 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational studies of the unusual water adduct [Cp2TiMe(OH2)]+: the roles of the solvent and the counterion†
Jörg
Saßmannshausen
UCL, Department of Chemistry, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: j.sassmannshausen@ucl.ac.uk
First published on 21st March 2014
Abstract
The recently reported cationic titanocene complex [Cp2TiMe(OH2)]+ was subjected to detailed computational studies using density functional theory (DFT). The calculated NMR spectra revealed the importance of including the anion and the solvent (CD2Cl2) in order to calculate spectra which were in good agreement with the experimental data. Specifically, two organic solvent molecules were required to coordinate to the two hydrogens of the bound OH2 in order to achieve such agreement. Further elaboration of the role of the solvent led to Bader's QTAIM and natural bond order calculations. The zirconocene complex [Cp2ZrMe(OH2)]+ was simulated for comparison.
Introduction
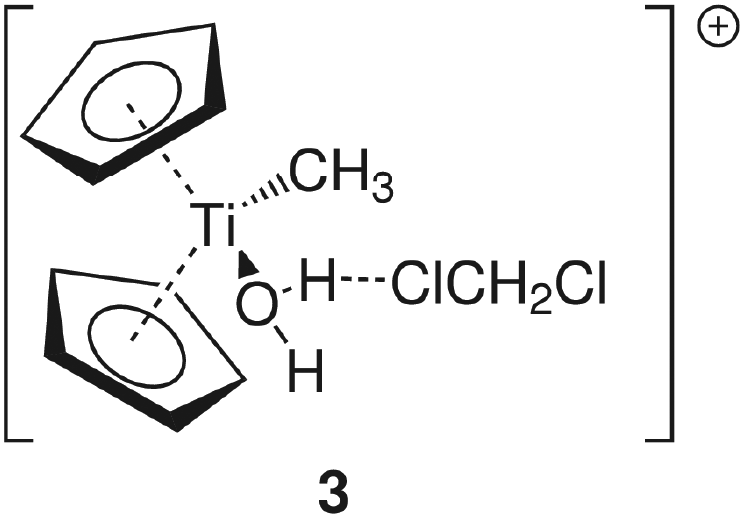
Group 4 metallocenes have been the workhorse for a number of reactions for some decades now. In particular the cationic compounds [Cp2MR]+ (M = Ti, Zr, Hf; R = Me, CH2Ph) are believed to be the active catalysts for a number of polymerization reactions such as Kaminsky type α-olefin,1–10 carbocationic11–15 or ring-opening lactide polymerization.16–18 For these highly electrophilic cationic compounds, water is usually considered a poison as it leads to catalyst decomposition. In this respect it was quite surprising that Baird recently reported the characterisation of the complex [Cp2TiMe(OH2)][B(C6F5)4] (I) using NMR spectroscopy.19 For some years now, we have been interested in the use of molecular modelling as a tool to predict NMR spectra and the use of NMR spectroscopy for confirming our theoretical results.20–27 For this reason, we modelled a number of possible compounds which are summarised in Chart 1 and compared these theoretical chemical shifts with the ones observed by Baird. The proposed compound [Cp2TiMe(OH2)]+ (1) together with the outer sphere ion pair [Cp2TiMe(OH2)]+[MeB(C6F5)3]− (2) served as the starting point for our investigations. Furthermore, the solvent adducts {[Cp2TiMe(OH2)](CH2Cl2)}+ (3) and {[Cp2TiMe(OH2)](CH2Cl2)2}+ (4), and for comparison, the zirconocene compounds [Cp2ZrMe(OH2)]+ (5), {[Cp2ZrMe(OH2)]CH2Cl2}+ (6) and {[Cp2ZrMe(OH2)](CH2Cl2)2}+ (7) were also modelled.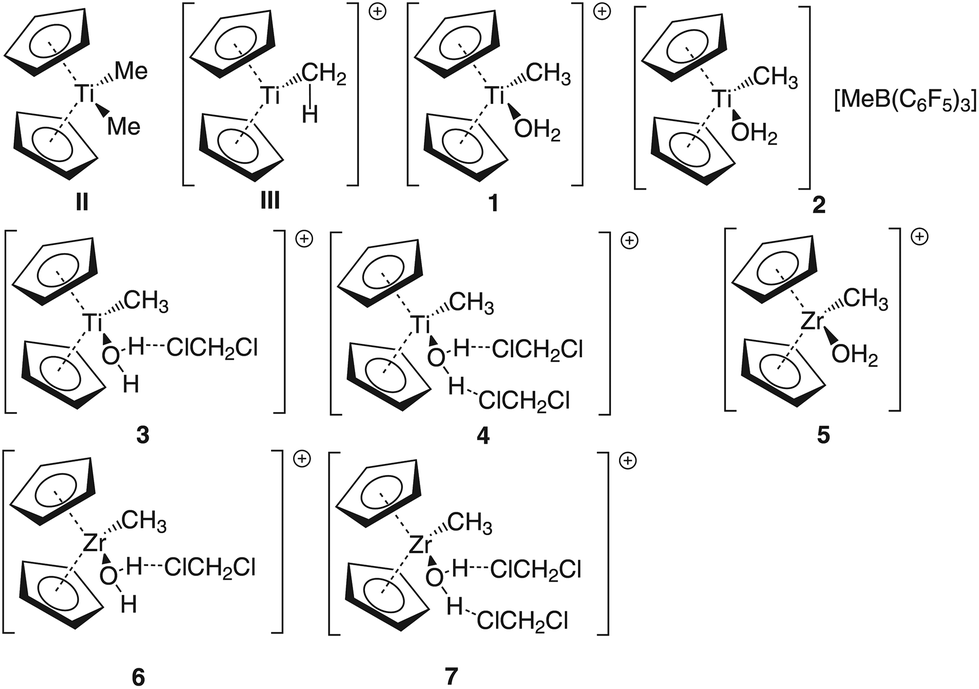 | ||
| Chart 1 Model compounds used for the calculation of the chemical shifts. | ||
In order to gain insight into the ‘acidity’ of the OH2 protons we calculated the pKa of 1, 3, 5 and 6. These models are summarised in Chart 2.
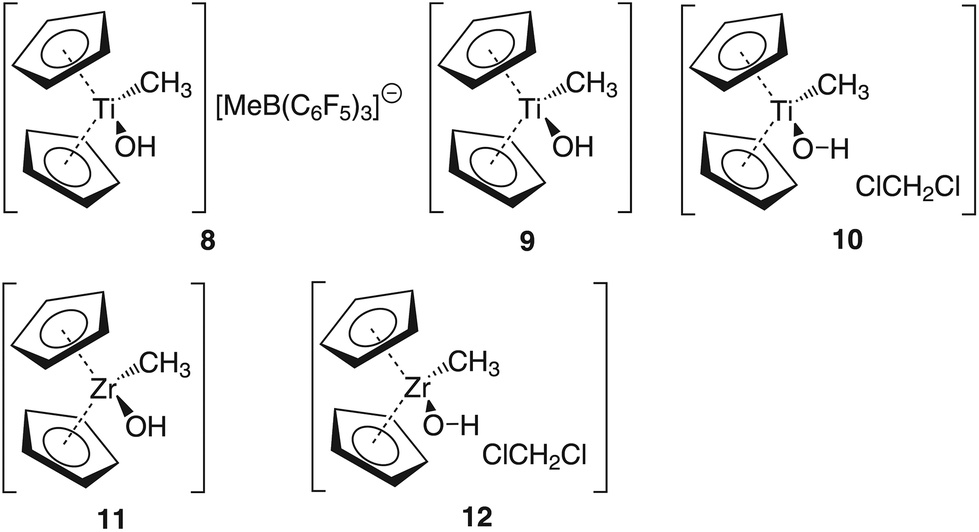 | ||
| Chart 2 Model compounds used for the calculation of the pKa. | ||
Improved reliability of the pKa data was achieved by employing the MP2 level of theory in calculations of certain species which are denoted by the suffix MP2.
To gain some insight into the ground state energy differences between the ‘naked’ cation [Cp2TiMe]+ (III) and the solvent adduct [Cp2TiMe(CH2Cl2)]+ (13) as well as between [Cp2TiMe(CH2Cl2)]+ [H2O] (14) and 3, these compounds have been modelled too (cf. Chart 3).
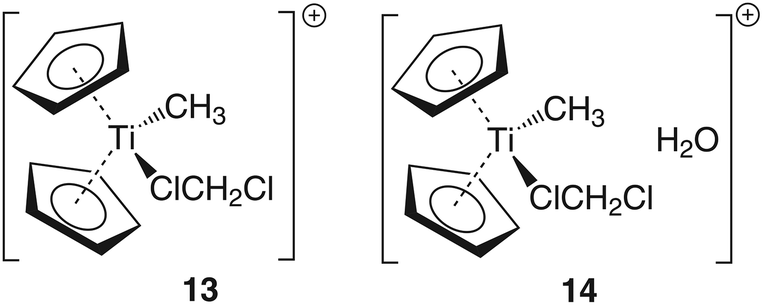 | ||
| Chart 3 Model compounds used for the calculation of the relative energies between III and 13 and 14. | ||
The methyl borate anion [MeB(C6F5)3]− was used instead of the borate [B(C6F5)4]− in the calculations in order to save computational cost.
Computational details
Density functional theory calculations were carried out using the GAUSSIAN03, Revision C.02, program package.28 For the larger compounds 2 and 8 and for the pKa calculations, GAUSSIAN09, Revision C.01, was utilised for the geometry calculations.29 Geometries have been fully optimized without symmetry constraints, involving the functional combinations according to Becke (hybrid)30 and Lee, Yang, and Parr31 (denoted B3LYP), with the corresponding effective core potential basis set for Ti and Zr (Stuttgart-Dresden, keyword SDD in Gaussian) and the standard 6-311G(d,p) basis set for C, H and Cl (denoted ECP11). For the larger compounds a smaller basis set was used which consists of the 6-31G(d) basis set for all elements except Ti for which the Stuttgart-Dresden basis set is again used (denoted ECP1). The stationary points were characterized as minima by analytical harmonic frequency (zero imaginary frequency).Magnetic shieldings σ have been evaluated for the B3LYP/ECP11 geometries by implementation of the GIAO (gauge included atomic orbitals)-DFT method, using the same B3LYP level of theory, together with the recommended IGLO II basis on C, H, F, and B.32 For Ti, an extended Wachters basis set was used.27,33 This combination is denoted NMR1. This approach with this particular combination of functionals and basis sets has proven to be quite effective for chemical shift computations for transition metal complexes.23 Chemical shifts of 1H and 13C have been calculated relative to benzene as a primary reference, with absolute shieldings for benzene σ(1H) 24.54 and σ(13C) 47.83 with the IGLO II basis set. The values for benzene were converted to the TMS scale using the experimental δ values of benzene (7.26 and 128.5, respectively).
The pKa calculations were performed using the PCM-SMD model as implemented in the program, with water as the solvent of choice. These calculations were done at the B3LYP/ECP11 level and additionally at the MP2/ECP11 level of theory. For the MP2 computation, GAMESS 2012, R2 was used.34 Here the B3LYP geometry was used as the starting point for the calculations at the MP2/ECP11 level of theory. We basically followed a procedure published in ref. 35. For the proton, an experimentally obtained Gibbs free energy value of −6.28 kcal mol−1 for the gas phase and −264.61 kcal mol−1 for the hydration was employed.
Tables of Cartesian coordinates of all calculated structures are available as ESI† in x, y, z format. QTAIM36 and NBO5 analyses37 were performed using the DZVP38 all-electron basis set on Ti and Zr and 6-311G(d,p) for C, H, F, and B, denoted DZVP1. Diagrams of the electron charge density plots were obtained using AIM2000.39,40 MOLDEN was used for the chemical representation of the calculated compounds.41
Results and discussion
Structural and NMR results
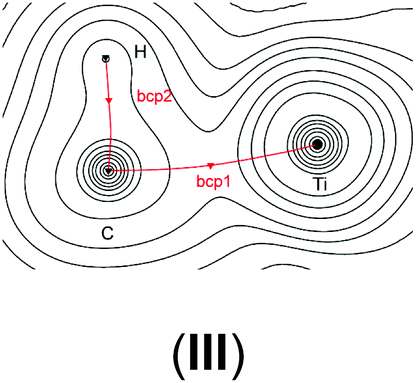
In order to verify our computational approach, we calculated the well-known structure of dimethyl titanocene (II) and the cationic methyl titanocene [Cp2TiMe]+ (III). For II, we computed a Ti–Me bond distance of 2.16 Å at the B3LYP/ecp11 level and a Me–Ti–Me angle of 91.7° which fits well with the experimentally observed values42 of 2.170(2) and 2.180(2) Å, and 91.3(1)°. Thus, we can be confident that our chosen level of theory is appropriate here.For the methyl group in III we observe two expected Ti–C–H angles of 120.8° and 121.3° with the third one rather acute (84.3°). These metric parameters and the reduced 1JCH coupling constant of 93.8 Hz, compared with the remaining two coupling constants of approximately 142 Hz, clearly indicate an agostic interaction here,43–45 while Bader analysis very clearly shows an absence of such a bonding path between the Ti and the C–H bond.46–51 This is, however, not unexpected as Lein pointed out recently.52 Indeed, detailed NBO analysis supports the α-agostic interaction in III (see Table 2 and ESI† for a plot of the electron density and NBO).
The starting geometry of the water adduct 1 was constructed by adding one molecule of water to III. Selected metric parameters, chemical shifts and CH coupling constants of III, 1, 3 and 4 are summarised in Table 1.
|
|
|
|
|
|
|---|---|---|---|---|
| d(Ti–C) | 2.08 (2.09) | 2.15 (2.19) | 2.15 (2.20) | 2.15 (2.20) |
| d(C–H1) | 1.12 (1.14) | 1.09 (1.10) | 1.09 (1.10) | 1.09 (1.10) |
| d(C–H2) | 1.09 (1.09) | 1.09 (1.10) | 1.09 (1.10) | 1.09 (1.10) |
| d(C–H3) | 1.09 (1.09) | 1.09 (1.10) | 1.09 (1.10) | 1.09 (1.10) |
| d(Ti–O) | 2.15 (2.14) | 2.13 (2.11) | 2.11 (2.09) | |
| d(O–H4) | 0.96 (0.97) | 0.96 (0.96) | 0.97 (0.97) | |
| d(O–H5) | 0.96 (0.97) | 0.97 (0.97) | 0.97 (0.97) | |
| d(H4–Cl) | 2.37 (2.29) | |||
| d(H5–Cl) | 2.31 (2.23) | 2.36 (2.24) | ||
| ∠(Ti–C–H1) | 84.3 (70.6) | 113.8 (114.7) | 114.0 (114.5) | 114.1 (114.7) |
| ∠(Ti–C–H2) | 120.8 (123.6) | 109.1 (107.8) | 108.8 (107.9) | 108.6 (107.6) |
| ∠(Ti–C–H3) | 121.3 (123.6) | 109.2 (108.8) | 109.0 (108.9) | 108.8 (108.6) |
| ∠(H4–O–H5) | 108.0 (106.2) | 107.3 (106.0) | 107.2 (105.8) | |
| δ(CH3) | H1 16.76 | 1.85 | 1.64 | 1.53 |
| H2 6.93 | ||||
| H3 6.80 | ||||
| δ(CH3) | 122.8 | 66.6 | 63.6 | 61.9 |
| 1 J CH1 | 93.8 | 131.5 | 131.4 | 130.0 |
| 1 J CH2 | 141.7 | 126.6 | 126.9 | 127.4 |
| 1 J CH3 | 141.9 | 126.5 | 126.7 | 126.7 |
| 1 J CH3 (average) | 125.8 | 128.2 | 128.3 | 128.0 |
| δ(OH2) | 2.79 | H4: 2.98 | H4: 5.03 | |
| H5: 5.10 | H5: 4.93 | |||
| δ(C5H5) | 6.29 | 6.24 | 6.25 | |
| δ(C5H5) | 118.8 | 117.8 | 117.2 |

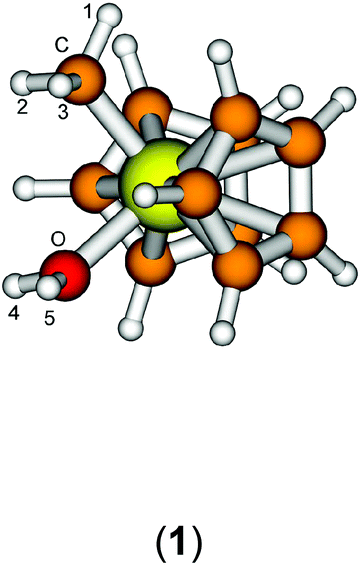
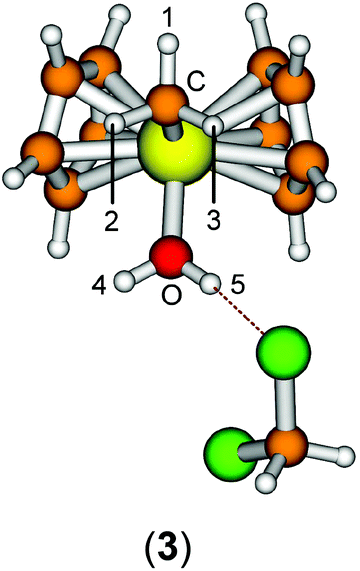
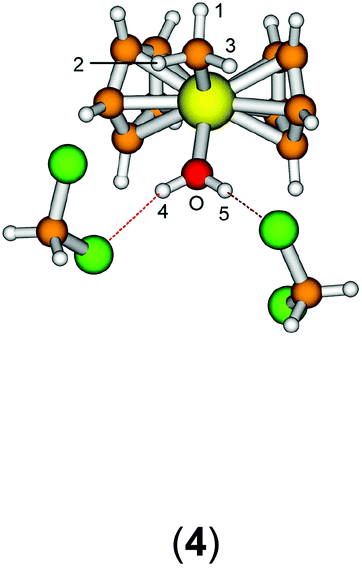
As is evident from Table 1, upon coordination of one molecule of water to III the rather acute Ti–C–H1 angle relaxes to around 114°, concomitant with a small elongation of the Ti–C bond and a more significant change of the 1JCH coupling constant. Addition of one or two molecules of dichloromethane does not substantially change the steric parameters of the methyl group.
A more dominant change upon dichloromethane coordination can be observed in the calculated proton and carbon NMR spectra. For the solvate free cation 1 we calculate δ(OH2) 2.79 ppm, which is in stark contrast with the experimentally observed value of 4.77 ppm. However, addition of one or two molecules of CH2Cl2 improves the situation significantly, with 5.10/2.98 ppm (3, one molecule of CH2Cl2) and 5.03/4.93 ppm (4, two molecules of CH2Cl2) which is in excellent agreement with the experimental data. For comparison, for the outer sphere ion pair (OSIP)532, we calculated an average chemical shift of δ(OH2) = 6.42 ppm. These findings very clearly indicate that the solvent adduct 4 is in fact the observed compound in the NMR. Similar findings have been reported before, most notably the coordination of dichloromethane to cationic zirconocene benzyl compounds.25
pKa computational results
One of the real advantages of molecular modelling is the possibility to investigate molecules which are, under normal experimental conditions, difficult to observe. This could be because these molecules of interest are either of fleeting existence or require special conditions like high pressure or simply would not exist as such in reality. With this in mind we were interested in knowing whether the above findings are mirrored in the ‘acidity’ of the coordinated water and thus we calculated the pKa values of compounds 1, 3, 5 and 6 (cf. Table 2). We do not claim that the calculated numbers are the ones which would be experimentally observed, as to the best of our knowledge to date there is no reliable method to compute the pKa without the use of a known base54 or the inclusion of the first solvation shell beyond the immediate ligand sphere.55 Thus, whilst we are confident that the computed numbers are reasonable they must be regarded as approximations.|
|
|
|
|
|
|---|---|---|---|---|
| B3LYP/ecp11 | 6.29 | 5.19 | 8.65 | 7.27 |
| MP2/ecp11 | 4.57 | 2.80 | 2.99 | 1.21 |
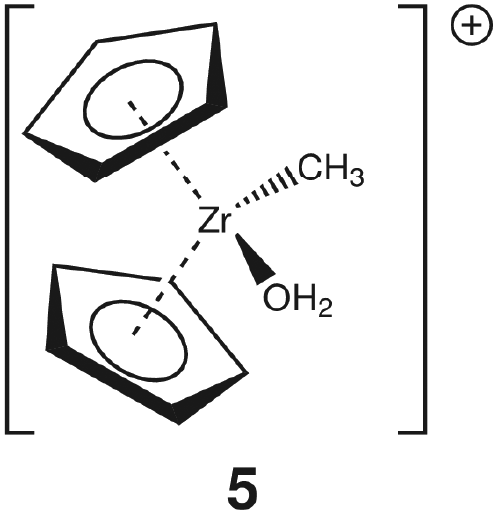
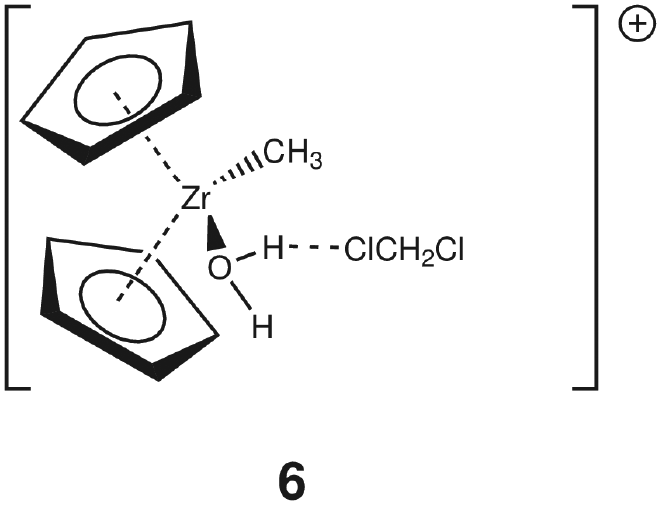
For 1, the calculated pKa of 4.57 at the MP2/ECP11 level of theory (6.29 for B3LYP/ECP11) is somewhat higher than that for the solvent adduct 3 (MP2: 2.80; B3LYP: 5.19). For comparison, the first pKa of the dicationic titanocene bis-water adduct [Cp2Ti(OH2)2]2+ (IV) was measured to be ∼3.51 as reported by Marks.56 For the zirconium congener 5 the calculated pKa of 2.99 (MP2) and 8.65 (B3LYP) is again somewhat higher than that for the solvent adduct 6 (MP2: 1.21; B3LYP: 7.27). Three observations can be made: (i) addition of one molecule of CH2Cl2 lowers the pKa, (ii) the zirconium congener is more acidic than the titanium compounds at the MP2/ecp11 level of theory, and (iii) the computed pKa values at the B3LYP level of theory appear to be rather high. For example, our calculated pKa values for 1 are closer to the observed value for IV at the MP2/ECP11 level of theory. Furthermore, the pKa of tris(allyl)amine (TAA) was determined to be 8.31.57 It is very difficult to believe that a cationic zirconocene compound has a similar pKa as TAA. Thus, we can conclude that the obtained figures at the B3LYP level of theory are probably incorrect. It is very difficult to establish the reason for this discrepancy, especially as the computed structures are very similar.
Another way to study the relative stability of the titanium and zirconium water adducts is to look at the isodesmic reaction between the water adducts and their corresponding bases:
With respect to the addition of one molecule of CH2Cl2 the calculated chemical shifts are a useful probe. The calculated chemical shift of the non-coordinated HO-H is 2.98 ppm whereas for the coordinated hydrogen it is 5.10 (Table 1). Thus, the coordinated hydrogen is deshielded, thus rendering it more positive, which in turn means that it should be easier to ionize. We will look into the bond properties in more detail in the next section. Compared with titanium the atomic radius of zirconium is larger (calculated radius for Zr: 206 pm; Ti: 176 pm);58 thus there is less steric congestion around the metal which leads to a tighter binding of the water to the cationic metallocene. As a result of the reduced congestion the zirconium compounds are stronger Lewis acids in general, which is mirrored here by a lower pKa value and, more generally, by a higher activity in, for example, α-olefin polymerization.
Electronic structural analysis: Bader and NBO
In order to gain a better insight into the electronic structure of the computed compounds III, 1, 3, 4, 5, and 6, we performed Bader's QTAIM and natural bond orbital analysis. We were particularly interested in the change of the O–H bond properties upon coordinating to the cationic metal centre and upon addition of one or two molecules of CH2Cl2. The results are summarised in Table 3.|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| a Bond critical point between O and H (not shown on the plots). b M = Ti or Zr. c Non-coordinated H if CH2Cl2 is present. d Coordinated H if CH2Cl2 is present. | ||||||
| bcp1 | ρ(r) = 0.1048 | ρ(r) = 0.0536 | ρ(r) = 0.0576 | ρ(r) = 0.3481 | ρ(r) = 0.0508 | ρ(r) = 0.0519 |
| ∇2ρ(r) = −0.031 | ∇2ρ(r) = −0.0804 | ∇2ρ(r) = −0.0861 | ∇2ρ(r) = 0.6173 | ∇2ρ(r) = −0.0641 | ∇2ρ(r) = −0.0685 | |
| bcp2 | ρ(r) = 0.2439 | ρ(r) = 0.0952 | ρ(r) = 0.3591 | ρ(r) = 0.3486 | ρ(r) = 0.3565 | ρ(r) = 0.3547 |
| ∇2ρ(r) = 0.178 | ∇2ρ(r) = −0.0222 | ∇2ρ(r) = 0.6387 | ∇2ρ(r) = 0.6187 | ∇2ρ(r) = 0.6374 | ∇2ρ(r) = 0.6265 | |
| bcp3 | ρ(r) = 0.3580 | ρ(r) = 0.3451 | ρ(r) = 0.0161 | ρ(r) = 0.3565 | ρ(r) = 0.3288 | |
| ∇2ρ(r) = 0.6394a | ∇2ρ(r) = 0.6125 | ∇2ρ(r) = −0.0126 | ∇2ρ(r) = 0.6374 | ∇2ρ(r) = 0.5505 | ||
| bcp4 | ρ(r) = 0.3580 | ρ(r) = 0.0180 | ρ(r) = 0.0155 | ρ(r) = 0.0174 | ||
| ∇2ρ(r) = 0.6394a | ∇2ρ(r) = −0.0136 | ∇2ρ(r) = −0.0123 | ∇2ρ(r) = −0.0138 | |||
| Mb | 1.761 (2.037) | 1.778 (1.976) | 1.761 (1.945) | 1.757 (1.958) | 2.032 (2.202) | 2.027 (2.234) |
| O | −0.364 (−0.907) | −1.118 (−0.927) | −1.114 (−0.939) | −1.125 (−0.932) | −0.939 (−0.953) | |
| H | 0.610 (0.517) | 0.603c (0.515)c | 0.602d (0.518)d | 0.614 (0.520) | 0.609c (0.516)c | |
| H2 | 0.610 (0.517) | 0.607d (0.518)d | 0.609d (0.517)d | 0.616 (0.520) | 0.616d (0.520)d | |
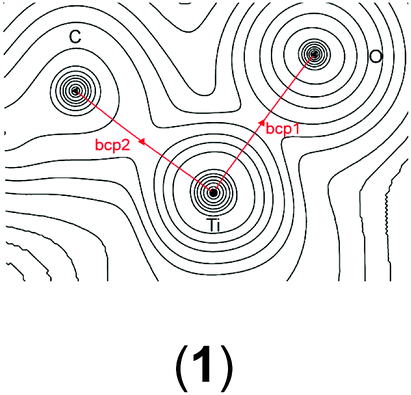
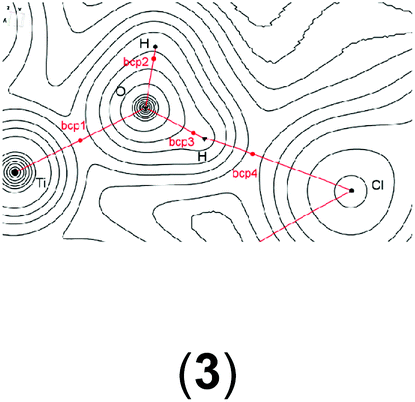
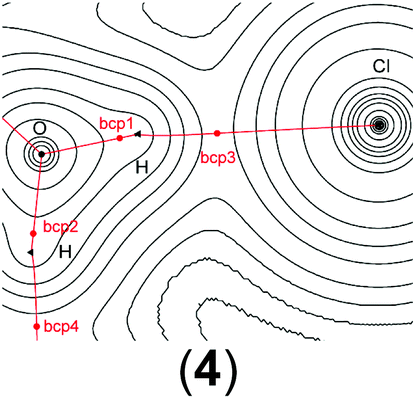
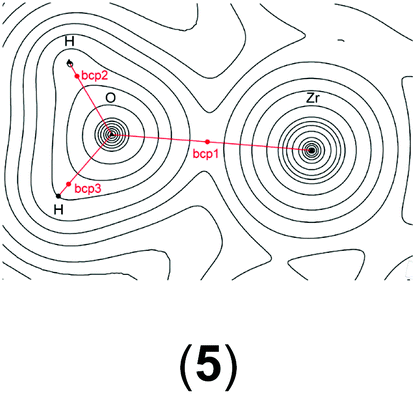

It is clear from Table 3 that addition of one or two molecules of CH2Cl2 changes the electronic properties of the metal–oxygen and oxygen–hydrogen bonds. These changes are in line with the calculated chemicals shifts. For example, the Ti–O electron density at the bond critical point (bcp) increases from ρ(r) = 0.0536 to ρ(r) = 0.0576 (ρ(r) = 0.0593 for two CH2Cl2) upon addition of one molecule CH2Cl2. There is a concomitant change of the O–H bond parameters. Upon addition of one CH2Cl2 the electron density of the coordinated H decreases to ρ(r) = 0.3451 from ρ(r) = 0.3580 whereas for the non-coordinated hydrogen it increases to ρ(r) = 0.3591. This observation is further reinforced by the (Natural) charges: upon addition of one CH2Cl2 the originally evenly charged hydrogens acquire a small charge imbalance: 0.515 for the ‘free’ hydrogen and 0.518 for the coordinate hydrogen. Thus, electron density is removed from the hydrogens upon coordination of even one molecule of CH2Cl2 and this electron density is pulled towards the more electronegative oxygen (change of (Natural) charge from −0.907 to −0.927). This change in the electronic properties of these hydrogens is further reflected in the change of the calculated chemical shifts: for the ‘free’ hydrogen we find a shift of δ(1H) = 2.98 ppm and for the coordinated hydrogen a shift of δ(1H) = 5.10 ppm. The addition of a second CH2Cl2 removes this small variation for the hydrogens and the calculated electronic parameters are again similar; however, the (Natural) charge for the oxygen has further decreased to 0.939. It should be noted that the Bader charges appear to be less sensitive to this and that these changes are small for the hydrogen atoms but more dominant for the oxygen. Similar observations can be made for the Zr analogue: the addition of one solvent molecule changes the electron density of the O–H bond from ρ = 0.3565 to ρ = 0.3547 (‘free’) and ρ = 0.3288 (coordinated) (see Table S15 in the ESI†).
Ground state energy calculations
In light of the different compounds already calculated, it is reasonable to look into the ground state energy between the naked cation III and its solvent adduct [Cp2TiMe(CH2Cl2)]+ (10). To save computational time and also to eliminate the need to search for a global minimum, we calculated CH2Cl2 at the B3LYP/6-311G(d,p) level of theory and added the electronic energies of that and III together. As expected, the ‘solvated’ cation 13 is around 323 kJ mol−1 lower in energy than the naked cation 3. Addition of one molecule of water to 13, which was optimized as 14, results in the formation of the water adduct 3 which is around 80 kJ mol−1 lower in energy than 14. Similar results can be obtained for the equilibrium between the Zr compounds [Cp2ZrMe(CH2Cl2)]+ [H2O] (15) and 6. Here we obtain a value of around 88 kJ mol−1.Conclusions
From the reported calculations it is clear that, in solution, the cationic titanocene water adduct [Cp2TiMe(OH2)]+ (1) does not exist as such. Our calculated chemical shifts very clearly indicate that at least two molecules of the solvent CH2Cl2 are coordinated to the water by means of hydrogen bonding. Thus, the observed chemical shifts actually belong to this solvated species, {[Cp2TiMe(OH2)](CH2Cl2)2}+ (4), and as such represent the outer sphere ion pair {[Cp2TiMe(OH2)](CH2Cl2)2}[B(C6F5)4]. The trends of our calculated chemical shifts are mirrored in the QTAIM model and the computed pKa values. As expected, the zirconium derivatives show a similar behaviour and are more acidic than the titanium ones. It would be interesting to determine whether these acidic protons could be utilised, for example in the carbocationic polymerization of isobutene.Acknowledgements
J.S. would like to thank Alexandra Simperler from the NSCCS/UK for useful discussions and help with the pKa calculations.References
- M. Bochmann, J. Chem. Soc., Dalton Trans., 1996, 255 RSC.
- H.-H. Brintzinger, D. Fischer, R. Mühlhaupt, B. Rieger and R. Waymouth, Angew. Chem., 1995, 107, 1255 CrossRef.
- M. Bochmann, Top. Catal., 1999, 7, 9 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS.
- R. Mülhaupt, Macromol. Chem. Phys., 2003, 204, 289 CrossRef.
- M. Bochmann, J. Organomet. Chem., 2004, 689, 3982 CrossRef CAS PubMed.
- W. Kaminsky, A. Funck and H. Hähnsen, Dalton Trans., 2009, 8803 RSC.
- M. Bochmann, Organometallics, 2010, 29, 4711 CrossRef CAS.
- J. J. Eisch, Organometallics, 2012, 31, 4917 CrossRef CAS.
- E. P. Talsi, K. P. Bryliakov, N. V. Semikolenova, V. A. Zakharov and M. Bochmann, Kinet. Catal., 2007, 48, 490 CrossRef CAS.
- A. G. Carr, D. M. Dawson and M. Bochmann, Macromolecules, 1998, 31, 2035 CrossRef CAS.
- A. G. Carr, D. M. Dawson and M. Bochmann, Macromol. Rapid Commun., 1998, 19, 205 CAS.
- K. R. Kumar, C. Hall, A. Penciu, M. J. Drewitt, P. J. McInely and M. C. Baird, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3302 CrossRef CAS.
- P. A. Wilson, M. H. Hannant, J. A. Wright, R. D. Cannon and M. Bochmann, Macromol. Symp., 2006, 236, 100 CrossRef CAS.
- J. Saßmannshausen, Dalton Trans., 2009, 9026 RSC.
- S.-d. Mun, Y. Hong and Y. Kim, Bull. Korean Chem. Soc., 2007, 28, 698 CrossRef CAS.
- A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804 RSC.
- D. Patel, S. T. Liddle, S. A. Mungur, M. Rodden, A. J. Blake and P. L. Arnold, Chem. Commun., 2006, 1124 RSC.
- A. F. Dunlop-Brière and M. C. Baird, Organometallics, 2010, 29, 6117 CrossRef.
- J. Saßmannshausen, Dalton Trans., 2009, 8993 RSC.
- A. A. Danopoulos, D. Pugh, H. Smith and J. Saßmannshausen, Chem. – Eur. J., 2009, 15, 5491 CrossRef CAS PubMed.
- J. Saßmannshausen, J. Klett, A. R. Kennedy, J. A. Parkinson and D. Armstrong, New J. Chem., 2013, 37, 494 RSC.
- J. Saßmannshausen and J. Baumgartner, Organometallics, 2008, 27, 1996 CrossRef.
- J. Saßmannshausen, A. Track and T. A. D. S. Diaz, Eur. J. Inorg. Chem., 2007, 16, 2327 CrossRef.
- J. Saßmannshausen, A. Track and F. Stelzer, Organometallics, 2006, 25, 4427 CrossRef.
- J. Saßmannshausen, J. C. Green, F. Stelzer and J. Baumgartner, Organometallics, 2006, 25, 2796 CrossRef.
- M. Bühl and J. Saßmannshausen, J. Chem. Soc., Dalton Trans., 2001, 79 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, in Gaussian 03, Revision C.02, 2004 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision C.01, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- W. Kutzelnigg, U. Fleischer and M. Schindler, in NMR Basic Principles and Progress, Berlin, 1990, vol. 23 Search PubMed.
- M. Bühl and F. T. Mauschick, Magn. Reson. Chem., 2004, 42, 737 CrossRef PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, Jr., J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- See: http://verahill.blogspot.de/2012/09/example-attempt-of-isodesmic-reactions.html.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.G. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560 CrossRef CAS.
- F. Biegler-König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545 CrossRef.
- F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489 CrossRef PubMed.
- G. Schaftenaar and J. H. Noordik, Molden: a pre- and post-processing program for molecular and electronic structures, J. Comput. - Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
- U. Thewalt and T. Wöhrle, J. Organomet. Chem., 1994, 464, C17 CrossRef CAS.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS.
- M. Brookhart, M. L. H. Green and L.-L. Wong, Prog. Inorg. Chem., 1988, 36, 1 CrossRef CAS.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- J. E. Barquera-Lozada, A. Obenhuber, C. Hauf and W. Scherer, J. Phys. Chem. A, 2013, 117, 4304 CrossRef CAS PubMed.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782 CrossRef CAS PubMed.
- W. Scherer, W. Hieringer, M. Spiegler, P. Sirsch, G. S. McGrady, A. J. Downs, A. Haarland and B. Pedersen, Chem. Commun., 1998, 2471 RSC.
- W. Scherer, P. Sirsch, D. Shorokhov, M. Tafipolsky, G. S. McGrady and E. Gullo, Chem. – Eur. J., 2003, 9, 6057 CrossRef CAS PubMed.
- J. Saßmannshausen, Dalton Trans., 2011, 40, 136 RSC.
- J. Saßmannshausen, Dalton Trans., 2012, 41, 1919 RSC.
- M. Lein, Coord. Chem. Rev., 2009, 253, 625 CrossRef CAS PubMed.
- F. Song, S. J. Lancaster, R. D. Cannon, M. Schormann, S. M. Humphrey, C. Zuccacia, A. Macchioni and M. Bochmann, Organometallics, 2005, 24, 1315 CrossRef CAS.
- See for example: M. W. Palascak and G. C. Shields, J. Phys. Chem. A, 2004, 108, 3692 CrossRef CAS; M. Namazian and S. Halvani, J. Chem. Thermodyn., 2006, 38, 1495 CrossRef PubMed.
- J. Li, C. L. Fisher, J. L. Chen, D. Bashford and L. Noodleman, Inorg. Chem., 1996, 35, 4694 CrossRef CAS.
- J. H. Toney and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 947 CrossRef CAS.
- N. M. Hext, J. Hansen, A. J. Blake, D. E. Hibbs, M. B. Hursthouse, O. V. Shishkin and M. Mascal, J. Org. Chem., 1998, 63, 6016 CrossRef CAS PubMed.
- Values taken from:http://www.webelements.com/zirconium/atom_sizes.html.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00310a |
| This journal is © The Royal Society of Chemistry 2014 |