 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The surprising lability of bis(2,2′:6′,2′′-terpyridine)chromium(III) complexes†
Edwin C.
Constable
*,
Catherine E.
Housecroft
*,
Markus
Neuburger
,
Jonas
Schönle
and
Jennifer A.
Zampese
Department of Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: edwin.constable@unibas.ch; catherine.housecroft@unibas.ch; Fax: +41 61 267 1018; Tel: +41 61 267 1008
First published on 31st March 2014
Abstract
The complex [Cr(tpy)(O3SCF3)3] (tpy = 2,2′:6′,2′′-terpyridine) is readily made from [Cr(tpy)Cl3] and is a convenient precursor to [Cr(tpy)2][PF6]3 and to [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 (4′-(4-tolyl)tpy = 4′-(4-tolyl)-2,2′:6′,2′′-terpyridine; 5,5′′-Me2tpy = 5,5′′-dimethyl-2,2′:6′,2′′-terpyridine); these are the first examples of heteroleptic bis(tpy) chromium(III) complexes. The single crystal structures of 2{[Cr(tpy)2][PF6]3}·5MeCN, [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3·3MeCN have been determined. Each cation contains the expected octahedral {Cr(tpy)2}3+ unit; in all three structures, the need to accommodate three anions per cation and the solvent molecules prevents the formation of a grid-like array of cations that is typical of many lattices containing {M(tpy)2}2+ motifs. Three reversible electrochemical processes are observed for [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3, consistent with those documented for [Cr(tpy)2]3+. At pH 6.36, aqueous solutions of [Cr(tpy)2][PF6]3 are stable for at least two months. However, contrary to the expectations of the d3 Cr3+ ion being a kinetically inert metal centre, the tpy ligands in [Cr(tpy)2]3+ are labile in the presence of base; absorption and 1H NMR spectroscopies have been used to monitor the effects of adding NaOH to aqueous and CD3OD solutions, respectively, of the homo- and heteroleptic complexes. Ligand dissociation is also observed when [Bu4N]F is added to CD3OD solutions of the complexes, but in aqueous solution, [Cr(tpy)2][PF6]3 is stable in the presence of fluoride ion.
Introduction
The coordination chemistry of 2,2′:6′,2′′-terpyridine (tpy) is a mature area which continues to receive widespread attention with a myriad of potential applications.1 Significantly, complexes of the group 8, 9 and 10 metals dominate the literature. We are particularly interested in the use of tpy complexes for applications related to energy conversion, where ruthenium(II) compounds are the state-of-the-art sensitizers in Grätzel-type photovoltaic solar cells.2,3 Ruthenium is one of the platinum-group metals, the total world accessible resources‡ of which are estimated to be of the order of 108 kg.4 The relative scarceness of the platinum-group metals contrasts with the terrestrial abundances of first row transition metals such as iron, chromium, copper and zinc, world resources‡ of which are estimated to be between 1.9 × 1012 (Zn) and >2.3 × 1014 (Fe) kg.4 As a part of our efforts towards developing a sustainable materials chemistry for application to energy conversion systems, we are currently focusing attention on applications of earth abundant metals in dye-sensitized solar cells (DSCs)5 and light-emitting electrochemical cells (LECs) and organic light emitting diodes (OLEDs).6We and other groups5 have developed molecular copper-based sensitizers with energy-to-electricity conversion efficiencies approaching 3% in Grätzel-type cells. While the lability of copper(I) can be conveniently utilized to assemble heteroleptic [Cu(L)(L′)]+ dyes by ligand exchange reactions on TiO2 surfaces,5 the same property may be disadvantageous in terms of ligand dissociation with concomitant dye degradation. We have recently reported that sensitizers incorporating a {Zn(tpy)2}2+ domain can be assembled in a stepwise fashion on TiO2; we adopted this method in response to the observation that ligand exchange at zinc(II) is unexpectedly slow,7 in contrast to faster exchange at copper(I). On the other hand, once assembled on the surface, the zinc(II) dyes are stable with respect to ligand dissociation under ambient conditions.
Chromium(III) (d3) is a text-book example of a kinetically inert metal ion,8 with the rate of water exchange in the complex [Cr(OH2)6]3+ being 2.4 × 10−6 s−1.9 This low rate of ligand exchange attracted us towards investigating the use of chromium(III) complexes, and in particular those containing its {Cr(tpy)2}3+ cores, as exceptionally stable materials for applications in DSCs and/or LECs. Surprisingly, of all of the tpy complexes of earth abundant first row transition metals, those of chromium(III) are relatively rarely investigated. The series of homoleptic complexes [Cr(tpy)2]n with n = +3, +2, +1, 0, 1 has been known for many years,10–15 and has recently been the subject of in-depth studies.16 A few investigations have probed electrochemical and photophysical properties of [Cr(tpy)2]3+ or homoleptic derivatives.16–25 To the best of our knowledge, no heteroleptic complexes with {Cr(tpy)2}3+ cores have been reported. We now report a convenient route to [Cr(tpy)2]3+ and heteroleptic derivatives, and the structural characterization of representative complexes. Solution studies reveal that under certain conditions, the chelating tpy ligands in these complexes are surprisingly labile.
Experimental section
General
FT IR spectra were recorded using either a Shimadzu FTIR 8400S or a Perkin Elmer Spectrum Two spectrophotometer with solid samples introduced in a Golden Gate ATR or UATR Two, respectively. Electronic absorption spectra were recorded on an Agilent 8453 spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance-400 spectrometer.Electrochemical measurements were made on a CH Instruments 900B potentiostat using glassy carbon, platinum wire and a silver wire as the working, counter, and pseudo reference electrodes, respectively. Samples were dissolved in HPLC grade MeCN (10−4 to 10−5 mol dm−3) containing 0.1 mol dm−3 [nBu4N][PF6] as supporting electrolyte; all solutions were degassed with argon. Cp2Fe was used as internal reference added at the end of experiments.
Reactions were carried out under N2. CrCl3 was obtained from Acros Organics and was used as received. The ligands tpy,26 4′-(4-tolyl)tpy27 and 5,5′′-Me2tpy26,28 were made by literature routes. The structures of ligands used are shown in Scheme 1.
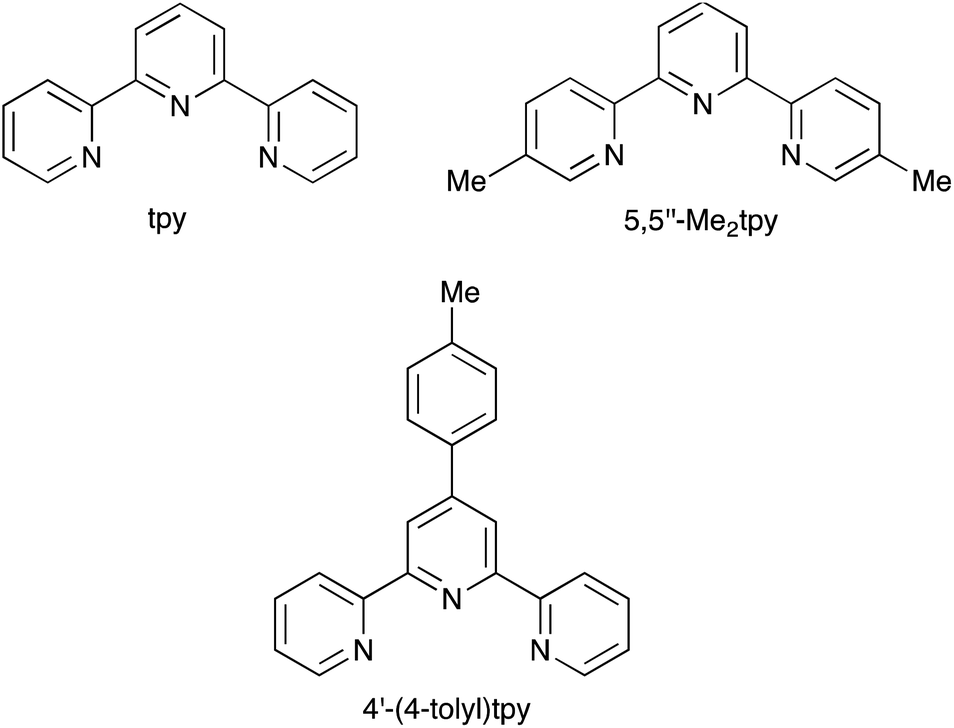 | ||
| Scheme 1 Ligand structures and abbreviations. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400), 282 sh (21700), 324 (14400), 334 (14900), 347 (15300), 363 (16700). Found C 38.18, H 2.58, N 8.86; C30H22CrF18N6P3 requires C 37.79, H 2.33, N 8.81.
700), 284 (23100), 347 (24150), 363 sh (20900). Found C 42.00, H 2.55, N 8.04; C37H28CrF18N6P3·MeOH requires C 42.43, H 3.00, N 7.81.
100), 286 sh (22300), 323 (16100), 334 (17050), 362 (14150), 362 (14150). Found C 38.73, H 3.21, N 8.53; C32H26CrF18N6P3·H2O requires C 38.45, H 2.82, N 8.41.
400), 282 sh (21700), 324 (14400), 334 (14900), 347 (15300), 363 (16700). Found C 38.18, H 2.58, N 8.86; C30H22CrF18N6P3 requires C 37.79, H 2.33, N 8.81.
700), 284 (23100), 347 (24150), 363 sh (20900). Found C 42.00, H 2.55, N 8.04; C37H28CrF18N6P3·MeOH requires C 42.43, H 3.00, N 7.81.
100), 286 sh (22300), 323 (16100), 334 (17050), 362 (14150), 362 (14150). Found C 38.73, H 3.21, N 8.53; C32H26CrF18N6P3·H2O requires C 38.45, H 2.82, N 8.41.
Reaction of [Cr(tpy)2][PF6]3 with [nBu4N]F
[Cr(tpy)2][PF6]3 (3.0 mg, 0.0031 mmol) was suspended in MeOH (2.5 ml) and [nBu4N]F (0.1 ml, 1 M in THF, ca. 33 fold excess) was added. The initially yellow mixture was stirred for 2 h, after which time a colourless solution was obtained.Crystallography
Data were collected on a Bruker-Nonius APEX2 diffractometer with data reduction, solution and refinement using the programs APEX2,29 SIR9230 and CRYSTALS31 or SHELXL97 or SHELX-13.32 The ORTEP diagrams were prepared and structure analyses carried out using Mercury v. 3.0.33,34 The structure of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN can be solved in C2/c, but is heavily disordered. The solution in Cc, refined as a twin by inversion (the Flack parameter refines to 0.416 with a su of 0.016), gives an ordered structure where the space that the tolyl group occupies on one side of the complex molecule is taken by an acetonitrile molecule on the other side. This allows for a near centrosymmetric arrangement of large parts of the structure.927(3) Å3, Z = 16, Dc = 1.654 Mg m−3, μ(Cu-Kα) = 4.390 mm−1, T = 123 K. Total 161369 reflections, 30417 unique, Rint = 0.1091. Refinement of 19918 reflections (2406 parameters) with I > 2σ(I) converged at final R1 = 0.0782 (R1 all data = 0.1232), wR2 = 0.1973 (wR2 all data = 0.2219), gof = 1.056. CCDC 981376.
498 reflections, 17525 unique, Rint = 0.032. Refinement of 12289 reflections (668 parameters) with I > 2σ(I) converged at final R1 = 0.0384 (R1 all data = 0.0509), wR2 = 0.0389 (wR2 all data = 0.0621), gof = 1.0984. CCDC 981377.
869 reflections, 8050 unique, Rint = 0.0469. Refinement of 7612 reflections (627 parameters) with I > 2σ(I) converged at final R1 = 0.0339 (R1 all data = 0.0356), wR2 = 0.0908 (wR2 all data = 0.0921), gof = 1.053. CCDC 981378.
Results and discussion
Syntheses of chromium(III) complexes
The chromium(III) complex [Cr(tpy)2][PF6]3 has previously been prepared by oxidation of [Cr(tpy)2][PF6]2 using AgPF6.16 We have found that [Cr(tpy)2][PF6]3 can be conveniently prepared by the strategy shown in Scheme 2, and this method is easily adapted to prepare heteroleptic chromium(III) complexes. The precursor [Cr(tpy)Cl3] was prepared by a modification of the method of Broomhead et al.;35,36 a similar method has been used to prepare [Cr(4′-(2-py)tpy)Cl3] (4′-(2-py)tpy = 4′-(2-pyridyl)-2,2′:6′,2′′-terpyridine).37 Treatment of [Cr(tpy)Cl3] with triflic acid results in the formation of a dark red precipitate which analyses as [Cr(tpy)(O3SCF3)3]. This compound was always freshly prepared for subsequent reactions. Treatment of [Cr(tpy)(O3SCF3)3] with tpy in MeCN at reflux followed by anion exchange (Scheme 2) leads to the homoleptic complex [Cr(tpy)2][PF6]3 in 57.0% yield. The heteroleptic complexes [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 were prepared in an analogous manner by treatment of [Cr(tpy)(O3SCF3)3] with the appropriate ligand (Scheme 2) and were isolated in 69.1 and 89.8% yields, respectively. Elemental analytical data for the complexes were satisfactory, but we were not able to obtain mass spectra by using MALDI-TOF (with or without matrix), electrospray ionization (ESI) or LC-ESI methods. All three complexes are soluble in acetonitrile, acetone, DMF, DMSO and water.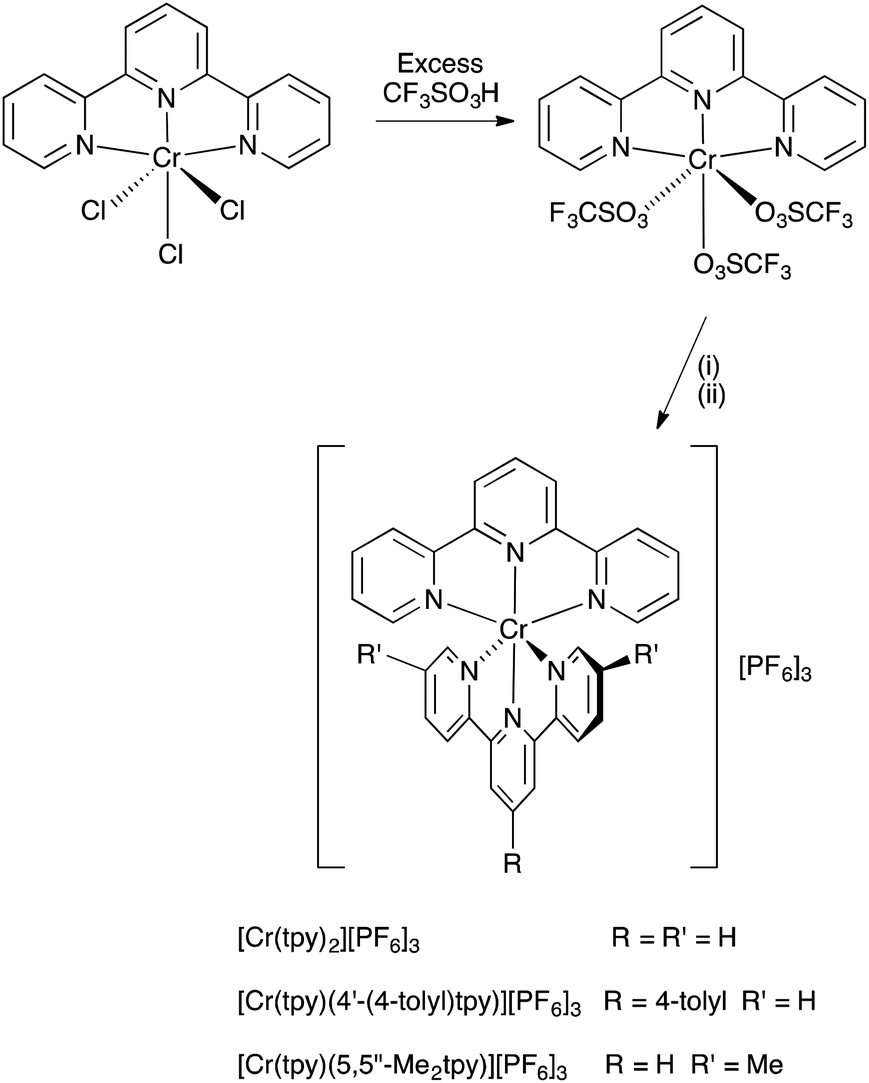 | ||
| Scheme 2 Synthetic route to homoleptic and heteroleptic bis(2,2′:6′,2′′-terpyridine)chromium(III) complexes. Conditions: (i) tpy, 4′-(4-tolyl)tpy or 5,5′′-Me2tpy, MeCN, reflux; (ii) NH4PF6. | ||
The solution absorption spectra of aqueous solutions of the complexes are shown in Fig. 1. They exhibit intense bands in the UV region arising from spin-allowed ligand-to-ligand transitions. The enhanced intensity of the band at 346 nm in the spectrum of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 compared to analogous absorptions for [Cr(tpy)2][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 is consistent with the extended conjugation onto the tolyl groups. The lower energy part of the spectrum of [Cr(tpy)2][PF6]3 corresponds to that previously reported,16 and the appearance of the spectrum of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 between 300 and 380 nm is similar to that reported for [Cr(4′-(4-tolyl)tpy)2]3+.19
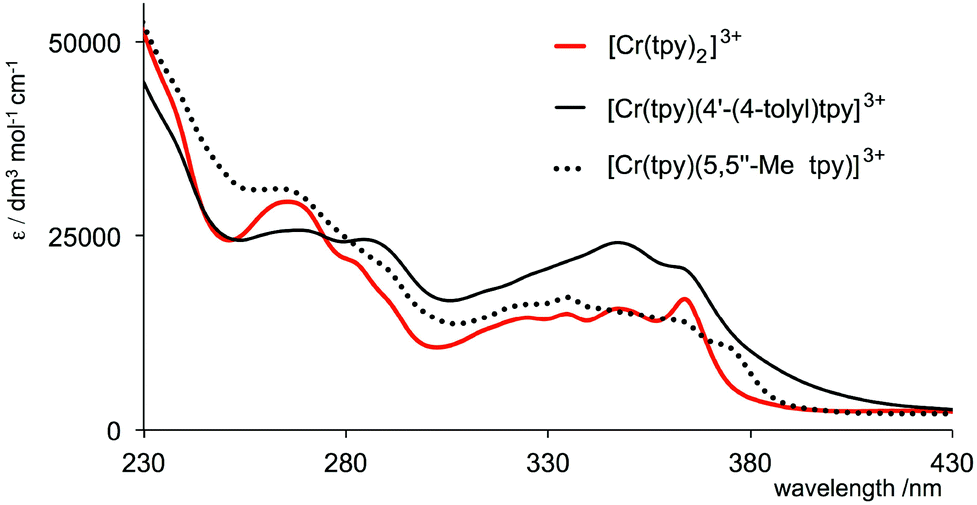 | ||
| Fig. 1 Absorption spectra of aqueous solutions (1.0 × 10−5 mol dm−3) of [Cr(tpy)2][PF6]3, [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3. | ||
Cyclic voltammetry was used to study the electrochemical processes exhibited by the complexes. Each complex shows three redox processes within the solvent accessible window (Table 1) which are fully reversible at a scan rate of 0.1 V s−1; the incorporation of the tolyl or methyl substituents has negligible effect on the redox potentials. This is consistent with trends observed between [Cr(tpy)2][ClO4]3 and [Cr(4′-Phtpy)2][ClO4]3,18 (4′-Phtpy = 4′-phenyl-2,2′:6′,2′′-terpyridine) and between [Cr(tpy)2][ClO4]3 and [Cr(4′-(4-tolyl)tpy)2][ClO4]3.19 The notation E1/23+/2+, E1/22+/1+ and E1/21+/0 used in Table 1 refers to the [Cr(tpy)2]3+/[Cr(tpy)2]2+, [Cr(tpy)2]2+/[Cr(tpy)2]+ and [Cr(tpy)2]+/[Cr(tpy)2] couples but does not imply that the formal oxidation state of chromium changes. Although the reversible waves observed in cyclic voltammograms were originally assigned to chromium-centred processes, recent elegant results from Sproules, Wieghardt and coworkers16 indicate that the redox processes are ligand-based and that chromium remains in oxidation +3 through the series. It has not been confirmed whether this formulation will hold for all complexes with substituted tpy ligands.
| Compound | E 1/2 3+/2+/V | E 1/2 2+/1+/V | E 1/2 1+/0/V |
|---|---|---|---|
| [Cr(tpy)2][PF6]3 | −0.533 | −0.953 | −1.469 |
| [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 | −0.544 | −0.946 | −1.463 |
| [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 | −0.537 | −0.957 | −1.457 |
Structural characterization
The Cambridge Structural Database38 (Conquest v. 1.16, CSD v. 5.35)33 contains only four complexes containing {Cr(tpy)2} units: [Cr(tpy)2][ClO4]3·H2O,39,40 [Cr(tpy)2][PF6]2·MeCN,16 [Cr(tpy)2][PF6]16 and [Cr(tpy)2].16 Single crystals of 2{[Cr(tpy)2][PF6]3}·5MeCN were grown by diffusion of Et2O into an MeCN solution of the complex. 2{[Cr(tpy)2][PF6]3}·5MeCN crystallizes in the orthorhombic space group Pbca with four independent [Cr(tpy)2]3+ cations in the asymmetric unit (Z′ = 4). Such an occurrence is rare. In 2008, an analysis of the CSD by Bond revealed only 71 structures in orthorhombic space groups with Z′ = 4, representing 0.19% of all entries in the CSD at that time.41Fig. 2 shows the structure of the cation containing Cr1A. Variation among the bond parameters within the octahedral coordination sphere of each of the four independent chromium(III) centres is small, as shown by the Cr–N bond distances listed in Table 2; N–Cr–N chelate angles lie in the range 77.39(15) to 79.59(17)°. These metrical parameters are consistent with those observed in [Cr(tpy)2][ClO4]3·H2O.40 Each tpy ligand is essentially planar and the angle between the least squares planes of the two ligands in each independent cation lies in the range 84.8° (cation A) to 89.5° (cation B).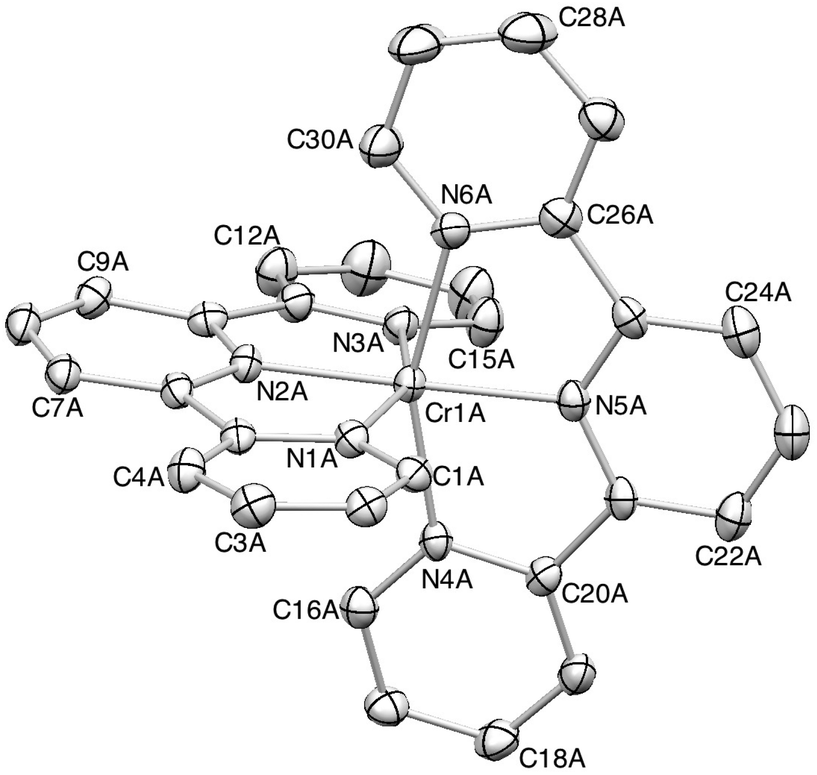 | ||
| Fig. 2 ORTEP representation of one of the four independent [Cr(tpy)2]3+ cations in 2{[Cr(tpy)2][PF6]3}·5MeCN (ellipsoids plotted at the 30% probability level, and H atoms omitted for clarity). | ||
| Bond | Distance/Å | Bond | Distance/Å |
|---|---|---|---|
| Cr1A–N1A | 2.054(3) | Cr1C–N1C | 2.099(4) |
| Cr1A–N2A | 1.985(3) | Cr1C–N2C | 2.003(4) |
| Cr1A–N3A | 2.058(4) | Cr1C–N3C | 2.035(4) |
| Cr1A–N4A | 2.042(3) | Cr1C–N4C | 2.063(4) |
| Cr1A–N5A | 1.987(3) | Cr1C–N5C | 1.983(3) |
| Cr1A–N6A | 2.062(4) | Cr1C–N6C | 2.049(3) |
| Cr1B–N1B | 2.055(3) | Cr1D–N1D | 2.039(4) |
| Cr1B–N2B | 1.976(3) | Cr1D–N2D | 1.981(4) |
| Cr1B–N3B | 2.057(3) | Cr1D–N3D | 2.049(4) |
| Cr1B–N4B | 2.062(3) | Cr1D–N4D | 2.053(4) |
| Cr1B–N5B | 1.989(3) | Cr1D–N5D | 1.978(4) |
| Cr1B–N6B | 2.042(3) | Cr1D–N6D | 2.057(4) |
It is noteworthy that packing of [Cr(tpy)2]3+ cations in 2{[Cr(tpy)2][PF6]3}·5MeCN (Fig. 3) does not involve a two-dimensional grid-like array arising from face-to-face and edge-to-face interactions between tpy domains of adjacent cations. Such arrangements are typical in salts of [M(tpy)2]2+ cations.42 However, we have previously noted that on going from [Co(tpy)2][PF6]2·2MeCN to 2{[Co(tpy)2][PF6]3}·5MeCN, the grid-like packing of the cations is lost.43 A similar observation has been made by Kuroda-Sowa and co-workers44 when comparing the packing in [Co(NCC6H4tpy)2][BF4]2·MeNO2 and [Co(NCC6H4tpy)2][BF4]3·1.5MeNO2 (NCC6H4tpy = 4′-(4-cyanophenyl)-2,2′:6′,2′′-terpyridine). Factors that contribute to the significant reorganization of the cations in the lattice are the accommodation of additional anions and inter-cation repulsion, both a consequence of an increase in the metal oxidation state, as well as accommodation of solvent molecules. Although not discussed in the original work, it is significant that [Cr(tpy)2]2+ cations in [Cr(tpy)2][PF6]2·MeCN16 (refcode BARRAP in the CSD38) pack in a two-dimensional grid-like array, whereas [Cr(tpy)2]3+ cations in [Cr(tpy)2][ClO4]3·H2O39,40 (refcodes TERPCR and TERPCR10 in the CSD38) form domains in which cations interact through tpy embraces,42 but this does not extend infinitely through the lattice.
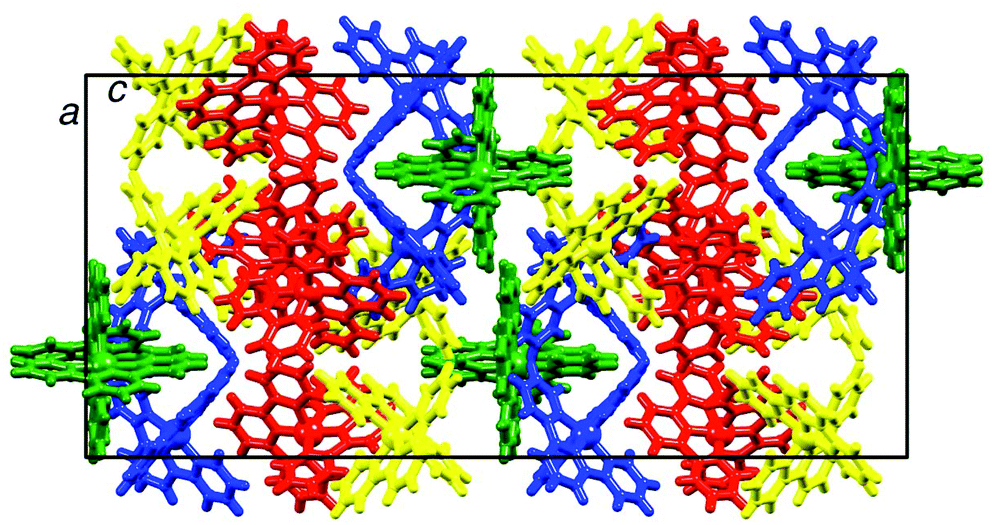 | ||
| Fig. 3 Packing of [Cr(tpy)2]3+ cations in 2{[Cr(tpy)2][PF6]3}·5MeCN. The four independent cations containing Cr1A, Cr1B, Cr1C and Cr1D are shown in blue, green, yellow and red, respectively. | ||
Orange blocks of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN grew by slow diffusion of Et2O into an MeCN solution of the complex. The compound crystallizes in the monoclinic space group Cc and Fig. 4 shows the structure of the [Cr(tpy)(4′-(4-tolyl)tpy)]3+ cation. The bond parameters in the octahedral coordination sphere (caption to Fig. 4) are similar to those in [Cr(tpy)2]3+, and the angle between the least squares planes through each near-to-planar tpy domain is 88.9°. Repulsions between the ortho-hydrogen atoms on the rings containing C23 and C31 are minimized by a twist angle of 24.4° between the rings. As in 2{[Cr(tpy)2][PF6]3}·5MeCN, the cations do not pack with the face-to-face and edge-to-face interactions. Instead, the tolyl-unit of one cation is accommodated within the cleft formed by the two pyridine rings containing N1i and N4i (symmetry code i = −1/2 + x, 1/2 − y, −1/2 + z) of the adjacent cation, resulting in the propagation of chains which run obliquely through the unit cell (Fig. 5). In Fig. 5, one interaction is represented in space-filling mode, while CH⋯π contacts are shown in red for the next analogous contact along the chain. The closest CH(methyl)⋯π contacts are 3.4 and 3.5 Å. All three anions and MeCN molecules are ordered, and extensive CH⋯F and CH⋯N interactions dominate the crystal packing forces.
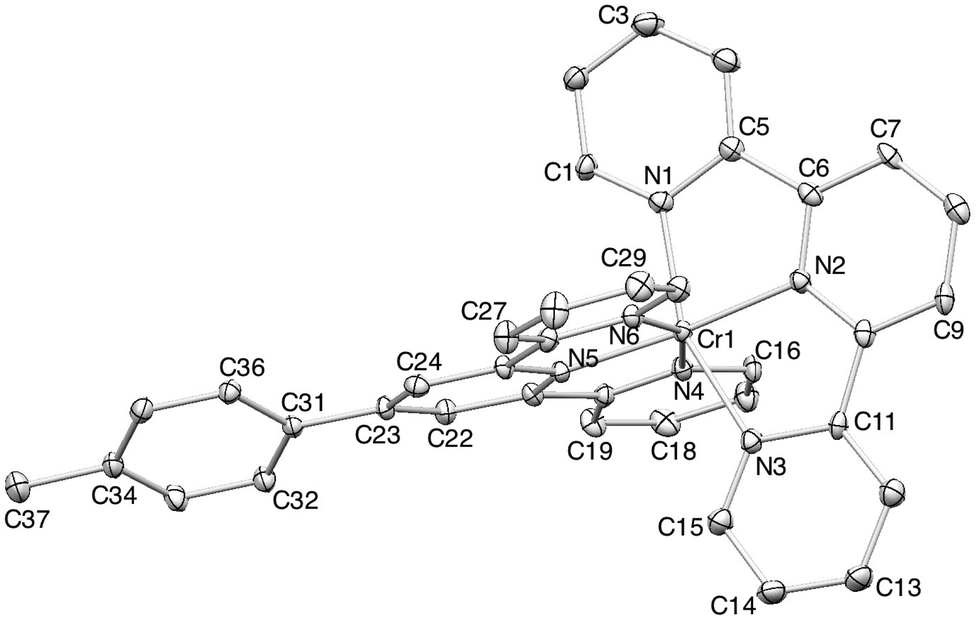 | ||
| Fig. 4 ORTEP representation of the [Cr(tpy)(4′-(4-tolyl)tpy)]3+ cation in [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN (ellipsoids plotted at the 40% probability level, and H atoms omitted). Important bond parameters: Cr1–N1 = 2.054(2), Cr1–N2 = 1.986(2), Cr1–N3 = 2.061(2), Cr1–N4 = 2.064(2), Cr1–N5 = 1.979(2), Cr1–N6 = 2.052(2) Å; N1–Cr1–N2 = 78.83(9), N2–Cr1–N3 = 78.73(9), N4–Cr1–N5 = 77.90(9), N5–Cr1–N6 = 78.67(9)°. | ||
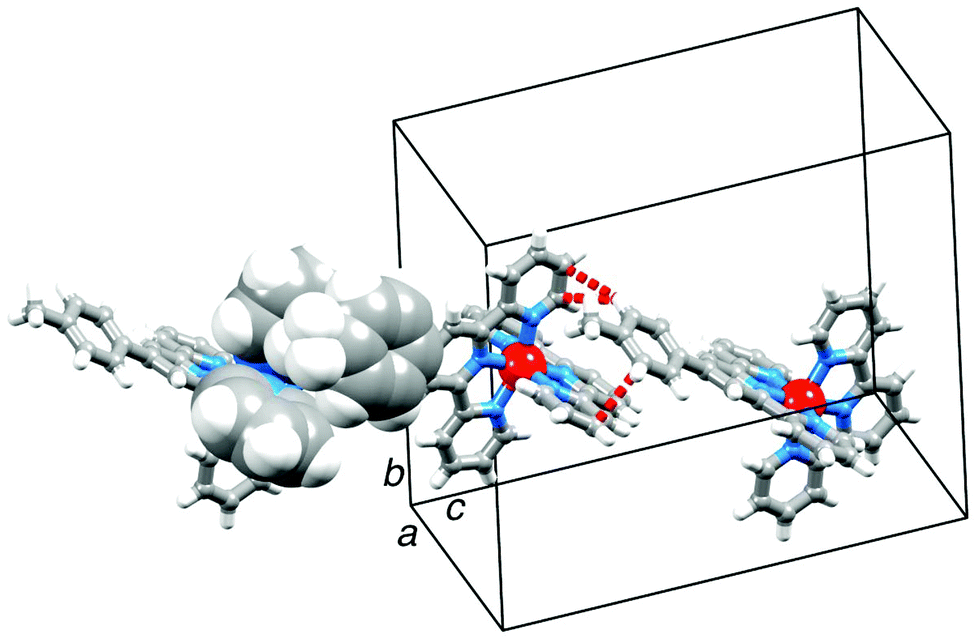 | ||
| Fig. 5 Intercation contacts in [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN. | ||
Orange blocks of [Cr(tpy)(5,5′′-Me2tpy)][PF6]3·3MeCN were grown by slow diffusion of Et2O into an MeCN solution of the compound. The structure of the [Cr(tpy)(5,5′′-Me2tpy)]3+ cation (Fig. 6) confirms the octahedral environment of the chromium(III) ion. The bond parameters given in the figure caption are in accord with those in [Cr(tpy)2]3+ and [Cr(tpy)(4′-(4-tolyl)tpy)]3+, as is the near orthogonal arrangement of the two tpy domains (angle between least squares planes = 85.8°). The 5,5′′-substitution pattern places the two methyl substituents well away from the central pyridine ring of the second ligand (closest CH(methyl)⋯centroid separation = 5.3 Å). The anions and solvent molecules are all ordered. As in the structures described above, accommodation of three [PF6]− anions and three MeCN solvent molecules per complex cation prevents the formation of inter-cation tpy embraces, and the principal packing interactions involve CH⋯F and CH⋯N contacts.
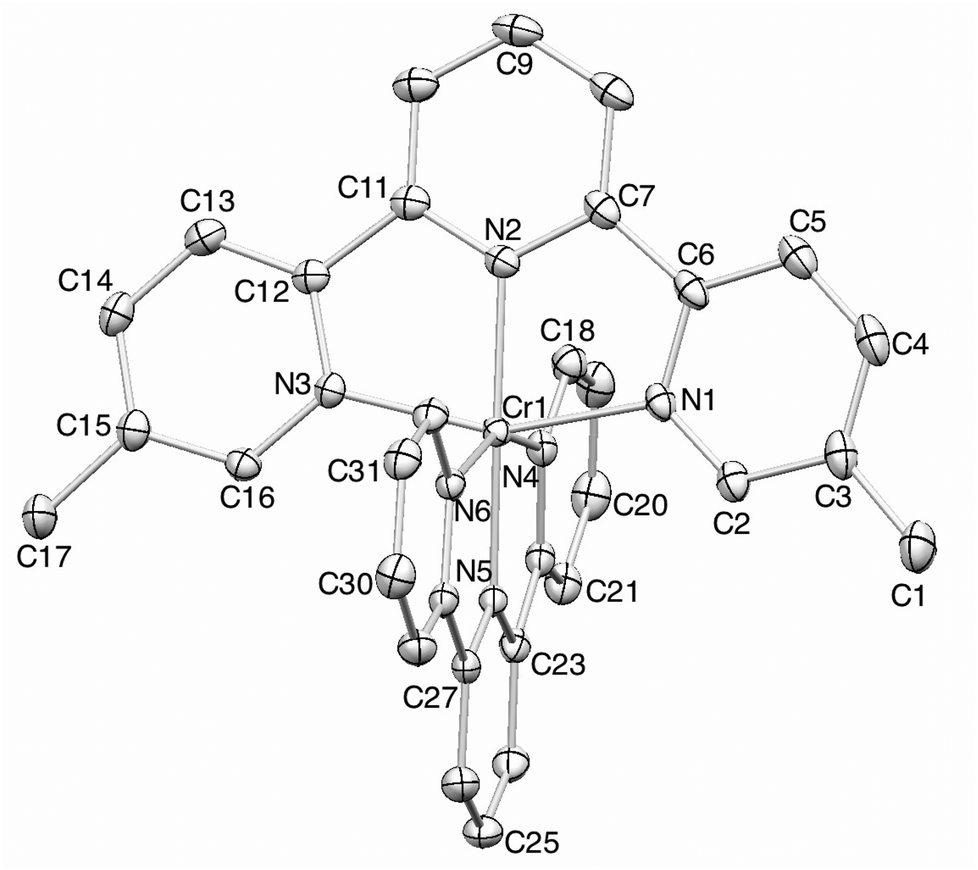 | ||
| Fig. 6 ORTEP representation of the [Cr(tpy)(5,5′′-Me2tpy)]3+ cation in [Cr(tpy)(5,5′′-Me2tpy)][PF6]3·3MeCN (ellipsoids plotted at the 40% probability level, and H atoms omitted). Selected bond parameters: Cr1–N1 = 2.0655(15), Cr1–N2 = 1.9887(15), Cr1–N3 = 2.0648(15), Cr1–N4 = 2.0715(15), Cr1–N5 = 1.9836(15), Cr1–N6 = 2.0579(15) Å; N2–Cr1–N1 = 78.53(6), N2–Cr1–N3 = 78.45(6), N5–Cr1–N4 = 78.49(6), N–Cr1–N6 = 78.65(6)°. | ||
Stability of complexes in aqueous solutions
Monitoring the absorption spectra indicates that aqueous solutions (deionized water, pH 6.36) of [Cr(tpy)2][PF6]3 are stable for at least two months (Fig. 7a). This parallels the behaviour of [Cr(bpy)3][ClO4]3 in aqueous solutions at pH < 7.45 Under basic conditions, however, it is known that [Cr(bpy)3]3+ undergoes base-catalysed ligand loss according to eqn (1). An associative mechanism has been proposed involving a seven-coordinate intermediate [Cr(bpy)3(OH2)]3+ which undergoes deprotonation at high pH, followed by loss of bpy; the proposed mechanism invokes a monodentate bpy ligand.45,46 The process under ambient conditions is the same as that of photoaquation, i.e. either the 4A2 ground or 2E excited state associates with H2O in the presence of [OH]− resulting in the overall reaction shown in eqn (1).47,48| [Cr(bpy)3]3+(aq) + 2[OH]−(aq) → [Cr(bpy)2(OH)2]+(aq) + bpy | (1) |
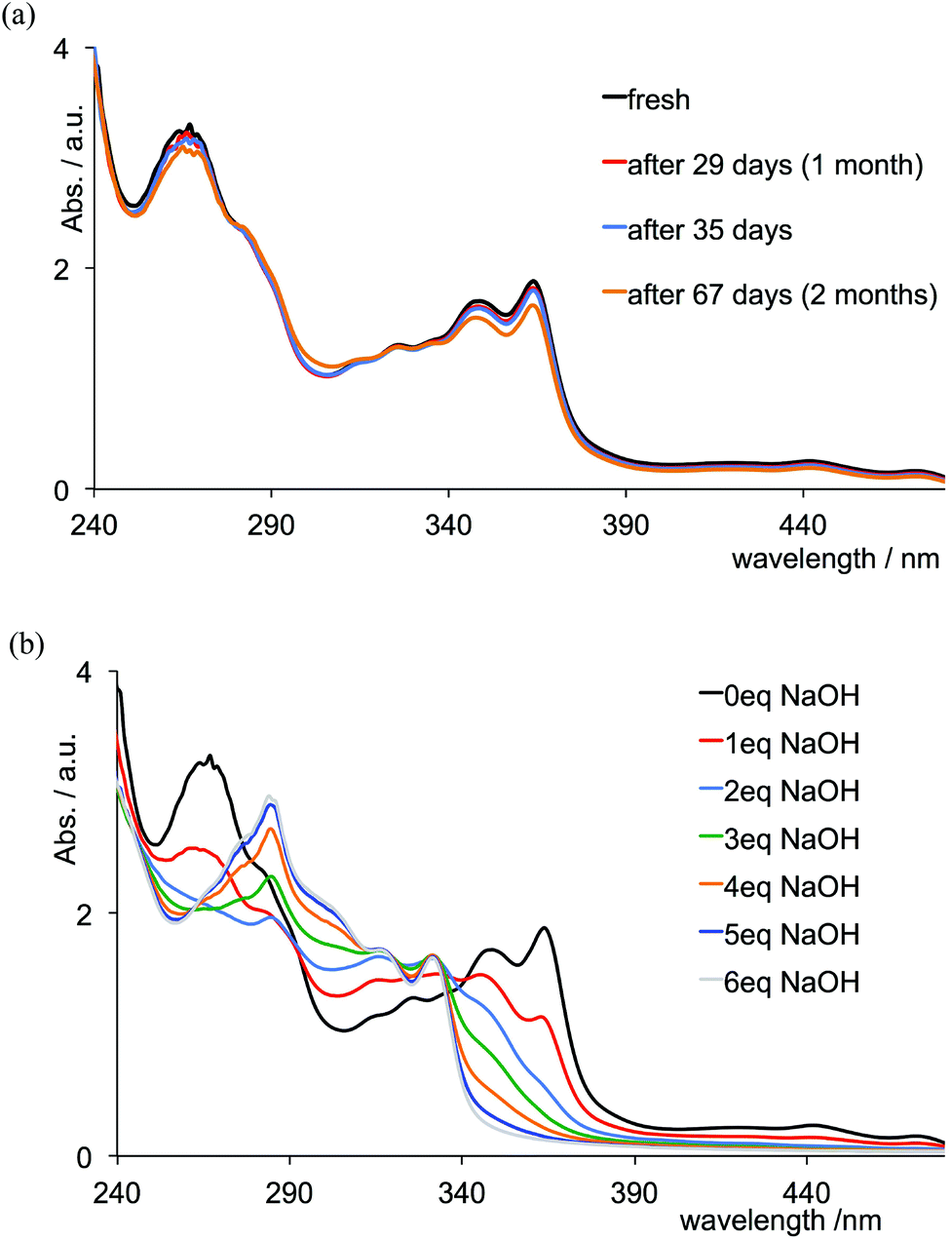 | ||
| Fig. 7 Absorption spectra of an aqueous solution of [Cr(tpy)2][PF6]3 (3.0 mL, 1.0 × 10−4 mol dm−3) (a) over a period of 67 day, and (b) during a titration of aqueous NaOH (0.03 mL aliquots, 0.01 mol dm−3) into the solution. | ||
Given that chromium(III) (d3) is usually considered8 to be kinetically inert with respect to ligand exchange, the above observations for the tris(chelate) are somewhat unforeseen. In [Cr(tpy)2]3+, the Cr3+ ion is bound by two tridentate chelating ligands, and we expected that this coupled with the d3 configuration would lead to a non-labile system. We were therefore surprised to observe significant changes in the absorption spectrum of [Cr(tpy)2][PF6]3 under aerobic aqueous alkaline conditions. A titration of aqueous [Cr(tpy)2][PF6]3 against six equivalents of aqueous NaOH in one equivalent steps was followed by UV-VIS spectroscopy. Fig. 7b shows that the absorption maxima at 363, 347 and 265 nm characteristic of aqueous [Cr(tpy)2][PF6]3 (compare Fig. 7 with the red curve in Fig. 1) decrease in intensity as new absorptions at λmax = 330 and 283 nm (with shoulders at 315, 301 and 275 nm) grow in. During the titration, the solution turned from yellow to pale green. After the addition of 5 equivalents of NaOH, conversion was essentially complete (Fig. 7b). The final absorption spectrum did not correspond to that of free tpy in aqueous solution at pH 9.5 (for which λmax = 290 and 226 nm), and the data were therefore ambiguous in terms of the presence of uncoordinated ligand. A similar titration was carried out with an aqueous solution of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and the changes in the absorption spectra over the addition of 6 equivalents of aqueous NaOH resemble those observed for [Cr(tpy)2][PF6]3 (compare Fig. 8 and 7b). Again, a colour change from yellow to pale green was observed. At the end of the titration, the solution was evaporated to dryness and the residue was extracted into chloroform. After filtration, the 1H NMR spectrum (in CDCl3) of the chloroform-soluble material was recorded. A comparison of the spectrum with those of authentic samples of tpy and 4′-(4-tolyl)tpy (Fig. S1†) confirmed the presence of both free ligands in an approximate ratio of 1:1, providing evidence that [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 is unstable in alkaline solution with respect to displacement of the two chelating ligands. The isolation of the free ligands also suggests that the site of attack of the hydroxide is at the metal rather than the coordinated ligand.49–51
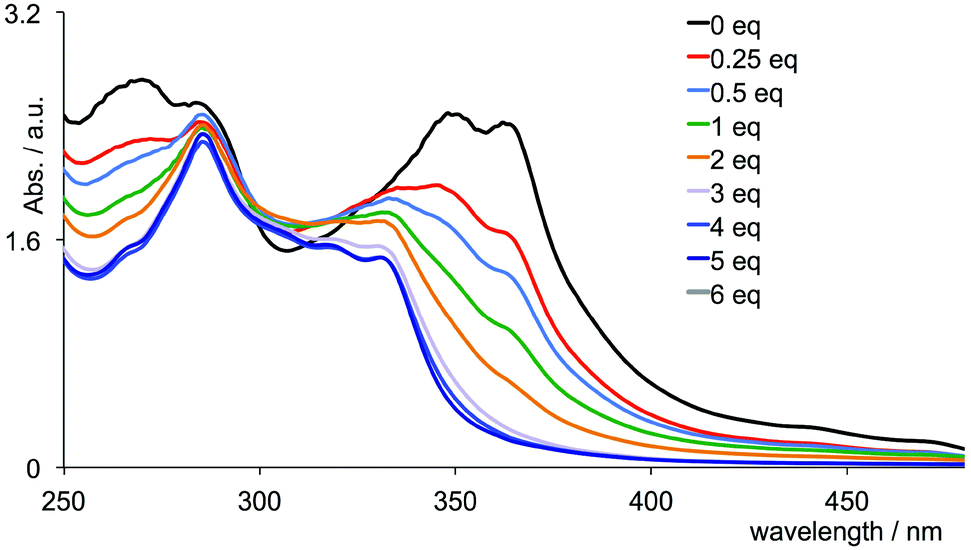 | ||
| Fig. 8 Absorption spectra of an aqueous solution of [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 (3.0 mL, 1.0 × 10−4 mol dm−3) during a titration with aqueous NaOH (0.03 mL aliquots, 0.01 mol dm−3). | ||
In an additional experiment, [Cr(tpy)2][PF6]3 was dissolved in CD3OD. In the 1H NMR spectrum, as expected, no signals were observed in the aromatic region of the spectrum. However, upon the addition of solid NaOH (a few mg) to the NMR tube, signals corresponding to free tpy were observed (Fig. S2a†). The 1H NMR spectra before and after adding NaOH are shown in Fig. S3.† Corresponding experiments were carried out with [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3, and in each case, the addition of NaOH to CD3OD solutions of the complexes resulted in the appearance of signals for the free ligands (Fig. S2b and S2c†). Note that the NMR spectroscopic data are consistent with both ligands being displaced from the metal centre in the heteroleptic complexes, but do not indicate whether they are both lost from the same or different metal centres.
The absorption spectra in Fig. 7b and 8 unambiguously show that [Cr(tpy)2][PF6]3 and [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 decay in basic aqueous media. The spectra recorded in the presence of 6 equivalents of NaOH are very similar to one another, and we propose that multinuclear chromium(III) aqua/hydroxo species52 are formed. We are not able to gain evidence for the formation of a simple hydroxo complex akin to the product of the aquation of [Cr(bpy)3]3+ (eqn (1)), and as far as we are aware, no reports appear in the literature of a mononuclear species such as [Cr(tpy)(OH)3]. On the other hand, [Cr(tpy)F3] has been described and structurally characterized,53 and we therefore hoped that interaction of fluoride ion with [Cr(tpy)2]3+ might provide further insight into the system.
The addition of [nBu4N]F to an aqueous solution of [Cr(tpy)2][PF6]3 resulted in no change in the absorption spectrum of the complex. However, when the reaction was performed in methanol (see Experimental section), the initially pale yellow solution became colourless. A decrease in the amount of hydrogen bonding between F− and solvent molecules on going from water to methanol is expected to render the F− ion a stronger nucleophile in methanol than in water. The absorption spectrum of the final solution (Fig. 9) exhibits a low intensity maximum in the visible region at 570 nm. This maximum agrees with that reported for [Cr(tpy)F3] (λmax = 581 nm in propan-2-ol and 556 in aqueous solution),53 suggesting that fluoride displaces one of the tpy ligands. To confirm this, the reaction of [nBu4N]F with [Cr(tpy)2][PF6]3 was repeated in CD3OD in an NMR tube. Fig. S4a† shows that signals assigned to free tpy indeed appear in the 1H NMR spectrum after [Cr(tpy)2][PF6]3 is treated with fluoride ion. Analogous experiments starting with [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 (Fig. S4b†) or [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 (Fig. S4c†) also confirmed that the tpy and substituted-tpy ligands are labile in the presence of fluoride ion.
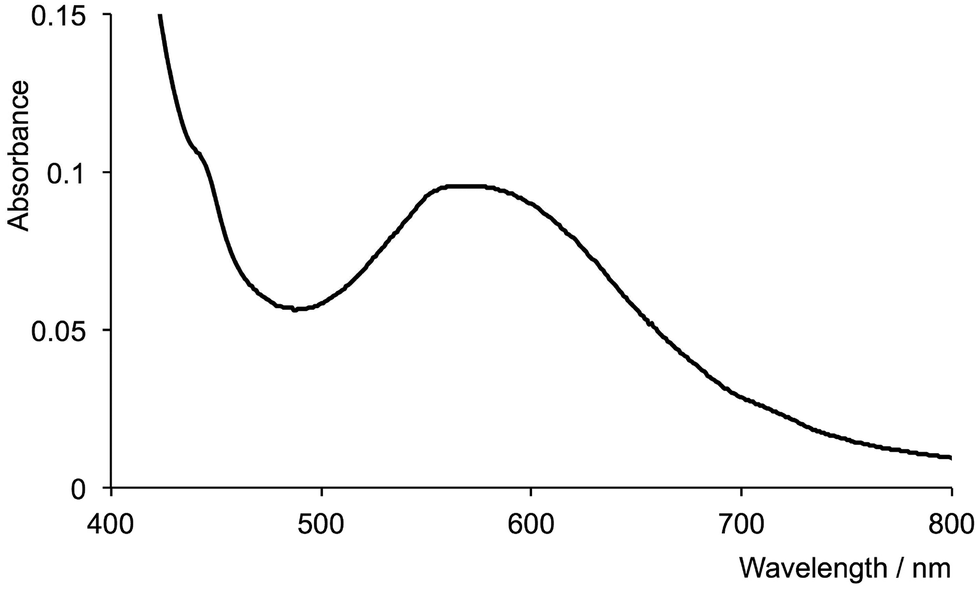 | ||
| Fig. 9 Absorption spectrum of a methanol solution of [Cr(tpy)2][PF6]3 after treatment with [nBu4N]F (λmax = 570 nm). | ||
Conclusions
We have reported the first examples of heteroleptic bis(tpy) chromium(III) complexes. [Cr(tpy)2][PF6]3, [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 are conveniently prepared by treating [Cr(tpy)(O3SCF3)3] with the appropriate tpy ligand. The cyclic voltammetric signatures of the heteroleptic complexes are similar to those of [Cr(tpy)2][PF6]3, indicating that the tolyl and methyl substituents have minimal influence on the gross redox behaviour of the complexes. Structural data for 2{[Cr(tpy)2][PF6]3}·5MeCN, [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3·3MeCN and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3·3MeCN reveal that the cations are not arranged in the grid-like manner that is ubiquitous among salts of {M(tpy)2}2+ complexes; this difference in packing is attributed to the consequences of the higher cationic charge and the concomitant space requirements of the anions and solvent molecules.At pH 6.36, aqueous solutions of [Cr(tpy)2][PF6]3 are stable for at least two months. However, despite the fact that the Cr3+ ion (d3) is widely recognized as a kinetically inert metal centre, the ligands in [Cr(tpy)2][PF6]3, [Cr(tpy)(4′-(4-tolyl)tpy)][PF6]3 and [Cr(tpy)(5,5′′-Me2tpy)][PF6]3 are labile in basic aqueous and basic methanol solutions. Ligand dissociation is also observed when [nBu4N]F is added to CD3OD solutions of the complexes, and, in the case of [Cr(tpy)2]3+, absorption spectroscopic data are consistent with the formation of [Cr(tpy)F3]. In the light of this study, the combination of chromium(III) with bis-chelating tpy ligands should not be expected to give non-labile complexes.
Acknowledgements
For financial support, we gratefully acknowledge the European Research Council (Advanced grant 267816 LiLo), the Swiss National Science Foundation and the University of Basel.Notes and references
- See for example: U. S. Schubert, A. Winter and G. R. Newkome, Terpyridine-based materials: For catalytic, optoelectronic and life science applications, Wiley-VCH, Weiheim, 2011 CrossRef CAS; R. Sakamoto, S. Katagiri, H. Maeda and H. Nishihara, Coord. Chem. Rev., 2013, 257, 1493 CrossRef CAS; E. C. Constable and C. E. Housecroft, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, Oxford, 2013, vol. 8, p. 1 CrossRef; E. C. Constable, in Comprehensive Coordination Chemistry II, ed. T. J. Meyer and J. A. McCleverty, Elsevier Ltd, Oxford, 2003, vol. 7, p. 263 CrossRef; J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef; E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147 RSC; A. Winter, M. D. Hager, G. R. Newkome and U. S. Schubert, Adv. Mater., 2011, 23, 5728 CrossRef PubMed; S. D. Cummings, Coord. Chem. Rev., 2009, 253, 449 CrossRef and references therein.
- M. Grätzel, Inorg. Chem., 2005, 44, 6841 CrossRef PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- http://minerals.usgs.gov/minerals/pubs/mcs/2013/mcs2013.pdf .
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089 CrossRef CAS.
- R. D. Costa, D. Tordera, E. Ortí, H. J. Bolink, J. Schönle, S. Graber, C. E. Housecroft, E. C. Constable and J. A. Zampese, J. Mater. Chem., 2011, 21, 16108 RSC.
- B. Bozic-Weber, E. C. Constable, N. Hostettler, C. E. Housecroft, R. Schmitt and E. Schönhofer, Chem. Commun., 2012, 48, 5727 RSC.
- See for example: C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Harlow, 4th edn, 2012, ch. 26 and references therein Search PubMed.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923 CrossRef CAS PubMed.
- M. C. Hughes and D. J. Macero, Inorg. Chim. Acta, 1970, 4, 327 CrossRef CAS.
- M. C. Hughes and D. J. Macero, Inorg. Chem., 1976, 15, 2040 CrossRef CAS.
- H. Behrens and A. Mueller, Z. Anorg. Allg. Chem., 1965, 341, 124 CrossRef CAS.
- H. Behrens and U. Anders, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1964, 19, 767 Search PubMed.
- H. Behrens, K. Meyer and A. Mueller, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1965, 20, 74 Search PubMed.
- S. Herzog and H. Aul, Z. Chem., 1966, 6, 382 Search PubMed.
- C. C. Scarborough, K. M. Lancaster, S. DeBeer, T. Weyhermüller, S. Sproules and K. Wieghardt, Inorg. Chem., 2012, 51, 3718 CrossRef CAS PubMed.
- M. C. Hughes, D. J. Macero and J. M. Rao, Inorg. Chim. Acta, 1981, 49, 241 CrossRef CAS.
- J. M. Rao, M. C. Hughes and D. J. Macero, Inorg. Chim. Acta, 1976, 18, 127 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, Eur. J. Inorg. Chem., 2004, 1840 CrossRef CAS.
- N. Serpone, M. A. Jamieson, M. S. Henry, M. Z. Hoffman, F. Bolletta and M. Maestri, J. Am. Chem. Soc., 1979, 101, 2907 CrossRef CAS.
- R. P. Bonomo, S. Musumeci, E. Rizzarelli and S. Sammartano, Gazz. Chim. Ital., 1974, 104, 1067 CAS.
- S. R. Allsopp, A. Cox, T. J. Kemp, W. J. Reed, S. Sostero and O. Traverso, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 162 RSC.
- P. B. Lutz, G. J. Long and W. A. Baker Jr., Inorg. Chem., 1969, 8, 2529 CrossRef CAS.
- H. Riesen and L. Wallace, PhysChemComm, 2003, 6, 9 RSC.
- C. K. Ryu, R. B. Lessard, D. Lynch and J. F. Endicott, J. Phys. Chem., 1989, 93, 1752 CrossRef CAS.
- D. L. Jameson and L. E. Guise, Tetrahedron Lett., 1991, 32, 1999 CrossRef CAS.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251 CAS.
- A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399 CrossRef CAS.
- Bruker Analytical X-ray Systems, Inc., 2006, APEX2, version 2 User Manual, M86-E01078, Madison, WI Search PubMed.
- A. Altomare, G. Cascarano, G. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- J. A. Broomhead, J. Evans, W. D. Grumley and M. Sterns, J. Chem. Soc., Dalton Trans., 1977, 173 RSC.
- N. Cloete, H. G. Visser and A. Roodt, Acta Crystallogr. Sect. E: Struct. Rep. Online, 2007, 63, M45 CAS.
- S. K. Padhi, D. Saha, R. Sahu, J. Subramanian and V. Madivannan, Polyhedron, 2008, 27, 1714 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- W. A. Wickramasinghe, P. H. Bird, M. A. Jamieson and N. Serpone, J. Chem. Soc., Chem. Commun., 1979, 798 RSC.
- W. A. Wickramasinghe, P. H. Bird and N. Serpone, Inorg. Chem., 1982, 21, 2694 CrossRef CAS.
- A. D. Bond, CrystEngComm, 2008, 10, 411 CAS.
- J. McMurtrie and I. Dance, CrystEngComm, 2005, 7, 216 RSC.
- E. C. Constable, K. Harris, C. E. Housecroft, M. Neuburger and J. A. Zampese, CrystEngComm, 2010, 12, 2949 RSC.
- Z. Yu, A. Nabei, T. Izumi, T. Okubo and T. Kuroda-Sowa, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m209 CAS.
- M. Maestri, F. Bolletta, N. Serpone, L. Moggi and V. Balzani, Inorg. Chem., 1976, 15, 2048 CrossRef CAS.
- M. A. Jamieson, N. Serpone and M. Maestri, Inorg. Chem., 1978, 17, 2432 CrossRef CAS.
- M. Maestri, F. Bolletta, L. Moggi, V. Balzani, M. S. Henry and M. Z. Hoffman, J. Am. Chem. Soc., 1978, 100, 2694 CrossRef CAS.
- M. A. Jamieson, N. Serpone, M. S. Henry and M. Z. Hoffman, Inorg. Chem., 1979, 18, 214 CrossRef CAS.
- E. C. Constable, Polyhedron, 1983, 2, 551 CrossRef CAS.
- R. D. Gillard, Coord. Chem. Rev., 1975, 16, 67 CrossRef CAS.
- N. Serpone, G. Ponterini, M. A. Jamieson, F. Bolletta and M. Maestri, Coord. Chem. Rev., 1983, 50, 209 CrossRef CAS.
- See for example: L. Spiccia, W. Marty and R. Giovanoli, Inorg. Chem., 1988, 27, 2660 CrossRef CAS.
- T. Birk, M. J. Magnussen, S. Piligkos, H. Weihe, A. Holten and J. Bendix, J. Fluorine Chem., 2010, 131, 898 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S4 NMR spectra showing ligand lability. CCDC 981376–981378. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00200h |
| ‡ ‘Resources’ are defined as: A natural deposit of a material in the Earth's crust in a form or amount that makes economic extraction viable, now or in the future. (http://minerals.usgs.gov/minerals/pubs/mcs/2009/mcsapp2009.pdf) |
| This journal is © The Royal Society of Chemistry 2014 |