DOI:
10.1039/C4DT00247D
(Paper)
Dalton Trans., 2014,
43, 8620-8632
Studies on the reactions of [AuCl4]− with different nucleophiles in aqueous solution†
Received
23rd January 2014
, Accepted 26th March 2014
First published on 23rd April 2014
Abstract
In order to distinguish between the different types of reactions that can occur between Au(III) species and simple nucleophiles, including iodide, bromide, nitrite, thiourea, pyridine and dimethyl sulfoxide, spectrophotometric techniques including stopped-flow and rapid-scan measurements were employed under specific reaction conditions. All experiments were performed in a 0.4 M NaCl aqueous solution to maintain a high chloride concentration and a constant ionic strength. The temperature dependence of the observed rate constants confirmed the associative nature of the ligand substitution reactions. The redox behaviour of the Au(III) species was studied by cyclic voltammetry and confirmed the reversible redox transitions at ca. 0.38 V (SCE, E = 0.1 V s−1). Results obtained during the reaction progress were attributed to the formation of Au0. This oxidation state was observed for the reactions with thiourea, iodide and nitrite, whereas pyridine showed a potential shift only to Au(I) formation, while bromide showed potential shifts typical of ligand substitution reactions. The reaction with dimethyl sulfoxide was studied using 1H NMR and ab initio (RMP2(full)/LANL2DZp) techniques, which revealed why Au(III) does not react with sulfoxide. The results are discussed in terms of the importance of the stability of the Au(III) species in aqueous solutions of the selected salts and bases. In this way, one could differentiate between a possible three-electron inner-sphere redox process and/or a substitution process during the rapid initial step of the reactions.
Introduction
Even though gold is known as the “king of the metals”, numerous uncertainties still remain concerning its chemical reactivity. One of the biggest challenges in the field of reactivity is the establishment of nucleophilicity scales that will assist in gaining a systematic understanding of the contributing factors. According to Pearson's theory, Au(III) is known to be a soft Lewis acid that prefers to bind to soft ligands,1 but the complication that appears is the reduction of Au(III) to Au(I) by softer nucleophiles.2 Burrell's publication on gold chemistry indicated that the stability of gold halogenide complexes is influenced by the relativistic theory and depends on the electronegativity of the ligand, viz. [AuF4]− > [AuCl4]− > [AuBr4]− > [AuI4]−, also supported by Hartree–Fock (HF) calculations.3 A similar trend was more recently explained by DFT calculations.4 Due to the high electronegativity of Au(III) in comparison to other d8 metal ions, it can be expected that reduction is much easier than in the case of Pt(II).5
Since Elding and Cattalini did not agree on whether the rate-determining step for reactions of Au(III) is a ligand substitution or a direct reduction process,6,7 we performed a series of studies to elucidate the interplay between substitution and redox processes in the reactions of Au(III) with different nucleophiles, such as iodide, bromide, nitrite, thiourea, pyridine and DMSO. Once again, substitution appeared to be faster than reduction, as found for the reaction between [AuCl4]− and thiocyanate and thiosulfate.8,9 Similar to the earlier reported reactions with sulfur donor ligands,10 concerted slow redox processes have a strong influence on the rate constants of the rapid initial substitution reaction.
Interestingly, spectral changes observed for the reaction with I− unexpectedly indicated that the reaction does not appear as a single step as was suggested earlier.11 Furthermore, this is not in agreement with the potentiometric titration for a mixture of [AuCl4]− and I−, reported by Robles et al.12 Since we are not aware of any cyclic voltammetric (CV) studies performed on the gold–halogenide system up to now, this study also deals with the electrochemical behaviour of Au(III) in such solutions.
Finally, we would like to draw attention to further interesting data on such interactions and to answer some important questions on gold chemistry, already nicely summarized by Puddephatt.13 In terms of our results, we report here new results on multistep reactions with iodide that were found, redox reactions that occur with pyridine and nitrite (not only substitution), whereas DMSO unexpectedly does not coordinate to Au(III). The results are supported by rate constants for substitution reactions with the studied nucleophiles. Obviously, even members of the same periodic group are not always comparable, whether it concerns d8 elements since Au(III) reacts somewhat differently than Pt(II)14 or concerns the copper group since gold is unexpectedly a smaller cation than silver.15
Experimental
Chemicals and solutions
All chemicals were used as received from commercial sources. The gold compound K[AuCl4] was purchased from Sigma-Aldrich. The ligands thiourea, KBr, NaNO2 (Acros Organic), KI (Fluka), pyridine, and dimethyl sulfoxide (DMSO) (Sigma-Aldrich) were used without further purification. 1H NMR measurements were conducted in D2O (Sigma-Aldrich). All other chemicals were of the highest purity commercially available. A stock solution of 0.4 M NaCl (pH 6.13) was used for all spectroscopic, kinetic and cyclic voltammetric measurements. Double distilled water was used throughout the study.
Instrumentation and measurements
Spectrophotometry.
The stopped-flow studies were conducted using a thermostated (±0.1 °C) SX-18MV, Applied Photophysics instrument coupled to an online data acquisition system. Reaction rates were measured under pseudo-first-order conditions ([Nu] ≫ [AuCl4]−), at a constant ionic strength (0.4 M NaCl). The reactions were started by mixing 2 × 10−4 M [AuCl4]− with 2 × 10−3 M nucleophile. The reported rate constants are the mean values of at least 5 kinetics runs. The working wavelengths were determined from UV-vis spectra obtained on Cary 5G and Hewlett-Packard 8452A diode-array spectrophotometers equipped with thermostated cell holders and a 0.88 cm path length tandem cuvette. Ambient temperature rapid scan spectra were recorded with the use of the stopped-flow spectrometer mentioned above coupled to a J&M TIDAS diode-array detector. Complete spectra were recorded between 280 and 650 nm with the integrated software J&M TIDAS-DAQ 2.3.7.4. The software used for the final data processing included Excel 7, Specfit and OriginLab 6. The pseudo-first-order rate constants, kobsd, were obtained from the fit of absorbance changes as a function of time, where k1 and k2 were calculated from the intercept and slope of the linear plots of kobsdversus the nucleophile concentration, respectively.
Cyclic voltammetry.
All electrochemical measurements were performed on an Autolab PGSTAT 30 unit at room temperature using a three electrode configuration. Working, counter and reference electrodes were glassy carbon (GC) (Metrohm) (disc diameter of 2.0 ± 0.1 mm), platinum wire (Metrohm) and a saturated calomel electrode (SCE, 0.24 V vs. NHE; Fisher Scientific), respectively. The reaction solution (10 mL) was deoxygenated by passing a stream of pure N2 prior to measurements. During the measurement the atmosphere above the solution was kept inert with a constant flow of N2. The reactions were performed in a 0.4 M NaCl solution as the supporting electrolyte. All voltammograms were recorded with a scan rate of 0.1 V s−1, and measured over the potential range +1.5 to −1.5 V or +1.5 to 0 V, for at least two cycles. The working electrode surface was polished with 0.05 μM alumina (Buechler) on a microcloth with water as a lubricant.
1H NMR.
1H NMR spectra were recorded for the reaction of [AuCl4]− with DMSO, in D2O. Different concentrations of the reactants were employed. The spectra were recorded on a Bruker AVANCE DRX 400WB spectrometer. The measurements were performed with a commercial 5 mm Bruker broadband probe thermostated with a Bruker B-VT 3300 (DPX)/3000 (DRX) variable-temperature unit. Sodium trimethylsilylpropane-3-sulfonate (TSP) was used as an internal reference. All NMR spectra were processed using the NMRnotebook software and the chemical shifts are reported in parts per million (ppm).
Quantum chemical methods.
We fully optimized the structures at the RMP2(full)/LANL2DZp level of theory16 with no constraints, while the influence of the bulk solvent was approximated using the CPCM-formalism17 as implemented (applying the default settings and water as a solvent) in RMP2(full)(CPCM)/LANL2DZp. The complexes were characterized as local minima by computation of vibration frequencies RMP2(full)(CPCM)/LANL2DZp in Gaussian 09.18 Note that the relative energies were corrected throughout for zero point vibrational energies (ZPE) (RMP2(full)(CPCM)/LANL2DZp).
Results and discussion
General information
Since [AuCl4]− species are not stable in aqueous solution and undergo spontaneous hydrolysis,19 we suggest that under the selected conditions Au(III) exists as [AuCl(OH)3]− that causes a decrease in pH as presented in Scheme 1. Since a 0.4 M NaCl solution was employed to maintain identical experimental conditions for kinetic as well as cyclovoltammetric studies, dissolving K[AuCl4] in neutral aqueous solution resulted in a decrease in pH to a final value of 4.28 in the reaction medium.
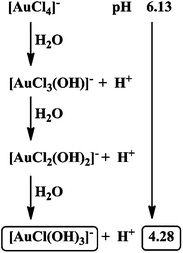 |
| | Scheme 1 Formation of reactive Au(III) species under the studied experimental conditions. | |
Reaction with thiourea
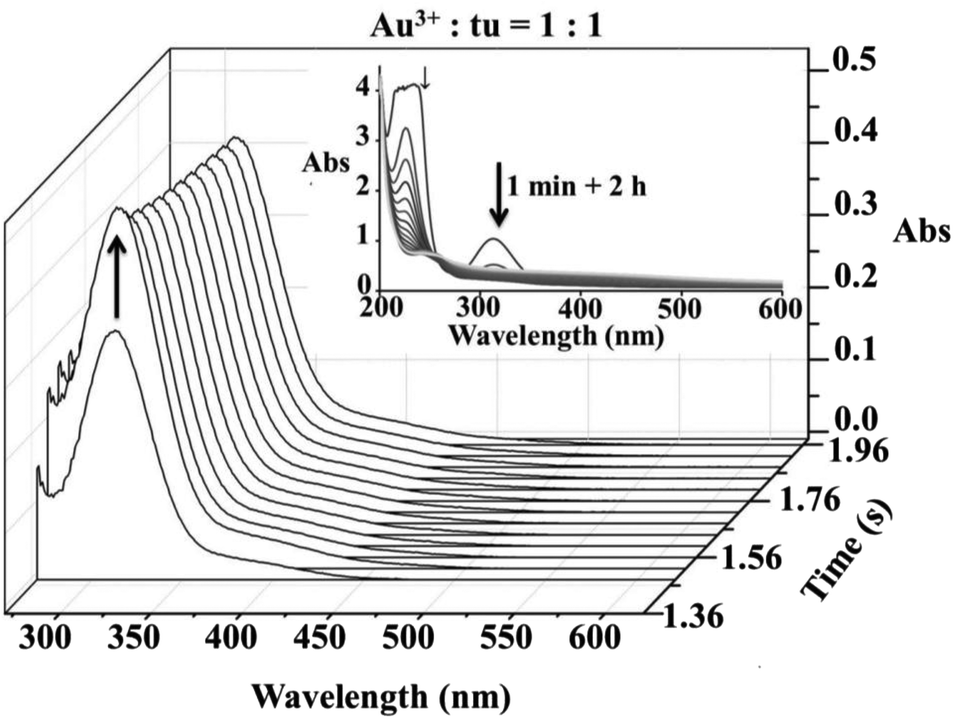
Spectrophotometric and cyclic voltammetric measurements show changes during the reaction of equimolar concentrations of [AuCl4]− and thiourea that correspond to the reduction of Au(III) to Au(I), whereas an excess of thiourea leads to complete reduction and formation of elemental gold. During these measurements the initial yellow colour of the solution faded and a suspension of a dark solid formed. However, in a very short time scale (up to 0.2 s), rapid-scan spectra indicated an initial substitution step accompanied by an increase in the absorbance band at 330 nm, see Fig. 1. The same spectral changes were observed under pseudo-first order reaction conditions during the typical stopped-flow measurements as shown in Fig. 2. The obtained kinetic data will be discussed later.
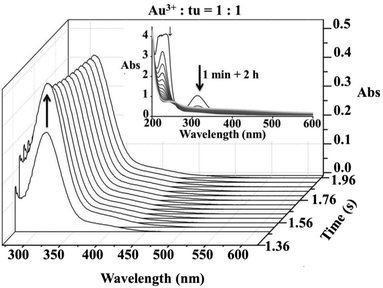 |
| | Fig. 1 Rapid-scan spectral changes observed during the first 2 s of the reaction between [AuCl4]− and thiourea in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio; inset – the same reaction progress followed on a longer time scale. :1 molar ratio; inset – the same reaction progress followed on a longer time scale. | |
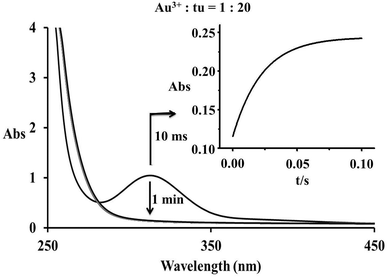 |
| | Fig. 2 Spectral changes observed for the redox reaction of 1 × 10−4 M [AuCl4]− and 2 × 10−3 M thiourea in 0.4 M NaCl aqueous solution; inset – kinetic trace recorded for the initial substitution step observed on a millisecond time-scale; T = 298.3 K; λ = 300 nm. | |
When using higher concentrations of the reactants in the electrochemical study, it was not possible to detect the initial substitution step anymore. Cyclic voltammetry appeared to be suitable for detecting the formation of Au(I). The cathodic peak observed at 0.32 V is known to be due to the reduction of Au(III) species. During the reaction of an equimolar amount of [AuCl4]− and thiourea, a new peak located at 0.46 V versus SCE formed in 30 min, see Fig. 3a. No further Au(III) species could be detected, whereas the observed shift to more positive potential is characteristic of the formation of Au(I).20 From the observed disappearance of all peak currents during the reaction with an excess of thiourea, it is obvious that complete reduction of Au(III) occurred, see Fig. 3b. At the same time the oxidation of thiourea probably leads to the formation of a disulfide bond.
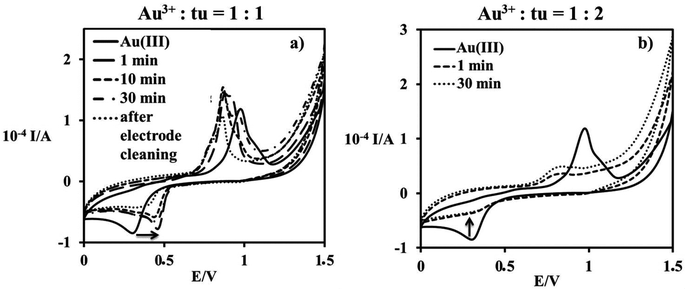 |
| | Fig. 3 Cyclic voltammograms for the reaction of 1 mM [AuCl4]− and different concentrations of thiourea: (a) 1 mM, (b) 2 mM, in 0.4 M NaCl; T = 298 K; GCE; E = 0.1 V s−1. | |
According to a study by Prudy et al., the oxidation of thiourea is pH-dependent21 such that different products can be formed. Since the detection of such products goes beyond the aims of this study, we suggest that the observed reactions can be summarized by eqn (1)–(5) based on available literature data:22,23
| | | AuCl4− + 3e− → Au0 + 4Cl−, E0 = 0.994 V | (1) |
| | | Au+ + e− → Au0, E0 = 1.2 V | (2) |
| | | 2CS(NH2)2 → NH2C(:NH)S·SC(:NH)NH2 + 2H+ + 2e−, E0 = 0.420 V | (3) |
| | | Au3+ + 3tu → [Au(tu)]+ + tu2 + 2H+ | (4) |
| | | [Au(tu)]+ → Au0 + 1/2tu2 + H+ | (5) |
Note that in situ produced formamidine disulfide can act as an active oxidant for further dissolution of gold according to the overall reaction:24
| Au0 + 1/2(SC(NH)NH2)2 + SC(NH2)2 + H+ → Au(SC(NH2)2)2+ |
A similar reaction with an excess of thiourea is to be expected: Au0 + 2tu → [Au(tu)2]+ + e−, while further oxidation of thiourea would lead to decomposition and formation of elemental sulfur.24
Reaction with bromide
Similar to the earlier results obtained for this reaction,25 absorbance changes observed for the reaction between [AuCl4]− and bromide indicate a single substitution step, see Fig. S1 (ESI),† that leads to the formation of [AuBr(OH)3]− as a result of partial hydrolysis as shown in Scheme 2.19
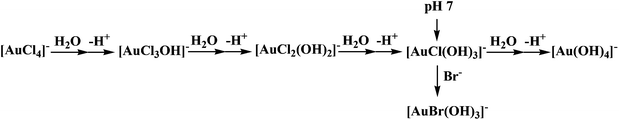 |
| | Scheme 2 Proposed reactive species and reaction products for the reaction of [AuCl4]− with Br− in 0.4 M NaCl. | |
Furthermore, cyclic voltammetric measurements did not show any meaningful shift during the reaction of Au(III) with lower bromide concentrations (1 and 2 mM) due to the influence of the large excess of chloride on the equilibrium speciation. In a series of experiments, addition of bromide caused a shift in the equilibrium towards the formation of the bromide complex of Au(III) (solution colour changes from yellow to orange), which electro-stability seems to be similar to that of the chloride complex, see Fig. S2.† A reaction with 20 mM bromide caused a shift of the electrode potential to a more negative value, from 0.48 to 0.43 V, followed by the appearance of a new reduction signal at 0.82 V, characteristic of free bromide in solution (Fig. S2†).
In terms of the stability of halogenide complexes, there is a well-known rule that “hard” metal cations form weak halogenide complexes in the order F− ≫ Cl− > Br− > I−, whereas “soft” metal ions form strong halogenide complexes with the stability order F− ≪ Cl− < Br− < I−.26,27 In the case of transition metal ions, the most important role is played by the d electron configuration.26 Furthermore, in comparison to dissolving elemental gold in halogenide solution, in the case of bromide it leads to the formation of the more stable [AuBr4]−.28 The lower electronegativity of iodide compared to the other halogenides could be a reasonable explanation for its redox activity toward gold(III).
Except for this rule, standard redox data available from the literature explain why bromide does not form a redox couple with Au(III).22 The standard redox potential for the reaction Br2 + 2e− → 2Br− is 1.087 V, which is higher than that for iodide (I2 + 2e− → 2I−, E0 = 0.535 V) which can be oxidized by Au3+ (AuCl4− + 3e− → Au0 + 4Cl−, E0 = 0.994 V). The more positive standard electrode potential of the Br2/Br− couple implies its tendency to be reduced, which at the same time stabilizes gold in the +3 oxidation state.
Reaction with iodide
The reaction of Au(III) and I− has already been studied as an outer-sphere redox reaction,11 but here for the first time, the initial substitution step could be observed. In Fig. 4b the observed absorbance increase during the first 2 s of the reaction indicates that the substitution step (r1) appears initially and is followed by reduction (r2, r3), Fig. 4c. Consecutive absorbance changes show several reaction steps and, therefore, the formation of different products, see Fig. 4a. Here r1 represents the suggested substitution reaction: [AuCl(OH)3]− + I− → [AuI(OH)3]− + Cl−, whereas r2 corresponds to reduction reaction: [AuI(OH)3]− + I− → [Au(OH)2]− + I2 followed by [Au(OH)2]− + I− → Au0 + I2. Finally, the characteristic spectral changes observed after 10 s of the reaction (r3) correspond to the reaction of I− and in situ formed I2, Fig. 4c. This step involves dissolution of iodine in iodide solution in the three-electron transfer step and formation of I3−,29 as a consequence of the first reduction step (r2), see Fig. 5.
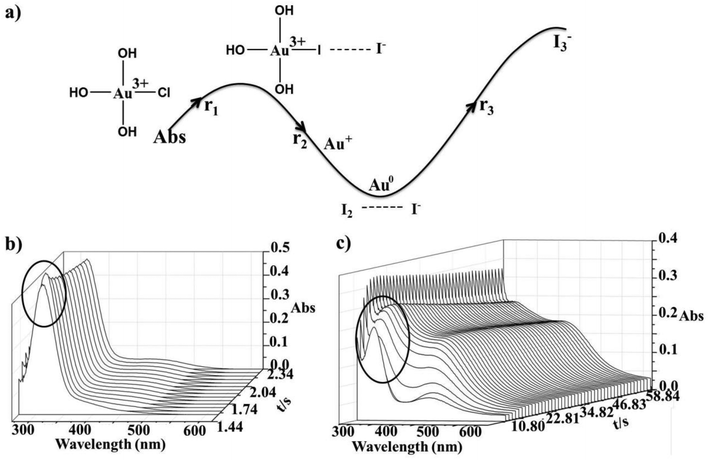 |
| | Fig. 4 Spectral changes recorded after rapid mixing of 1 × 10−4 M [AuCl4]− and an equimolar concentration of iodide; (a) trend in the absorbance changes upon formation of different reaction products, λ = 285 nm, (b) during the first 3 s of the reaction, and (c) the next 60 s. | |
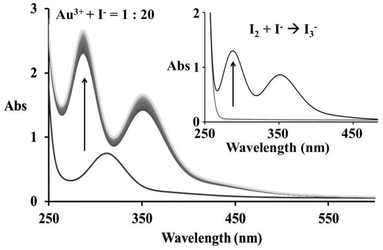 |
| | Fig. 5 Absorbance spectra recorded before and after addition of 2 × 10−3 M iodide to 1 × 10−4 M [AuCl4]−; inset – characteristic spectral changes for the reaction of I2 and I− leading to the formation of I3−. | |
Further results were obtained from the CV measurements. For the same concentration of the employed reactants, the observed peak current was similar to that obtained for thiourea, see Fig. S3 (ESI),† where a positive potential shift appeared during the redox reaction. In the presence of an excess of iodide, the CV curves show well-defined peaks, which indicate the electro-catalytic reduction of I− ions present in the studied solution. This demonstrates the formation of Au(I) complexes, but a further comparison with the Au(III) signal was not possible due to it being covered by the signal of I−. Furthermore, at the end of electrochemical measurements, the working electrode was covered by a deposit that was either yellow or black, but could be successfully removed.
According to the values of the standard electrode potentials,22 it can be expected that Au(III) is able to oxidize iodide. All relevant electrochemical half reactions are presented in eqn (6)–(13):22,30
| | | [AuCl4]− + 3e− → Au0 + 4Cl−, E0 = 0.994 V | (6) |
| | | [AuCl4]− + 2e− → [AuCl2]− + 2Cl−, E0 = 0.926 V | (7) |
| | | Au3+ + 3e− → Au0, E0 = 1.42 V | (8) |
| | | Au3+ + 2e− → Au+, E0 = 1.29 V | (9) |
| | | Au+ + e− → Au0, E0 = 1.68 V | (10) |
| | | I2 + 2e− → 2I−, E0 = 0.535 V | (11) |
| | | 3I2 + 2e− → 2I3−, E0 = 0.489 V | (12) |
| | | I3− + 2e− → 3I−, E0 = 0.5338 V | (13) |
Note: Iodide can react as an efficient nucleophile as well as a reductant in the reaction with Au(III) complexes, but iodine can participate in the opposite oxidation toward Au(0), which supports the disproportionation of Au(I) and formation of many more intermediate species.31 For example, 2Au0 + 2KI + I2 → 2KAuI2.32
Reaction with nitrite
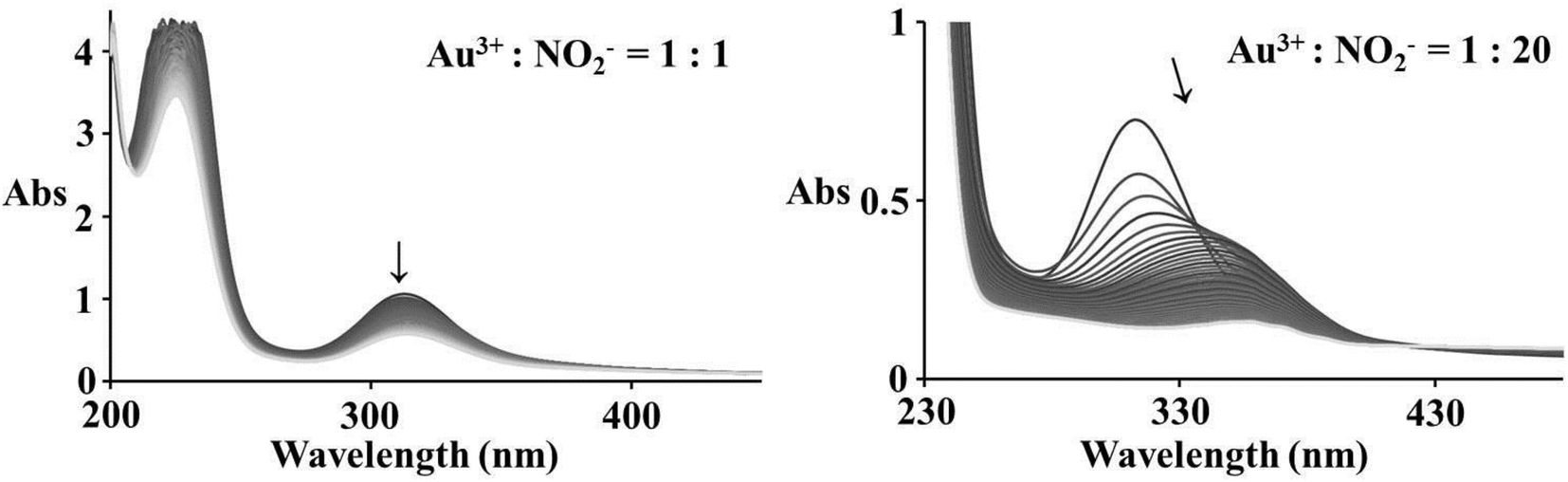
Spectrophotometric measurements show significant changes during the reaction of equimolar amounts of Au(III) and nitrite, corresponding to a slow reduction reaction, whereas an excess of nucleophile indicates first a substitution followed by a slower reduction leading to the complete decrease of all absorbance bands, see Fig. 6. Since nitrite appears to be not a strong enough nucleophile for Au(III), substitution seems to be very slow in the case of equimolar amounts of Au(III) and NO2−. On the other hand, coordinated chloride is capable of reacting as an efficient electron transfer bridge,31 and is suggested to be responsible for a dominant redox reaction with nitrite.
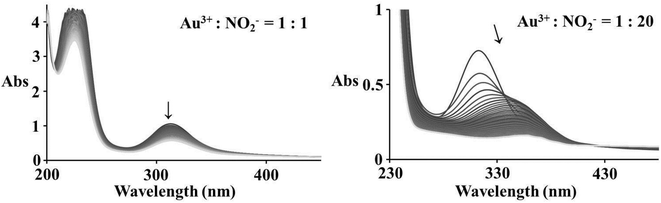 |
| | Fig. 6 Spectral changes observed during the reaction of 2 × 10−4 M [AuCl4]− and different concentrations of NO2− in 0.4 M NaCl aqueous solution; the arrows show the reaction progress during 2 h (left) and 1 h (right). | |
The results obtained from the CV measurements are not relevant due to the formation of a yellow precipitate on the Pt-wire. Even upon stirring and backward scanning, most of the precipitate associated with the peak was still observed. Since higher concentrations of the reactants were employed in the electrochemical measurements, a dark precipitate of elemental gold was visible during these measurements.
The obtained results also agree with the values of the standard electrode potentials. Au(III) with a bit more positive standard electrode potential ([AuCl4]− + 3e− → Au0 + 4Cl−, E0 = 0.994 V) shows the ability to oxidize nitrite, characterized by a more negative value of E0, NO2− + H2O → NO3− + 2H+ + 2e−, E0 = −0.94 V.33 Furthermore, since it is well known that nitrite disproportionates in aqueous solution, HNO2(aq.) → H3O+ + NO3− + 2NO, the reaction is forced to proceed in the direction of nitrate formation. Furthermore, the cathodic peak observed at −1 V during the reaction, Fig. S4 (ESI),† characteristic of a decrease in pH,34 proves the participation of water molecules in the redox process leading to formation of H3O+ ions. The decrease in pH value was also observed during the reaction of 1 mM [AuCl4]− with 20 mM NO2− leading to the final pH of ca. 3.4.
In comparison to the other employed nucleophiles, nitrite appears to be the weakest in the studied series, tu ≈ I− > py > Br− > NO2−, which is to be expected in the terms of the “hardness” of nitrogen donor nucleophiles.
Reaction with pyridine
The most surprising results were obtained for the reaction of Au(III) with pyridine. The reaction of equimolar amounts of [AuCl4]− and py is accompanied by an absorbance change with a sharp isosbestic point at 295 nm, corresponding to a substitution reaction, see Fig. 7. Further experiments in the presence of an excess of nucleophile resulted in a two step reaction. The first observed substitution reaction was followed by a reduction step, clearly presented in Fig. 7. Spectral changes follow the same direction of an absorbance decrease in the range >280 nm, but this time followed by a reduction.
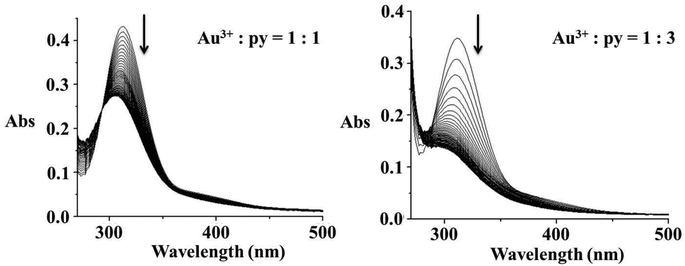 |
| | Fig. 7 Rapid-scan spectral changes recorded for the reaction between [AuCl4]− and pyridine in different molar ratios over 50 s; left – the observed substitution reaction, right – substitution followed by redox reaction. | |
CVs show a similar result. The starting signal at 0.31 V of Au(III) shifted to a more negative value, 0.15 V, upon the formation of [Au(OH)3py]. Measurements repeated after 30 min showed the formation of a possible equilibrium due to the presence of a large excess of chloride, Fig. 8a:
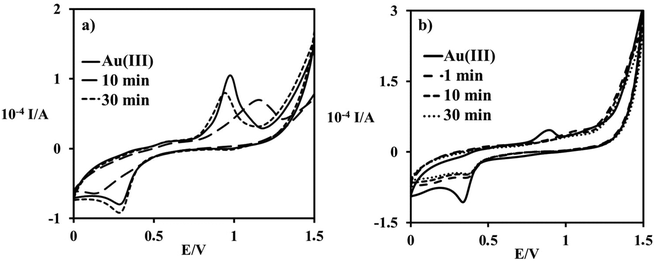 |
| | Fig. 8 Cyclic voltammograms recorded during the first 30 min of the reaction between 1 mM [AuCl4]− and (a) 1 mM pyridine; (b) 20 mM pyridine, in 0.4 M NaCl aqueous solution; GCE; E = 0.1 V s−1. | |
The CVs recorded in the presence of an excess of pyridine are very similar to each other, and a representative example is shown in Fig. 8b. During the reaction the signal of Au(III) species disappears, while the new species formed show an irreversible reduction peak in a potential range higher than 0.35 V, corresponding to reduced gold: [Au(OH)3py] + excess py → [Au(OH)2]− + pyN–O− + 2H+. Due to the fact that significant signals of Au(III) (315 nm detected by UV-vis and 0.31 V detected by CV) disappear during the progress of the reaction, and the formation of elemental gold was not observed, we conclude that reduction of Au(III) proceeds to the formation of Au(I).
It is noteworthy that in the case of higher concentration of the reactants, the formed precipitate prevented NMR measurements and the detection of N-oxidized pyridine.35
Reaction with DMSO
Although Potts et al. suggested that there could be formation of a mixed valence complex between Au and DMSO, or even a complex with Au(II),36 we did not observe any evidence for a substitution reaction between Au(III) and DMSO: neither absorbance changes indicative of a substitution reaction nor potential changes caused by reduction. Obviously, Au(III) cannot coordinate to sulfoxide and does not form a redox couple.
Spectral changes indicative of a time-dependent instability of Au(III) aqueous solutions were observed during the first 12 h of the reaction between [AuCl(OH)3]− and DMSO (1:1), see Fig. S5 (ESI),† which suggested that ca. 6% degradation leading to the formation of Au(I) had occurred. Furthermore, this induced the slight oxidation of DMSO to DMSO2 observed by 1H NMR measurements, see Fig S6 (ESI).† In the expanded 1H NMR spectra, the singlet observed at 3.1 ppm is evidence for dimethylsulfone formation.37 The same signal appeared in the case of an excess of Au(III) and also when an excess of DMSO was employed. Furthermore, a similar observation was made for the reaction of [AuBr4]− with DMSO as a solvent.38
Ab initio calculations
To gain more insight into the behaviour of [AuCl4]− and DMSO, we investigated three possible aggregates: (i) DMSO O-bonded to AuCl3 and a chloride in the second coordination sphere [Cl−⋯AuCl3–OSMe2]; (ii) DMSO S-bonded to AuCl3 and a chloride in the second coordination sphere [Cl−⋯AuCl3–SOMe2]; (iii) AuCl4− separated from DMSO [AuCl4−⋯OSMe2]. The most stable species is AuCl4− separated from DMSO [AuCl4−⋯OSMe2] (see Scheme 3). Whereas the methyl groups are oriented towards the AuCl4− moiety, the gold ion and the DMSO sulfur atom are separated by 4.6 Å. The oxygen bound substitution product [Cl−⋯AuCl3–OSMe2] is ca. 15 kcal mol−1 higher in energy, demonstrating the high stability of the Au–Cl-bond.39 The S-bonded DMSO–[AuCl4]− adduct is an additional 8 kcal mol−1 higher in energy. It is interesting to note that the cis-influence of DMSO is negligible and independent of whether it is S- or O-bonded, whereas the difference in the trans-influence is clearly visible between the uncharged S- and O-bound DMSO and the coordinated chloride (see Fig. 9).
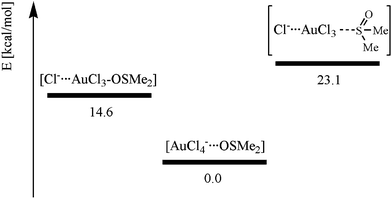 |
| | Scheme 3 Energetic differences (RMP2(full)(CPCM)/LANL2DZp) of the three possible aggregates formed between DMSO and [AuCl4]−. | |
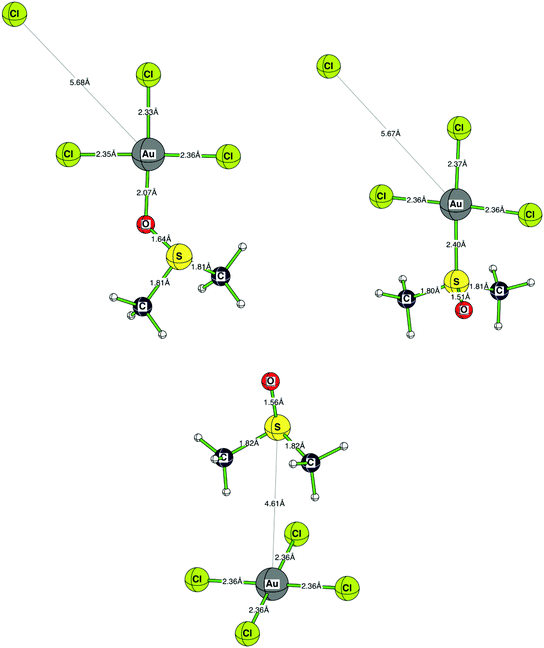 |
| | Fig. 9
Ab initio calculated (RMP2(full)(CPCM)/LANL2DZp) structures of the three possible aggregates formed between DMSO and [AuCl4]−. | |
Since [AuCl(OH)3]− is the predominant species in solution, we also investigated this species being well aware of the fact that this system is severely hampered by intermolecular hydrogen bonding in the calculations. Most probably, hydrogen bonding patterns in aqueous solution will not be the same as in the gas phase calculations due to the surrounding water molecules. The order of the three investigated species is: the most stable state [AuCl(OH)3]− and DMSO hydrogen bonded, DMSO O-bonded to Au(OH)3 and Cl− bound by hydrogen bonding (+5.2 kcal mol−1), and DMSO S-bonded to Au(OH)3 and Cl− chelated by H-bonding (+13.5 kcal mol−1) are identical. The absolute energy values between these systems are somewhat smaller than in the [AuCl4]− case, most probably due to the different hydrogen bonding in the gold-hydroxy species and that no strong Au–Cl bonds are anymore involved39 (see Scheme 4). In the most stable species, [AuCl(OH)3]− and DMSO are separated by 3.81 Å. This significantly shorter distance than in [AuCl4]− is due to the hydrogen bonds binding DMSO to the OH− ligands of 1.87 Å and 1.91 Å. As expected, the Au–S distance (2.37 Å) is much longer than the Au–O distance (2.10 Å) in the gold–DMSO complexes. In contrast, the bond length to the trans-OH− ligands (1.98 Å and 1.99 Å) and to the cis-OH− ligands (2.01 Å/2.02 Å and 2.01 Å/2.03 Å) are nearly identical and show no large cis- or trans effects. We attribute this again to the involved hydrogen bonds. In contrast, the distance between the gold(III) cation and the chloride anion is significantly different (4.13 Å as compared to 4.07 Å), see Fig. 10.
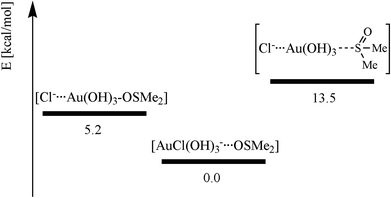 |
| | Scheme 4 Energetic differences (RMP2(full)(CPCM)/LANL2DZp) between the three possible aggregates formed between DMSO and [AuCl(OH)3]−. | |
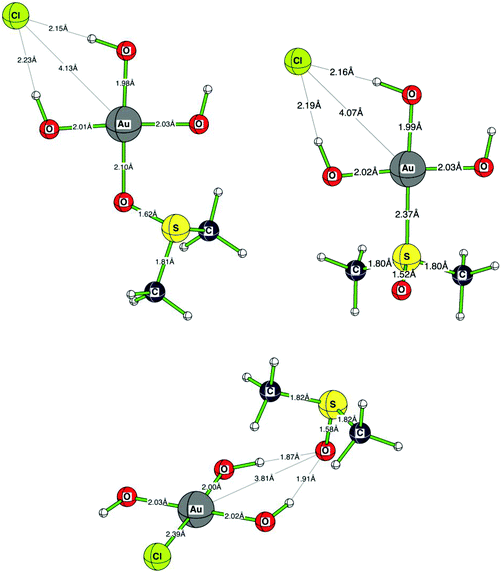 |
| | Fig. 10
Ab initio calculated (RMP2(full)(CPCM)/LANL2DZp) structures of the three possible aggregates formed between DMSO and [AuCl(OH)3]−. | |
Kinetic measurements
The kinetic traces obtained as absorbance versus reaction time plots were fitted to a double exponential function because of the influence of the subsequent redox reaction on the substitution process (for an example see Fig. 11). In both cases, whether the redox reaction results in an opposite absorbance change as in the case of thiourea, see Fig. 11, or an absorbance change in the same direction as in the case of iodide and nitrite, calculations of the rate constants gave excellent fits (a typical example is shown in Fig. S7, ESI†).
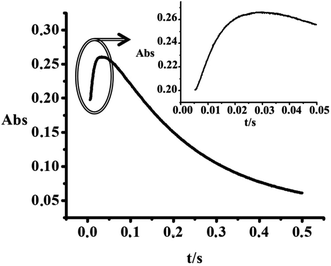 |
| | Fig. 11 Kinetic trace obtained for the reaction of 1 × 10−4 M [AuCl4]− with a hundred-fold excess of thiourea, T = 287.9 K; λ = 300 nm. | |
The obtained pseudo-first-order rate constants for the first reaction step, kobsd (data are given in Tables S1–S5, ESI†), were plotted against the concentration of the entering nucleophiles. From Fig. 12 it follows that a linear dependence on the nucleophile concentration with a significant intercept was observed in most cases.
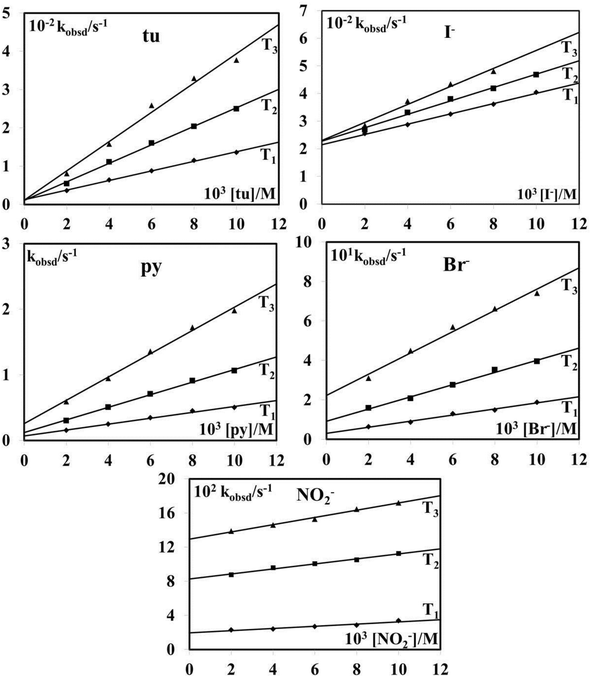 |
| | Fig. 12 Pseudo-first-order rate constants as a function of nucleophile concentration for the reaction of [AuCl4]− with thiourea, iodide, bromide, pyridine and nitrite in 0.4 M NaCl aqueous solution at three different temperatures: T1 (288 K), T2 (298 K), T3 (310 K). | |
The rate constants k1 and k2, summarized in Table 1, represent the intercept and slope of the plots, respectively, and were measured at three different temperatures, viz. 288, 298 and 310 K. Due to different reactions that can contribute to the studied substitution process, the rate law could not be defined in a general form for all studied reactions. The temperature dependence of the obtained rate constants enabled the calculation of the activation enthalpies (ΔH≠) and entropies (ΔS≠) for k2 using the Eyring equation, which are included in Table 1. The significantly negative activation entropies found for the studied reactions support the operation of an associative substitution mechanism.
Table 1 Rate constants and activation parameters for the reactions of [AuCl4]− with the studied nucleophiles in 0.4 M NaCl aqueous solution
| |
λ (nm) |
T (K) |
k
2/M−1 s−1 |
ΔH≠2/kJ mol−1 |
ΔS≠2/J K−1 mol−1 |
k
1/s−1 |
| Thiourea |
300 |
287.9 |
(12.5 ± 0.3) × 103 |
35 ± 4 |
−60 ± 15 |
(1.2 ± 0.2) × 101 |
| 298.3 |
(24.2 ± 0.7) × 103 |
(1.1 ± 0.5) × 101 |
| 310.0 |
(38 ± 3) × 103 |
(1 ± 2) × 101 |
| Iodide |
380 |
287.9 |
(18.7 ± 0.5) × 103 |
16.1 ± 0.3 |
−122 ± 1 |
(21.4 ± 0.4) × 101 |
| 298.2 |
(24 ± 1) × 103 |
(22.8 ± 0.8) × 101 |
| 310.1 |
(33 ± 3) × 103 |
(23 ± 2) × 101 |
| Pyridine |
310 |
288.0 |
(4.5 ± 0.3) × 101 |
43 ± 4 |
−77 ± 13 |
(7 ± 2) × 10−2 |
| 298.3 |
(9.6 ± 0.3) × 101 |
(12 ± 2) × 10−2 |
| 310.2 |
(17.8 ± 0.8) × 101 |
(26 ± 5) × 10−2 |
| Bromide |
350 |
288.2 |
(1.5 ± 0.1) × 101 |
40 ± 4 |
−100 ± 13 |
(3.0 ± 0.7) × 10−2 |
| 298.2 |
(3.1 ± 0.2) × 101 |
(9 ± 1) × 10−2 |
| 310.2 |
(5.4 ± 0.4) × 101 |
(22 ± 2) × 10−2 |
| Nitrite |
310 |
288.6 |
1.3 ± 0.2 |
40 ± 11 |
−118 ± 36 |
(2.0 ± 0.1) × 10−2 |
| 298.6 |
2.9 ± 0.2 |
(8.3 ± 0.1) × 10−2 |
| 310.0 |
4.2 ± 0.2 |
(12.9 ± 0.2) × 10−2 |
The only reaction observed as a substitution process without any doubt is the reaction with an excess of Br−. Since [AuCl4]− undergoes hydrolysis and exists in equilibrium with other species (see Scheme 1), we expect to have [AuCl(OH)3]− as the main reactive species under these conditions (pH 6.13). The observed pseudo-first-order rate constant as a function of the nucleophile concentration is described by eqn (14), and the rate constants can be determined from the slopes (k2) and intercepts (k1) of the plots, see Table 1. The observed intercept suggests the presence of a back reaction, since Cl− and Br− show similar nucleophilicities. The overall reaction can be formulated as:
for which the rate expression is given by
| | | kobsd = k1[Cl−] + k2[Br−] | (14) |
The weakest influence of the subsequent redox reaction was found for the reaction of Au(III) with thiourea due to the clearly separated first step, presented in Fig. 2. Similar to the reaction with Br−, the observed rate constants for the initial substitution reaction with thiourea can be expressed as given in eqn (14), and is followed by the redox process. For comparison, a typical observed rate constant for the substitution step, which appears as an absorbance increase (see Fig. 11), is kobsd/subst = 245.51 ± 0.01 s−1, whereas a typical value for the subsequent redox step, observed as an absorbance decrease, is kobsd/redox = 4.370 ± 0.002 s−1. Thus the substitution process is ca. 60 times faster than the subsequent redox reaction.
For the reaction with iodide, the absorbance spectra clearly indicate that the reaction does not occur in a single step, but as two subsequent steps, see Fig. 4. Due to the large absorbance change at 285 nm for the reaction with excess iodide, kinetic measurements were conducted at 380 nm. From the double exponential fit, the first observed rate constant was assigned to the pseudo-first-order rate constant for the substitution step, kobsd, see Fig. S7, ESI.† Furthermore, from Fig. 13 it is clearly seen how with increasing the I− concentration, the initial substitution step dominates the reaction.
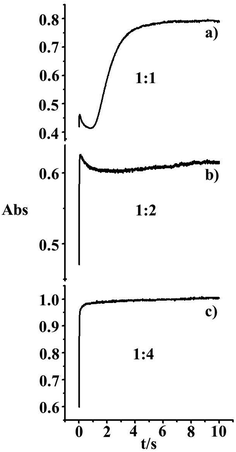 |
| | Fig. 13 Kinetic traces recorded for the reaction of 1 × 10−4 M [AuCl4]− with different concentrations of I−: (a) 1 × 10−4, (b) 2 × 10−4, (c) 4 × 10−4 M at 298 K. | |
From the fact that numerous side reactions can occur at the same time, it follows that the interpretation of the values for the rate constant k1 (see Table 1) is not as easy as in the case of the reaction with Br−. Except for the earlier described electrochemical support for this redox reaction, instability of the [AuI4]− complex is in agreement with the dissociation energy, [AuI4]− → Au0 + 3I + I−, which is lower in comparison to [AuCl4]−.39 Furthermore, Hu and Huang showed via DFT and high-level ab initio calculations that the ligand binding energy in [AuI4]− is 63 kcal mol−1 lower compared to [AuCl4]−, while the bond length is 0.339 Å longer in the case of the iodido complex.4 The iodido complex [AuI4]− has never been isolated, which is in agreement with the negative value calculated at the non-relativistic and relativistic levels of theory. This complex is characterized by the longest bond distance, the lowest dissociation energy, as well as the most negative value of ΔUSO0 for the series of [AuX4]− (X = F−, Cl−, Br−, I−) complexes (ΔUSO0 is the spin–orbit corrected ΔU0: [AuX4]− → [AuX2]− + 2X + ΔU0).39
In the case of the reaction with pyridine, the absorbance at 310 nm decreases during the first minute of the reaction, which is in line with the reduction of Au(III). However, since the substitution process shows absorbance changes in the same direction, the kobsd values were calculated in a similar way as in the case of iodide described above, and show that the rate constant of the redox reaction is controlled by ligand substitution, see Fig. S8.† Since nitrogen donor nucleophiles are not ‘soft’ enough compared to Au(III), the substitution step is slow and is followed by a fast inner-sphere electron transfer reaction, see Scheme 5.
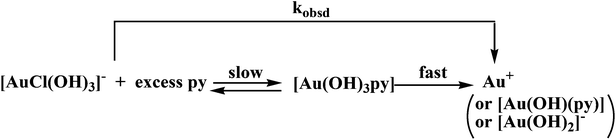 |
| | Scheme 5 Proposed reaction pathway for the reaction of Au(III) with an excess of pyridine. | |
In terms of all the results discussed above, the reaction with NO2− is also characterised as an initial substitution process with a concerted slower inner-sphere electron transfer reaction, see Fig. 14. Since nitrite is not a strong nucleophile, a competitive substitution reaction between NO2− and Cl− was observed, where the large intercept is interpreted in terms of an efficient back reaction, see Fig. 12.
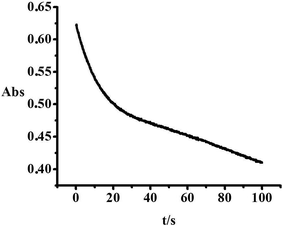 |
| | Fig. 14 Typical reaction trace obtained for the reaction of 1 × 10−4 M [AuCl4]− with the forty-fold excess of NO2−, T = 298.6 K; λ = 310 nm. | |
The kinetic parameters for the ligand substitution reactions show that the nucleophilicity as well as polarizability do not play such an important role in Au(III) chemistry as in the case of Pt(II).14,40 Apparently, the standard redox potential determines whether the reaction occurs as a reduction or only as a substitution process. In the light of this fact we can discuss the influence of the reduction process and its dependence on the nucleophilicity of the reaction partner. Based on the obtained results, the order of reactivity can be formulated as: tu ≈ I− > py > Br− > NO2−. Comparing the values of the standard electrode potential, 0.42 (tu), 0.535 (I−), 0.54 (py),41 1.087 (Br−) and 0.94 V (NO2−), the most positive value allows bromide to undergo only a substitution reaction, whereas nitrite induces a slow reduction due to the similar E0 value compared to Au(III). On the other side, tu, I− and py induce fast reduction due to the most negative value and ability to be the strongest reducing agent. Furthermore, due to their similar E0 values, the order of the nucleophiles in the substitution reaction is influenced by their “softness” and polarizability. The highest reactivity of thiourea is to be expected since it falls in the group of “soft” bases which favour “soft” substrates. The high reactivity of iodide is quite expected due to its higher polarizability compared to bromide, and iodide is, therefore, “softer” than bromide. Furthermore, the rate of redox reaction obviously depends on the electronegativity,5 where sulfur and iodide easily and rapidly reduce gold(III) due to their lower electronegativity compared to the high electronegativity of nitrogen (in pyridine and nitrite), which causes a slower reaction.
The results discussed above undoubtedly demonstrate that it is difficult to establish a nucleophilicity scale due to different oxidation states that represent gold. Therefore, it is not possible to make a direct comparison between Pt and Au.40,42
Conclusions
From the results presented here, it can be concluded that some of the studied nucleophiles undergo only substitution reactions, whereas in other cases substitution appears as a fast initial step followed by reduction. According to the kinetic results, the rapid initial step in the reaction of Au(III) with tu, I− and NO2− is a ligand substitution process. In a slower reductive elimination step, attack by excess free nucleophile leads to the final formation of elemental gold. Nucleophiles that undergo a dominant redox reaction, such as py, display similar spectral changes, consisting of an absorbance decrease and complete disappearance of the characteristic band, due to the disappearances of Au(III) species. Studies that have been carried out on the reactions with Br− yielded [AuBr(OH)3]− as the only product. The standard redox potential allows bromide not to undergo a redox reaction with Au(III). There appeared to be no direct evidence for the reaction with DMSO, which was supported by ab initio calculations.
Additional steps for Au(I) formation were detected by cyclic voltammetry on the GC electrode versus SCE, using a Pt counter electrode. Indeed, comparison of the current peak before and after addition of a nucleophile suggested that in the reactions with I−, tu, py and NO2−, there are characteristic positive peak shifts due to the formation of Au(I), whereas with Br− the detected species is still Au(III). Furthermore, some compounds are observed to undergo decomposition on the electrode surface with formation of a gold film, which inhibits the regeneration of an active electrode surface.
An associative ligand substitution mechanism seems to be common for the [AuCl4]− square-planar complex. A possible mechanism for the reduction of Au(III) to Au(I) was also discussed. The order of nucleophilicity is the one that was expected, viz. tu ≈ I− > py > Br− > NO2−. In spite of these results, it is worth mentioning that the chemical behaviour of Au(III) is not defined only by the periodic group affiliation, but also by the solvent, pH, redox potential and nucleophilicity of the coordinated ligands.
Acknowledgements
The authors gratefully acknowledge financial support from the Ministry of Science and Technological Development of the Republic of Serbia (Project no. 172011), the Deutsche Forschungsgemeinschaft and DAAD. We are also thankful to Dr Achim Zahl (University of Erlangen-Nürnberg) for his help with NMR measurements and data presentation. In addition, we thank Prof. Tim Clark for hosting this work at the CCC and the Regionales Rechenzentrum Erlangen (RRZE) for a generous allotment of computer time.
References
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- L. H. Skibsted, Adv. Inorg. Bioinorg. Mech., 1986, 4, 137 CAS.
- P. Schwerdtfeger, P. D. W. Boyd, S. Brienne and A. K. Burrell, Inorg. Chem., 1992, 31, 3411 CrossRef CAS.
- W.-P. Hu and C.-H. Huang, J. Am. Chem. Soc., 2001, 123, 2340 CrossRef CAS PubMed.
-
T. L. Brown, H. E. LeMay, B. E. Bursten and C. J. Murphy, Chemistry-The Central Science, Pearson Education, Inc., USA, 2009, ch. 8.4 Search PubMed.
- L. I. Elding and L. F. Olsson, Inorg. Chem., 1982, 21, 779 CrossRef CAS.
- L. Cattalini, A. Orio and M. L. Tobe, J. Am. Chem. Soc., 1967, 89, 3130 CrossRef CAS.
- L. I. Elding, A.-B. Gröning and Ö. Gröning, J. Chem. Soc., Dalton Trans., 1981, 1093 RSC.
- S. Elmroth, L. H. Skibsted and L. I. Elding, Inorg. Chem., 1989, 28, 2703 CrossRef CAS.
- M. D. Đurović, Ž. D. Bugarčić, F. W. Heinemann and R. van Eldik, Dalton Trans., 2014, 43, 3911 RSC.
-
M. L. Tobe and J. Burgess, Inorganic Reaction Mechanisms, Longman, England, 1999, p. 88 Search PubMed.
- L. C. Robles, C. Garcia-Olalla and A. J. Aller, Fresenius. J. Anal. Chem., 1993, 345, 441 CrossRef CAS.
-
R. J. Puddephatt, The Chemistry of Gold, Elsevier, Amsterdam, 1978 Search PubMed.
- M. Đurović, J. Bogojeski, B. Petrović, D. Petrović, F. Heinemann and Ž. D. Bugarčić, Polyhedron, 2012, 41, 70 CrossRef PubMed.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006 CrossRef CAS.
-
(a)
W. J. Hehre, L. Random, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed;
(b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS PubMed;
(c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef PubMed;
(d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed.
-
(a) V. Barone and M. Cossi, J. Phys. Chem. A, 1988, 102, 1995 CrossRef;
(b) M. Cossi, N. Rega, G. Scalmani and V. Brone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- S. Ivanova, C. Petit and V. Pitchon, Appl. Catal., A, 2004, 267, 191 CrossRef CAS PubMed.
- S. S. Seo, X. Wang and D. Murray, Ionics, 2009, 15, 67 CrossRef CAS.
- J. Kirchnerová and W. C. Prudy, Anal. Chim. Acta, 1981, 123, 83 CrossRef.
-
R. C. Weast, Handbook of Chemistry and Physics, CRS Press, Inc., USA, 57th edn, 1976 Search PubMed.
- T. Groenewald, J. Appl. Electrochem., 1975, 5, 71 CrossRef CAS.
- J. Li and J. D. Miller, Hydrometallurgy, 2002, 63, 215 CrossRef CAS.
- A. J. Hall and D. P. N. Satchel, J. Chem. Soc., Dalton Trans., 1977, 1403 RSC.
-
G. P. Haight Jr., Halogen Chemistry, ed. V. Gutman, Academic Press, New York, 1967, vol. 2, p. 351 Search PubMed.
-
C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Essex, England, 2001, ch. 16.4 Search PubMed.
-
R. B. E. Trindade, P. C. P. Rocha and J. P. Barbosa, Hydrometall., Pap. Int. Symp., Chapman & Hall, London, UK, 1994, p. 527 Search PubMed.
- P. H. Qi and J. B. Hiskey, Hydrometallurgy, 1991, 27, 47 CrossRef CAS.
- Y. A. Yaraliyev, Electrochim. Acta, 1984, 29, 1213 CrossRef.
- L. I. Elding and L. H. Skibsted, Inorg. Chem., 1986, 25, 4084 CrossRef CAS.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151 CrossRef CAS PubMed.
- T. L. Broder, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. B, 2007, 111, 7778 CrossRef CAS PubMed.
- S. Roy, T. L. Groy and A. K. Jones, Dalton Trans., 2013, 42, 3843 RSC.
- Y. Ding, W. Zhao, W. Song, Z. Zhang and B. Ma, Green Chem., 2011, 13, 1486 RSC.
- B. A. Schoenfelner and R. A. Potts, J. Inorg. Nucl. Chem., 1981, 43, 1051 CrossRef CAS.
- U. F. H. Engelke, A. Tangerman, M. A. A. P. Willemsen, D. Moskau, S. Loss, S. H. Mudd and R. A. Wevers, NMR Biomed., 2005, 18, 331 CrossRef CAS PubMed.
- R. Ettorre and A. S. Gonzáles, Inorg. Chim. Acta, 1990, 168, 221 CrossRef CAS.
- P. Schwerdtfeger, J. Am. Chem. Soc., 1989, 111, 7261 CrossRef CAS.
- G. Nardin, L. Randaccio, G. Annibale, G. Natile and B. Pitteri, J. Chem. Soc., Dalton Trans., 1980, 220 RSC.
- L. D. Burke and P. F. Nugent, Gold Bull., 1998, 31, 39 CrossRef CAS.
- H. B. Gray, J. Am. Chem. Soc., 1962, 84, 1548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00247d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence