 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed-ligand hydroxocopper(II)/pyridazine clusters embedded into 3D framework lattices†
Anna S.
Degtyarenko
a,
Marcel
Handke
b,
Karl W.
Krämer
c,
Shi-Xia
Liu
*c,
Silvio
Decurtins
c,
Eduard B.
Rusanov
d,
Laurence K.
Thompson
e,
Harald
Krautscheid
b and
Konstantin V.
Domasevitch
*a
aInorganic Chemistry Department, Taras Shevchenko National University of Kyiv, Volodimirska Street 64/13, Kyiv 01601, Ukraine. E-mail: dk@univ.kiev.ua
bInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany
cDepartement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: liu@iac.unibe.ch
dInstitute of Organic Chemistry, Murmanskaya Str. 4, Kyiv 253660, Ukraine
eDepartment of Chemistry, Memorial University of Newfoundland, St. John's, A1B 3X7, Canada
First published on 24th March 2014
Abstract
Rational combination of pyridazine, hydroxo and carboxylate bridging ligands led to the assembly of three types of mixed-ligand polynuclear Cu(II) clusters (A: [Cu2(μ-OH)(μ-pdz)(μ-COO)]; B: [Cu4(μ3-OH)2(μ-pdz)2]; C: [Cu5(μ-OH)2(μ-pdz)4(μ-COO)2(μ-H2O)2]) and their integration into 3D framework structures. Mixed-ligand complexes [Cu2(μ-OH){TMA}(L)(H2O)] (1), [Cu4(μ3-OH)2{ATC}2(L)2(H2O)2]·H2O (2) [Cu4(μ3-OH)2{TDC}3(L)2(H2O)2]·7H2O (3) (L = 1,3-bis(pyridazin-4-yl)adamantane; TMA3− = benzene-1,3,5-tricarboxylate, ATC3− = adamantane-1,3,5-tricarboxylate, TDC2− = 2,5-thiophenedicarboxylate) and [Cu5(μ-OH)2{X}4(L)2(H2O)2]·nH2O (X = benzene-1,3-dicarboxylate, BDC2−, n = 5 (4) and 5-hydroxybenzene-1,3-dicarboxylate, HO-BDC2−, n = 6 (5)) are prepared under hydrothermal conditions. Trigonal bridges TMA3− and ATC3− generate planar Cu(II)/carboxylate subtopologies further pillared into 3D frameworks (1: binodal 3,5-coordinated, doubly interpenetrated tcj-3,5-Ccc2; 2: binodal 3,8-coordinated tfz-d) by bitopic pyridazine ligands. Unprecedented triple bridges in 1 (cluster of type A) support short Cu⋯Cu separations of 3.0746(6) Å. The framework in 3 is a primitive cubic net (pcu) with multiple bis-pyridazine and TDC2− links between the tetranuclear nodes of type B. Compounds 4 and 5 adopt uninodal ten-coordinated framework topologies (bct) embedding unprecedented centrosymmetric open-chain pentanuclear clusters of type C with two kinds of multiple bridges, Cu(μ-OH)(μ-pdz)2Cu and Cu(μ-COO)(μ-H2O)Cu (Cu⋯Cu distances are 3.175 and 3.324 Å, respectively). Magnetic coupling phenomena were detected for every type of cluster by susceptibility measurements of 1, 3 and 4. For binuclear clusters A in 1, the intracluster antiferromagnetic exchange interactions lead to a diamagnetic ground state (J = −17.5 cm−1; g = 2.1). Strong antiferromagnetic coupling is relevant also for type B, which consequently results in a diamagnetic ground state (J1 = −110 cm−1; J2 = −228 cm−1, g = 2.07). For pentanuclear clusters of type C in 4, the exchange model is based on a strongly antiferromagnetically coupled central linear trinuclear Cu3 group (J1 = −125 cm−1) and two outer Cu centers weakly antiferromagnetically coupled to the terminal Cu ions of the triad (J2 = −12.5 cm−1).
Introduction
The study of metal–organic framework (MOF) structures adopted by linkage of polynuclear metal ion clusters is a special topic in coordination and materials chemistry and crystal engineering, and has attracted rapidly growing interest during the last decade.1 In view of framework topologies,2 the design of such solids offers a particularly successful approach. It allows to avoid the common limitations for the node connections imposed by the typical coordination numbers,3 and therefore a diversity of extremely highly-connected frameworks became accessible by propagation of coordination geometries established by polynuclear clusters.4 An even more important aspect considers the utility of the latter for functionalization of the MOFs towards specific applications in magnetism and catalysis,5 while imprinting the inherent properties of the clusters into the extended lattices.6 Therefore, the development of polynucleating ligand systems, which combine the abilities for generation of polynuclear fragments7 and their further integration into the framework, has received a special value.8Different types of azole and azine 1,2-dinitrogen donors are applicable to assemble clusters of variable nuclearity and establish pathways for magnetic exchange between paramagnetic metal ions, cf. copper(II) and cobalt(II). In this series, the pyridazine linker is a particular paradigm, while offering a large magnetic superexchange interaction and thus mediating magnetic exchange most efficiently in comparison with corresponding phthalazine, 1,2,4-triazole, 1,2,4-triazolate and pyrazolate analogs.9 The ability of pyridazine to generate polynuclear complexes with tunable spin states of the metal ions is also known.10 The attractiveness of polypyridazinyl ligands, however, was limited until now because of their low chemical accessibility. Actually, the preparative chemistry involving pyridazine relies on only one general method which is applicable for facile functionalization of different substrates, namely a very simple click reaction11 involving inverse electron demand cycloadditions of 1,2,4,5-tetrazine.12 Recent developments have made this key intermediate readily available,12,13 thus offering flexible pathways towards pyridazine ligands.13,14 A second potential drawback one might find is the inherent coordination ability of pyridazine.13–16 It manifests a pronounced affinity towards d10 Cu(I) and Ag(I) ions, commonly generating either single, double or triple bridges between the metallic centres involved in the desired and peculiar polynuclear and polymeric motifs.14,17 In the case of divalent d-metal ions, bridging coordination of this electron deficient and low basic ligand (pKa = 2.24 for pyridazine vs. pKa = 5.25 for pyridine) is less characteristic and predictable, compared with a simple monodentate pyridine-like function.15,18 Nevertheless, double coordination of pyridazine may be stabilized using suitable complementary short-distance co-bridges, such as hydroxo,10,13,19 halogenido,20 nitrato,15 isothiocyanato,10,21 and saccharinato,22 and actually all of the reports for pyridazine bridges between 3d-metal ions consider such a multiple heteroligand linkage, which is best illustrated by a Cu(II) series (Table 1). Thus a promising approach towards the development of nanosized molecular magnets may rely on a synergism of the ligands in multicomponent systems24 based upon bifunctional pyridazines, which could be well suited for the synthesis of extended frameworks incorporating polynuclear cluster units.
| Complex | co-Bridge(s) | Cu⋯Cu/Å | Structural pattern and magnetic features | Ref. |
|---|---|---|---|---|
| a sac = saccharinate; bpdz = 4,4′-bipyridazine; pp = pyridazino[4,5-d]pyridazine; L = 1,3-bis(pyridazin-4-yl)adamantane; BDC = isophthalate; HO-BDC = 5-hydroxyisophthalate; TDC = thiophene-2,5-dicarboxylate; TMA = benzene-1,3,5-tricarboxylate; ATC = adamantane-1,3,5-tricarboxylate. | ||||
| [Cu(OH)(sac)(μ-pdz)]n | μ-OH, μ-sac-N,O | 3.360 | 1D chain of triply bridged Cu2+ ions; strong AFM, μeff (RT) = 0.99 μB | 22 |
| [Cu(μ-pdz)Cl2]n | μ-Cl (×2) | 3.378 | 1D chain of triply bridged Cu2+ ions; μeff (RT) = 1.5–1.7 μB; low-temperature AF coupling | 20a, 23 |
| [Cu(OH)(μ-pdz)(NO3)]n | μ-OH, μ-NO3-O,O′ | 3.322 | 1D chain of triply bridged Cu2+ ions; strong AFM, μeff (RT) = 1.08 μB | 19 |
| [Cu3(μ-pdz)4(pdz)2(μ-NO3)2(NO3)4] | μ-NO3-O,O | 3.406 | Triple bridges [M(μ-pdz)2(μ-NO3)M]; Discrete trinuclear; strong AFM, μeff (RT) = 1.51 μB | 15, 19 |
| [Cu3(OH)2(μ-bpdz)3(H2O)2{CF3CO2}2]2+ | μ-OH | 3.236 | Discrete trinuclear, triple bridges [M(μ-pdz)2(μ-OH)M] | 13 |
| [Cu(OH)(pp)](H2NSO3)·H2O | μ-OH | 3.394 | 1D chain of triply bridged Cu2+ ions | 14a |
| [Cu2(OH){TMA}(L)(H2O)] | μ-OH, μ-RCO2-O,O′ | 3.075 | Discrete binuclear cluster with triply bridged Cu2+ ions | This work |
| [Cu4(OH)2{ATC}2(L)2(H2O)2]·H2O | μ3-OH | 3.272 | Discrete tetranuclear cluster | This work |
| [Cu4(OH)2{TDC}3(L)2(H2O)2]·7H2O | μ3-OH | 3.334 | The same | This work |
| [Cu5(OH)2{BDC}4(L)2(H2O)2]·5H2O | μ-OH | 3.175 | Discrete pentanuclear cluster with a trinuclear triply-bridged skeleton M{(μ-pdz)2(μ-OH)M}2 | This work |
| [Cu5(OH)2{HO-BDC}4(L)2(H2O)2]·6H2O | μ-OH | 3.187 | The same | This work |
In this work we report the construction of unprecedented discrete di-, tetra- and pentanuclear copper(II)/pyridazine clusters and their integration into the 3D MOFs structures by the concerted action of representative di- and tricarboxylate linkers (H2BDC = isophthalic acid; H2HO-BDC = 5-hydroxyisophthalic acid; H2TDC = thiophene-2,5-dicarboxylic acid; H3TMA = trimesic acid; H3ATC = adamantane-1,3,5-tricarboxylic acid) and prototypical pyridazine tecton 1,3-bis(pyridazin-4-yl)adamantane (L) (Scheme 1), which features a double ligand functionality established at a rigid alicyclic molecular platform.
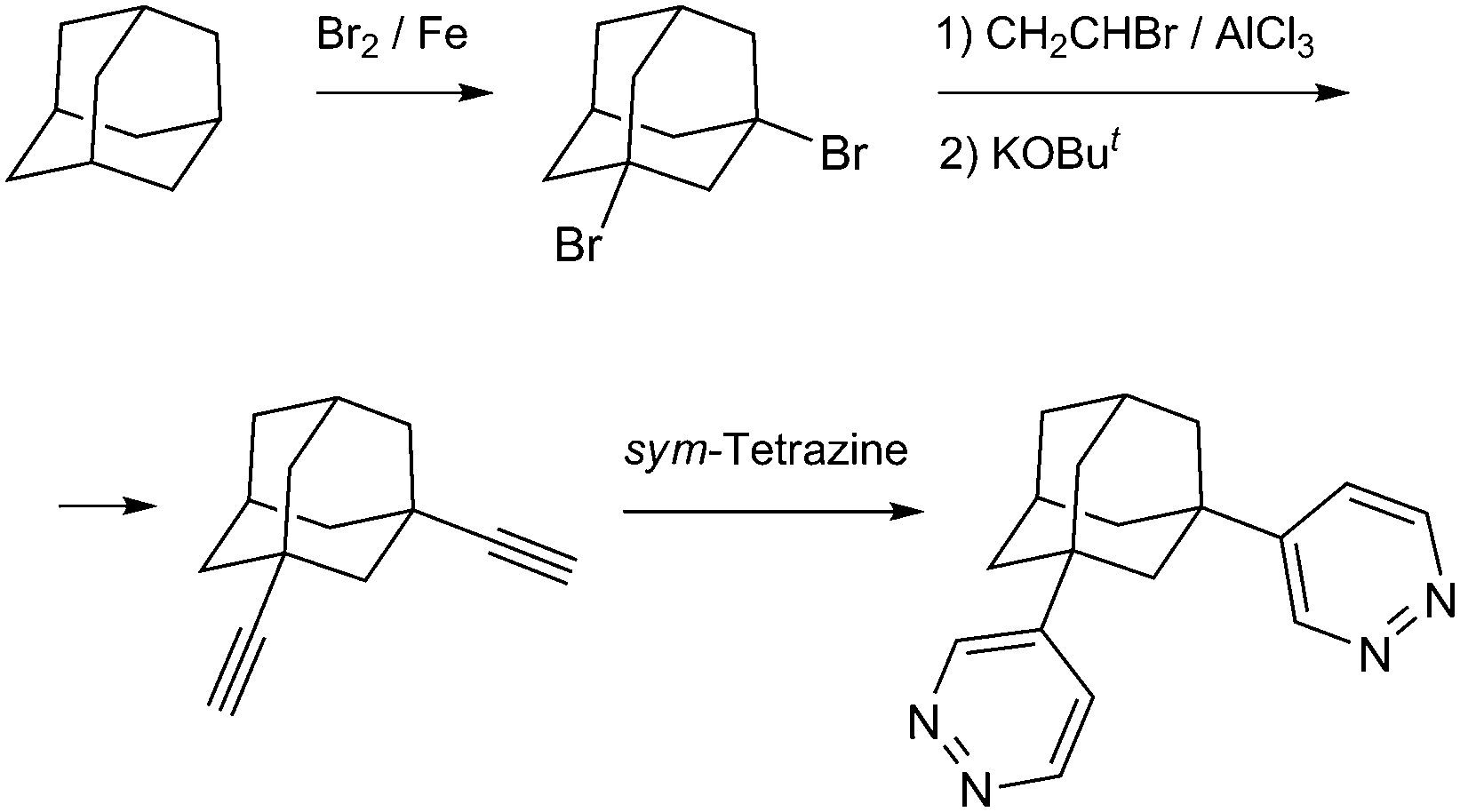 | ||
| Scheme 1 Synthesis of the bis-pyridazine ligand by a stepwise functionalization of the adamantane core. | ||
Results and discussion
Structure of the coordination compounds
All the compounds adopt 3D framework structures based upon multinuclear heteroligand clusters, which represent topological net nodes and thus provide a primary factor for the complicated connectivities. Three types of the observed clusters, such as binuclear in 1, tetranuclear in 2, 3 and pentanuclear units in 4 and 5 (Scheme 2), originate from a combination of three kinds of ligand bridges (pyridazine, hydroxo and carboxylate), while preserving a common simple submotif in the form of Cu ions linked by a double pdz/OH bridge. This suggests perfect compatibility of μ-pdz and μ-OH linkers, which are commonly concomitant and act in a synergetic manner.10,13,19 In fact, the significance of the hydroxo bridges is most crucial for the present systems since the bidentate coordination of pyridazine itself is less applicable for the chemistry of transition metal dications, as stated above. In this view, the clusters demonstrate a clear structural hierarchy, which implies a basic hydroxocopper(II) core, pyridazine co-bridges, auxiliary carboxylate ligands and additional monodentate pyridazine and aqua donors. The function of the carboxylate is most important for compounds 4 and 5, providing expansion of the trinuclear [Cu3(μ-pdz)4(μ-OH)2] skeleton to a pentanuclear architecture. Especially close structural relationships may be found for binuclear (A) and tetranuclear (B) motifs. Doubling the cluster nuclearity is made possible by elimination of the carboxylate bridge and a simple dimerization along the Cu–OH sides with formation of two edge-sharing [Cu3(μ3-OH)] fragments. Such a “dimer”, often encountered for different combinations of bridging ligands, is particularly characteristic of the hydroxocopper systems.25 Further intercluster connection with generation of frameworks occurs by the interplay of bipyridazine (as a bitopic linker between the clusters, establishing three or four Cu–N bonds) and dicarboxylate bridges (TDC2− (3), BDC2− (4), HO-BDC2− (5)), which are equally important in view of the resulting topologies. This is contrary to the tricarboxylate (TMA3− (1), ATC3− (2)) frameworks. Trifunctional anions, as the three-coordinated nodes, provide an additional origin of connectivity and generate distinct planar Cu-carboxylate subtopologies, further pillared with bitopic bipyridazine ligands (Table 2).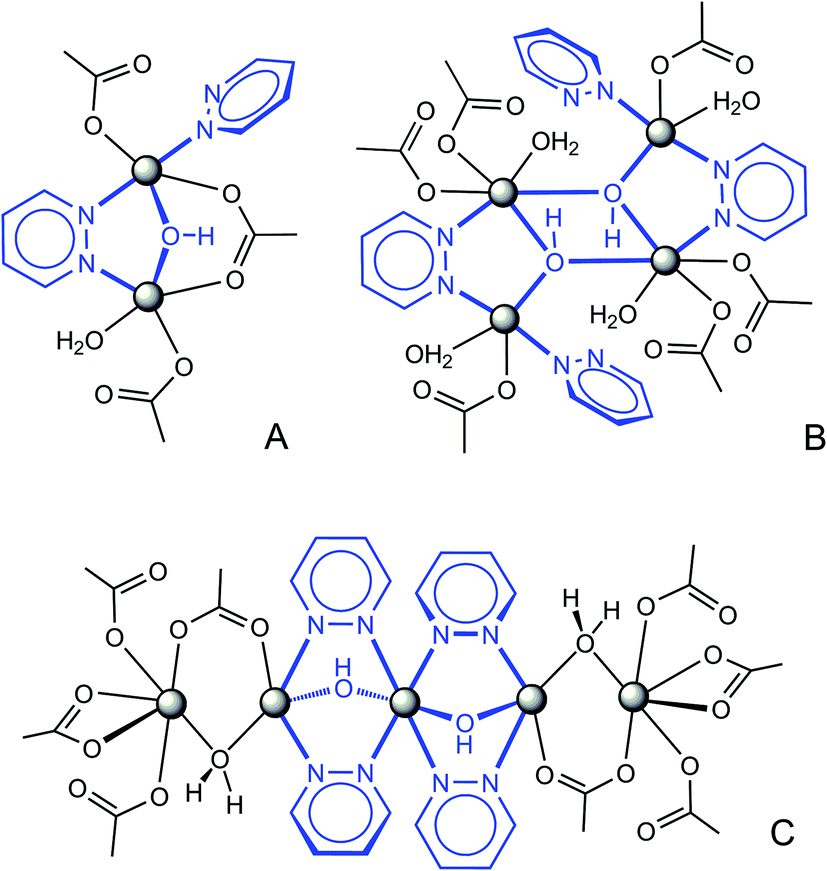 | ||
| Scheme 2 Three kinds of polynuclear Cu/OH/pyridazine clusters, representing the structures of the reported complexes: A – binuclear motif in 1; B – tetranuclear clusters in 2 and 3; C – pentanuclear units in 4 and 5, with a central trinuclear hydroxo/pyridazine core. | ||
| Complex | Cluster type | Ligand coordination | Dimensionality | Net nodes (coordination) | Schläfli symbol | Topological type |
|---|---|---|---|---|---|---|
| a Dimensionality of the Cu/carboxylate subtopology and the overall dimensionality of the resulting heteroligand frameworks. | ||||||
| 1 | A | 3 | 2D → 3Da | [Cu2(μ-OH)] (5), TMA (3) | {63}{69·8} | tcj-3,5-Ccc2 |
| 2 | B | 3 | 2D → 3Da | [Cu4(μ3-OH)2] (8), ATC (3) | {43}2{46·618·84} | tfz-d (UO3) |
| 3 | B | 3 | 3D | [Cu4(μ3-OH)2] (6) | {412·63} | pcu (α-Po) |
| 4 | C | 4 | 3D | [Cu5(μ-OH)2] (10) | {312·428·55} | bct |
| 5 | C | 4 | 3D | [Cu5(μ-OH)2] (10) | {312·428·55} | bct |
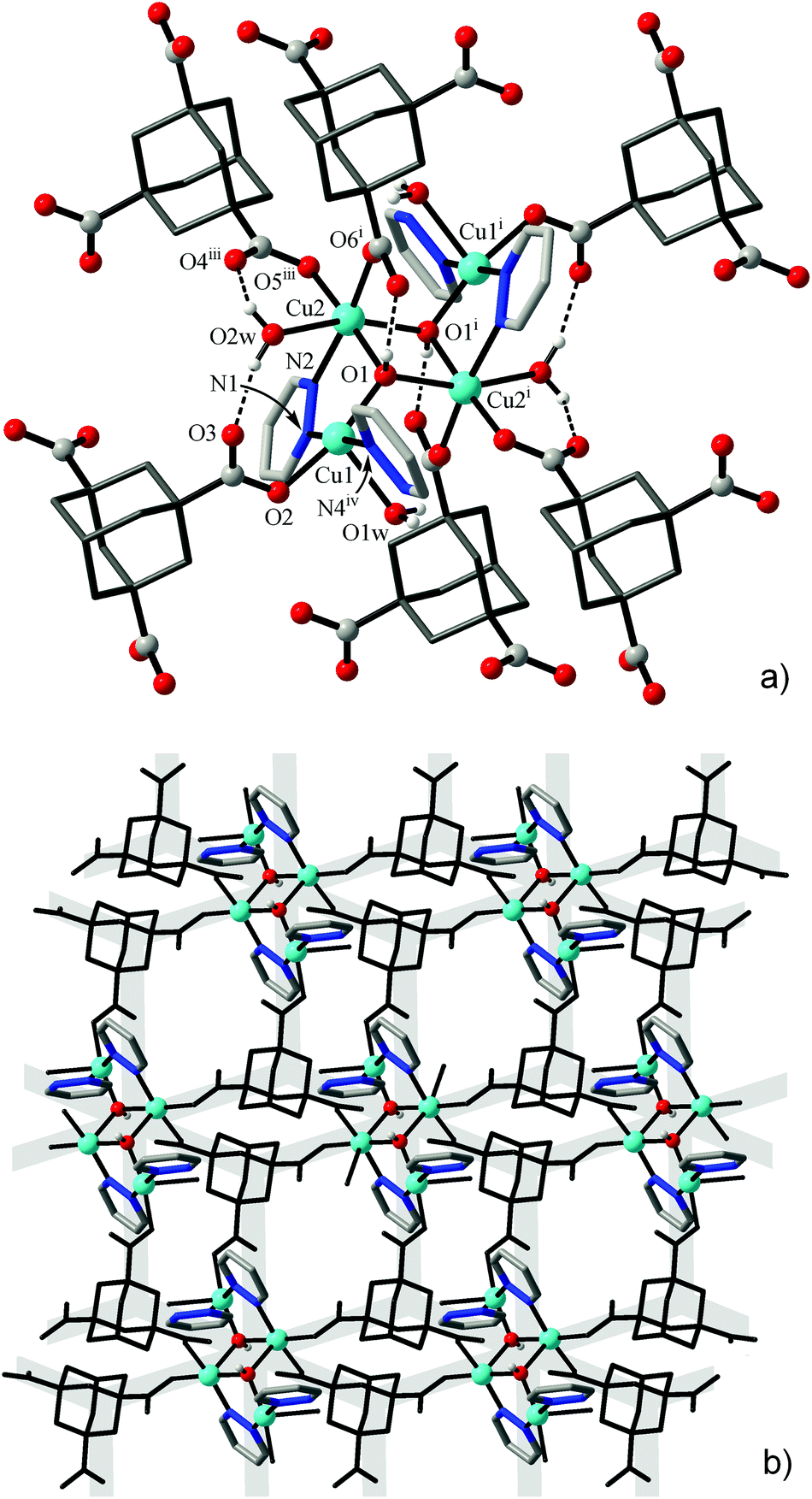
A simpler binuclear cluster A was observed in [Cu2(μ-OH){TMA}(L)(H2O)] (1), with an unprecedented mixed-ligand pdz/OH/carboxylate triple bridge connecting two Cu ions at very short distances of Cu1⋯Cu2A 3.0746(6) Å and Cu1⋯Cu2B 3.1270(7) Å (for two orientations of the disordered fragment). The environments of metal ions are complete with monodentate carboxylate and pyridazine groups and aqua ligands (Fig. 1, Table 3). As was indicated by the values of Addison τ parameters,26 the geometries of the coordination polyhedra are intermediate between idealized trigonal-bipyramidal (τ = 1) and square-pyramidal (τ = 0) extremes: for orientation A, τ = 0.66 (Cu1) and 0.39 (Cu2A); for orientation B, τ = 0.53 (Cu1) and 0.32 (Cu2B).
 | ||
| Fig. 1 (a) Binuclear cluster in the environment of carboxylate and pyridazine ligands in the structure of 1; (b) 2D Cu/carboxylate subtopology in the form of hexagonal net, with the pyridazine-N donors accommodated at two sides of the plane. Only one orientation of the disordered cluster is shown. | ||
| a Symmetry codes: (i) −0.5 + x, −0.5 + y, z; (ii) x, −y, 0.5 + z; (iii) x, 1 − y, 0.5 + z. | |||
|---|---|---|---|
| Cu1–O3 | 2.0418(18) | O3–Cu1–N1 | 89.10(8) |
| Cu1–N4i | 2.043(2) | O3–Cu1–O7ii | 115.76(8) |
| Cu1–N1 | 2.053(2) | N4i–Cu1–O7ii | 89.91(9) |
| Cu1–O7ii | 2.1577(16) | N1–Cu1–O7ii | 84.24(7) |
| O3–Cu1–N4i | 91.75(9) | ||
| N4i–Cu1–N1 | 173.85(9) | ||
| Disordered fragment | |||
| Orientation A | Orientation B | ||
| Cu1–O1A | 1.873(3) | Cu1–O1B | 1.913(3) |
| Cu2A–O1A | 1.906(3) | Cu2B–O1B | 1.895(4) |
| Cu2A–O5iii | 1.946(2) | Cu2B–O6iii | 1.9264(19) |
| Cu2A–O4 | 1.984(2) | Cu2B–O8ii | 1.9307(18) |
| Cu2A–O2A | 1.991(3) | Cu2B–O2B | 1.952(4) |
| Cu2A–N2A | 2.207(4) | Cu2B–N2B | 2.262(4) |
| O1A–Cu1–N4i | 91.62(12) | O1B–Cu1–N4i | 86.79(12) |
| O1A–Cu1–N1 | 93.80(12) | O1B–Cu1–N1 | 96.27(12) |
| O1A–Cu2A–O5iii | 87.56(13) | O1B–Cu2B–O6iii | 88.57(13) |
| O1A–Cu2A–O4 | 88.78(14) | O1B–Cu2B–O8ii | 91.98(12) |
| O5iii–Cu2A–O4 | 155.29(11) | O6iii–Cu2B–O8ii | 159.17(9) |
| O1A–Cu2A–O2A | 178.92(17) | O1B–Cu2B–O2B | 178.52(14) |
| O1A–Cu2A–N2A | 88.73(14) | O1B–Cu2B–N2B | 87.98(15) |
| O5iii–Cu2A–N2A | 109.02(13) | O6iii–Cu2B–N2B | 104.15(12) |
| O4–Cu2A–N2A | 95.32(13) | O8ii–Cu2B–N2B | 96.68(12) |
| Cu2A–O1A–Cu1 | 108.89(16) | Cu2B–O1B–Cu1 | 110.43(16) |
The low nuclearity of the cluster is likely a consequence of topology limitations imposed by the rigid trigonal triple-charged TMA3− connector: the planar Cu/carboxylate subtopology in the form of a hexagonal net (Fig. 1b) clearly implies a three-fold coordination of the hydroxocopper nodes and their complementary charge of +3 [e.g. Cu2(μ-OH)]. The bipyridazine ligands are accommodated on both axial sides of this plane, and they connect pairs of clusters at a distance of 13.56 Å and expand the structure in a third dimension as pillars between the successive hexagonal layers. The result is a rare binodal three- and five-connected {63}{69·8} net (three-letter notation is tcj-3,5-Ccc2), which appears to be very open and, therefore, two identical nets interpenetrate (Fig. 2). That is a class IIa interpenetration, Z = 2, with a full symmetry element ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .27,28
.27,28
 | ||
| Fig. 2 Interpenetration of two inversion-related 3D frameworks (marked with blue and grey bonds) in the structure of 1. The hydroxocopper-carboxylate layers are orthogonal to the drawing plane. | ||
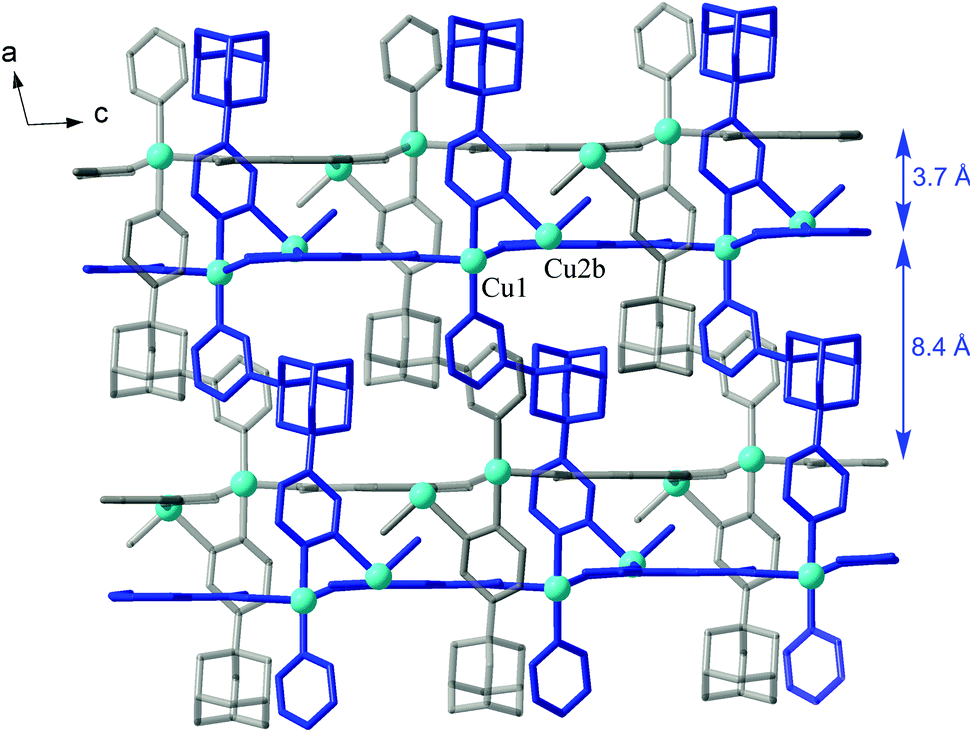
A comparable morphology of the framework is observed for the aliphatic analog of trimesic acid, 1,3,5-adamantanetricarboxylate (ATC3−) in [Cu4(μ3-OH)2{ATC}2(L)2(H2O)2]·H2O (2). That is a first coordination polymer generated by this ligand and we introduce ATC3− as a novel geometrically rigid tripodal linker, potentially complementing and expanding the chemistry of functional frameworks based upon TMA3− (such as HKUST-1).29 Both metal ions adopt typical Jahn–Teller polyhedra in the form of square pyramids (Cu1; Addison parameter26τ = 0.18) or axially elongated octahedra (Cu2) (Table 4), with pyridazine-N donors positioned at the equatorial planes. In the metal–carboxylate subtopology, the net nodes exist as the above six-connected tetranuclear clusters [Cu4(μ3-OH)2] and three-connected ATC3− (in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 proportion), giving rise to a 2D structure of the CdI2 or Mg(OH)2 type (Kagomé dual net, {43}2{46·66·83}, three-letter notation “kgd”) (Fig. 3). Pairs of bipyridazine ligands, as double links, interconnect the clusters from the successive layers (separated at 12.83 Å), yielding a 3D eight- and three-coordinated framework, with a Schläfli point symbol {43}2{46·618·84} (tfz-d, topological type of UO3) (Table 2). There are a limited number of structural precedents for such a linkage, most of which also incorporate polynuclear cluster nodes and trigonal TMA3− links.30
:2 proportion), giving rise to a 2D structure of the CdI2 or Mg(OH)2 type (Kagomé dual net, {43}2{46·66·83}, three-letter notation “kgd”) (Fig. 3). Pairs of bipyridazine ligands, as double links, interconnect the clusters from the successive layers (separated at 12.83 Å), yielding a 3D eight- and three-coordinated framework, with a Schläfli point symbol {43}2{46·618·84} (tfz-d, topological type of UO3) (Table 2). There are a limited number of structural precedents for such a linkage, most of which also incorporate polynuclear cluster nodes and trigonal TMA3− links.30
 | ||
| Fig. 3 (a) Tetranuclear cluster in 2 showing the hydrogen bonding interactions with μ3-OH and aqua ligands; (b) Cu/carboxylate topology in the form of six- and three-connected CdI2-like network. Note the intrinsic topological significance of the tricarboxylate linker. Symmetry codes: (i) –x, 2 − y, 1 − z; (ii) −1 + x, y, z; (iii) 1 − x, 1 − y, 1 − z; (iv) x, y, −1 + z. | ||
| a Symmetry codes for 2: (i) −x, 2 − y, 1 − z; (ii) −1 + x, y, z; (iii) 1 − x, 1 − y, 1 − z; (iv) x, y, −1 + z. For 3: (i) 1 − x, 2 − y, −z; (ii) −0.5 + x, 2.5 − y, −0.5 + z; (iii) 1.5 − x, 0.5 + y, 0.5 − z; (iv) −x, 2 − y, −z. | |||
|---|---|---|---|
| [Cu4(μ3-OH)2{ATC}2(L)2(H2O)2]·H2O (2) | |||
| Cu1–O1 | 1.931(2) | Cu2–O6ii | 1.948(3) |
| Cu1–O2 | 1.952(3) | Cu2–O5iii | 1.953(3) |
| Cu1–N4iv | 2.026(3) | Cu2–O1 | 1.962(2) |
| Cu1–N1 | 2.068(3) | Cu2–N2 | 2.016(3) |
| Cu1–O1w | 2.359(3) | Cu2–O1i | 2.423(2) |
| O1–Cu1–O2 | 167.55(11) | O6ii–Cu2–O5iii | 85.36(12) |
| O1–Cu1–N4iv | 94.38(12) | O6ii–Cu2–O1 | 97.84(11) |
| O2–Cu1–N4iv | 93.50(12) | O5iii–Cu2–O1 | 176.64(12) |
| O1–Cu1–N1 | 86.13(12) | O6ii–Cu2–N2 | 173.27(12) |
| O2–Cu1–N1 | 86.29(12) | O5iii–Cu2–N2 | 90.36(12) |
| N4iv–Cu1–N1 | 178.20(13) | O1–Cu2–N2 | 86.36(12) |
| O1–Cu1–O1w | 100.77(10) | O6ii–Cu2–O1i | 99.00(10) |
| N4iv–Cu1–O1w | 90.72(12) | O1–Cu2–O1i | 85.52(10) |
| N1–Cu1–O1w | 87.49(11) | N2–Cu2–O1i | 86.53(11) |
| Cu1–O1–Cu2 | 114.38(12) | Cu2–O1–Cu2i | 94.48(10) |
| Cu1–O1–Cu2i | 120.24(12) | ||
| [Cu4(μ3-OH)2{TDC}3(L)2(H2O)2]·7H2O (3) | |||
| Cu1–O1 | 1.920(2) | Cu2–O1 | 1.919(2) |
| Cu1–O1i | 2.352(2) | Cu2–O6 | 1.968(2) |
| Cu1–O2 | 1.942(11) | Cu2–N4iii | 2.007(3) |
| Cu1–O8ii | 1.952(2) | Cu2–N2 | 2.058(3) |
| Cu1–O5iv | 1.959(12) | Cu2–O10 | 2.236(2) |
| Cu1–N1 | 2.052(3) | ||
| O1–Cu1–O2 | 160.6(3) | O1–Cu2–O6 | 172.67(10) |
| O1–Cu1–O8ii | 97.53(10) | O1–Cu2–N4iii | 91.96(10) |
| O2–Cu1–O8ii | 84.5(4) | O6–Cu2–N4iii | 87.77(11) |
| O1–Cu1–N1 | 88.00(10) | O1–Cu2–N2 | 88.41(10) |
| O2–Cu1–N1 | 90.8(4) | O6–Cu2–N2 | 89.29(10) |
| O8ii–Cu1–N1 | 174.25(11) | N4iii–Cu2–N2 | 159.51(12) |
| O5iv–Cu1–N1 | 83.1(3) | O1–Cu2–O10 | 93.71(9) |
| O1–Cu1–O1i | 85.37(9) | O6–Cu2–O10 | 93.25(10) |
| N1–Cu1–O1i | 94.72(10) | N2–Cu2–O10 | 90.06(10) |
| Cu1–O1–Cu1i | 94.63(9) | Cu2–O1–Cu1i | 110.79(10) |
| Cu2–O1–Cu1 | 120.57(12) | ||
Similar tetranuclear net nodes are also observed for [Cu4(μ3-OH)2{TDC}3(L)2(H2O)2]·7H2O (3). However, substitution of the carboxylate portion of the structure for dibasic thiophenedicarboxylate leads to simplification of the entire array. This concerns the elimination of three-connected nodes and an increased carboxylate/cluster ratio (3:1), which in effect increases the dimensionality of the carboxylate subconnectivity, existing in the form of a primitive cubic net (pcu, Schläfli point symbol {412·63}). The bipyridazine bridges did not generate additional intercluster links, but rather repeat the existing links already established by the TDC2− bridges. In this way, four out of six topological links are doubled as a result of the concerted action of both sorts of ligands (Fig. 4). Two CuII ions adopt square-pyramidal environments (Addison parameters26τ are 0.23 for Cu1 and 0.22 for Cu2) (Table 4).
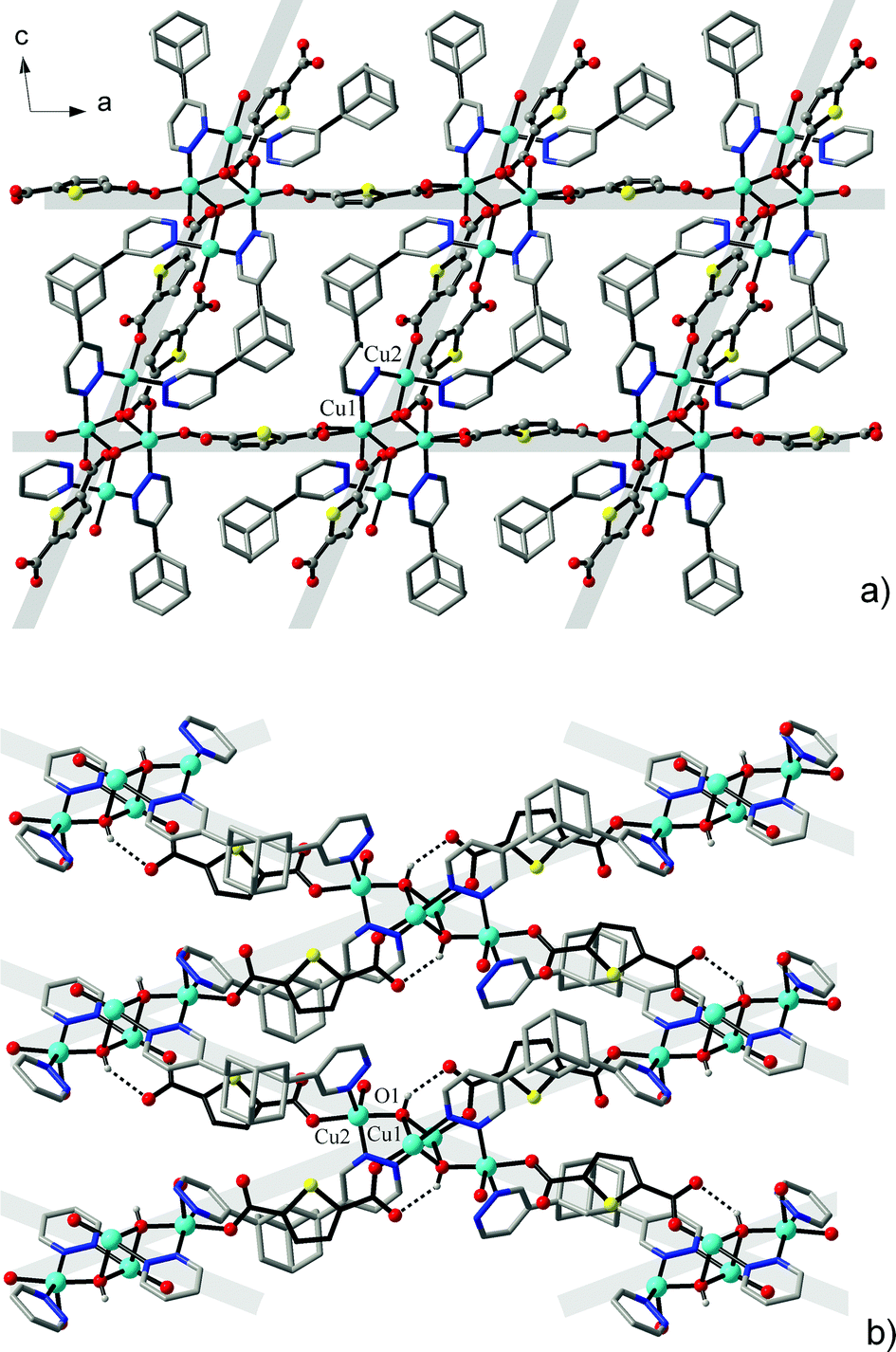 | ||
| Fig. 4 (a) Primitive cubic framework of 3 viewed down the b direction showing interconnection of heteroligand carboxylate/pyridazine planes by additional carboxylate linkers. (b) The heteroligand planes with double organic bridges between the tetranuclear clusters constituting the framework nodes. | ||
The structures of the two isotypic compounds, [Cu5(μ-OH)2{X}4(L)2(H2O)2]·nH2O (4: X = BDC2−, n = 5; 5: X = HO-BDC2−, n = 6), are more complicated and they include pentanuclear net nodes (Fig. 4). The present open-chain centrosymmetric Cu5 units are unprecedented yet comprising a central trinuclear core [Cu3(μ-OH)2(pdz)4] built up with two triple bis-pyridazino/hydroxo bridges, which itself is known for copper(II)13 and cobalt(II)10 pyridazine complexes. These units are extended to the pentanuclear pattern by connecting two Cu3 ions as annexes through carboxylate bridges and distal aqua ligands (Fig. 5). These outer ions have a typically distorted [4 + 2] octahedral coordination, whereas the environment geometry of Cu2 is close to a square pyramid with N3 in the apex position, with Addison parameters26τ = 0.41 (4a at 296 K), τ = 0.37 (4b at 105 K) and τ = 0.42 (5) (Table 5).
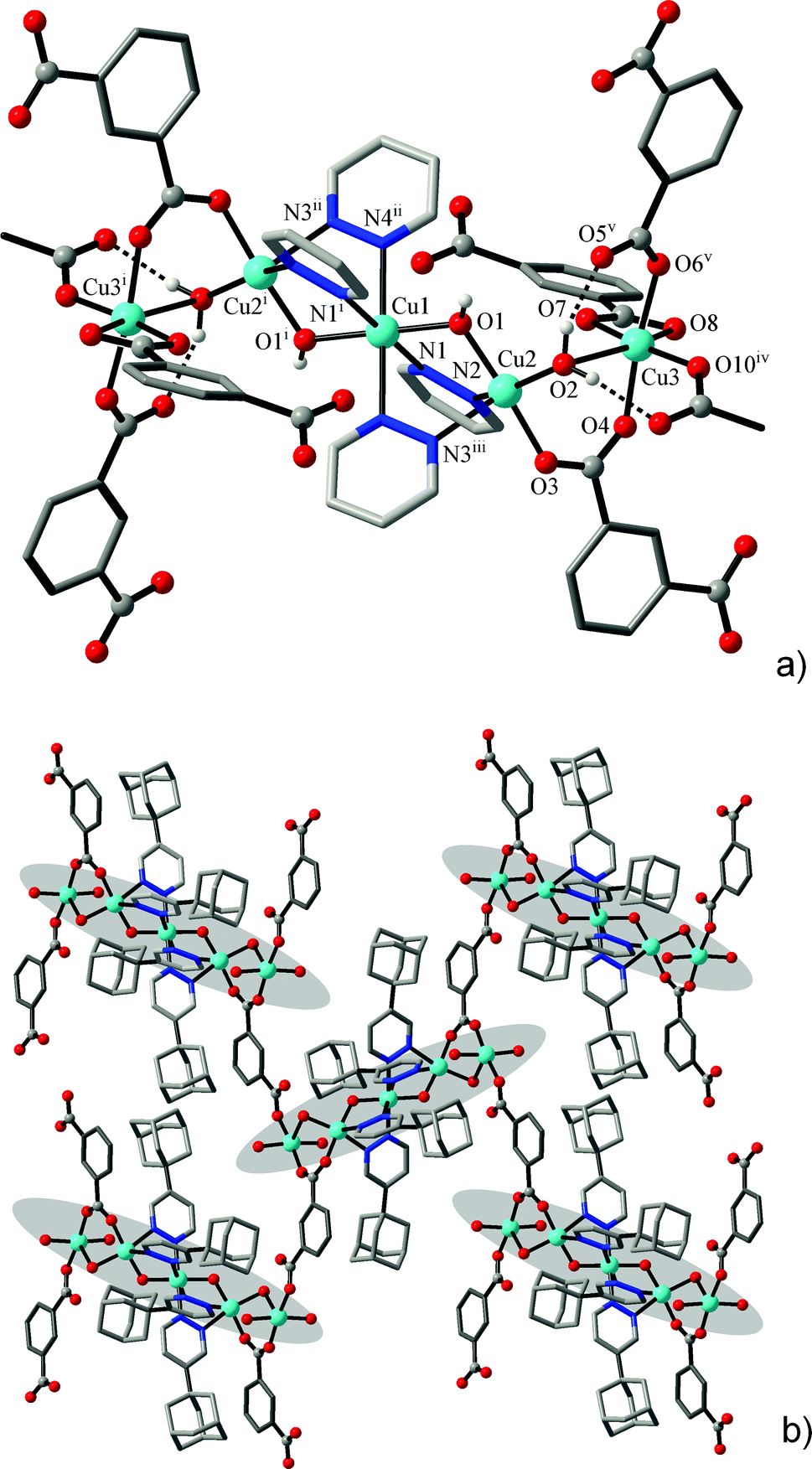 | ||
| Fig. 5 (a) Pentanuclear centrosymmetric clusters observed in 4 and 5. Note the compressed octahedral geometry around the Cu1 ion. (b) Mode of interconnection of the clusters by dicarboxylate links. Symmetry codes: (i) −x, −y, 2 − z; (ii) 0.5 − x, 0.5 + y, 2.5 − z; (iii) −0.5 + x, −0.5 − y, −0.5 + z; (iv) −1 + x, y, z; (v) −0.5 − x, 0.5 + y, 2.5 − z. | ||
| a Symmetry codes for 4: (iii) −0.5 + x, −0.5 − y, −0.5 + z; (iv) −1 + x, y, z; (v) −0.5 − x, 0.5 + y, 2.5 − z. For 5: (ii) x, 0.5 − y, 0.5 + z; (iv) −x, −0.5 + y, 0.5 − z; (v) 1 + x, y, z. | |||
|---|---|---|---|
| [Cu5(μ-OH)2{BDC}4(L)2(H2O)2]·5H2O (4a, 296 K) | |||
| Cu1–O1 | 1.9086(16) × 2 | Cu3–O10iv | 1.9085(17) |
| Cu1–N1 | 2.221(2) × 2 | Cu3–O6v | 1.9593(19) |
| Cu1–N4 | 2.252(2) × 2 | Cu3–O7 | 1.9614(16) |
| Cu3–O4 | 1.9685(18) | ||
| Cu2–O1 | 1.8800(17) | Cu3–O8 | 2.482(2) |
| Cu2–O3 | 1.9286(18) | Cu3–O2 | 2.733(2) |
| Cu2–O2 | 2.0040(18) | ||
| Cu2–N2 | 2.195(2) | ||
| Cu2–N3iii | 2.224(2) | ||
| [Cu5(μ-OH)2{BDC}4(L)2(H2O)2]·5H2O (4b, 105 K) | |||
| Cu1–O1 | 1.9162(15) × 2 | Cu3–O10iv | 1.9139(16) |
| Cu1–N1 | 2.300(2) × 2 | Cu3–O6v | 1.9616(16) |
| Cu1–N4 | 2.1733(18) × 2 | Cu3–O7 | 1.9615(14) |
| Cu3–O4 | 1.9715(16) | ||
| Cu2–O1 | 1.8784(15) | Cu3–O8 | 2.4799(18) |
| Cu2–O3 | 1.9245(15) | Cu3–O2 | 2.7019(18) |
| Cu2–O2 | 2.0093(16) | ||
| Cu2–N2 | 2.1671(19) | ||
| Cu2–N3iii | 2.227(2) | ||
| [Cu5(μ-OH)2{HO-BDC}4(L)2(H2O)2]·6H2O (5) | |||
| Cu1–O1 | 1.901(2) × 2 | Cu3–O8 | 1.915(2) |
| Cu1–N1 | 2.138(3) × 2 | Cu3–O6iv | 1.927(2) |
| Cu1–N4 | 2.344(3) × 2 | Cu3–O4 | 1.949(2) |
| Cu3–O10v | 1.964(2) | ||
| Cu2–O1 | 1.882(2) | Cu3–O11v | 2.541(3) |
| Cu2–O3 | 1.933(2) | Cu3–O2 | 2.667(3) |
| Cu2–O2 | 2.000(3) | ||
| Cu2–N2 | 2.154(3) | ||
| Cu2–N3ii | 2.230(3) | ||
The most striking features of the local coordination geometries are the appreciably compressed octahedral environments adopted by the central Cu1 ions lying on the centre of inversion. For both of the compounds, these involve two pairs of long “equatorial” Cu–N bonds (2.1733(18)–2.300(2) Å) accompanied by two very short “axial” Cu–O1 bonds (1.901(2)–1.9086(16) Å). The only documented example of the [Cu{(μ-OH)(μ-pdz)2Cu}2] pattern, in a 4,4′-bipyridazine complex, also implies such a kind of coordination octahedron adopted by the central Cu ion (Cu–O 1.887(2); Cu–N 2.146(2)–2.340(3) Å).13 Although these observations are suggestive of Jahn–Teller compression (indicating the unusual {d(z2)} electronic ground state of Cu1 ions), the present geometries, most likely, may be attributed to the dynamic disorder of an axis of Jahn–Teller elongation and consequent librational effects.31 First, the examination of complex 4 at different temperatures (4a: 296 K, 4b: 105 K, using the same single crystal) reveals an appreciable temperature dependence of the Cu1–N bond lengths. Being actually equivalent at r.t. (2.221(2) × 2 and 2.252(2) × 2 Å), two pairs of bonds became essentially differentiated upon cooling (2.1733(18) × 2 and 2.300(2) × 2 Å) (Table 5). Second, mean-square displacement amplitude (MSDA)32 analysis clearly indicates the presence of librational disorder involving all four Cu1–N bonds, at both temperatures (for Cu1–O1, Cu1–N1 and Cu1–N4 bonds, <d2> (×104 Å2) = 11(11), 95(12) and 78(12) at 296 K, and 9(8), 74(11) and 41(10) at 105 K, respectively), with the corresponding <d2> values being particularly high at r.t. Importantly, the bonds adopted by Cu2 and Cu3 ions are actually temperature invariant (Table 5). The application of such an approach towards “compressed” coordination geometry of Cu2+ ions was extensively discussed by Halcrow.31
The fact that the cluster units accommodate in total eight carboxylate and four pyridazine groups allows a very high, up to twelve, connectivity at the net nodes.4 The latter is decreased in view of the generation of two double carboxylate links. This produces only a six-connected cluster/carboxylate subtopology (primitive cubic net), while further cross-linking of the nodes by bipyridazine ligands yields a rarely encountered33 uninodal ten-coordinated framework with a Schläfli point symbol {312·428·55} (identified by a “bct” notation in the Reticular Chemistry Structure Resource database).34
Magnetic properties
Compounds 1, 3 and 4, representing the cluster types A, B and C, respectively, were selected for an investigation of their magnetic properties. The χmT(T) plot of a polycrystalline sample of [Cu2(μ-OH){TMA}(L)(H2O)] (1) (scaled per Cu2 unit) exhibits decreasing χmT values on cooling, starting from 0.91 cm3 K mol−1 at 300 K and approaching a value of 0.05 cm3 K mol−1 at 1.9 K (Fig. 6). This plot is typical of intracluster antiferromagnetic exchange interactions leading to a diamagnetic ground state. This behaviour may be compared with simpler binuclear systems sustained by the mixed hydroxo + syn–syn carboxylate bridge between the Cu ions, which exhibit a strong ferromagnetic coupling.35 The small low temperature χmT value originates from a minor paramagnetic phase; this is reflected in the Curie tail as seen in the χm(T) plot (Fig. S24†).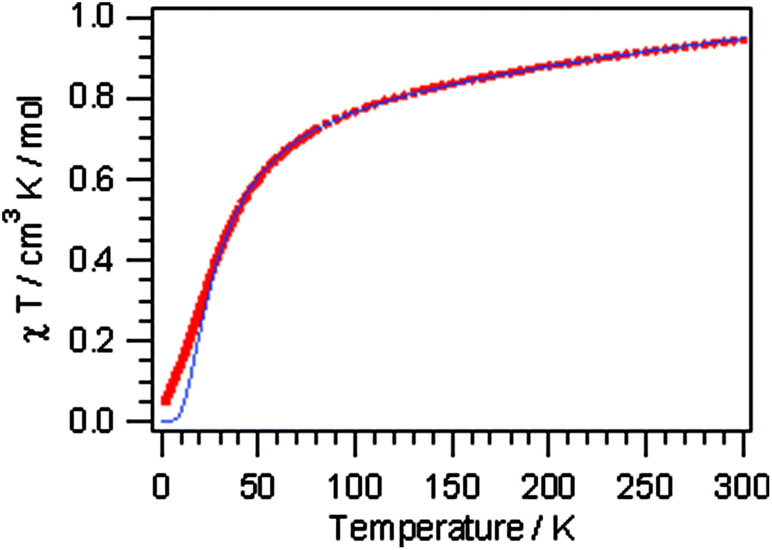 | ||
| Fig. 6 Thermal variation of χmT for 1 (the solid line is drawn based on the Bleaney–Bowers equation). | ||
The inverse magnetic susceptibility curve (Fig. S25†) shows a linear behavior in the temperature range of 50 to 300 K, which results in a Weiss constant θ = −37 K, in good agreement with the negative slope of the χmT versus T curve. We can use the following isotropic Hamiltonian to describe the intracluster magnetic exchange interactions:
| H = −2J(S1S2) |
The magnetic susceptibility data were least-squares fit to the Bleaney–Bowers equation36 for isotropic exchange in the Cu(II) pair. A good simulation of the data is achieved with J = −17.5 cm−1 (g = 2.1). The fact that compound 1 shows a very complex bonding pattern with a heteroligand pyridazine/OH/carboxylate triple bridge connecting the two Cu(II) ions prevents a detailed structure–property correlation, and thus the experimentally determined J parameter comprises all possible exchange pathways between the spin centers.
The χmT(T) plot of a polycrystalline sample of [Cu4(μ3-OH)2{TDC}3(L)2(H2O)2]·7H2O (3) (scaled per Cu4 unit) exhibits strongly decreasing χmT values on cooling, starting from 0.34 cm3 K mol−1 at 300 K and approaching a zero value around 90 K (Fig. 7). The high temperature χmT values are much smaller than the spin-only value of 1.5 cm3 K mol−1 for four non-interacting spins with S = 1/2 (g = 2.0) which is due to strong intracluster antiferromagnetic exchange interactions. The low temperature χmT values indicate a diamagnetic ground state of the tetranuclear complex.
 | ||
| Fig. 7 Thermal variation of χmT for 3 (solid line is a fit according to eqn (1)). | ||
Given the butterfly-type arrangement of the tetranuclear cluster fragment (Scheme 3) and leaving aside the asymmetry of the bonding pattern on the wings, we can use it as an approximation which is justified below. The following isotropic Hamiltonian describes the intracluster magnetic exchange interactions [eqn (1)]:
| H = −2J1(S2S2i) − 2J2(S1S2 + S1S2i + S2S1i + S2iS1i) | (1) |
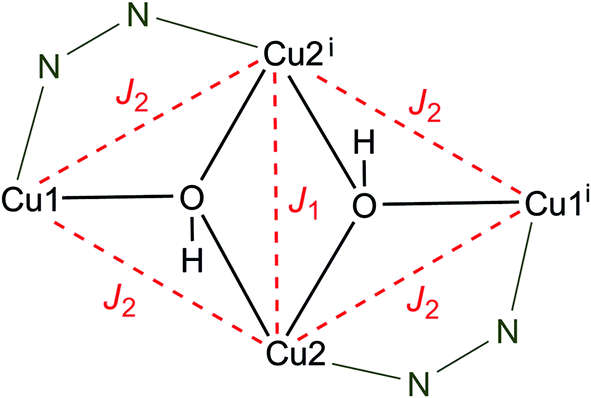 | ||
| Scheme 3 {Cu4(μ3-OH)2} core fragment of 3 and the corresponding magnetic coupling scheme. | ||
This model approximates the [Cu4] core with a rhombic symmetry while differentiating two magnetic exchange pathways, namely Cu2–Cu2i (J1) vs. Cu1–Cu2, Cu1–Cu2i, Cu2–Cu1i, Cu2i–Cu1i (J2). Consequently, this Hamiltonian gives rise to six spin states comprising the total spin values (ST) of 2, 1, 0 with the corresponding energy levels in terms of the magnetic coupling constants as given below:
| E1(ST = 2) = −½J1 − 2J2 |
| E2(ST = 1) = −½J1 + 2J2 |
| E3(ST = 1) = −½J1 |
E4(ST = 1) = +![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) J1 J1 |
| E5(ST = 0) = −½J1 + 4J2 |
| E6(ST = 0) = +J1 |
Applying these energy values to the van Vleck equation gives the following analytical expression [eqn (2)]:
| χm = 2Nβ2g2/kT(A/B); A = 5exp(−E1/kT) + exp(−E2/kT) + exp(−E3/kT) + exp(−E4/kT) B = 5exp(−E1/kT) + 3exp(−E2/kT) + 3exp(−E3/kT) + 3exp(−E4/kT) + exp(−E5/kT) + exp(−E6/kT) | (2) |
To reiterate at this stage, the linking pattern at the wing sides of the {Cu4(μ3-OH)2} core differs since on two sides a chelating pyridazine is involved, a fact which would principally ask for two different exchange parameters, say J2 and J3. This would result in six energy levels in the function of three J parameters.25b However, in view of the rather smooth curve of the χmT(T) plot one can understand that any trials to fit the data to a three parameter model are not conclusive due to overparametrization. Next, it clearly turned out best to fix the J1 parameter to a reasonable value of −110 cm−1; this coupling strength results from an analogous core structure25a and then using eqn (2), the experimental data were fitted satisfactorily in the temperature range 300–30 K with J2 = −228 cm−1 (g = 2.07). Both values of the coupling constants express the fairly strong antiferromagnetic intracluster coupling which consequently results in a diamagnetic ground state. One has to bear in mind, however, that the J2 parameter now represents an average coupling value on the wing sides.
The χmT(T) and χm(T) plots of a polycrystalline sample of [Cu5(μ-OH)2{BDC}4(L)2(H2O)2]·5H2O (4) (scaled per Cu5 unit) are shown in Fig. 8. The high-temperature χmT value of 1.75 cm3 K mol−1 is smaller than the spin-only value of 2.06 cm3 K mol−1 for five non-interacting spins with S = 1/2 (g = 2.2) and they decrease to a value approaching zero at 2 K. Both are indicative of strong antiferromagnetic exchange interactions within the pentameric unit.
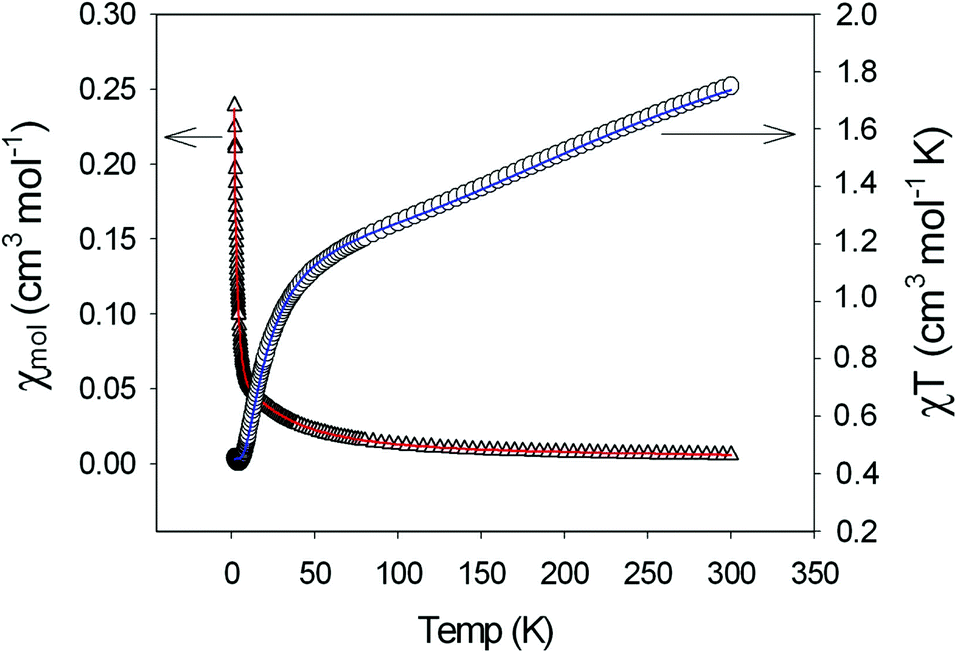 | ||
| Fig. 8 Thermal variation of χm and χmT for 4 (the solid lines are a fit to the data). | ||
A structural analysis reveals that the central Cu3 subunit involves Cu1 bound to two equivalent inversion related Cu2 via two bridges, hydroxo and pyridazine. The pyridazine bridges can be considered to be orthogonal connections, because of the short–long bonds to Cu2 and Cu2′ within each pair. Cu–O connections to the bridging OH− groups are short and since the Cu–OH–Cu angle (114.3°) is large, one would anticipate strong antiferromagnetic exchange between Cu1 and the two Cu2 atoms through these bridges (equatorial–equatorial connections). The Cu2–Cu3 connection is really just a 1,3-carboxylate with short contacts to both copper centers (the aqua O2 donor is just bonded to Cu2 with a short contact, whereas the secondary Cu3–O2 interactions are only very distal and weak, Table 6). Therefore both Cu2 atoms are going to be antiferromagnetically coupled to Cu3, but with a much smaller J value. The corresponding magnetic coupling scheme is given as
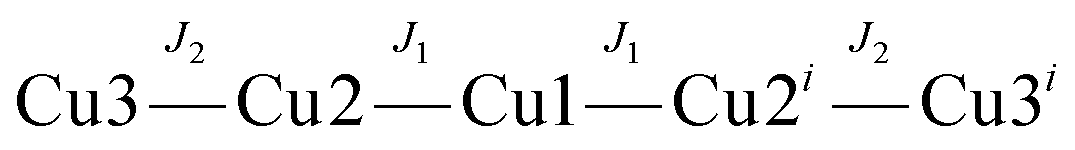
| H = −2J1(S1S2 + S1S2i) − 2J2(S2S3 + S2iS3i) |
| 1 | 2 | 3 | 4a | 4b | 5 | |
|---|---|---|---|---|---|---|
| Formula | C27H26Cu2N4O8 | C62H78Cu4N8O19 | C54H66Cu4N8O23S3 | C68H72Cu5N8O25 | C68H72Cu5N8O25 | C68H74Cu5N8O30 |
| T, K | 213 | 296 | 173 | 296 | 105 | 296 |
| M | 661.60 | 1493.48 | 1545.49 | 1719.04 | 1719.04 | 1801.05 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group, Z | C2/c, 8 |
P, 1 |
P21/n, 2 | P21/n, 2 | P21/n, 2 | P21/c, 2 |
| a/Å | 24.8536(18) | 11.1208(9) | 13.3933(4) | 10.0517(7) | 10.0322(6) | 10.0552(5) |
| b/Å | 10.8716(7) | 11.5078(10) | 9.9035(3) | 18.4855(9) | 18.4439(9) | 18.7840(8) |
| c/Å | 18.6837(14) | 13.8841(13) | 23.7661(8) | 19.1143(13) | 19.0354(12) | 19.1757(9) |
| α | 90 | 83.981(6) | 90 | 90 | 90 | 90 |
| β | 101.581(8) | 67.507(6) | 98.036(2) | 104.721(5) | 104.897(5) | 104.829(2) |
| γ | 90 | 62.065(5) | 90 | 90 | 90 | 90 |
| V/Å3 | 4945.5(6) | 1444.3(2) | 3121.39(17) | 3435.1(4) | 3403.8(3) | 3501.2(3) |
| μ(Mo-Kα)/mm−1 | 1.783 | 1.541 | 1.530 | 1.613 | 1.627 | 1.591 |
| D c/ g cm−3 | 1.777 | 1.717 | 1.644 | 1.662 | 1.677 | 1.708 |
| θ max/° | 25.96 | 26.94 | 27.10 | 28.54 | 28.54 | 27.48 |
| Meas/ Unique reflns | 16964/4789 |
15505/6248 |
29009/6862 |
28251/8582 |
29556/8509 |
31226/8020 |
| R int | 0.041 | 0.061 | 0.085 | 0.042 | 0.046 | 0.088 |
| Parameters refined | 433 | 419 | 477 | 502 | 502 | 502 |
| R 1 [I > 2σ(I)] | 0.033 | 0.055 | 0.049 | 0.036 | 0.032 | 0.047 |
| wR2 [all data] | 0.084 | 0.098 | 0.104 | 0.095 | 0.081 | 0.104 |
| Goof on F2 | 0.917 | 0.916 | 0.990 | 0.965 | 0.901 | 1.010 |
| Max, min peak/e Å−3 | 0.44, −0.78 | 0.58, −0.59 | 0.59, −0.51 | 0.64, −1.10 | 0.54, −0.55 | 0.47, −0.53 |
The exchange model is therefore based on a strongly antiferromagnetically coupled central linear trinuclear Cu3 group, with the Cu3 centers weakly antiferromagnetically coupled to the terminal coppers (Cu2) of the triad.
In the data fitting J1 and J2 were represented as a ratio (J1/J2) and after varying the ratio, fitting the data, and observing and minimizing the fitting coefficient, a satisfactory fit was obtained using MAGMUN4.11.37 This program calculates the total spin states and their energies based on the exchange Hamiltonian, and determines the fitted parameters internally through weighted non-linear least squares procedures. The optimum ratio was found to be J1/J2 = 10, with the best fit parameters at J1 = −125(3) cm−1, J2 = −12.5(3) cm−1 and g = 2.18, providing the eminently sensible model based on the regression statistics and the structure.
Thermal stability
The thermal behavior of compounds 1–5 was examined by complementary TG/DTA-MS and temperature-dependent powder X-ray diffractometry (TD PXRD) techniques. With the exception of complex 4, the stages of dehydration and further weight losses due to the thermal destructions are not separated and proceed above 150 °C, with the crystallization of an inorganic product (CuO) observed above 350 °C. Compound 1 is somewhat more stable. It does not show any weight loss until a temperature of 260 °C. In the range 260–340 °C, with a DTG peak maximum at 300 °C, it decomposes (−29.7%) with dehydration (m/z 18) and release of CO2 (m/z 44) due to decarboxylation. This is accompanied by a loss of crystallinity at 260 °C, as evidenced by PXRD patterns. The stability of the aliphatic analog 2 is comparable. Dehydration starts at 160 °C, and above 220 °C it was accompanied by decarboxylation which results in one unresolved stage of 12.3% weight loss in the range of 160–270 °C (maximum at 250 °C). The PXRD patterns are also indicative of the dehydration process (160–170 °C) since the interlayer spacing is sensitive to elimination of the guest molecules located between the coordination layers. Disintegration of the structure was observed at 230 °C. Complex 3 experiences partial dehydration above 60 °C, with the release of 5 water molecules in the temperature range of 60–170 °C (5.60% observed; 5.82% calculated). This results in a phase transition at 170 °C, and at 195 °C the compound gets amorphous due to decomposition with the release of CO2. For the closely related 4 and 5, disintegration of the frameworks occurs at an identical temperature of 235 °C. However, due to a more hydrophobic nature of the isophthalate framework in 4, vs. the 5-hydroxyisophthalate analog 5, the initial dehydration in the first case proceeds more readily. The TG curve indicates two insufficiently separated stages at 110–200 °C and 200–250 °C, with a total weight loss of 7.3% corresponding to the release of coordinated and outer sphere water molecules (calculated 7.32%). Further thermal decomposition proceeds at 270–340 °C. In the case of 5, the dehydration begins at 160 °C. Progressive release of water molecules (m/z 18) coincides with beginning of decarboxylation (m/z 44) at 265 °C. The weight loss of 8.5% in the temperature range 160–260 °C corresponds to the elimination of 8 water molecules (calculated 8.0%).IR spectra
The IR spectra of complexes 1–5 exhibit strong and broad absorption bands in the region of 3230–3500 cm−1, which are attributed to the ν(OH) vibrations of the aqua and hydroxo ligands. Bands at 2850–2933 cm−1 correspond to ν(CH) vibrations of adamantane and aromatic moieties. The absorptions for the carbonyl group, ν(CO), appear as very strong bands at 1590–1627 cm−1 (see the Experimental section). The absence of bands at 1730–1690 cm−1, where ν(CO) of COOH is expected to appear, confirms full deprotonation of the polycarboxylate ligands in 1–5.38 Strong absorption bands at 1358–1382 cm−1 are characteristic of all the compounds; they could be assigned to ν(CN) of the pyridazine rings.39Conclusions
The present results are important for providing innovative strategies for the construction of extended coordination lattices incorporating polynuclear metal–organic clusters. A combination of pyridazine, carboxylate and hydroxo ligands is especially well suited for sustaining coordination patterns of different nuclearities and connectivities: this kind of bridges reveals a perfect compatibility and they readily complement each other and act in a synergistic manner. In cooperation with μ-carboxylate and, especially μ-hydroxo groups, pyridazine typically behaves as a short-distance diatomic bridge. Therefore, the present heteroligand system unites and extends the structural and functional potential of such common types of organic and inorganic bridges for the generation of discrete polynuclear arrangements of metal ions. At the same time, a multiplication of the ligand functionality, as it occurs for di- and tricarboxylate and bipyridazine ligands, allows the integration of the clusters into polymeric arrays, which could be anticipated for a broad range of transition metal ions and different kinds of organic linkers. Our study suggests also a new approach and attractive preparative sequence towards bridge-head heteroaryl C-functionalization of adamantane, which could find wider applications for the development of multivalent geometrically rigid molecular building blocks incorporating “nanodiamond scaffolds”.40 Moreover, the study of magnetic properties of coordination network compounds is a very topical issue in the field of molecular magnetism.41 In the present case, the magnetic susceptibility data for clusters 1, 3 and 4 reveal substantial antiferromagnetic coupling strengths, however, with varying ratios of the coupling parameters. In particular, common to all three structure types A, B and C is the quite complex nature of bonding patterns including heteroligand multiple bridges connecting the spin centers, which prevents a discussion of more detailed structure–property correlations.Experimental
All starting materials were chemicals of reagent grade and used as received without further purification. Adamantane-1,3,5-tricarboxylic acid was synthesized in 62% yield by the Koch–Haaf carboxylation of 1,3,5-adamantanetriol with 100% HCOOH in a 15% oleum medium.42 The 1,2,4,5-tetrazine was prepared and freshly purified by sublimation as described previously.13,14cSynthesis of 1,3-bis(pyridazin-4-yl)adamantane (L) (Scheme 1). A solution of 1.24 g (15.1 mmol) 1,2,4,5-tetrazine and 1.26 g (6.8 mmol) 1,3-diethynyladamantane43 in 40 ml of dry 1,4-dioxane was stirred at 90 °C over a period of 25 h. The reaction proceeded smoothly and the evolution of dinitrogen gas ceased after 15–16 h. The precipitate was filtered off, washed with 1,4-dioxane and diethyl ether and dried in air. It was dissolved in boiling methanol and the solution was decolorized by 15 min reflux with charcoal, then it was filtered and evaporated yielding 1.41 g (71%) of a colorless crystalline product. 1H NMR (400 MHz, dmso-d6): δ 9.38–9.28 (m, 2H), 9.06 (d, J = 5.4 Hz, 2H), 7.58 (dd, J = 5.5, 2.6 Hz, 2H), 2.42–2.32 (m, 2H), 2.10 (s, 2H), 1.98 (t, J = 3.7 Hz, 8H), 1.82 (d, J = 3.5 Hz, 2H). Anal. Calcd for C18H20N4: C, 73.94; H, 6.90; N, 19.17. Found: C, 74.06; H, 6.88; N, 19.04.
Preparation of the coordination compounds
The complexes were prepared under hydrothermal conditions as follows. A mixture of the starting compounds and distilled water were placed in a 20 mL Teflon-lined stainless steel autoclave, stirred for 10–30 min, and heated at 140 °C for 40–70 h in an oven, with further cooling to room temperature. In each case, the excess of bipyridazine ligand was essential for partial hydrolysis of Cu(II) ions and generation of the desired hydroxo-bridged species. Under these conditions and absence of di- or tricarboxylate components, the reactions of Cu(OAc)2·H2O and the bipyridazine ligand did not afford insoluble products.
Synthesis of [Cu2(μ-OH){TMA}(L)(H2O)] (1). A mixture of 6.8 mg (0.034 mmol) Cu(OAc)2·H2O, 3.1 mg (0.015 mmol) trimesic acid and 10.0 mg (0.034 mmol) ligand in a 1:0.44:1 molar ratio with 4 mL water was stirred for 30 min in a Teflon vessel, and then it was heated at 140 °C for 70 h. Slow cooling to r.t. over a period of 48 h (cooling rate 2.5 °C h−1) afforded a pure product as green prisms, which were washed with 3 mL of water and dried in air for 1 h (yield: 7.9 mg, 80%). Anal. Calcd for C27H26Cu2N4O8: C, 49.01; H, 3.96; N, 8.47. Found: C, 48.89; H, 4.01; N, 8.35. IR (KBr discs, selected bands, cm−1): 568w, 717s, 761m, 977w, 1022w, 1091w, 1359vs, 1407m, 1433s, 1558vs, 1607vs, 2852w, 2900m, 3233mbr, 3409m.
Synthesis of [Cu4(μ3-OH)2{ATC}2(L)2(H2O)2]·H2O (2). The compound was prepared in a similar manner, starting with 6.8 mg (0.034 mmol) Cu(OAc)2·H2O, 3.5 mg (0.013 mmol) adamantane-1,3,5-tricarboxylic acid, 10.0 mg (0.034 mmol) ligand (1:0.38:1 molar ratio) and 4 mL of water. Small green prismatic crystals of the product were obtained in 80% yield. Anal. Calcd for C62H78Cu4N8O19: C, 49.86; H, 5.26; N, 7.50. Found: C, 50.03; H, 5.19; N, 7.59. IR (KBr discs, selected bands, cm−1): 568w, 710m, 832w, 1084w, 1209w, 1274m, 1328s, 1358vs, 1442w, 1590vs, 2850m, 2930s, 3044m, 3269mbr, 3471mbr.
Synthesis of [Cu4(μ3-OH)2{TDC}3(L)2(H2O)2]·7H2O (3). An equimolar mixture of 5.0 mg (0.025 mmol) Cu(OAc)2·H2O, 4.3 mg (0.025 mmol) 2,5-thiophenedicarboxylic acid, 7.4 mg (0.025 mmol) ligand were placed in a Teflon vessel, and 5 mL of water was added. The mixture was stirred for 30 min and then it was heated at 140 °C for 24 h. Small blue prisms of the pure product were obtained in 60% yield after slow cooling to r.t. for a period of 72 h. Anal. Calcd for C54H66Cu4N8O23S3: C, 41.96; H, 4.30; N, 7.25. Found: C, 42.11; H, 4.22; N, 7.37. IR (KBr discs, selected bands, cm−1): 556w, 689w, 774m, 808w, 973w, 1022w, 1072w, 1111w, 1314s, 1358vs, 1452w, 1527s, 1561s, 1597vs, 2852m, 2913m, 3407sbr.
Synthesis of [Cu5(μ-OH)2{BDC}4(L)2(H2O)2]·5H2O (4). A mixture of 6.8 mg (0.034 mmol) Cu(OAc)2·H2O, 5.7 mg (0.034 mmol) isophthalic acid, 10.0 mg (0.034 mmol) ligand, all in an equimolar ratio, and 4 mL of water was stirred for 10 min and then heated at 140 °C for 24 h in a Teflon vessel. After cooling to r.t. for a period of 72 h, large blue-green crystals of the pure product were collected by filtration (yield: 9.3 mg, or 80%, based on Cu). Anal. Calcd for C68H72Cu5N8O25: C, 47.51; H, 4.22; N, 6.52. Found: C, 47.59; H, 4.19; N, 6.63. IR (KBr discs, selected bands, cm−1): 548w, 716m, 748m, 806w, 1084w, 1156w, 1268w, 1368s, 1390vs, 1450m, 1476w, 1560s, 1622vs, 2856m, 2922m, 3400sbr, 3456sbr.
Synthesis of [Cu5(μ-OH)2{HO-BDC}4(L)2(H2O)2]·6H2O (5). The compound was prepared similarly as for 4 from a mixture of 6.8 mg (0.034 mmol) Cu(OAc)2·H2O, 6.2 mg (0.034 mmol) 5-hydroxyisophthalic acid and 10.0 mg (0.034 mmol) ligand, giving large blue-green prisms (yield 75%). Anal. Calcd for C68H74Cu5N8O30: C, 45.34; H, 4.14; N, 6.22. Found: C, 45.47; H, 4.13; N, 6.39. IR (KBr discs, selected bands, cm−1): 558w, 646w, 678w, 721s, 777s, 985m, 1001m, 1021m, 1102w, 1128w, 1195m, 1218w, 1265m, 1280m, 1296m, 1382vs, 1420s, 1449m, 1472m, 1545s, 1593s, 1627s, 2856m, 2899m, 2933s, 3370sbr, 3500s, 3586m.
Measurements
Thermogravimetric/differential thermal analysis mass spectrometry (TG/DTA-MS) was performed on a Netzsch F1 Jupiter device connected to an Aeolos mass spectrometer. The sample was heated at a rate of 10° min−1. The temperature-dependent X-ray measurements were carried out on a Stoe STADIP with a high-temperature attachment and a image-plate detector system. PXRD was carried out on a Stoe STADIP (Cu Kα1) using a linear PSD detector and on a Shimadzu XRD-6000 (Cu Kα radiation). Elemental analysis was carried out with a Vario EL-Heraeus microanalyzer. IR spectra (400–4000 cm−1, KBr disks) were collected using a Perkin-Elmer FTIR spectrometer.Magnetic susceptibility measurements were made on a Quantum Design MPMS SQUID-XL magnetometer under an applied magnetic field of 103 Oe between 300 and 1.9 K. The samples were prepared in a gelatine capsule. Diamagnetic corrections were made for the samples using the approximation −0.45 × molecular weight × 10−6 cm3 mol−1 and the sample holder was corrected for by measuring directly the susceptibility of the empty capsule.
X-Ray crystallography
The diffraction data were collected with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) (Table 6). Measurements for 1 at 213 K and for 4 (4a: 296 K, 4b: 105 K) were made using a Stoe Image Plate Diffraction System, φ oscillation scans (numerical absorption correction using X-RED and X-SHAPE).44 Measurements for 2, 3 and 5 were performed at 173 K on a Bruker APEXII CCD area-detector diffractometer (ω scans). The data were corrected for Lorentz-polarization effects and for the effects of absorption (multi-scans method). The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELX-97 package.45 The CH-hydrogen atoms were added geometrically, with Uiso = 1.2Ueq (C) and OH-hydrogen atoms were located and included with fixed d (O–H) = 0.85 Å and Uiso = 1.5Ueq (O). Part of the solvate water molecules in 2–4 is disordered. They were refined anisotropically and the hydrogen atoms were not added. In 1, the binuclear cluster is equally disordered over two positions (Cu ion, μ-OH and aqua ligand; one of the pyridazine cycles is also disordered adopting two orientations). Attempted refinements in space groups of lower symmetry did not afford an ordered model. This fragment was freely refined anisotropically and the hydrogen atoms were added as stated above with partial contributions of 0.5. In 3, one of the TDC2− ligands is equally disordered over two positions across a center of inversion. The disorder was resolved without restraints in geometry, but with restrained parameters for thermal motion of the carbon atoms. The topological analysis was performed using TOPOS 4.046 and Graphical visualization of the structures was made using the program Diamond 2.1e.47Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft, grant KR 1675/4-3 (HK and KVD) and by the Swiss National Science Foundation (grant 200021-147143) is gratefully acknowledged.References
- D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC; J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC; H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed; J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef CAS PubMed.
- P. Hubberrstey, X. Lin, N. R. Champness and M. Schröder, in Metal-Organic Frameworks: Design and Applications, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010 Search PubMed.
- Q. Lin, T. Wu, X. Bu and P. Feng, Dalton Trans., 2012, 41, 3620 RSC; J. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schröder, Chem. Commun., 2007, 840 RSC; G.-H. Cui, C.-H. He, C.-J. Jiao, J.-C. Geng and V. A. Blatov, CrystEngComm, 2012, 14, 4210 RSC; X.-M. Zhang, R.-Q. Fang and H.-S. Wu, J. Am. Chem. Soc., 2005, 127, 7670 CrossRef CAS PubMed; D. Li, T. Wu, X.-P. Zhou, R. Zhou and X.-C. Huang, Angew. Chem., Int. Ed., 2005, 44, 4175 CrossRef PubMed; J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757 RSC.
- B. Gil-Hernández, P. Gili, J. K. Vieth, C. Janiak and J. Sanchiz, Inorg. Chem., 2010, 49, 7478 CrossRef CAS PubMed; J. K. Maclaren, J. Sanchiz, P. Gilib and C. Janiak, New J. Chem., 2012, 36, 1596 RSC; H. H. Monfared, J. Sanchiz, Z. Kalantari and C. Janiak, Inorg. Chim. Acta, 2009, 362, 3791 CrossRef PubMed.
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC; D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123 CrossRef CAS PubMed.
- S. S. Tandon, L. K. Thompson and R. C. Hynes, Inorg. Chem., 1992, 31, 2210 CrossRef CAS; C. Li, N. Kanehisa, Y. Miyagi, Y. Nakao, S. Takamizawa, W. Mori and Y. Kai, Bull. Chem. Soc. Jpn., 1997, 70, 2429 CrossRef.
- T. Yi, C. Ho-Chol, S. Gao and S. Kitagawa, Eur. J. Inorg. Chem., 2006, 1381 CrossRef CAS.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC.
- A. T. M. Marcelis and H. C. van der Plas, J. Heterocycl. Chem., 1987, 24, 545 CrossRef CAS; J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, Eur. J. Org. Chem., 1998, 2885 CrossRef.
- K. V. Domasevitch, I. A. Gural'skiy, P. V. Solntsev, E. B. Rusanov, H. Krautscheid, J. A. K. Howard and A. N. Chernega, Dalton Trans., 2007, 3140 RSC.
- (a) I. A. Gural'skiy, P. V. Solntsev, H. Krautscheid and K. V. Domasevitch, Chem. Commun., 2006, 4808 RSC; (b) K. V. Domasevitch, P. V. Solntsev, I. A. Gural'skiy, H. Krautscheid, E. B. Rusanov, A. N. Chernega and J. A. K. Howard, Dalton Trans., 2007, 3893 RSC; (c) A. S. Degtyarenko, P. V. Solntsev, H. Krautscheid, E. B. Rusanov, A. N. Chernega and K. V. Domasevitch, New J. Chem., 2008, 32, 1910 RSC.
- T. Otieno, S. J. Rettig, R. C. Thompson and J. Trotter, Inorg. Chem., 1995, 34, 1718 CrossRef CAS.
- K. V. Domasevitch, J. A. Rusanova, I. A. Gural'skiy and P. V. Solntsev, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, m295 CAS.
- C. Näther and I. Jeß, Inorg. Chem., 2003, 42, 2968 CrossRef CAS PubMed; P. V. Solntsev, J. Sieler, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2004, 1153 RSC; K. V. Domasevitch, P. V. Solntsev, H. Krautscheid, I. S. Zhylenko, E. B. Rusanov and A. N. Chernega, Chem. Commun., 2012, 48, 5847 RSC; L. Plasseraud, H. Maid, F. Hampel and R. W. Saalfrank, Chem. – Eur. J., 2001, 7, 4007 CrossRef; A. S. Batsanov, M. Begley, M. W. George, P. Hubberstey, M. Munakata, C. Russell and P. H. Walton, J. Chem. Soc., Dalton Trans., 1999, 4251 RSC; M. Maekawa, M. Munakata, T. Kuroda-Sowa and Y. Nozaka, J. Chem. Soc., Dalton Trans., 1994, 603 RSC; M. J. Begley, P. Hubberstey, C. E. Russell and P. H. Walton, J. Chem. Soc., Dalton Trans., 1994, 2483 RSC; D. Hagrman, C. Sangregorio, C. J. O'Connor and J. Zubeita, J. Chem. Soc., Dalton Trans., 1998, 3707 RSC.
- F. Lloret, G. De Munno, M. Julve, J. Cano, R. Ruiz and A. Caneschi, Angew. Chem., Int. Ed., 1998, 37, 135 CrossRef CAS; A. Escuer, F. A. Mautner, N. Sanz and R. Vicente, Inorg. Chim. Acta, 2002, 340, 163 CrossRef; T. Otieno, S. J. Rettig, R. C. Thompson and J. Trotter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 2067 CrossRef; L. Pazderski, E. Szlyk, A. Wojtczak, L. Kozerski and J. Sitkowski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, 1270 Search PubMed; E. Krupicka and A. Lentz, Z. Kristallogr. - New Cryst. Struct., 2001, 216, 289 Search PubMed; A. S. Degtyarenko and K. V. Domasevitch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 219 Search PubMed.
- L. Carlucci, G. Ciani, M. Moret and A. Sironi, J. Chem. Soc., Dalton Trans., 1994, 2397 RSC.
- (a) T. Fetzer, A. Lentz, T. Debaerdemaeker and O. Abou-El-Wafa, Z. Naturforsch., Teil B, 1990, 45, 199 CAS; (b) L. Pazderski, E. Szlyk, A. Wojtczak, L. Kozerski, J. Sitkowski and B. Kamieński, J. Mol. Struct., 2004, 697, 143 CrossRef CAS PubMed.
- J. Cano, G. De Munno, F. Lloret and M. Julve, Inorg. Chem., 2000, 39, 1611 CrossRef CAS.
- T. V. Yilmaz, E. Senel and C. Kazak, Aust. J. Chem., 2008, 61, 634 CrossRef.
- K. Hyde, G. F. Kokoszka and G. Gordon, J. Inorg. Nucl. Chem., 1969, 31, 1993 CrossRef CAS; J. R. Ferraro, J. Zipper and W. Wozniak, Appl. Spectrosc., 1969, 23, 160 CrossRef; J. R. Allan, G. A. Barnes and D. H. Brown, J. Inorg. Nucl. Chem., 1971, 33, 3765 CrossRef; S. Emori, M. Inoue and M. Kubo, Bull. Chem. Soc. Jpn., 1972, 45, 2259 CrossRef.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC; B. Wisser, Y. Lu and C. Janiak, Z. Anorg. Allg. Chem., 2007, 633, 1189 CrossRef CAS; H. A. Habib, A. Hoffmann, H. A. Höppe and C. Janiak, Dalton Trans., 2009, 1742 RSC; H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734 RSC.
- (a) G. A. Senchyk, A. B. Lysenko, H. Krautscheid, E. B. Rusanov, A. N. Chernega, K. W. Krämer, S.-X. Liu, S. Decurtins and K. V. Domasevitch, Inorg. Chem., 2013, 52, 863 CrossRef CAS PubMed; (b) H. A. Habib, J. Sanchiz and C. Janiak, Inorg. Chim. Acta, 2009, 362, 2452 CrossRef CAS PubMed; (c) G. A. Senchyk, A. B. Lysenko, H. Krautscheid, J. Sieler and K. V. Domasevitch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, m246 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC.
- I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, J. Solid State Chem., 2005, 178, 2452 CrossRef CAS PubMed; I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2008, 10, 1822 RSC; I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, Cryst. Growth Des., 2008, 8, 519 Search PubMed.
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS; L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. D. Vos, Chem. – Eur. J., 2006, 12, 7353 CrossRef PubMed.
- G.-X. Liu, K. Zhu, H. Chen, R.-Y. Huang, H. Xu and X.-M. Ren, Inorg. Chim. Acta, 2009, 362, 1605 CrossRef CAS PubMed; K.-H. He, Y.-W. Li, Y.-Q. Chen, W.-C. Song and X.-H. Bu, Cryst. Growth Des., 2012, 12, 2730 Search PubMed; H. Li, W. Shi, K. Zhao, Zh. Niu, H. Li and P. Cheng, Chem. – Eur. J., 2013, 19, 3358 CrossRef PubMed; H.-H. Li, W. Shi, N. Xu, Zh.-J. Zhang, Zh. Niu, T. Han and P. Cheng, Cryst. Growth Des., 2012, 12, 2602 Search PubMed.
- M. A. Halcrow, Dalton Trans., 2003, 4375 RSC; M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784 RSC.
- L. R. Falvello, J. Chem. Soc., Dalton Trans., 1997, 4463 RSC; J. D. Dunitz, V. Schomaker and K. N. Trueblood, J. Phys. Chem., 1988, 92, 856 CrossRef CAS.
- W.-C. Song, Q. Pan, P.-C. Song, Q. Zhao, Y.-F. Zeng, T.-L. Hu and X.-H. Bu, Chem. Commun., 2010, 46, 4890 RSC.
- (a) Reticular Chemistry Structure Resource (RCSR), http://rcsr.anu.edu.au; (b) V. A. Blatov and A. P. Shevchenko, TOPOS 4.0, Samara State University, Russia, 1999 Search PubMed.
- H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 4877 RSC.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- MAGMUN4.11/OW01.exe is available as a combined package free of charge from the authors (http://www.ucs.mun.ca/~lthomp/magmun). MAGMUN was developed by Dr Zh. Xu (Memorial University), and OW01.exe by Dr O. Waldmann.
- W. Brzyska and P. Sadowski, Pol. J. Chem., 1987, 61, 273 Search PubMed; W. Brzyska and W. Wolodkiewicz, Pol. J. Chem., 1986, 60, 697 Search PubMed; K. Wieghardt, J. Chem. Soc., Dalton Trans., 1973, 2548 RSC; L. J. Bellamy, The Infrared Spectra of Complex Molecules, Wiley, New York, 1958 Search PubMed.
- S. Breda, I. D. Reva, L. Lapinski, M. J. Nowak and R. Fausto, J. Mol. Struct., 2006, 786, 193 CrossRef CAS PubMed; J. Vázquez, J. J. L. Gozález, F. Márquez and J. E. Boggs, J. Raman Spectrosc., 1998, 29, 547 CrossRef.
- M. A. Gunawan, J.-C. Hierso, D. Poinsot, A. A. Fokin, N. A. Fokina, B. A. Tkachenko and P. R. Schreiner, New. J. Chem., 2014, 38, 28 RSC.
- (a) O. Kahn, Molecular Magnetism, VCH, Weinheim, Gemany, 1993 Search PubMed; (b) L. K. Thompson (ed.) Magnetism: Molecular and Supramolecular Perspectives, Coord. Chem. Rev., 2005, 249, 2549; (c) F. Bonadiao, M.-C. Senna, J. Ensling, A. Sieber, A. Neels, H. Stoeckli-Evans and S. Decurtins, Inorg. Chem., 2005, 44, 969 CrossRef; (d) M. Pilkington and S. Decurtins, Chimia, 2000, 54, 593 CAS.
- G. A. Senchyk, A. B. Lysenko, I. Boldog, E. B. Rusanov, A. N. Chernega, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2012, 41, 8675 RSC.
- T. G. Archibald, A. A. Malik and K. Baum, Macromolecules, 1991, 24, 5261 CrossRef CAS.
- (a) Stoe & Cie, X-SHAPE, Revision 1.06, Stoe & Cie GmbH, Darmstadt, Germany, 1999 Search PubMed; (b) Stoe & Cie, X-RED, Version 1.22, Stoe & Cie GmbH, Darmstadt, Germany, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef CAS; G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- V. A. Blatov, TOPOS, IUCr CompComm Newsletter, 2006, 7, 4 CrossRef CAS; V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- K. Brandenburg, Diamond 2.1e, Crystal Impact GbR, Bonn, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details for organic synthesis and characterization; IR spectra, TGA and thermo-XRPD patterns; X-ray structure refinement. CCDC 967343–967348. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00174e |
| This journal is © The Royal Society of Chemistry 2014 |