each of the four waves in the Fe(btzp)22+ voltammogram is a single electron couple. Titration of Fe(BF4)2·6H2O into a solution of btzp, monitored by CV, tied all the solution characterization data together (Fig. S9†), illustrating both the emergence of the four wave CV profile of Fe(btzp)22+ and its facile formation in solution.
Spectra of the reduced complexes and reversibility of the redox changes
UV-Vis spectroelectrochemistry (UV-Vis SEC) was carried out on a solution of Fe(btzp)22+ (Fig. 7) to characterize the electronic spectral features of the complex through the first three reduction peaks shown in the CV in Fig. 5. Upon reduction, peaks initially present at 418 and 580 nm showed a red shift. Additionally, the intensity of the peak originating at 418 nm increased while the peak at 580 nm decreased. Titration with progressive aliquots of cobaltocene (Cp2Co) (Fig. 8) revealed changes to visible peaks consistent with the data generated by UV-Vis SEC, with good correspondence between peak positions using both methods. An additional very broad peak was observed (see ESI†) stretching across the NIR into the IR band, growing in intensity with the addition of equivalents of Cp2Co, although the nature of this transition remains unknown. The solution itself underwent a progressive color change with the addition of equivalents of Cp2Co, going from a dull purple in the unreduced state to green, orange and ultimately a brown or tan color at higher equivalents. A separate titration with decamethylferrocene, which has only sufficient reducing power to obtain the doubly reduced product Fe(btzp)20, duplicates the Fe(btzp)20 spectra collected using UV-Vis SEC and Cp2Co.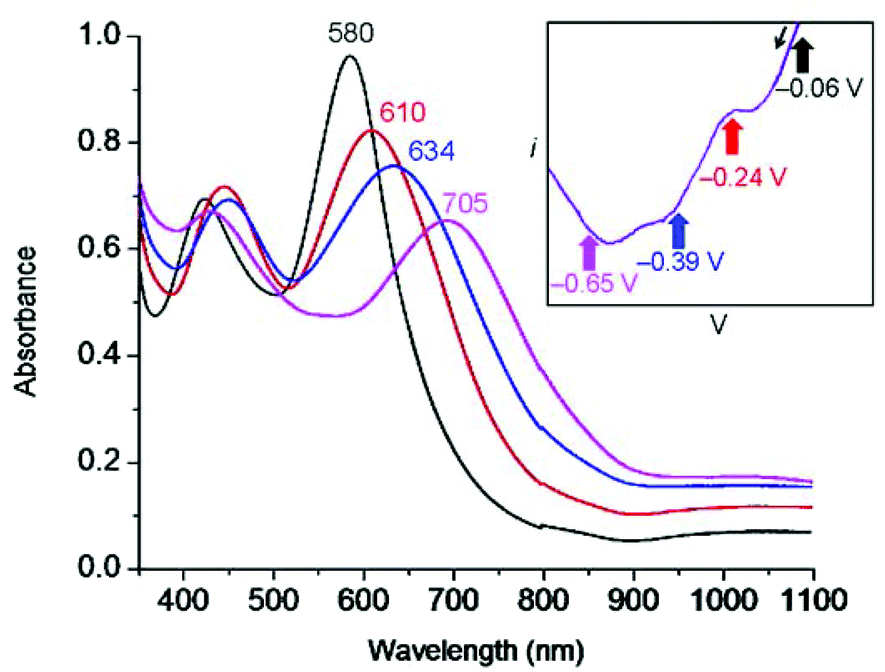 | ||
| Fig. 7 UV/Vis SEC of Fe(btzp)22+ showing the dication and its three sequentially reduced products at potentials marked on the cathodic sweep of the slow scan-rate CV (0.2 mV s−1) shown in the inset (black = 0 V, red = −0.24 V, blue = −0.39 V, purple = −0.65 V). SEC experiment was carried out on a 1.18 mM solution of Fe(bisTzP)22+ in a 1 mm thin-layer SEC cell with a platinum mesh working electrode, a platinum wire counter electrode, and a Ag/AgCl pseudoreference electrode. | ||
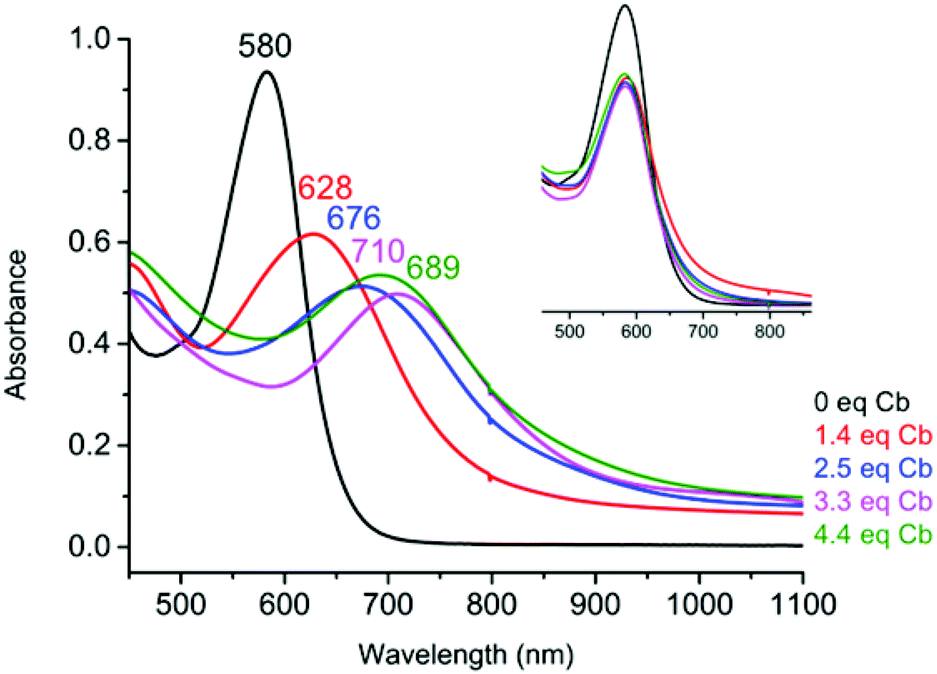 | ||
| Fig. 8 UV/Vis spectra after chemical reduction of Fe(btzp)22+ with successive equivalents of Cp2Co. Inset shows dilution-corrected spectra after [NO]PF6 reoxidation of each of the four reduction experiments. | ||
Each reduction product generated in the Cp2Co titration was reoxidized (inset, Fig. 8) with an excess of an acetonitrile solution of nitrosonium tetrafluoroborate to determine the stability of the reduced products and the reversibility of the chemical reduction processes. In addition to a visible return of color to that of Fe(btzp)22+, the spectra for these samples were collected for comparison. The original UV-Vis peak positions are recovered in all reduction products up to ∼4 equivalents of Cp2Co. Correcting peak heights for dilution, reoxidation of each sample shows that the spectrum for each reductive process returns approximately 90% of original peak intensity. This suggests that the reduction products of Fe(btzp)22+ in any of its reduced forms are persistent on the timescale of these experiments (approximately 15 min to completion of the spectra) and that reoxidation is reversible.
EPR characterization of the location of the electron upon reduction
Equimolar Cp2Co upon reduction reacts with btzp in MeCN in time of mixing to give Cp2Co+ (verified by 1H NMR spectroscopy) but shows no 1H NMR signal for the radical anion pincer btzp−1 in the chemical shift range +200 to −100 ppm. The resulting radical produced by cobaltocene reduction was shown to have an approximate half-life of 10 minutes in MeCN and 30 minutes in a 0.1 M solution of TBABF4 in MeCN. The EPR spectrum of this solution (Fig. 9) taken immediately after production, shows a well resolved 9 line pattern with g = 2.0040 due to four nitrogens coupling with AN = 5.1 G; this value is comparable to that observed in the radical anion of symmetrical 1,2,4,5 tetrazine, C4N2H2, where all four symmetry equivalent nitrogens couple equally.30 Additional coupling to three I = 1/2 particles with AH = 1.3 G is resolved in the spectrum of [Cp2Co][btzp] and can be accurately simulated. For comparison, reduction of btzp in THF by Na, Na(anthracene), K mirror, KC8, or Mg all show a structured pattern at essentially the same g value and with the analogous hyperfine structure and AN values, but not as well resolved (broader lines) as with the non-interacting Cp2Co+ cation. We attribute the broadening to a combination of dynamic effects and possible quadrupolar broadening by each of these cations coordinated to the btzp radical anion. EPR simulations show that coupling to eight nitrogens of two tetrazines would give line intensities observably different from the experiment, so the assignment of detectable coupling to four tetrazine nitrogens is most consistent with observation. No four nitrogens in (all planar) btzp are symmetry equivalent, so we did EPR simulations to establish how different the AN values could be and still have the differences unresolved experimentally. This showed that a 15% difference would observably change the spectra. Simulations showed that additional coupling to a pair of nitrogens larger than 0.8 G would be resolved in our spectra. Likewise, coupling to the unique pyridyl nitrogen larger than 0.8 G would be resolved in our spectra. | ||
| Fig. 9 Observed (red) and simulated (blue) X-band EPR spectrum of [Cp2Co][btzp] in MeCN at 25 °C. EPR parameters, X-band; microwave frequency, 9.855; microwave power 2.002 mW; modulation amplitude, 0.50 G; time constant, 20.960 ms; scan time, 42 s. | ||
The question unresolved by these observations is whether the four nitrogens are in one tetrazine arm or are two nitrogens (only) in each of the two arms. This is central to learning whether the spin is delocalized over two arms, or localized in one. If our assignment of the smaller hyperfine component to only one methyl group is correct, this argues against delocalization over both tetrazines; such delocalization would show hyperfine to six methyl protons. To resolve this, we have employed the radical with only one arm and formed [Cp2Co][3-pyridyl 6-methyl tetrazine] (Fig. S10†), and recorded its EPR spectrum. This also shows a nine line pattern, each line of which shows coupling to three I = 1/2 nuclear spins. In short, the EPR spectrum of [Cp2Co][3-pyridyl 6-methyl tetrazine] is very similar to that of btzp−1, which would indicate spin localization in btzp−1. We additionally tested our assignment of the three I = 1/2 hyperfine coupling to one methyl group by forming [Cp2Co][1,2,4,5-dimethyltetrazine, C4N2Me2], (Fig. 10). This highly structured spectrum is well simulated by coupling to four 14N spins (AN = 5.1 G) and six methyl protons (AH = 1.5 G). Overall, the hyperfine assignments are strongly supported by these comparison radical spectra, and the observed number of hyperfine lines strongly supports the conclusion that [Cp2Co][btzp] radical is localized within one tetrazine arm, with no resolvable participation of the pyridyl nitrogen in the SOMO.
 | ||
| Fig. 10 X-band EPR spectrum of [Cp2Co][dimethyl-1,2,4,5-tetrazine] in MeCN at 25 °C. EPR parameters, see Fig. 11. | ||
Having established that the spin in the radical anion in numerous (cation)[btzp] species in the polar and Lewis basic solvents THF or MeCN is localized in one tetrazine arm, the remaining question is whether localization originates in symmetry-breaking of the structure by location of the cation, or whether localization is intrinsic to the unperturbed anion. While nesting of Na+, K+ or Mg2+ in the center of the three inwardly directed nitrogens of the pincer moiety is possible, it should not break twofold symmetry equivalence of the two tetrazine arms, and structures of many arene/alkali metals ion pairs show that the electrophilic cation often sits above the arene ring; given that the tetrazine is the reduced arene here, this would break the symmetry equivalence of the two arms in (cation)[btzp]. We cannot establish this structural feature based on the available experimental evidence.
DFT characterization of btzp−1 and its interaction with counterions
We first carried out a survey of possible structures of the isolated anion btzp−1 using DFT methods (Table 1). We find that the inclusion of implicit solvation strongly influences the most stable structure of this anion.31 Without consideration of solvent effects, a delocalized (twofold symmetric) state is the minimum; all attempts to converge to a localized state through initial geometries and wavefunction manipulation collapse back to the delocalized state with two half-reduced tetrazine arms. However, when the continuum solvent is included during the geometry optimization, the delocalized twofold symmetric structure is found to be a transition state (TS). Displacement away from this TS along the mode with an imaginary frequency in either direction leads to a state with a localized SOMO on one of the two tetrazine arms (see Fig. 11). This localized radical anion is the lowest energy minimum we could locate, but lies less than 1 kcal mol−1 lower in energy than the delocalized anion (a transition state). Such a low barrier would suggest rapid interconversion between the localized anions, in contrast to the multiplicity of the observed nitrogen hyperfine structure. Thus, the cation–anion interactions that are not included in these calculations are likely significant. The SOMO of the localized anion radical shows coplanarity of the three rings, but negligible contribution from the pyridyl and neutral tetrazine rings which is consistent with the experimental EPR data. Fig. 11 also shows that the dianion btzp2− is twofold symmetric, but the spin is still primarily at the tetrazines. | ||
| Fig. 11 Spin density isosurface plots (0.002 au) of doublet bztp1− (left) and triplet bztp2− (right). | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Gas-phase | Solution-phase | |||||||
| bztp0 singlet | bztp1− doublet | bztp2− singlet | bztp2− triplet | bztp0 singlet | bztp1− doublet | bztp2− singlet | bztp2− triplet | |
| Npy–Cpy | 1.337 | 1.341 | 1.352 | 1.349 | 1.338 | 1.340–1.343 | 1.346 | 1.344 |
| Cpy–Ctet | 1.489 | 1.497 | 1.503 | 1.495 | 1.490 | 1.501–1.487 | 1.510 | 1.506 |
| Ctet–N1 | 1.348 | 1.344 | 1.345 | 1.347 | 1.344 | 1.339–1.348 | 1.334 | 1.338 |
| Ctet–N3 | 1.343 | 1.342 | 1.341 | 1.346 | 1.347 | 1.336–1.348 | 1.336 | 1.338 |
| N1–N2 | 1.317 | 1.362 | 1.404 | 1.405 | 1.321 | 1.397–1.327 | 1.404 | 1.402 |
| N3–N4 | 1.320 | 1.347 | 1.375 | 1.399 | 1.315 | 1.384–1.317 | 1.371 | 1.396 |
| N1–Ctet–Cpy–Npy | 13.5 | 0.0 | 0.0 | 19.4 | 0.0 | 0.0–0.0 | 0.0 | 0.3 |

DFT calculations were carried out to see whether alkali metal cations are consistent with the localization observed experimentally in bptz−1. Two basic binding motifs were tested as starting geometries for the computational optimization of Na(bztp) with a continuum solvation model: (i) with the metal η6-bound to one of the tetrazine rings and (ii) with the metal κ3-bound to two tetrazine and one pyridine nitrogens. For the Na complex, geometry optimizations from different starting geometries converged to two different minima. The localized structure shown in Fig. S6 in ESI† has Na+ η2-bound to the reduced tetrazine ring of bztp (Na–N = 2.32 and 2.44 Å). The Na–Npyr separation is large at 3.97 Å, ruling out any interaction between Na+ and the other rings. A second structure has κ3 connectivity, but the metal ion binds to only one N of each tetrazine. Expanding our study to Li and K revealed (Fig. 12) minima for both η2 and κ3, and the κ3 was more stable by 4–10 kcal mol−1 for all three alkali metals. Significantly, all three κ3 structures have a highly asymmetric unpaired spin (Fig. 12), located in the tetrazine with the shorter M/N distances. For completeness, we also minimized a κ3 structure (Fig. 13) for reduced species Mg(btzp)+, to show the effect of a small dication. This too has Mg2+ closer to the tetrazine which carries the majority of the spin density, so is wholly consistent with the picture of asymmetric (localized) radical character, but κ3 connectivity.
 | ||
| Fig. 12 Optimized geometries with short M–N bond lengths (left) and spin density isosurface plots (right, 0.002 au) for the [M(bztp)]0 species (M = Li (top), Na (middle), K (bottom)). | ||
 | ||
| Fig. 13 Optimized geometry with short Mg–N bond lengths in angstrom (left) and the spin density isosurface plot of [Mg(bztp)]+ (right, 0.002 au). | ||
Taken together, these results show that none of these metals is “too large” to fit in the plane of the pincer rings (note the distortion of the MNCCN rings at left in Fig. 12 as M gets larger), so any localization of unpaired spin in a κ3 structure is due to intrinsic electronic preferences. Also based on the bond distances in the calculated species, metal radii increase in the order Li+ < Mg2+ < Na+ < K+.
In summary, we find, with DFT, a localized structure that agrees with experiment for M(btzp)−1.
Na[dimethyltetrazine] structure and spin density
How can chemically inequivalent nitrogens fail to show a resolvably distinct hyperfine coupling in btzp−1? Our EPR hyperfine multiplicity establishes that Na[dimethyltetrazine] also has equal coupling to all four ring nitrogens. DFT geometry optimizations of neutral doublet Na[dimethyltetrazine] are informative of structural preferences in the absence of a second tetrazine and the donor pyridyl. In the gas-phase two minima were found, one (Fig. 14 left) where the Na+ remains bound to the face of the ring (which is highly ruffled – Na/N distances of 2.42 twice and 2.60 Å twice) and the other (Fig. 14 right) where the Na+ is η2 bound to two adjacent nitrogens in the ring. The ruffled structure is surely symptomatic of loss of aromaticity upon reduction. The η2 bound form is more stable by over 30 kcal mol−1. During solution-phase optimizations (continuum model, so no explicit solvent molecules are binding to sodium), both starting structures (η2 and η6) converge to the η2 form. Fig. 15 shows the spin density of the solution-phase η2 structure, showing that all four nitrogens retain nearly equal spin; in spite of symmetry-breaking by sodium, the Mulliken charge on each of the four nitrogens is identical at −0.4. These agree with EPR spectral evidence for delocalization of spin to all four nitrogens in this species. Interestingly, optimizations (from both starting geometries) of the cation singlet species Na(btzp)+ yield only the η2 bound form so we can confidently say sodium has a preference to bind to the harder Lewis base, nitrogen, rather than the softer filled π system of the arene ring (i.e., η6). A sodium cation here behaves like a hard Lewis acid, even though it is also known32,33 to interact with the π system of purely hydrocarbon radicals and non-radicals when an alternative nitrogen donor is absent. | ||
| Fig. 14 Two minimum energy structures for Na[dimethyltetrazine] from gas phase calculations. | ||
 | ||
| Fig. 15 Spin densities from solution phase minimum energy structure of Na[dimethyltetrazine], showing equal spin over all four nitrogens (i.e., centrosymmetric), in spite of asymmetric location of the Na+. | ||
Bond lengths in the η2-bound form of Na[dimethyltetrazine] are C2v symmetric. In the solution-phase structure, the Na–N bond lengths are 2.36 Å; the N–N for those bound to Na is 1.40 Å while those on the opposite side of the ring have a N–N distance of 1.40 Å. This would suggest that the presence of a metal cation has relatively little impact on the ligand geometry but the C–N bond lengths do show a difference of 0.01 Å. The C–N bonds on the side with Na bound are 1.340 vs. 1.331 Å for those on the opposite side of the ring.
EPR characterization of Fe(btzp)2+
Several approaches were employed to further investigate the redox properties of the Fe(btzp)22+ and prove that products are independent of the reducing agent. A reduction of Fe(btzp)22+ with excess zinc powder occurred over 48 hours at RT in MeCN to give an olive green solution from which the product was isolated by filtration and evaporation to dryness. The resulting Fe(btzp)2+ exhibited absorptions at 449 nm and 637 nm in MeCN, and shows (Fig. 16) an EPR spectrum at RT in MeCN with g = 1.998 and a multiplet pattern which is consistent with coupling to 4 nitrogens with AN = 6.0 G. The UV-Vis spectrum of the reduced solution was restored to peaks consistent with dicationic Fe(btzp)22+ by treatment with excess NO[PF6] in MeCN. | ||
| Fig. 16 X-band EPR spectrum of Fe(btzp)2+ in MeCN at 25 °C. EPR parameters, see Fig. 11. | ||
Following reduction of Fe(btzp)22+ with equimolar Cp2Co in CH3CN, then vacuum removal of solvent, and washing of the green solid with toluene to remove any residual Cp2Co, the 1H NMR spectrum of Fe(btzp)2+ was recorded in CD2Cl2. This showed signals at +19.9 and −3.3 ppm, intensity ∼2:1, together with the Cp2Co+ singlet at 5.75 ppm, in correct intensity for 1:1 reaction stoichiometry Co:Fe; the third btzp signal is presumably too broad and weak to be detected.
In three separate solutions, we reduced Fe(btzp)22+ with 1.0, 1.5 and 2.0 equivalents of outer sphere reductant Cp2Co in MeCN and recorded UV/Vis spectra, EPR, and then UV/Vis again (to confirm persistence of the product in solution). At the UV/Vis positions described above, we observed the growth (1 equiv.), and then progressive decrease in EPR intensity of the 9-line signal assigned to Fe(btzp)2+. In each case, the UV/Vis absorption was unchanged after the EPR collection, showing that Fe(btzp)2+ persists under these conditions for at least 2 h. Consistent with the DFT result that the neutral complex has two unpaired electrons, titration of the second electron into the dication diminishes the EPR intensity characteristic of the singly reduced (S = 1/2) species. This follows since triplet states generally relax fast and thus give no EPR signal until exceptionally low temperatures, if at all. Addition of oxidant [NO]PF6 to a reduced solution recovered Fe(btzp)22+, as judged by both spectroscopies, consistent with no loss of btzp from iron upon reduction. The g and AN values are both consistent with ligand centered reduction to give a species of formula Fe(btzp)2+.
Separately, a product with this same UV-Vis signature was produced by addition of increasing aliquots (up to 1:1) of (C5Me5)2Fe to a MeCN solution of Fe(btzp)22+. In contrast to the slower heterogeneous reduction with zinc powder, this solution-based outer sphere electron transfer reaction appears to proceed to completion within time of mixing, accompanied by the growth of a modest absorption at ∼750 nm, due to (C5Me5)2Fe+.34–36 Addition of a solution of NO[PF6] to the reduced species largely returns the original UV-Vis spectrum of Fe(btzp)22+, consistent with the reversibility of the reduction and the retention of the structure of unaltered btzp in the reduced product.
We wanted to evaluate whether a slender and polar molecule like MeCN would be the cause of localization of spin in one tetrazine in Fe(btzp)2+. This monocation was therefore synthesized using Cp2Co in MeCN, and then the resulting solid pumped in vacuum for several hours, to remove all MeCN. A sample of THF was then saturated with the resulting solid, to yield a faintly colored solution. The EPR spectrum of this sample at 25 °C showed a multiplet with the same g value and AN value as in MeCN, although the lines were broader and signal strength was inferior due to low solubility of the compound in this solvent. We conclude that the spin localization in Fe(btzp)2+ is the same in both MeCN and in THF.
Fe(btzp)2n+ characterization by DFT
Gas-phase geometry optimizations were performed on Fe(btzp)2n+ with n = 2, 1 and 0 in order to determine where electrons are added. Singlet, triplet, and quintet states were optimized for the 2+ species and the singlet was found to be lowest in energy (using B3LYP, a density functional known for overstabilizing high spin states),37 in agreement with the experiment. The triplet and quintet were approximately 7 and 12 kcal mol−1 higher in energy, respectively. The ground state singlet Fe(btzp)22+ is best described as FeII with two neutral bztp ligands. The HOMO is mostly due to tetrazine lone pairs pointing both towards and away from Fe, and the LUMO is composed of tetrazine π* orbitals. The monocation was optimized as a doublet starting from both the optimized dication structure and a localized structure with elongated N–N bonds in only one of the tetrazine arms. Both starting structures optimized to the structure with delocalized spin across the four tetrazines of both btzp ligands, as shown in Fig. 17. Attempts were made to optimize these structures with implicit solvation included, but a delocalized structure was obtained in both cases. Uncharged species Fe(btzp)20 was optimized as both a triplet and singlet; the triplet was lower in energy by ∼10 kcal mol−1. The additional electron is again spread over both ligands (Fig. 17), which is evidenced by the highly delocalized spin density for the neutral species. It is worth noting that the larger spin density lobes in the neutral are indicative of two spins. Thus, DFT predicts a redox sequence of [FeII(bztp)2]2+,S=0 → [FeII(bztp−1/2)2]1+,S=1/2 → [FeII(bztp−1)2]0,S=1.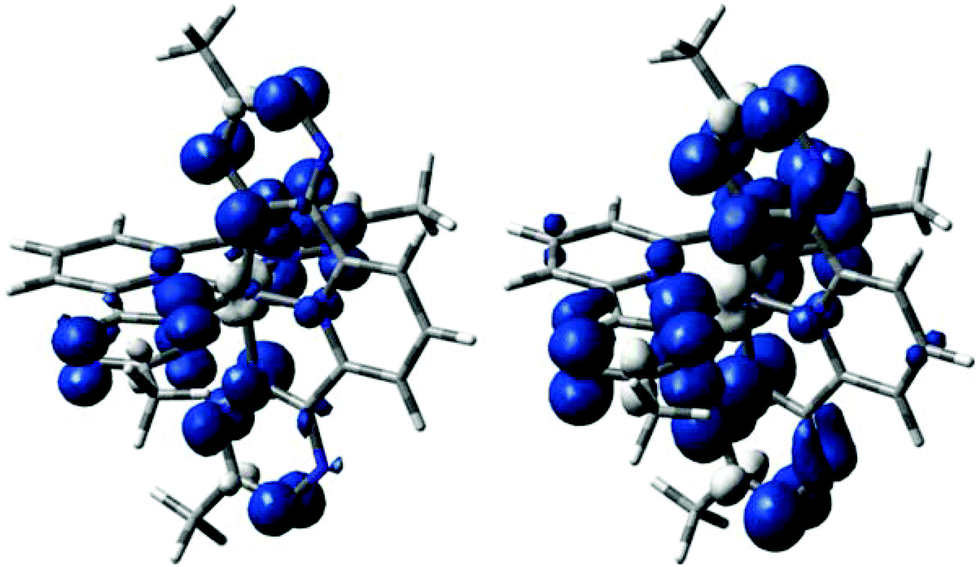 | ||
| Fig. 17 Spin density isosurface plots (0.002 au) for Fe(btzp)21+ (left) and Fe(btzp)2 (right) species. | ||
The metal–nitrogen, nitrogen-bridging carbon, and inter-ring C–C bond lengths are established38–40 to be quite diagnostic of redox loci in polypyridyl (and related) species. A summary of the most important bond lengths in Fe(btzp)2n+ is included in Table 2.
| Species | Fe–Npy | Fe–Ntet | Npy–Cpy | Cpy–Ctet | Ctet–Ntet |
|---|---|---|---|---|---|
| 2+, S = 0 | 1.923 | 2.000 | 1.341 | 1.469 | 1.355 |
| 1+, S = 1/2 | 1.915 | 1.977 | 1.342 | 1.467 | 1.364 |
| 0, S = 1 | 1.912 | 1.963 | 1.342 | 1.467 | 1.369 |
Large Fe–N(tetrazine) bond length contractions are observed with each reduction, presumably due to the electrostatic attraction between the cationic metal and anionic ligands; this Fe/N bond strengthening supports the experimental evidence that the −850 mV potential process observed by CV (Fig. 6b) cannot be due to reduction of free btzp from ligand loss. The smaller Fe/N decrease to pyridyl N than to tetrazine N upon reduction also supports the development of anionic charge selectively at tetrazines. The individual Fe/N distances show that S4 symmetry holds for all three charge states. The Npy–Cpy and Cpy–Ctet bond lengths are essentially invariant, reinforcing the fact that the central pyridine plays only a small role in the acceptor behavior of bztp. The Ctet–Ntet bond elongates from 1.355 to 1.364 to 1.369 Å, consistent with occupation of an orbital with tetrazine π* character. The relatively modest elongation of 0.014 Å is because the one or two electron reduction is distributed over all four tetrazine arms. In summary, the calculations yield spin delocalized over both pincer arms first in one, and then in the second btzp.
Discussion
We have focused our efforts on confirming the emergence and assignments of extra redox peaks. The iron complex shows four reductive steps, while the free ligand shows only one reductive process (in spite of containing two redox-active tetrazines). Each ligand goes from having a single electron redox process when free of a metal center to being capable of two one electron processes in the complex. This we term redox multiplication: distinctly different ligand behavior (capacity) dependent upon the ligand coordination environment. Our DFT results may serve to clarify the reason for this behavior: upon complexation with iron, extensive remixing of orbitals occurs, giving the ligand access to the previously unattainable second reduction process at greatly reduced potentials.The question of localized vs. delocalized is clearly influenced by subtle factors (e.g., solvent, counter ions and hydrogen bonding), as has already been established41–43 in the case of intervalence charge transfer in M2q species where q is an odd number. Hydrogen bonding to a solvent is indeed one mechanism which has been seriously evaluated for converting between localized and delocalized ground states of mixed valence systems.44 In the present case, some factors that the DFT calculations have not modeled are interactions with redox innocent counter anions and specific solvation (rather than dielectric effects) with any charge dense regions of the reduced complex. The outwardly directed nitrogens are sterically available as is the π face of the tetrazines. Furthermore also consider the fact that, in contrast to a terpyridyl ligand, the inner coordination sphere surrounding the metal ion center involving the btzp ligand is very exposed. First, the lone pairs on the nitrogen α to the M–N, may display greater negative charge density than the other pendant nitrogens due to the polarizing influence of the Fe(II) center, akin to the electrostatic effect of the Na+ ion; this is another location for counter ion or solvent effects to be felt. Second, where terpy CH bonds would be, the tetrazine α nitrogens provide steric access to the Fe(II) center28 providing another site for specific counter ion or solvation effects for localizing the redox orbitals.
Electrostatic potential (ESP) maps can be used to establish where an anion or a nucleophile would be attracted following reduction. An ESP map of the ligand in its neutral (ESI†) and singly reduced form (Fig. 18) was used to evaluate the charge distributions. Consistent with the geometry-optimized preference of the sodium cation to the complex with the reduced half of the btzp−1 ligand in an η2 fashion, the ESP shows its largest negative value coincident with the location of the nitrogen lone pairs, rather than the π-orbital location of the unpaired spins.
 | ||
| Fig. 18 ESP plot for bztp− with the reduced ring shown on left (isovalue = 0.02 au, range: +0.31 au in green to −0.31 au in red) showing ESP values in the regions indicated. | ||
Returning to the observed redox doubling, we prepared [Na(bztp)2]+, containing a redox inactive cation, to compare its behavior to the iron(II) complex. 1H NMR titration shows that it forms a tight 2:1 complex at 1 mM (ESI). Unlike the iron(II) complex the CV data show just two reduction peaks at −0.64 and −0.97 V (Fig. S2†) demonstrating the typical8 types of stabilization expected when reducible ligands are coupled to cationic centers: there is a direct correspondence between the number of redox processes and the number of ligands. This observation unambiguously shows that the iron(II) is involved synergistically with the ligands to multiply the storage of the redox activity of the complex.
Conclusion
The successful synthesis and redox chemistry of the btzp ligand and its iron(II) bis-ligand complex have been described. The highly π-acidic character of the ligand was verified and originates from the low-lying LUMO on the tetrazines. Facile complexation with iron(II) in solution leads to multiple redox states becoming accessible at modest potentials. With the iron(II) state being highly stabilized in this environment, (unobserved) oxidation of FeIII was shifted anodically by at least 900 mV to beyond +2.2 V vs. Ag/AgCl compared to an analogous bis-terpyridine complex. If, as some hope,45 we are able to confer electronic “nobility” onto first row transition metals, redox properties like those demonstrated here would be highly advantageous, as they make it possible to access multiple low-energy reductions as observed in the CV profiles reported here.A currently unsolved question for Fe(btzp)2n+ is charge localization/delocalization in mixed valence species, which most generally are odd-electron species with two compositionally identical halves. What we have discovered here is that the answer may be dependent on surrounding environment, and that even DFT calculations will need to model explicit partners, once these are identified with certainty.
Notes and references
- S. C. Bart, K. Chlopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901–13912 CAS.
- W. Kaim, Coord. Chem. Rev., 1987, 76, 187–235 CAS.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CAS.
- A. M. Tondreau, C. C. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CAS.
- E. Billig, R. Williams, I. Bernal, J. H. Waters and H. B. Gray, Inorg. Chem., 1964, 3, 663–666 CAS.
- H. B. Gray, R. Williams, I. Bernal and E. Billig, J. Am. Chem. Soc., 1962, 84, 3596–3597 CAS.
- C. K. Jorgensen, Coord. Chem. Rev., 1966, 1, 164–178 Search PubMed.
- L. J. K. Cook, F. Tuna and M. A. Halcrow, Dalton Trans., 2013, 42, 2254–2265 Search PubMed.
- B. De Bruin, E. Bill, E. Bothe, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 2000, 39, 2936–2947 CAS.
- K. A. McNitt, K. Parimal, A. I. Share, A. C. Fahrenbach, E. H. Witlicki, M. Pink, D. K. Bediako, C. L. Plaisier, N. Le, L. P. Heeringa, D. A. Vander Griend and A. H. Flood, J. Am. Chem. Soc., 2009, 131, 1305–1313 CAS.
- K. Parimal, E. H. Witlicki and A. H. Flood, Angew. Chem., Int. Ed., 2010, 49, 4628–4632 CAS.
- A. I. Share, K. Parimal and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 1665–1675 CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 CAS.
- V. Lyaskovskyy and B. De Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CAS.
- K. G. Caulton, Eur. J. Inorg. Chem., 2012, 2012, 435–443 CrossRef CAS.
- W. Kaim, Coord. Chem. Rev., 2002, 230, 127–139 CrossRef CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef.
- G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299–3314 CrossRef CAS PubMed.
- J. R. Rebek, S. Gu and S. Biros, Novel Scaffolds for Beta-Helix Mimicry, U.S. Patent, 20060205728A1, 2006 Search PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- We gratefully acknowledge Paul Chirik for his assistance in providing these data.
- E. Baggio-Saitovitch and M. A. De Paoli, Inorg. Chim. Acta, 1978, 27, 15–20 CrossRef CAS.
- H. Oshio, H. Spiering, V. Ksenofontov, F. Renz and P. Gutlich, Inorg. Chem., 2001, 40, 1143–1150 CrossRef CAS PubMed.
- B. M. Wile, R. J. Trovitch, S. C. Bart, A. M. Tondreau, E. Lobkovsky, C. Milsmann, E. Bill, K. Wieghardt and P. J. Chirik, Inorg. Chem., 2009, 48, 4190–4200 CrossRef CAS PubMed.
- P. S. Braterman, J.-I. Song and R. D. Peacock, Inorg. Chem., 1992, 31, 555–559 CrossRef CAS.
- C. W. Machan, M. Adelhardt, A. A. Sarjeant, C. L. Stern, J. Sutter, K. Meyer and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 16921–16924 CrossRef CAS PubMed.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szlyk, Magn. Reson. Chem., 2011, 49, 237–241 CrossRef CAS PubMed.
- E. W. Stone and A. H. Maki, J. Chem. Phys., 1963, 39, 1635–1642 CrossRef CAS PubMed.
- R. K. Hocking, R. J. Deeth and T. W. Hambley, Inorg. Chem., 2007, 46, 8238–8244 CrossRef CAS PubMed.
- J. J. Brooks and G. D. Stucky, J. Am. Chem. Soc., 1972, 94, 7333–7338 CrossRef CAS.
- S. Krieck, H. Goerls and M. Westerhausen, Organometallics, 2010, 29, 6790–6800 CrossRef CAS.
- H. B. Gray, D. N. Hendrickson and Y. S. Sohn, Inorg. Chem., 1971, 10, 1559–1563 CrossRef CAS.
- R. Prins, J. Chem. Soc., Chem. Commun., 1970, 280–281 RSC.
- M. C. Schnitzler, A. S. Mangrich, W. A. A. Macedo, J. D. Ardisson and A. J. G. Zarbin, Inorg. Chem., 2006, 45, 10642–10650 CrossRef CAS PubMed.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chim. Acta, 2001, 107, 48–55 CrossRef CAS PubMed.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyermueller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- R. L. Lord, M. M. Allard, R. A. Thomas, O. S. Odongo, H. B. Schlegel, Y.-J. Chen and J. F. Endicott, Inorg. Chem., 2013, 52, 1185–1198 CrossRef CAS PubMed.
- C. C. Scarborough and K. Wieghardt, Inorg. Chem., 2011, 50, 9773–9793 CrossRef CAS PubMed.
- A. Bencini, I. Ciofini, C. A. Daul and A. Ferretti, J. Am. Chem. Soc., 1999, 121, 11418–11424 CrossRef CAS.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- P. H. Dinolfo, R. D. Williams and J. T. Hupp, Chem. Phys., 2005, 319, 28–38 CrossRef CAS PubMed.
- A. B. Wragg, S. Derossi, T. L. Easun, M. W. George, X.-Z. Sun, F. Hartl, A. H. Shelton, A. J. H. M. Meijer and M. D. Ward, Dalton Trans., 2012, 41, 10354–10371 RSC.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic, synthetic, DFT, and spectroscopic data, as well as results from supporting experiments. CCDC 955991 and 955992. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00341a |
| This journal is © The Royal Society of Chemistry 2014 |