 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NHC-coordinated silagermenylidene functionalized in allylic position and its behaviour as a ligand†
Anukul
Jana
,
Moumita
Majumdar
,
Volker
Huch
,
Michael
Zimmer
and
David
Scheschkewitz
*
Krupp-Chair of General and Inorganic Chemistry, Saarland University, 66125 Saarbrücken, Germany. E-mail: scheschkewitz@mx.uni-saarland.de
First published on 15th January 2014
Abstract
Vinylidenes are common in transition metal chemistry with catalytic applications in alkene and alkyne metathesis. We report here the isolation of a heavier analogue of vinylidene, an α-chlorosilyl functionalized silagermenylidene stabilized by an N-heterocyclic carbene (NHC). Silagermenylidene (Tip2Cl)Si-(Tip)Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ge·NHCiPr2Me2 (4-E/Z; Tip = 2,4,6-iPr3C6H2; NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) is available as an E/Z-equilibrium mixture from Tip2SiSi(Tip)Li and NHCiPr2Me2·GeCl2. Reaction of 4-E/Z with Fe2(CO)9 affords a silagermenylidene Fe(CO)4 complex, which slowly isomerizes to its E-isomer at 25 °C. A rearranged Fe(CO)3 complex with an allylic SiGeSi ligand is obtained as a side product at 65 °C.
Ge·NHCiPr2Me2 (4-E/Z; Tip = 2,4,6-iPr3C6H2; NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) is available as an E/Z-equilibrium mixture from Tip2SiSi(Tip)Li and NHCiPr2Me2·GeCl2. Reaction of 4-E/Z with Fe2(CO)9 affords a silagermenylidene Fe(CO)4 complex, which slowly isomerizes to its E-isomer at 25 °C. A rearranged Fe(CO)3 complex with an allylic SiGeSi ligand is obtained as a side product at 65 °C.
Introduction
The chemistry of low-coordinate germanium has received considerable attention in recent years.1 Important bonding motifs experimentally realized include two-coordinate germylenes2 and digermynes,3 as well as three-coordinate digermenes,4 silagermenes,5 and germachalcogenones6 on the other hand. Since Robinson et al. reported the NHC-stabilized disilicon(0) species Ia,7 the use of strong donors for the isolation of highly reactive low-valent species by raising the coordination number has drastically increased.8 In germanium chemistry, germylene-type compounds (e.g. dihalogermylenes,9 digermanium(0) Ib10), and inherently polar/polarizable multiple bonds (e.g. germachalcogenones,11 digermynes12) are prominent examples that are stabilized by base-coordination under retention of remarkable reactivity. Very recently, we reported on a N-heterocyclic carbene stabilized silagermenylidene, Tip2SiGe·NHCiPr2Me2II (NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene, Scheme 1).13 With the SiGe bond, the lone pair of electrons and the coordination site of the NHC, compound II offers various potential sites for further manipulation. Initially, we demonstrated the clean [2 + 2] cycloaddition of an alkyne to the SiGe bond.13 In view of the prominent role of carbon-based vinylidene complexes in catalysis,14 an open question remains the coordination behavior of isolable heavier vinylidenes towards transition metals.15 In the case of heavier analogues of carbenes, transition metal coordination compounds are known.16
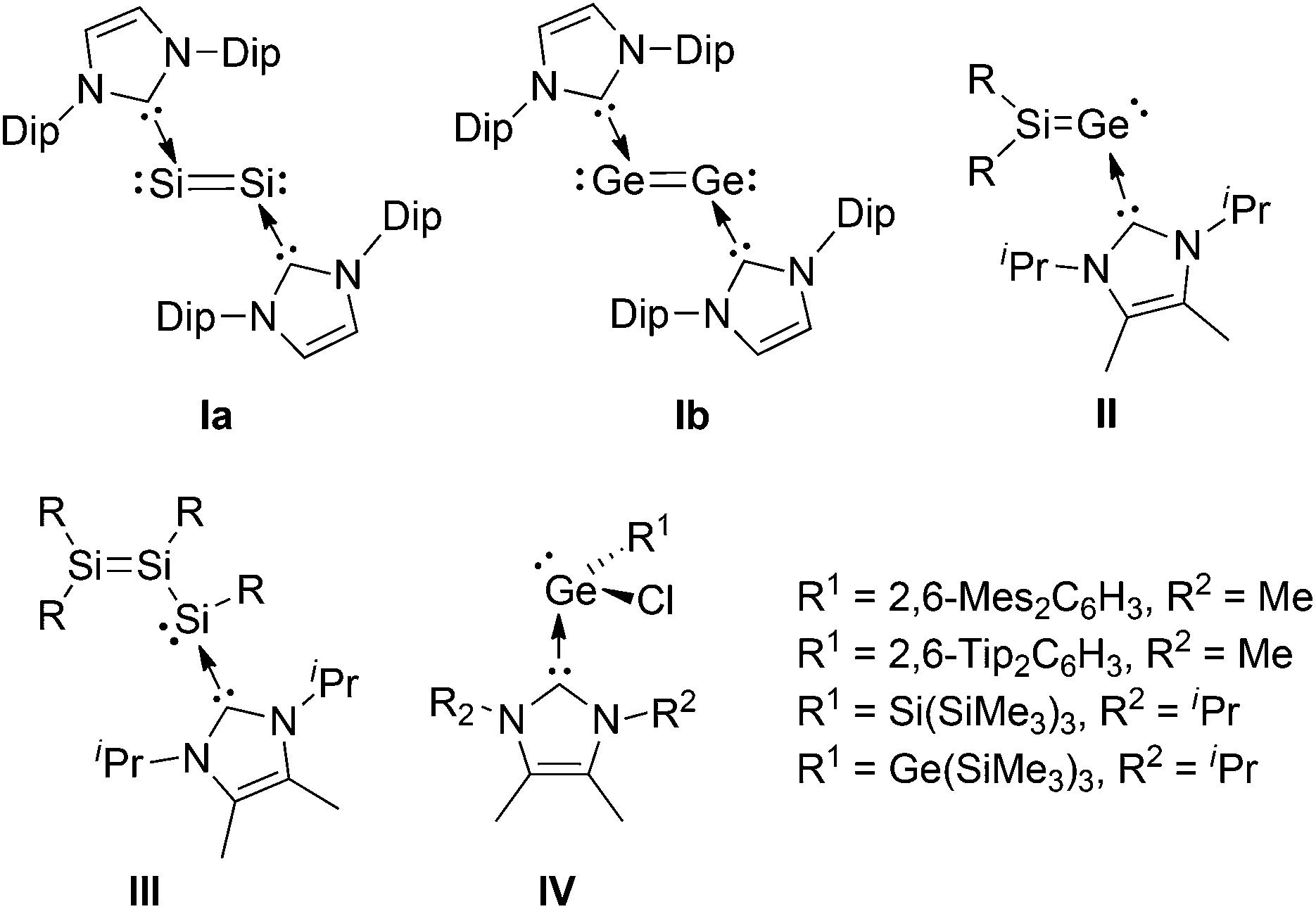 | ||
| Scheme 1 Chemical structures of Ia, Ib, II, III, and IV (Dip = 2,6-iPr2C6H3, R = Tip = 2,4,6-iPr3C6H2, Mes = 2,4,6-Me3C6H2). | ||
Our recent isolation of a stable NHCiPr2Me2-stabilized aryl(disilenyl)silylene III17 encouraged us to target the corresponding disilenyl-substituted chlorogermylene 3. We thus reacted disilenide 118 and NHC-coordinated germanium(II)chloride, NHCiPr2Me2·GeCl229c (Scheme 2). Monosubstituted NHC-coordinated chlorogermylenes IV (Scheme 1) have been prepared via similar approaches.19
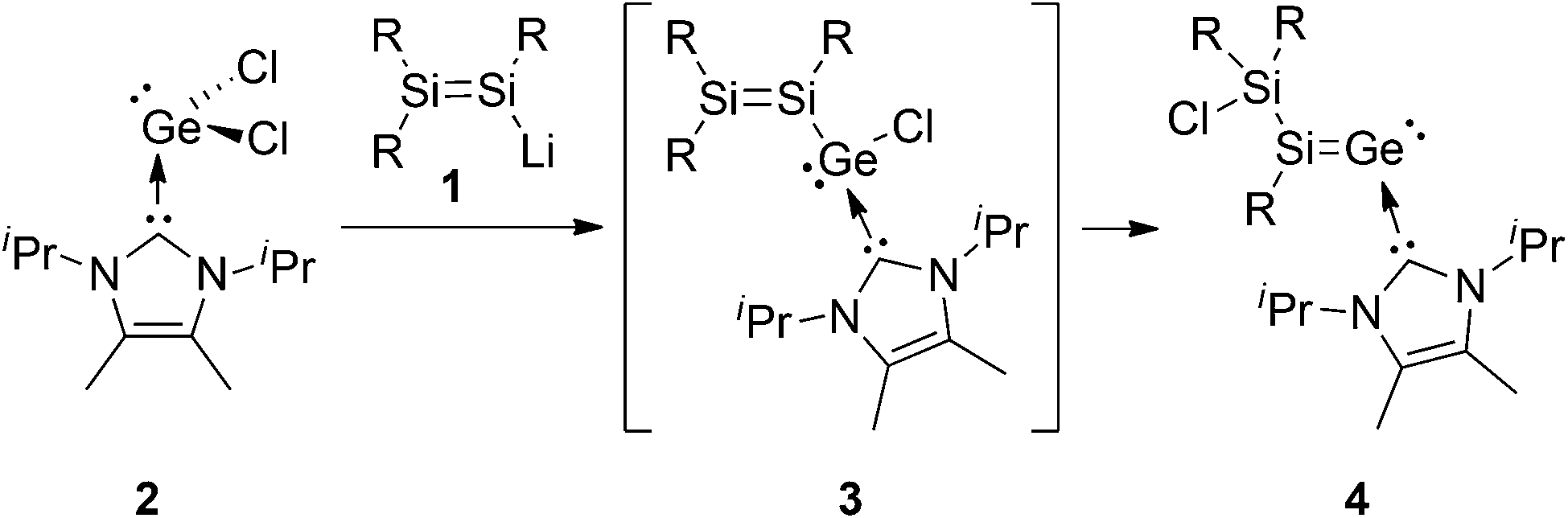 | ||
| Scheme 2 Synthesis of 4 (R = Tip = 2,4,6-iPr3C6H2). | ||
Results and discussion
Surprisingly, instead of the targeted 3 the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of 118 and 29c in toluene at −78 °C affords the NHC-coordinated silagermenylidene 4 in 62% yield (mp. 126–128 °C) (Scheme 2) with an additional peripheral Si–Cl functionality (see the Experimental section). The reaction plausibly proceeds through the NHC-stabilized chloro(disilenyl)germylene 3 as a transient followed by subsequent 1,3-migration of chlorine from germanium to the β-silicon. In solution, the 29Si resonances of 4 at 162.5 and 7.3 ppm served as the first indication of the formation of a silagermenylidene due to the close similarity to the low-field resonance of II (158.9 ppm).13 The red color of 4 is due to the longest wavelength absorption in the UV/vis spectrum at λmax = 451 nm (Table 1, ε = 9220 L mol−1 cm−1), which almost matches with that of compound II (λmax = 455 nm). In contrast to II, however, the second absorption of 4 appears as a shoulder (4: λmax = 389 nm, II: λmax = 365 nm). To gain more information about the origins of the UV/vis absorptions, we performed TD-DFT calculations of the silagermenylidene II on the basis of the experimentally determined molecular structure in the solid state. Solvent effects were approximated using the Tomasi's polarized continuum model (PCM) at the B3LYP/6-31G(d,p) level of theory.† The calculated lowest-energy excitation of II at 439 nm is predominantly associated with the π–π* transition (HOMO → LUMO), in very good agreement with the experimental value of 455 nm. The second experimental absorption band at λmax = 365 nm is due to various excitations, but does contain a significant component originating from the n–π* transition (HOMO−1 → LUMO) as suspected in our previous communication.13
:1 reaction of 118 and 29c in toluene at −78 °C affords the NHC-coordinated silagermenylidene 4 in 62% yield (mp. 126–128 °C) (Scheme 2) with an additional peripheral Si–Cl functionality (see the Experimental section). The reaction plausibly proceeds through the NHC-stabilized chloro(disilenyl)germylene 3 as a transient followed by subsequent 1,3-migration of chlorine from germanium to the β-silicon. In solution, the 29Si resonances of 4 at 162.5 and 7.3 ppm served as the first indication of the formation of a silagermenylidene due to the close similarity to the low-field resonance of II (158.9 ppm).13 The red color of 4 is due to the longest wavelength absorption in the UV/vis spectrum at λmax = 451 nm (Table 1, ε = 9220 L mol−1 cm−1), which almost matches with that of compound II (λmax = 455 nm). In contrast to II, however, the second absorption of 4 appears as a shoulder (4: λmax = 389 nm, II: λmax = 365 nm). To gain more information about the origins of the UV/vis absorptions, we performed TD-DFT calculations of the silagermenylidene II on the basis of the experimentally determined molecular structure in the solid state. Solvent effects were approximated using the Tomasi's polarized continuum model (PCM) at the B3LYP/6-31G(d,p) level of theory.† The calculated lowest-energy excitation of II at 439 nm is predominantly associated with the π–π* transition (HOMO → LUMO), in very good agreement with the experimental value of 455 nm. The second experimental absorption band at λmax = 365 nm is due to various excitations, but does contain a significant component originating from the n–π* transition (HOMO−1 → LUMO) as suspected in our previous communication.13
| 4-Eb | 4-Zb | 5-Zc | 5-Ec | 6 | |
|---|---|---|---|---|---|
| a Calculated values in parentheses. b In d6-benzene. c In d8-toluene. d In THF. | |||||
| δ29Si (SiTip) | 162.5 (159.5) | 134.0 (90.4) | 100.7 | 98.1 | 113.7 (138.3) |
| δ29Si (SiTip2) | 7.3 (1.7) | −0.2 (−9.8) | 3.0 | −3.9 | 91.5 (110.7) |
| δ13C (NCN) | 178.4 (168.4) | 178.3(165.1) | 165.8 | 167.0 | — |
| δ13C Fe(CO)4 | — | 217.6 | 216.8 | 219.0, 216.0 | |
| λ max [nm] | 451, 389 | 503 | 512; 427 | 368d | |
Crystals of 4 suitable for X-ray diffraction analysis were obtained from pentane at 25 °C. The structure in the solid state (Fig. 1) confirmed the constitution of 4 as the sterically most favorable E-stereoisomer. The Ge1–Si1 bond length is by 2.2757(10) Å slightly longer than in II (2.2521(5) Å),13 whereas it is almost identical with that of the bulkily substituted silagermene (tBu3Si)2SiGeMes2 (2.2769(8) Å; Mes = 2,4,6-Me3C6H2).5c As in II, the NHC coordinates to germanium in a near-orthogonal manner with respect to the Si1–Ge1 bond vector (C46–Ge1–Si1 101.90(10)°). The Ge1–C46 distance in 4-E (2.061(4) Å) is between that of the simple silagermenylidene II (2.0474(18) Å) and the GeCl2 precursor 2 (2.106(3) Å).9c
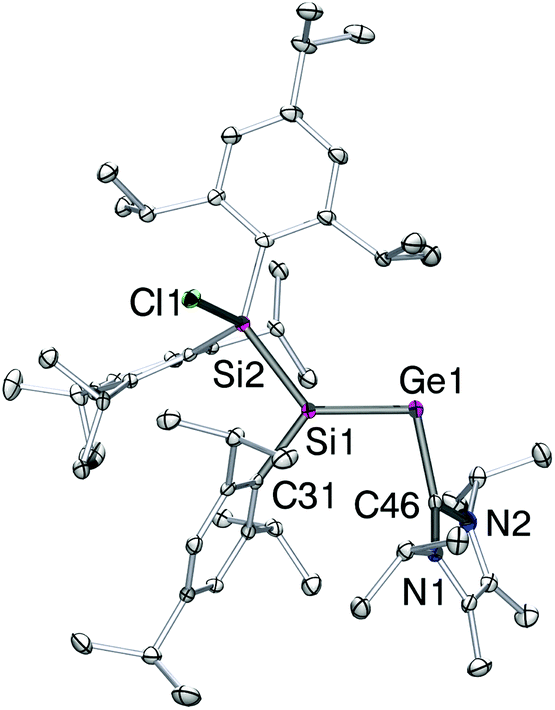 | ||
| Fig. 1 Structure of 4-E in the solid state (thermal ellipsoids at 30%). Hydrogen atoms are omitted. Selected bond lengths [Å]: Si1–Ge1 = 2.2757(10), Ge1–C46 = 2.061(4), Si1–Si2 = 2.3776(13), Si2–Cl1 = 2.1179(13). | ||
Interestingly, in solution, 4-E slowly converts to a new compound with 29Si NMR resonances at 134.0 and −0.2 ppm, which we assign to stereoisomer 4-Z (Scheme 3). Equilibrium is reached after approximately 4 h in benzene-d6 at an E/Z ratio of 0.85:0.15, essentially unaffected by temperature (+70 to −60 °C) or the presence of excess NHCiPr2Me2.20
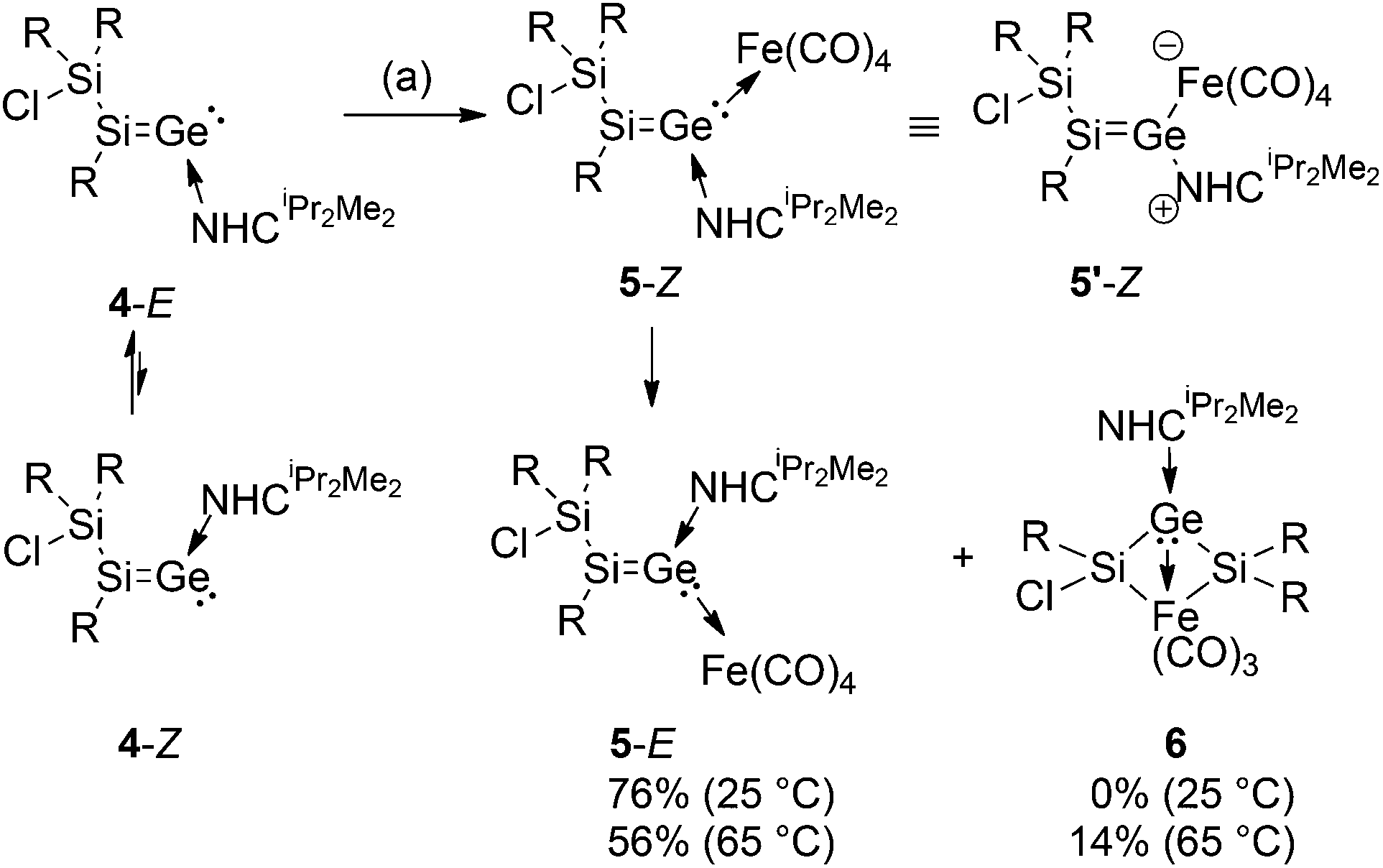 | ||
| Scheme 3 Equilibrium of 4-E to 4-Z and synthesis of transition metal complexes 5-Z, 5-E and 6 (a: THF, Fe2(CO)9; R = Tip = 2,4,6-iPr3C6H2; NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). | ||
The calculated 29Si shifts [GIAO/B3LYP/6-31G(d,p) for H, C, N, 6-311+G(2d,p) for Si, Ge, Cl] of the truncated model systems for both isomers 4Dip-E and 4Dip-Z (R = Dip = 2,6-iPr2C6H3 instead of Tip) are 159.5, 1.7 and 90.4, −9.8 ppm, respectively.† Although the experimental trend is reproduced, the absolute agreement of the calculated and the experimental values is better for the major isomer 4-E. The deviations presumably arise from the neglect of dispersive forces that should affect the sterically unfavorable isomer 4-Z considerably more than 4-E.17
Mills et al. had obtained the first structurally characterized transition metal complexes of diphenylvinylidene from diphenylketene and Fe(CO)5.21 The reaction of 4-E/Z with Fe2(CO)9 in THF at room temperature initially affords only the Z-stereoisomer of the silagermenylidene complex 5, which corresponds to the relative orientation of the chlorosilyl group and the NHCiPr2Me2 ligand in 4-E (Scheme 3) (see the Experimental section). The iron germenylidene complex 5-Z was isolated as a red-brown solid (mp. 140–142 °C) in 63% yield. In the case of germylenes, similar complexes have been reported.16b In the 13C NMR of 5-Z, the two downfield resonances at 216.8 and 167.0 ppm were assigned to the carbonyl ligands at the Fe-center and coordinated NHCiPr2Me2, respectively.22 The 29Si NMR exhibits signals at 100.7 and 3.0 ppm; the formal sp2-Si is substantially upfield shifted compared to that of the free ligand 4-E (Table 1). As shown by the new 29Si signals at 98.1 and −3.9 ppm in a 1:1 ratio, 5-Z slowly – but in this case irreversibly – rearranges to the stereoisomer, 5-E (mp. 158–160 °C) in solution. This is in contrast to the steric preferences of the NHCiPr2Me2 ligand in 4-E/Z, which, however, can readily be explained by the comparatively larger Fe(CO)4 moiety (Scheme 3) (see the Experimental section). In the IR, the most intense carbonyl stretching bands of 5-Z and 5-E are observed at 2018, 2010, 1923, 1899 cm−1 and 2011, 2008, 1917, 1897 cm−1, respectively. Apparently, silagermenylidenes 4-E/Z are somewhat stronger σ-donors compared to, for instance, the N-heterocyclic carbene, NHCDip (NHCDipFe(CO)4:23aν = 2035, 1947, 1928, 1919 cm−1) (NHCDip = C{N(Ar)CH}2, Ar = 2,6-diisopropylphenyl) and intramolecularly base-coordinated germylene LGeOH23b (CO stretching frequencies of LGe(OH)Fe(CO)4: ν = 2039, 1956, 1942 cm−1) (L = CH{(CMe)2(2,6-iPr2C6H3N)2}). Incidentally, the carbonyl stretching frequencies of 5-Z and 5-E are very similar to those of the carbon-based vinylidene complex H(CHO)CCFe(CO)2(P(OMe)3)2 (ν = 2015, 2007 cm−1).23c
The iron complex 5-E crystallizes from concentrated pentane solution (Fig. 2). The structural model confirmed that the SiGe bond is retained upon coordination (Ge1–Si1 2.2480(10) Å). Both formally sp2-hybridized heavier atoms, Si and Ge, are pyramidalized (∑ of angles: Ge1 354.74°; Si1 351.25°). The SiGe bond adopts a strongly trans-bent geometry (Si1: 30.27 (1)°, Ge1: 21.07 (8)°). Another noteworthy feature of 5-E is the twisting angle of 17.09(1)° between the planes of C1–Si–Si2 and Fe1–Ge1–C16. The bond length of Ge1–C16 is by 2.020(3) Å significantly shorter than in the free ligand (4-E: 2.061(4) Å). The Ge1–Fe1 bond length (2.3780(6) Å) is slightly longer than in germylene-coordinated iron(0)tetracarbonyl complexes (LGeOH Å 2.330(1) Å,23b LGeF 2.3262(7) Å,23d L = CH{(CMe)2(2,6-iPr2C6H3N)2}). The C16–Ge1–Si1 bond angle is by 115.59(10)° much wider than that in silagermenylidenes (II,13 C31–Ge–Si 98.90(5)° and 4-E, C(46)–Ge(1)–Si(1) 101.90(10)°). These structural parameters are reminiscent of the η1-vinyl coordination mode in Tip2SiSiTip-(Cl)ZrCp2.24 In the light of the current discussion on the use of arrows in the context of donor–acceptor interactions25 it should be noted that obviously the formulation of 5-Z as zwitterionic complex 5′-Z is equally valid.
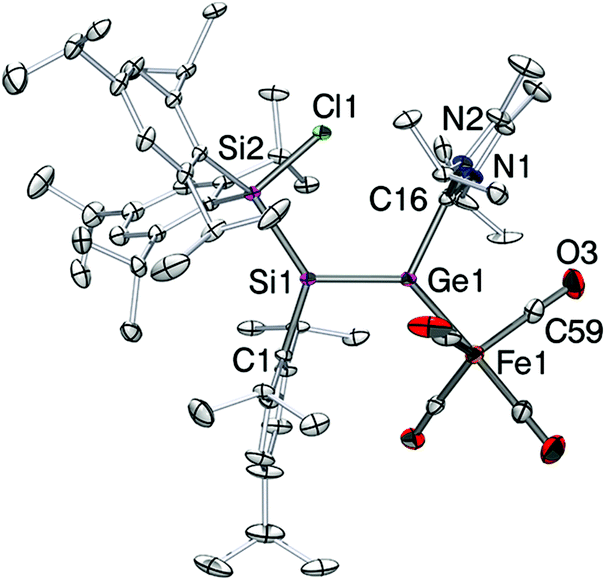 | ||
| Fig. 2 Structure of 5-E in the solid state (thermal ellipsoids at 30%). Hydrogen atoms are omitted. Selected bond lengths [Å]: Si1–Ge1 = 2.2480(10), Ge1–C16 = 2.020(3), Ge1–Fe1 = 2.3780(6), Fe1–C59 = 1.772(4), C59–O3 = 1.162(5), Si1–Si2 = 2.3767(12), Si2–Cl1 = 2.1118(11). | ||
When the isomerization process of 5-Z was carried out at 65 °C, an additional product 6 is formed in 14% yield (Scheme 3) along with the major product 5-E (56%) (see the Experimental section). Notably, 6 cannot be obtained by heating an isolated sample of 5-E. Spatial proximity between the Fe(CO)4 and SiTip2Cl moieties seems to be required for the isomerization under loss of one CO ligand. By fractional crystallization, we isolated 6 as yellow blocks (mp. 197–199 °C). In 29Si NMR, both resonances of silicon appear at 113.7 (SiTip) and 91.5 (SiTip2) ppm, which hints at the absence of saturated silicon atoms, such as in the chlorosilyl side chain of 5-Z/E.
An X-ray diffraction study on single crystals of 6 revealed a bicyclo[1.1.0]butane-like butterfly structure with the Fe1 and Ge1 in the bridgehead positions (Fig. 3). Apparently, a chlorine migration from the SiTip2 moiety to the SiTip moiety took place during conversion from 5-Z to 6. The 29Si NMR shifts of 6 are close to those of Ogino's alkoxy- and amido-bridged bis(silylene)iron complexes 7 (Scheme 4).26 In the present case, however, the bridging unit is the NHCiPr2Me2-stabilized germylene moiety so that an analogous electronic description (6′, Scheme 4) probably contributes less than a zwitterionic resonance form of allylic type (6′′).
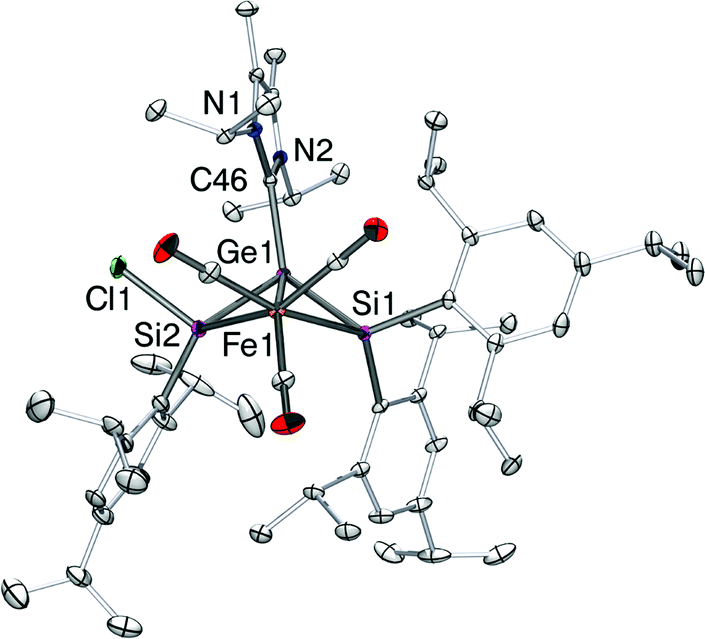 | ||
| Fig. 3 Structure of 6 in the solid state (thermal ellipsoids at 30%). Hydrogen atoms are omitted. Selected bond lengths [Å]: Ge1–C46 = 2.053(2), Ge1–Si1 = 2.3870(7), Ge1–Si2 = 2.3249(7), Fe1–Si1 = 2.3520(7), Fe1–Si2 = 2.3166(8), Ge1–Fe1 = 2.6875(4). | ||
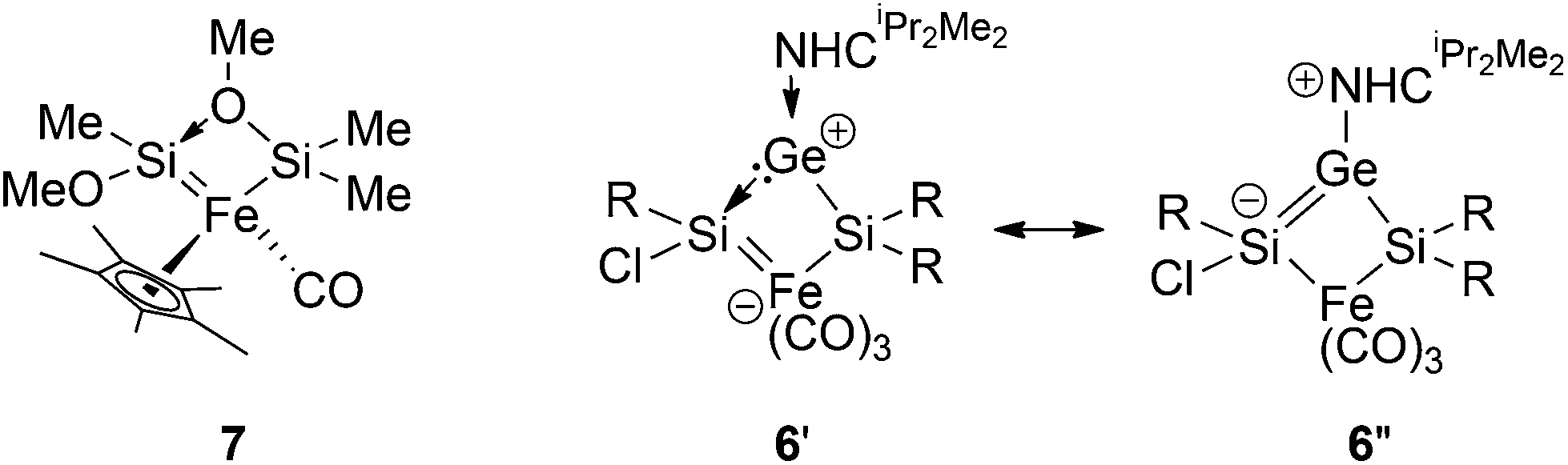 | ||
| Scheme 4 Chemical structures of 7 and canonical forms of 6 (R = Tip = 2,4,6-iPr3C6H2). | ||
This assertion finds support in the pertinent structural features of 6. The averaged distance between Fe1 and Si1/Si2 in 6 is 2.3343(7) Å, shorter than that in the tetracarbonyl iron complexes of a Z-1,2-dichlorodisilene (2.4358(6) Å, average distance),27 but longer than the Si–Fe distances in silylene–iron complexes ((CO)4FeSi(Me)2·HMPA,28a 2.280(1) and 2.294(1) Å for two crystallographic independent molecules; (CO)4FeSi(StBu)2·HMPA28b (2.278(1) Å) (HMPA = (NMe2)3PO/hexamethylphosphoric triamide). The average distance between silicon and germanium in 6 is 2.3560(7) Å, considerably longer than that of the reported 2-germadisilaallene (2.2370(7) Å, average distance).29 The mechanism for formation of 6 remains obscure. However, the intramolecular activation of a silicon–silicon bond in oligosilyl iron complexes has been reported30 and recently Marschner et al. demonstrated the Lewis acids catalyzed shuttling of germanium atoms into branched polysilanes.31
Experimental section
General remarks
All experiments were carried out under a protective atmosphere of argon applying standard Schlenk techniques or in a glove box. All the solvents were refluxed over sodium/benzophenone, distilled and stored under argon. Benzene-d6, toluene-d8, and THF-d8 were dried and distilled over potassium under argon. 1H and 13C{1H} NMR spectra were referenced to the peaks of residual protons of the deuterated solvent (1H) or the deuterated solvent itself (13C). 29Si{1H} NMR spectra were referenced to external SiMe4. UV/vis spectra were acquired using a Perkin-Elmer Lambda 35 spectrometer using quartz cells with a path length of 0.1 cm. IR spectra were recorded using a Varian 2000 FT-IR FTS 2000 spectrometer. Melting points were determined under argon in closed NMR tubes and are uncorrected. Elemental analyses were performed using a Leco CHN-900 analyzer.Synthesis of 4-E: A precooled (–78 °C) solution of 1 (2.70 g, 3.17 mmol, in 30 mL of toluene) was transferred by cannula to a suspension of 2 (1.02 g, 3.17 mmol, in 15 mL of toluene) at −78 °C. The reaction mixture was allowed to warm slowly to room temperature and stirred overnight. All the volatiles were removed under vacuum and the solid residue dissolved in 30 mL of hexane. After filtration, the solution was concentrated to about 10 mL and kept overnight at room temperature. The red precipitate was separated from the supernatant solution, washed with 5 mL of pentane and dried under vacuum to yield 1.90 g (62%) of 4-E. Single crystals suitable for X-ray diffraction were obtained from a saturated pentane solution after keeping for 2 days at room temperature. Mp: 126–128 °C. 1H NMR (300 MHz, benzene-d6, TMS): δ 7.08 (s, 4H, TipH), 7.03 (s, 2H, TipH), 5.38 (2H, sept, NiPr–CH), 4.47–3.96 (m and br, altogether 6H, o-iPr–CH), 2.83–2.65 (m, 3H, p-iPr–CH), 1.52 (s, 6H, CH3CC), 1.33–1.11 (br and m, altogether 48H, iPr–CH3), 1.01 (d, 6H, iPr–CH3), 0.85 (d, 12 H, NiPr–CH3) ppm. 13C NMR (75.4 MHz, benzene-d6, TMS): δ 178.45 (NCN), 154.41, 153.96, 149.51, 149.17, 142.48, 138.44 (TipCquart), 126.43 (NCCN), 122.51, 121.64 (TipCH), 53.92 (NiPr–CH), 35.74, 34.79, 34.53, 34.42 (iPr–CH), 25.64, 25.25, 24.95, 24.29, 24.09, 24.05 (iPr–CH3), 20.69 (N iPr–CH3), 10.05 (CH3CC) ppm. 29Si NMR (59.5 MHz, benzene-d6, TMS): δ 162.50 (SiTip), 7.3 (SiTip2) ppm. UV/vis (hexane): λmax(ε) = 451 nm (9220 L mol−1 cm−1), 389 nm (sh). Anal. Calcd for C56H89ClGeN2Si2 (954.58): C, 70.46; H, 9.40; N, 2.93. Found: C, 70.47; H, 9.47; N, 2.93. Crystallographic data: C56H89ClGeN2Si2, Mr = 954.51, monoclinic, space group P2(1)/c, a = 20.3816(11), b = 10.9027(6), c = 25.5288(13) Å, β = 90.840(3)°, V = 5672.3(5) Å3; Z = 4, ρc = 1.118 g cm−3, T = 133(2) K, λ = 0.71073 Å, 48361 reflections, 14017 independent (Rint = 0.1264), R1 = 0.0619 (I > 2σ(I)) and wR2(all data) = 0.1332, GooF = 0.970, max/min residual electron density: 0.827/−0.923 e Å−3.
Equilibrium between 4-E and 4-Z and NMR data of 4-Z: In solution 4-E isomerizes to 4-Z reaching equilibrium after about 4 h. The ratio of the two isomers was approximately 0.84:0.16 (4-E/4-Z). 4-Z: 1H NMR (300 MHz, benzene-d6, TMS): δ 7.24 (br, 2H, TipH), 7.21 (br, 1H, TipH), 6.97 (br, 1H, TipH), 6.77 (br, 1H, TipH), 5.67 (2H, sept, NiPr–CH), 4.75–4.54 (m and br, altogether 2H, o-iPr–CH), 3.76–3.64 (m, 2H, o-iPr–CH), 1.80 (d, 3H, iPr–CH3), 1.76–1.69 (m, 6H, iPr–CH3), 1.61 (s, 6H, CH3CC), 0.61 (d, 3H, iPr–CH3), 0.47 (d, 3H, iPr–CH3), 0.40 (d, 3H, iPr–CH3), 0.30 (d, 3H, iPr–CH3) ppm. 13C NMR (75.4 MHz, [D6]benzene, TMS): δ 178.33 (NCN) ppm. 29Si NMR (59.5 MHz, [D6]benzene, TMS): δ 134.02 (SiTip), −0.20 (SiTip2) ppm (minor isomer).
Synthesis of 5-Z: Dry and degassed THF (30 mL) was added to a Schlenk flask containing compound 4-E (1.90 g, 1.99 mmol) and Fe2(CO)9 (0.80 g, 2.19 mmol) at room temperature. The color of the reaction mixture changed immediately to deep red. The reaction mixture was stirred for another 4 h and all the volatiles were removed under vacuum. The solid residue was extracted with 80 mL of hexane and the resulting solution was filtered to remove insoluble impurities. The hexane was distilled off under vacuum. After addition of 20 mL of pentane, 1.40 g (63%) of 5-Z were isolated as a dark-red solid. Mp: 140–142 °C. 1H NMR (300 MHz, toluene-d8, TMS): δ 7.26, 7.18, 6.86, 6.81, 6.65 (d, each having 1H, TipH), another Tip-H signal is masked by residual proton signals of toluene-d8, 5.83 (sept, 1H, iPr–CH), 5.09 (sept, 1H, iPr–CH), 5.00 (sept., 1H, iPr–CH), 4.55 (sept, 1H, iPr–CH), 4.07 (sept, 1H, iPr–CH), 3.80–3.62 (m, 2H, iPr–CH), 2.78 (sept, 1H, iPr–CH), 2.70–2.53 (m, 2H, iPr–CH), 2.48 (sept, 1H, iPr–CH), 1.73 (d, 3H, iPr–CH3), 1.67–1.51 (s and m, altogether 15H, iPr–CH3 and CH3CC), 1.45–1.39 (s and m, altogether 6H, iPr–CH3 and CH3CC), 1.34–1.25 (m, 9H, iPr–CH3), 1.22 (d, 6H, iPr–CH3), 1.17–1.16 (m, 9H, iPr–CH3), 1.09–1.03 (m, 6H, iPr–CH3), 0.48 (d, 3H, iPr–CH3), 0.43 (d, 3H, iPr–CH3), 0.37 (d, 3H, iPr–CH3), 0.20–0.11 (m, 9H, iPr–CH3) ppm. 13C NMR (75.4 MHz, toluene-d8, TMS): δ 217.56 (CO), 165.81 (NCN) ppm. (We were unable to assign other signals correctly, because they overlap with its isomer 5-E.) 29Si NMR (59.5 MHz, toluene-d8, TMS): δ 100.67 (SiTip), 2.98 (SiTip2) ppm. Mp.: 140–142 °C. UV/vis (hexane): λmax(ε) = 503 nm (8260 Lmol−1 cm−1). IR (KBr, cm−1): ν = 2018 (s), 2010 (s), 1923 (s), 1899 (s). Anal. Calcd for C60H89ClFeGeN2O4Si2 (1122.47): C, 64.20; H, 7.99; N, 2.50. Found: C, 64.10; H, 7.74; N, 2.63.
Synthesis of 5-E: A solution of 5-Z (1.00 g, 0.89 mmol) in toluene (60 mL) was stirred for five days at room temperature. All volatiles were removed under vacuum and the solid residue extracted with 70 mL of hexane. The solution was concentrated to about 20 mL and after keeping at −20 °C for a week afforded brown-red crystals of 5-E (0.77 g, 76%). Mp: 158–160 °C. 1H NMR (300 MHz, toluene-d8, TMS): δ 7.27–7.23 (m, 2H, TipH), 7.21 (d, 1H, TipH), 7.05 (1H, TipH, masked by toluene-d8), 6.96 (d, 1H, TipH), 6.73 (d, 1H, TipH), 5.72 (sept, 1H, NiPr–CH), 5.51 (sept, 1H, NiPr–CH), 4.62 (sept, 1H, iPr–CH), 4.54 (sept, 1H, iPr–CH), 4.30 (sept, 1H, iPr–CH), 3.67 (sept, 1H, iPr–CH), 3.57 (sept, 1H, iPr–CH), 2.88–2.62 (m, 4H, iPr–CH), 2.00 (d, 3H, iPr–CH3), 1.79 (d, 3H, iPr–CH3), 1.68–1.46 (s and m, altogether 30H, iPr–CH3 and CH3CC), 1.27–1.21 (m, 12H, iPr–CH3), 1.19–1.15 (m, 12H, iPr–CH3), 0.56 (d, 3H, iPr–CH3), 0.43 (d, 3H, iPr–CH3), 0.34 (d, 3H, iPr–CH3), 0.24 (d, 3H, iPr–CH3) ppm. 13C NMR (75.4 MHz, toluene-d8, TMS): δ 216.85 (CO), 166.99 (NCN), 156.72, 155.76, 155.69, 154.05, 153.23, 152.37, 151.32, 150.41, 137.67, 133.07, 130.55, 127.35, 127.00 (TipCquart and NCCN), 124.07, 123.37, 122.53, 122.40, 122.12, 120.96 (TipCH), 55.53, 54.84 (NiPr–CH), 38.48, 38.11, 37.20, 35.24, 34.81, 34.62, 34.47, 33.97, 31.04 (iPr–CH), 32.00, 30.66, 27.80, 27.30, 26.00, 25.36, 25.16, 24.83, 24.30, 24.16, 24.02, 23.97, 23.92, 23.87, 23.05, 22.50, 22.43, 22.40, 21.87, 20.83, 14.29 (iPr–CH3) 10.17, 9.92 (CH3CC) ppm. 29Si NMR (59.5 MHz, toluene-d8, TMS): δ 98.14 (SiTip), −3.89 (SiTip2) ppm. UV/vis (hexane): λmax(ε) = 512 nm (7050 Lmol−1 cm−1), 427 nm (6670) nm (L mol−1 cm−1). IR (KBr, cm−1): ν = 2011 (s), 2008 (s), 1917 (s) 1897 (s). Anal. Calcd for C60H89ClFeGeN2O4Si2 (1122.47): C, 64.20; H, 7.99; N, 2.50. Found: C, 64.15; H, 7.90; N, 2.26. Crystallographic data: C60H89ClFeGeN2O4Si2·0.25C5H12, Mr = 1140.44, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.2910(4), b = 19.9842(5), c = 24.8636(7) Å, α = 89.1430(10), β = 95.311(2), γ = 76.122(2)°, V = 6407.6(3) Å3; Z = 4, ρc = 1.182 g cm−3, T = 123(2) K, λ = 0.71073 Å, 105034 reflections, 28114 independent (Rint = 0.0436), R1 = 0.0619 (I > 2σ(I)) and wR2 (all data) = 0.1744, GooF = 1.427, max/min residual electron density: 2.142/−0.789 e Å−3.
, a = 13.2910(4), b = 19.9842(5), c = 24.8636(7) Å, α = 89.1430(10), β = 95.311(2), γ = 76.122(2)°, V = 6407.6(3) Å3; Z = 4, ρc = 1.182 g cm−3, T = 123(2) K, λ = 0.71073 Å, 105034 reflections, 28114 independent (Rint = 0.0436), R1 = 0.0619 (I > 2σ(I)) and wR2 (all data) = 0.1744, GooF = 1.427, max/min residual electron density: 2.142/−0.789 e Å−3.
Synthesis of 6: A solution of 5-Z (0.50 g, 0.44 mmol) in toluene (30 mL) was stirred in a sealed Schlenk flask overnight at 65 °C. All the volatiles were removed under vacuum and the solid residue extracted with 40 mL of hexane. The solution was concentrated to about 20 mL and after keeping at room temperature for two days afforded yellow blocks of 6 (0.075 g, 14%). Keeping the mother liquor at −20 °C for a week afforded brown-red crystals of 5-E (0.28 g, 56%). 6: Mp: 197–199 °C. 1H NMR (300 MHz, toluene-d8, TMS): δ 7.23 (d, 1H, TipH), 7.13 (1H, TipH, masked by toluene-d8), 7.02 (sept, 1H, NiPr–CH), 7.01 (1H, TipH, masked by toluene-d8), 6.98–6.95 (m, 2H, TipH), 6.87 (d, 1H, TipH), 6.41 (sept, 1H, NiPr–CH), 5.25 (sept, 1H, iPr–CH), 4.86 (sept, 1H, iPr–CH), 4.35 (sept, 1H, iPr–CH), 4.22 (sept, 1H, iPr–CH), 3.61 (sept, 1H, iPr–CH), 3.44 (sept, 1H, iPr–CH), 2.79 (sept, 1H, iPr–CH), 2.77 (sept, 1H, iPr–CH), 2.65 (sept, 1H, iPr–CH), 1.87–1.78 (m, 6H, iPr–CH3), 1.68 (s, 3H, CH3CC), 1.66 (s, 3H, CH3CC), 1.56–1.37 (m, altogether 18H, iPr–CH3), 1.31–1.04 (m, altogether 30H, iPr–CH3), 0.72 (d, 3H, iPr–CH3), 0.48 (d, 3H, iPr–CH3), 0.45 (d, 3H, iPr–CH3), 0.19 (d, 3H, iPr–CH3) ppm. 1H NMR (300 MHz, thf-d8, TMS): δ 7.03 (d, 1H, TipH), 6.96 (d, 1H, TipH), 6.95 (sept, 1H, NiPr–CH), 6.80 (d, 1H, TipH), 6.76 (d, 1H, TipH), 6.71 (d, 1H, TipH), 6.67 (d, 1H, TipH), 6.19 (sept, 1H, NiPr–CH), 4.82 (sept, 1H, iPr–CH), 4.61 (sept, 1H, iPr–CH), 4.15 (sept, 1H, iPr–CH), 3.82 (sept, 1H, iPr–CH), 3.34–3.18 (m, 2H, iPr–CH), 2.87–2.68 (m, 2H, iPr–CH), 2.63 (sept, 1H, iPr–CH), 2.41 (s, 3H, CH3CC), 2.35 (s, 3H, CH3CC), 1.85 (d, 3H, iPr–CH3), 1.62 (d, 3H, iPr–CH3), 1.58–1.49 (m, altogether 9H, iPr–CH3), 1.44 (d, 3H, iPr–CH3), 1.24 (d, 3H, iPr–CH3), 1.20–1.11 (m, altogether 21H, iPr–CH3), 1.08–1.03 (m, altogether 9H, iPr–CH3), 0.88 (d, 3H, iPr–CH3), 0.45 (d, 3H, iPr–CH3), 0.18–0.14 (m, altogether 6H, iPr–CH3), −0.20 (d, 3H, iPr–CH3) ppm. 13C NMR (75.4 MHz, thf-d8, TMS): δ 218.99, 215.98 (CO), 157.19, 156.89, 155.09, 154.75, 154.08, 152.28, 150.90, 150.71, 149.48, 143.26, 142.18, 136.52 ((TipCquart), 129.52, 129.26 (NCCN), 123.68, 122.72, 122.66, 122.06, 121.88, 121.43 (TipCH), 56.24, 54.08 (NiPr–CH), 38.20, 36.65, 36.46, 35.29, 35.39, 34.98, 34.83, 34.16, 30.24 (iPr–CH), 28.21, 26.76, 26.28, 25.97, 25.65, 25.48, 24.91, 24.38, 24.36, 24.28, 24.18, 23.99, 23.90, 23.84, 23.40, 22.88, 22.23, 21.98 (iPr–CH3), 11.21, 10.73 (CH3CC) ppm (we did not observe the carbenic carbon resonance). 29Si NMR (59.5 MHz, toluene-d8, TMS): δ 113.66 (SiTip), 91.46 (SiTip2) ppm. 29Si NMR (59.5 MHz, [D8]THF, TMS): δ 111.36 (SiTip), 88.27 (SiTip2) ppm. UV/vis (THF): λmax(ε) = 368 nm (6820 Lmol−1 cm−1). IR (KBr, cm−1): ν = 1982 (s), 1920 (s), 1916 (s). Anal. Calcd for C59H89ClFeGeN2O3Si2 (1094.46): C, 64.75; H, 8.20; N, 2.56. Found: C, 65.35; H, 8.08; N, 2.38. Crystallographic data: C59H89ClFeGeN2O3Si2, Mr = 1094.39, orthorhombic, space group Pbca, a = 19.6288(5), b = 24.6607(7), c = 24.6974(7) Å, V = 11955.0(6) Å3; Z = 8, ρc = 1.216 g cm−3, T = 132(2) K, λ = 0.71073 Å, 104743 reflections, 14312 independent (Rint = 0.0577), R1 = 0.0442 (I > 2σ(I)) and wR2 (all data) = 0.1131, GooF = 1.022, max/min residual electron density: 1.184/−0.536 e Å−3.
Conclusions
We have shown an efficient method for the synthesis of side chain-functionalized silagermenylidene stabilized by coordination of an N-heterocyclic carbene. Its suitability as a ligand for transition metal complexes was demonstrated by coordination to the Fe(CO)4 fragment. Moreover, the resulting silagermenylidene iron complex thermally rearranges to an apparently more stable complex of unprecedented allylic structure, which is undoubtedly a consequence of the ease of migration of the residual chlorine functionality.Acknowledgements
Support for this study was provided by the EPSRC (EP/H048804/1), the Alfried Krupp von Bohlen und Halbach Foundation. We thank Dr Carsten Präsang for assistance with the IR measurement.Notes and references
- (a) J. Barrau, J. Escudié and J. Satgé, Chem. Rev., 1990, 90, 283–319 CrossRef CAS; (b) J. Barrau and G. Rima, Coord. Chem. Rev., 1998, 178–180, 593–622 CrossRef CAS; (c) V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-Coordinate Si, Ge, Sn, Pb: From Phantom Species to Stable Compounds, Wiley, Chichester, U.K., 2010 Search PubMed; (d) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (e) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- (a) D. H. Harris and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1974, 895–896 RSC; (b) P. B. Hitchcock, M. F. Lappert, B. J. Samways and E. L. Weinberg, J. Chem. Soc., Chem. Commun., 1983, 1492–1494 RSC; (c) A. Meller and C. P. Gribe, Chem. Ber., 1985, 118, 2020–2029 CrossRef CAS; (d) T. Fjeldberg, P. B. Hitchcock, M. F. Lappert, S. J. Smith and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1986, 939–941 Search PubMed; (e) P. Jutzi, A. Becker, H. G. Stammler and B. Neumann, Organometallics, 1991, 10, 1647–1648 CrossRef CAS; (f) W. A. Herrmann, M. Denk, J. Behm, W. Scherer, F.-R. Klingan, H. Bock, B. Solouki and M. Wagner, Angew. Chem., Int. Ed. Engl., 1992, 31, 1485–1488 CrossRef.
- (a) M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785–1787 CrossRef CAS; (b) Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023–1031 CrossRef CAS PubMed; (c) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (d) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622–18625 CrossRef CAS PubMed.
- (a) P. B. Hitchcock, M. F. Lappert, S. J. Miles and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1984, 480–482 RSC; (b) D. E. Goldberg, P. B. Hitchcock, M. F. Lappert, K. M. Thomas, A. J. Thorne, T. Fjeldberg, A. Haaland and B. E. R. Schilling, J. Chem. Soc., Dalton Trans., 1986, 2387–2394 RSC; (c) V. Y. Lee, K. McNeice, Y. Ito and A. Sekiguchi, Chem. Commun., 2011, 47, 3272–3274 RSC.
- (a) K. M. Baines and J. A. Cooke, Organometallics, 1992, 11, 3487–3488 CrossRef CAS; (b) M. Ichinohe, Y. Arai and A. Sekiguchi, Organometallics, 2001, 20, 4141–4143 CrossRef CAS; (c) M. Igarashi, M. lchinohe and A. Sekiguchi, Heteroat. Chem., 2008, 19, 649–653 CrossRef CAS.
- (a) M. C. Kuchta and G. Parkin, J. Chem. Soc., Chem. Commun., 1994, 1351–1352 RSC; (b) R. Okazaki and N. Tokitoh, Acc. Chem. Res., 2000, 33, 625–630 CrossRef CAS PubMed; (c) L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Ta-naka and K. Tamao, Nat. Chem., 2012, 4, 361–365 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- (a) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (b) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326–12337 CrossRef CAS PubMed; (c) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS PubMed; (d) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- (a) P. Jutzi, H. J. Hoffmann, D. J. Brauer and C. Krüger, Angew. Chem., Int. Ed. Engl., 1973, 12, 1002–1003 CrossRef; (b) T. Fjeldberg, A. Haaland, B. E. R. Schilling, M. F. Lappert and A. J. Thorne, J. Chem. Soc., Dalton Trans., 1986, 1551–1556 RSC; (c) P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138–15139 CrossRef CAS PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701–9704 CrossRef CAS PubMed.
- (a) M. Veith, S. Becker and V. Huch, Angew. Chem., Int. Ed. Engl., 1989, 28, 1237–1238 CrossRef; (b) Y. Ding, Q. Ma, I. Usón, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, J. Am. Chem. Soc., 2002, 124, 8542–8543 CrossRef CAS PubMed; (c) S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 6466–6468 RSC; (d) A. Jana, D. Ghoshal, H. W. Roesky, I. Objartel, G. Schwab and D. Stalke, J. Am. Chem. Soc., 2009, 131, 1288–1293 CrossRef CAS PubMed.
- (a) S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978–3980 RSC; (b) S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459–5463 CrossRef CAS; (c) C. Jones, S. J. Bonyhady, N. Holzmann, G. Frenking and A. Stasch, Inorg. Chem., 2011, 50, 12315–12325 CrossRef CAS PubMed.
- A. Jana, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2013, 52, 12179–12182 CrossRef CAS PubMed.
- (a) M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS; (b) M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 CrossRef CAS PubMed; (c) A. M. Lozano-Vila, S. Monsaert, A. Bajek and F. Verpoort, Chem. Rev., 2010, 110, 4865–4909 CrossRef CAS PubMed.
- For a report on the trapping of a base-stabilized stannavinylidene in an iron complex, see: W.-P. Leung, W.-K. Chiu and T. C. W. Mak, Inorg. Chem., 2013, 52, 9479–9486 CrossRef CAS PubMed.
- (a) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed; (b) K. E. Litz, J. E. Bender IV, J. W. Kampf and M. M. B. Holl, Angew. Chem., Int. Ed., 1997, 36, 496–498 CrossRef CAS; (c) P. G. Hayes, C. W. Gribble, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 4606–4607 CrossRef CAS PubMed; (d) M. E. Fasulo and T. D. Tilley, Chem. Commun., 2012, 48, 7690–7692 RSC; (e) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed.
- M. J. Cowley, V. Huch, H. S. Rzepa and D. Scheschkewitz, Nat. Chem., 2013, 5, 876–879 CrossRef CAS PubMed.
- D. Scheschkewitz, Angew. Chem., Int. Ed., 2004, 43, 2965–2967 CrossRef CAS PubMed.
- (a) A. C. Filippou, O. Chernov, B. Blom, K. W. Stumpf and G. Schnakenburg, Chem.–Eur. J., 2010, 16, 2866–2872 CrossRef CAS PubMed; (b) N. Katir, D. Matioszek, S. Ladeira, J. Escudié and A. Castel, Angew. Chem., Int. Ed., 2011, 50, 5352–5355 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- O. S. Mills and A. D. Redhouse, Chem. Commun., 1966, 444–445 RSC.
- In 13C NMR, the carbenic carbon of NHCiPr2Me2·GeCl22 appears at δ = 169.11 ppm in [d6]-benzene, see Fig. S5 in the ESI.†.
- (a) S. Warratz, L. Postigo and B. Royo, Organometallics, 2013, 32, 893–897 CrossRef CAS; (b) L. W. Pineda, V. Jancik, J. F. Colunga-Valladares, H. W. Roesky, A. Hofmeister and J. Magull, Organometallics, 2006, 25, 2381–2383 CrossRef CAS; (c) C. Löwe, H.-U. Hund and H. Berke, J. Organomet. Chem., 1989, 372, 295–309 CrossRef; (d) A. Jana, P. P. Samuel, H. W. Roesky and C. Schulzke, J. Fluorine Chem., 2010, 131, 1096–1099 CrossRef CAS.
- T.-l. Nguyen and D. Scheschkewitz, J. Am. Chem. Soc., 2005, 127, 10174–10175 CrossRef CAS PubMed.
- D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370–374 CrossRef CAS PubMed.
- (a) K. Ueno, H. Tobita, M. Shimoi and H. Ogino, J. Am. Chem. Soc., 1988, 110, 4092–4093 CrossRef CAS; (b) H. Tobita, K. Ueno, M. Shimoi and H. Ogino, J. Am. Chem. Soc., 1990, 112, 3415–3420 CrossRef CAS; (c) K. Ueno, S. Ito, K. Endo, H. Tobita, S. Inomata and H. Ogino, Organometallics, 1994, 13, 3309–3314 CrossRef CAS.
- H. Hashimoto, K. Suzuki, W. Setaka, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2004, 126, 13628–13629 CrossRef CAS PubMed.
- (a) C. Leis, D. L. Wilkinson, H. Handwerker and C. Zybill, Organometallics, 1992, 11, 514–529 CrossRef CAS; (b) L. Christian and C. Zybill, Polyhedron, 1991, 10, 1163–1171 CrossRef.
- T. Iwamoto, H. Masuda, C. Kabuto and M. Kira, Organometallics, 2005, 24, 197–199 CrossRef CAS.
- H. K. Sharma and K. H. Pannell, Chem. Rev., 1995, 95, 1351–1374 CrossRef CAS.
- H. Wagner, J. Baumgartner, T. Müller and C. Marschner, J. Am. Chem. Soc., 2009, 131, 5022–5023 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV/vis spectra of all new compounds, X-ray crystallographic data (CIF) for 4-E, 5-E, and 6, and computational details. CCDC 953520–953522. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00094c |
| This journal is © The Royal Society of Chemistry 2014 |