 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble exchange in a mixed-valent octanuclear iron cluster, [Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4]−†
Ekaterina M.
Zueva
ab,
Radovan
Herchel
c,
Serguei A.
Borshch
d,
Evgen V.
Govor‡
e,
W. M. C.
Sameera
a,
Ross
McDonald
f,
John
Singleton
f,
Jurek
Krzystek
g,
Zdeněk
Trávníček
c,
Yiannis
Sanakis
h,
John E.
McGrady
*a and
Raphael G.
Raptis‡
*e
aDepartment of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: john.mcgrady@chem.ox.ac.uk
bDepartment of Inorganic Chemistry, Kazan National Research Technological University, Kazan, K. Marx 68, 420015, Russia
cRegional Centre of Advanced Technologies and Materials, Department of Inorganic Chemistry, Faculty of Science, Palacký University, 17. listopadu 12, CZ-771 46 Olomouc, Czech Republic
dLaboratoire de Chimie, UMR 5182, Ecole Normale Supérieure de Lyon, 46 allée d'Italie, 69364 Lyon Cedex 07, France
eDepartment of Chemistry and the Institute for Functional Nanomaterials, University of Puerto Rico, San Juan, PR 00936-8377, USA. E-mail: rraptis@fiu.edu
fNational High Magnetic Field Laboratory, Los Alamos National Laboratories, Los Alamos, NM 87545, USA
gNational High Magnetic Field Laboratory, Tallahassee, FL 32310, USA
hInstitute of Materials Science, NCRS “Demokritos”, 15310 Aghia Paraskevi, Athens, Greece
First published on 25th February 2014
Abstract
A combination of SQUID and pulsed high-field magnetometry is used to probe the nature of mixed valency in an FeIIFe7III cluster. DFT-computed spin Hamiltonian parameters suggest that antiferromagnetic coupling dominates, and that electron transfer both between the four irons of the cubane core (t1) and between a cubane and three neighboring irons (t2) is significant. Simulations using the computed parameters are able to reproduce the key features of the measured effective magnetic moment, μeff(T), over the 2 < T < 300 K temperature range. In contrast, the field dependence of the molar magnetization, Mmol, measured at 0.4 K is inconsistent with substantial electron transfer: only values of t2 ∼ 0 place the separation between ground and first excited states in the region indicated by experiment. The apparent quenching of the cubane–outer electron transfer at very low temperatures indicates that vibronic coupling generates one or more shallow minima on the adiabatic potential energy surfaces that serve to trap the itinerant electron in the cubane core.
Introduction
The magnetic properties of clusters of transition metals with partially filled paramagnetic d shells are dominated by Mott physics in the sense that on-site Coulomb repulsion dominates direct overlap between the metal centers, such that the salient material properties can be described by magnetic exchange interactions between localized electron spins. Antiferromagnetic coupling between the moments then leads to a complex hierarchy of spin states and associated structure (steps and plateaux) in the field dependence of the magnetization. In contrast, mixed-valent clusters, where isotropic exchange and electron transfer,1–3 intrinsic site asymmetry (e.g., in the ligand environment)1,3 and/or intercenter Coulomb repulsion3 and vibronic coupling1–5 all play a significant role, are rather less well understood. These clusters exhibit spin-dependent electron transfer, which can be formulated as double exchange: the interaction between the localized electron spins through the itinerant electrons.6–13 The majority of studies of double exchange have focused on binuclear {dn+1 + dn} complexes where eigenvalues take the form:1–3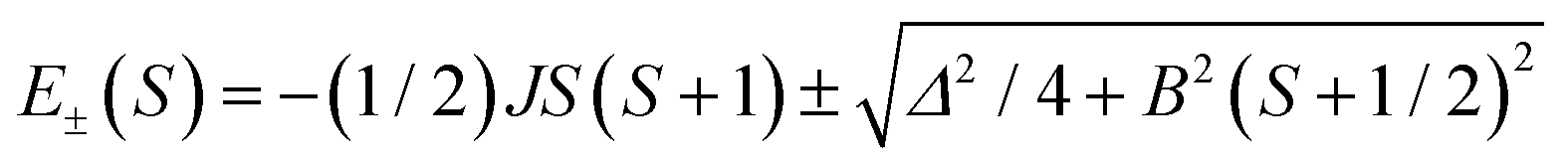 | (1) |
In a series of recent papers, we have discussed the structural, spectroscopic and magnetic properties of a family of octanuclear iron(III) complexes of the general formula [Fe8(μ4-O)4(μ-4-R-pz)12X4] (= [Fe8]0) and their redox-modified, mixed-valent [Fe8]n− derivatives, where R = H, Me, Cl, Br, I, X = Cl, Br, NCS, n = 1–4.27–29 All of these complexes contain an Fe8(μ4-O)4 core featuring an Fe4O4 cubane unit and four outer iron centers (Fec and Feo, respectively, Fig. 1). The Fe8(μ4-O)4-motif is identical to that found in the structures of the minerals maghemite,30 magnetite31 and ferrihydrite32 and has also recently been proposed as the cluster motif produced at the oxireductase site of ferritin, prior to storage as ferrihydrite in the ferritin central cavity.33 SQUID magnetometry and density functional theory indicate that antiferromagnetic coupling between the cubane and outer iron centers dominates in these all-ferric clusters. The corresponding anionic [Fe8]n− complexes with n = 1–4 are all accessible electrochemically, and the singly-reduced anions of the R = H, Cl and X = Cl species have also been prepared chemically via reduction with a stoichiometric amount of [BH4]−.28 The picture of mixed valency that has emerged depends strongly on the timescale of the chosen experiment.28 X-ray crystallography (293 K) indicates that the structure of the Fe8 cluster is almost totally unaffected by reduction: the bond lengths of neutral and anionic species are identical within experimental error. Analysis of 57Fe-Mössbauer spectra (78 K) offers a different perspective, suggesting that the reduction is delocalized over all four Fec sites of the Fe4O4-cubane, although a detailed comparison of observed and DFT-computed 57Fe-Mössbauer parameters was unable to rule out partial delocalization onto the Feo sites.29 On the very short timescale of X-ray photoelectron spectroscopy (10−17 s), however, the electron appears localized. The profile of the near-infrared intervalence charge transfer (IVCT) band confirms that the [Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4]− cluster is an example of a Robin–Day Class II complex.34 In this paper, we report magnetic measurements for the [Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4]− cluster performed at both low and high fields. Pulsed high magnetic field measurements have proved to be a powerful tool for the study of multinuclear homovalent complexes, including octanuclear ferric species.35–38 We also use density functional theory (DFT) to develop a spin Hamiltonian including the effects of isotropic exchange (J), electron transfer (t) and intrinsic site asymmetry (Δ). The computation of isotropic exchange using DFT is a well-established discipline, and there is an extensive body of literature on the extent to which values depend on methodology. The computation of transfer parameters is, in contrast, less common and the performance of different functionals is therefore less certain.39 A particularly well-studied example is the Class III delocalized iron(II,III) complex, [LFe(μ-OH)3FeL]2+ (L = N,N′,N′′-trimethyl-1,4,7-triazacyclononane), which has J < 0, but an S = 9/2 ground state as a result of the effect of double exchange. The value of B = 1366 cm−1 computed by Barone and co-workers using the VWN-Stoll functional is very similar to experiment (1350 cm−1).17 Shoji and co-workers have also shown that the computed value of B for sulphide-bridged Fe(μ-S)2Fe clusters was largely unaffected by the amount of exact Hartree–Fock exchange used in the functional.40
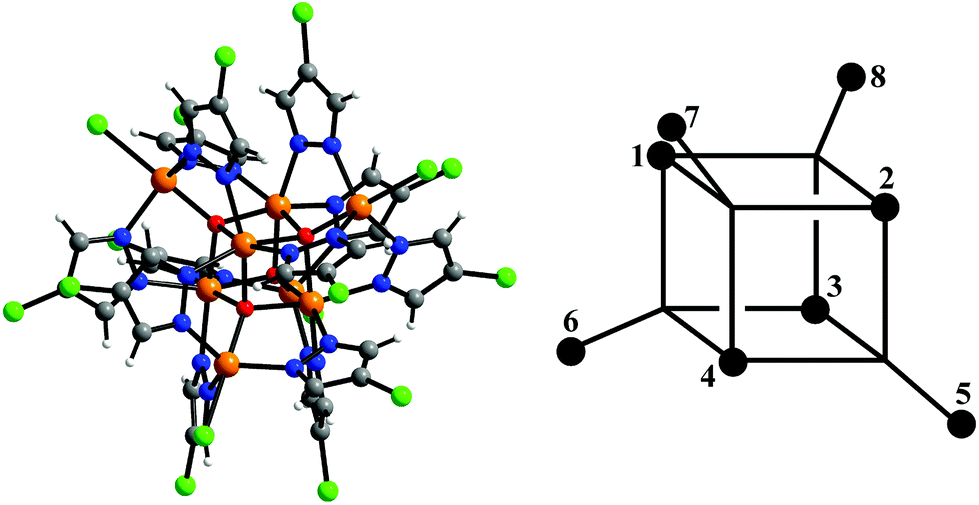 | ||
| Fig. 1 Structure of the [Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4]− cluster – O, red; Fe, orange; N, blue; Cl, green – and the numbering scheme used in eqn (2) and (3). Fec = 1–4, Feo = 5–8. | ||
Comparison with the new experimental data shows that the temperature dependence of μeff can only be reproduced if electron transfer between the cubane and outer iron centers (t2) is included in the spin Hamiltonian. The field dependence of Mmol (measured at 0.4 K), in contrast, can only be reproduced if t2 is close to zero, suggesting that localization of the itinerant electron at low temperatures effectively quenches the cubane–outer electron transfer.
Materials and methods
[Bu4N][Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4] was prepared by reduction of its all-ferric precursor with a stoichiometric amount of [Bu4N][BH4], as described previously.28 The identity of the sample of [Bu4N][Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4] used here was confirmed by satisfactory elemental analysis, infrared and 57Fe-Mössbauer spectroscopy.SQUID magnetometry
The temperature dependent magnetization was measured with an MPMS XL-7 Quantum Design SQUID magnetometer on the polycrystalline sample in the range of T = 2 to 300 K at B = 0.1 T, and the isothermal magnetization was measured at T = 2 and 4.6 K up to magnetic field B = 7 T. Experimental data were corrected for the diamagnetism of the constituent atoms by using the Pascal constants and for the diamagnetism of the sample holder.Pulsed magnetic field measurements
Pulsed high-field magnetization measurements were performed using a 1.5 mm long susceptometer coil with a 1.5 mm diameter bore made of 50 gauge, high-purity copper wire of approximately 1500 turns.41 Approximately the first 1000 inner turns are in the opposite direction to the final outer 500 turns such that the coil is compensated, and when a magnetic field is passed through the coil with sample absent, the induced emf and current in the inner turns is cancelled by that induced in the outer turns. The sample is placed inside the coil in a 1.3 mm diameter ampoule and the induced voltage . Numerical integration with respect to time, t, yields M. In order to ensure accurate values of M, the sample can be moved in and out of the coil, allowing empty coil data to be subtracted from sample data under identical conditions. Samples are placed within a 3He cryostat to allow low-temperature studies down to approximately 0.4 K to be performed. A ten-turn coil accompanies the susceptometer into the cryostat to measure the applied field, B. The voltage induced in this coil is
. Numerical integration with respect to time, t, yields M. In order to ensure accurate values of M, the sample can be moved in and out of the coil, allowing empty coil data to be subtracted from sample data under identical conditions. Samples are placed within a 3He cryostat to allow low-temperature studies down to approximately 0.4 K to be performed. A ten-turn coil accompanies the susceptometer into the cryostat to measure the applied field, B. The voltage induced in this coil is  and numerical integration yields B. Calibration of the instruments is accomplished using the de Haas–van Alphen oscillations of the belly orbits of the copper of the susceptometer. The method has been calibrated using all-ferric [Fe8]0 compounds (ESI, S5†).
and numerical integration yields B. Calibration of the instruments is accomplished using the de Haas–van Alphen oscillations of the belly orbits of the copper of the susceptometer. The method has been calibrated using all-ferric [Fe8]0 compounds (ESI, S5†).
DFT calculations
All calculations described in this paper were done using spin-unrestricted DFT as implemented in the Gaussian 09 program.42 The B3LYP functional was used throughout,43,44 in conjunction with the TZV basis set of Ahlrichs et al.45 In all cases, the data correspond to single point calculations performed at the crystallographically determined geometry (293 K). In order to extract values of the various parameters in a spin Hamiltonian, we mapped DFT-computed energies of selected microstates onto their spin Hamiltonian counterparts.46 The set of single determinants in question (Scheme 1) represent a delocalized S = 39/2 state (HS) and localized and pair-delocalized broken-symmetry states (BS1–4 and BS5–6, respectively). Convergence on a given BS state was achieved using the converged all-ferric BS-state density as an initial guess and, in some cases, the guess = alter keyword. The all-ferric BS-state density was obtained using the guess = fragment keyword. In the HS state, there are no restrictions on the location of the itinerant electron (spin-β by construction). In the BS states, in contrast, the alignment of the spin vectors on the core electrons determines the localization of the itinerant electron (spin-α by construction). Thus, in BS1 and BS2 (MS = 31/2), the itinerant spin-α electron must be localized on the single iron center where the core electrons are spin-β. For BS3 and BS4 (MS = 21/2), there is no direct spin block to delocalization over the two spin-β iron centers (centers 1 and 5 in Scheme 1), but the spatial separation between the two iron centers effectively prevents delocalization. There are, in principle, four versions of each of BS1, BS2, BS3 and BS4, generated by cyclic permutation around the cubane and outer iron centers (for example, BS1 could also be generated with the iron(II) site at center 2, 3 or 4). The energies of these differ only marginally due to the very slight difference of the bond lengths in the X-ray structure, so the averaged energies over all microstates (which are necessarily equivalent in the limit of perfect T point symmetry) were taken. The energies of these microstates can be mapped onto the diagonal elements of the Heisenberg components of the spin Hamiltonian matrix computed in products of single-center spin functions (basis functions of localized configurations). Using the expressions for the energy differences between any two of these microstates, we estimate the isotropic exchange and asymmetry parameters, which are characteristics of localized configurations. The transfer parameters are estimated from the pair-delocalized BS states with MS = 21/2, BS5 and BS6 (of which there are 6 and 12 almost identical versions, respectively, obtained by cyclic permutation) in Scheme 1, where the itinerant electron is trapped either in a single Fec–Fec dimer unit or a single Fec–Feo dimer unit. Complete trapping is guaranteed in BS5 and BS6 because the itinerant electron (spin-α by construction) must be localized on iron centers where the alignment of the other five electrons is spin-β. | ||
| Scheme 1 HS and BS states used in developing a spin Hamiltonian. Black and white circles denote the d5-core with spin-α and spin-β electrons, respectively. | ||
Results and discussion
The results of the magnetometry experiments are shown in Fig. 2 and 3: the temperature dependence of the effective magnetic moment, μeff, at a field of 0.1 T in Fig. 2 and the field dependence of the molar magnetization, Mmol, in Fig. 3 (an expanded view of the low-field region is shown on the right). In Fig. 3, circles show the data measured by SQUID magnetometry (2 K, 0 < B < 7 T) while triangles show the pulsed high-field data (0.4 K, 0 < B < 65 T). In Fig. 2, the sharp decrease in μeff on cooling offers strong evidence for dominant antiferromagnetic exchange. Moreover, the low-temperature limit of μeff = 2.34 μB is consistent with an S = 1/2 ground state as is the saturation value of 1.12 NAμB for Mmol in the region 5–60 T in Fig. 3 (minor deviations from the spin-only values of 1.73 μB and 1.0 NAμB being due to a very small amount of paramagnetic impurity). The pulsed high-field experiment, however, reveals a step in Mmol at ∼63 T (the crossover field Bc, shown in Fig. 3) where the Zeeman splitting causes the MS = −3/2 component of the first excited quartet state to cross the MS = −1/2 component of S = 1/2. Assuming a value of 2.0 for g, the corresponding energy gap between the S = 1/2 and S = 3/2 states at zero field can be estimated as ΔE(1/2–3/2) = gμBBc ∼ 85 K (59 cm−1). | ||
| Fig. 2 Plot of μeff(T) at B = 0.1 T for the hexanuclear spin Hamiltonian, H(6): J1 = −5.9 cm−1, J2 = −10.1 cm−1, J3 = J4 = −55.1 cm−1, t1 = −1438 cm−1 (all fixed), t2 varies from 0 to 2000 cm−1 in steps of 250 cm−1, g = 2.0, Δ is set to 0. | ||
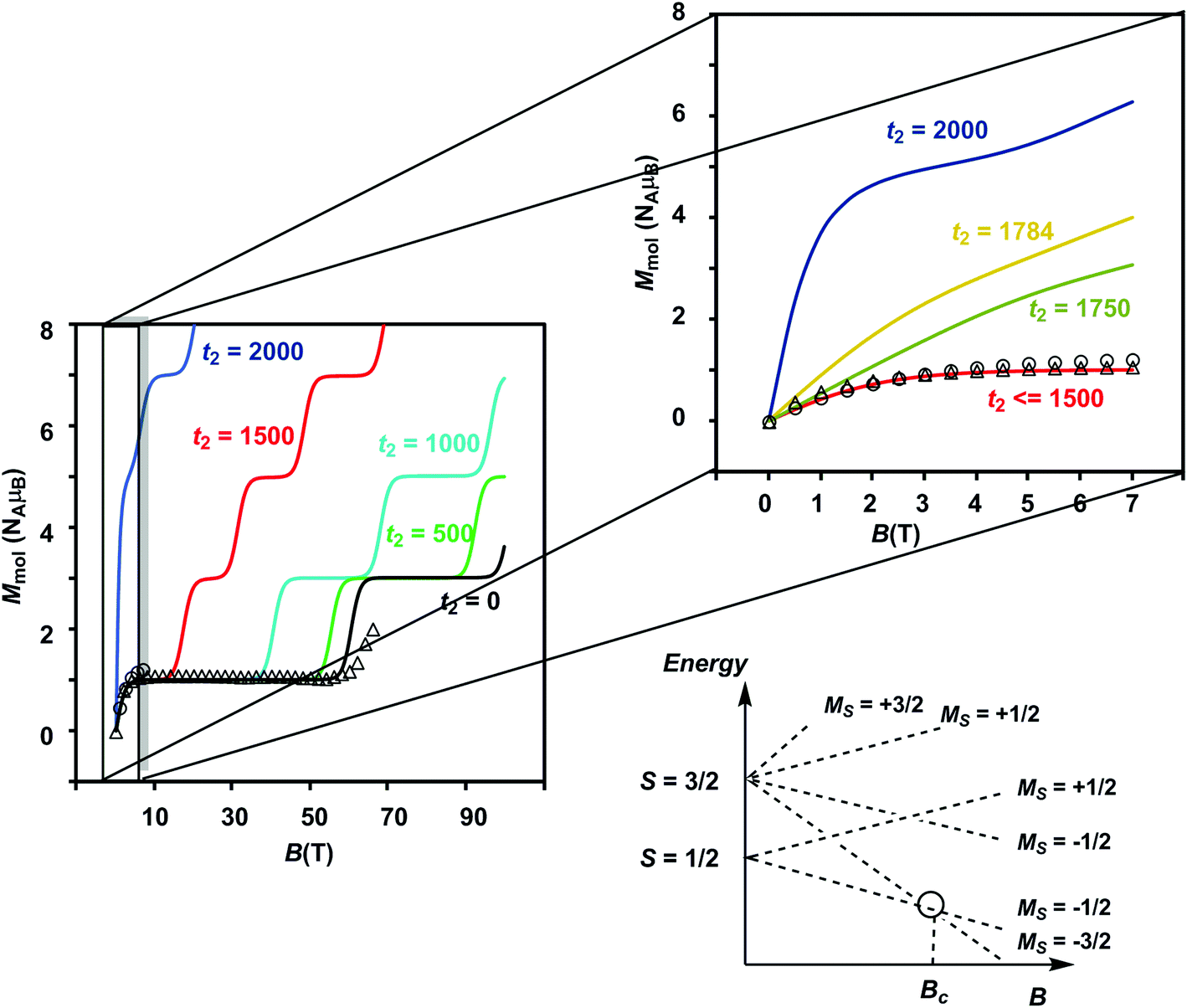 | ||
| Fig. 3 Plot of Mmol(B) showing data measured by SQUID (2 K, circles) and pulsed high-field (0.4 K, triangles) magnetometry. To the right is an expanded view of the low-field region. The parameters used for the simulations are the same as in Fig. 2. The schematic energy level diagram shows the relationship between the Zeeman splitting and the crossover field. | ||
The magnetic data can be interpreted in terms of an exchange + transfer Hamiltonian, H(8), summarized in eqn (2) and (3).
| H(8) = Hex + Htr |
 | (2) |
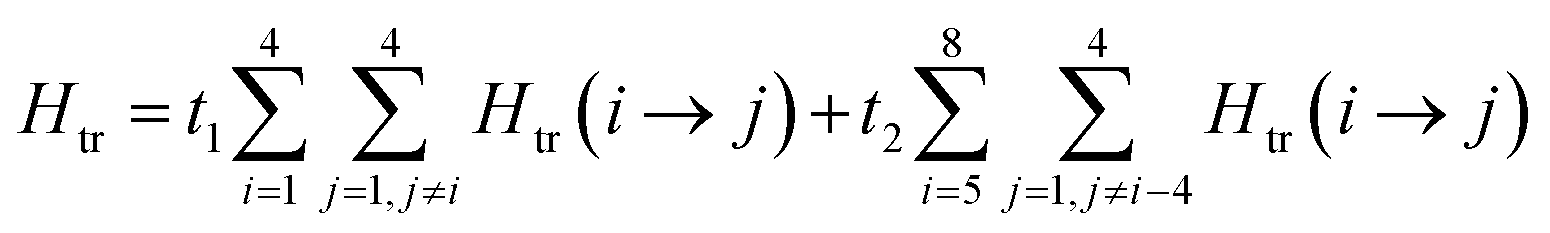 | (3) |
The Heisenberg part, Hex, consists of eight components describing the energy levels of localized configurations – four of the FecII(FecIII)3(FeoIII)4 type (ED = Ec = 0) and four of the (FecIII)4FeoII(FeoIII)3 type (ED = Eo = Δ). The transfer Hamiltonian, Htr, contains two distinct transfer parameters, t1 for cubane–cubane (FecII→FecIII) transfer and t2 for cubane–outer (FecII/III↔FeoIII/II) transfer. The Heisenberg and transfer parts of H(8) are therefore both dependent on the position of the itinerant electron. No attempt has been made to incorporate the effects of anisotropy into the spin Hamiltonian because high-field EPR measurements give no indication that it is significant (ESI, S1†).
The isotropic exchange and asymmetry parameters, J1 (FecIII–FecIII), J2 (FecII–FecIII), J3 (FecIII–FeoIII), J4 (FecII/III–FeoIII/II) and Δ, of eqn (2) can be estimated by mapping the DFT-computed energies of the four localized microstates identified in Scheme 1, BS1, BS2 (MS = 31/2) and BS3, BS4 (MS = 21/2), onto the diagonal elements of the Hex-matrices computed in products of single-center spin functions:


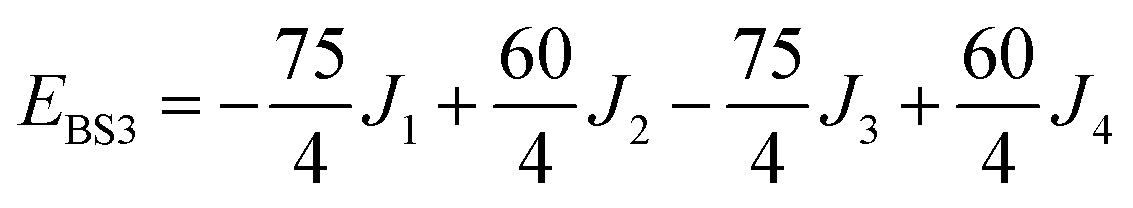

Taking pairwise differences between the energies of these microstates gives rise to a system of linear equations, which can be solved for four unknowns, J1, J2, J3 and Δ. Note that the four equations above in fact contain five unknowns, but J4 appears with the same coefficient (+60/4) in all four expressions and can therefore be eliminated, allowing J1, J2, J3 and Δ to be determined uniquely.
The pair-delocalized BS states with MS = 21/2, BS5 and BS6 in Scheme 1, define ‘effective dimer’ models where the itinerant electron is trapped in a single Fec–Fec dimer unit (BS5) or in a single Fec–Feo dimer unit (BS6). In each case, there are two separate states where the itinerant electron occupies the in-phase (EBS+) and out-of-phase (EBS−) combinations of the d orbitals involved in the transfer pathway. The energies of these BS states can be associated with the eigenvalues of the [2 × 2] spin Hamiltonian matrices computed in two products of single-center spin functions (one where the itinerant electron is localized on the first center of the pair, and the other on the second center of the pair). For the BS5 states, the matrix and its eigenvalues take the form:



The expression for t1 is identical to the analytical form shown in eqn (1) for symmetric clusters 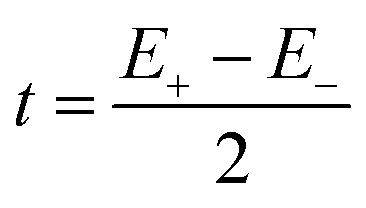 where E+ and E− are the eigenvalues corresponding to symmetric and antisymmetric combinations of basis functions. The energy difference E+ − E− can be approximated as the difference between the Kohn–Sham eigenvalues for the in-phase and out-of-phase combinations of the relevant d orbitals in the isovalent ferromagnetically coupled state (the all-ferric BS5 state in this case).40
where E+ and E− are the eigenvalues corresponding to symmetric and antisymmetric combinations of basis functions. The energy difference E+ − E− can be approximated as the difference between the Kohn–Sham eigenvalues for the in-phase and out-of-phase combinations of the relevant d orbitals in the isovalent ferromagnetically coupled state (the all-ferric BS5 state in this case).40
The corresponding matrix for t2 (using the BS6 states) is:
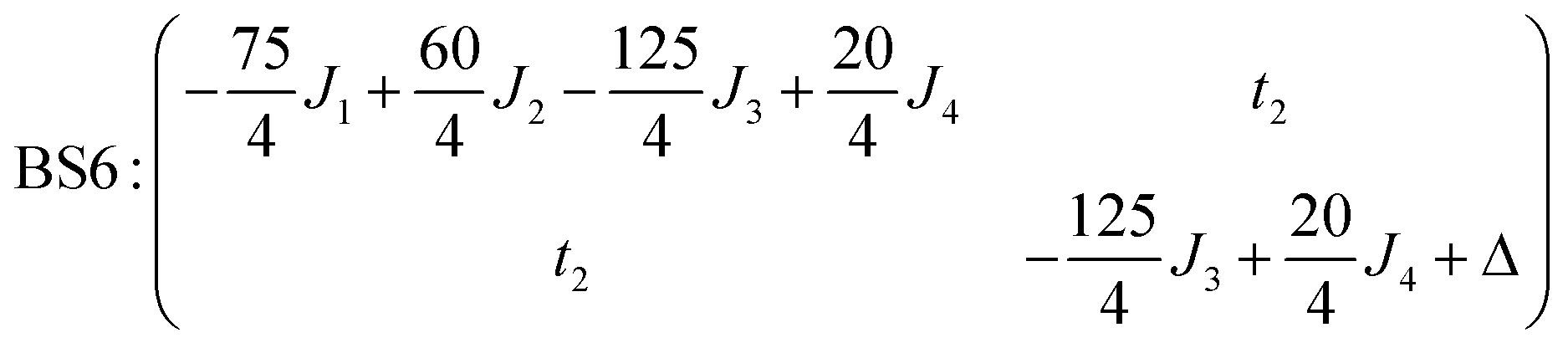
The full set of spin Hamiltonian parameters that emerges from this analysis is (further details are given in ESI, S2†):

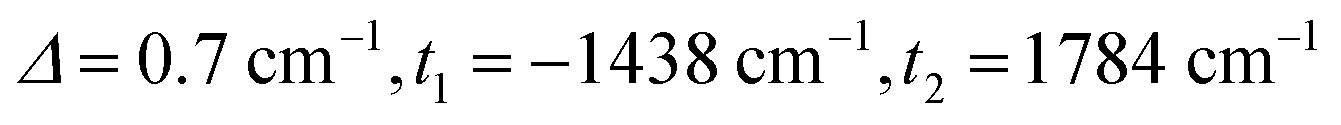
The J1 and J3 values, corresponding to the FecIII–FecIII and FecIII–FeoIII interactions, are very similar to those reported previously for the all-ferric analogue, both from DFT (−6.3 cm−1, −52.8 cm−1) and from best fit to the SQUID magnetometry data (−2.1 cm−1, −50.6 cm−1).27
Clemente-Juan et al. have noted that the exchange part of the H(8) Hamiltonian (eqn (2)) can be made independent of the position of the itinerant electron by assuming the isotropic exchange to be independent of the oxidation state (i.e., J1 = J2 and J3 = J4).26 The values of J1 and J2 that emerge from our DFT analysis are indeed quite similar, fully justifying the first of these assumptions. As emphasized above, J4 is unavailable from our DFT analysis, so we are unable to assess the validity of the second approximation, which we nevertheless adopt in the subsequent modelling of the magnetic data. The imposed symmetry of the isotropic exchange parameters does nothing, however, to alleviate the intrinsic problem that the dimension of the largest submatrix of H(8) is such that exact diagonalization is intractable. This problem can be avoided if electron transfer between the cubane and outer iron centers is neglected (t2 = 0 in eqn (3)), effectively localizing the itinerant electron on the cubane core and reducing the number of localized configurations to four (of the FecII(FecIII)3(FeoIII)4 type). Our computed value of t2 = 1784 cm−1, however, suggests that such an approximation will miss much of the important mixed valency physics. Alternatively, diagonalization of a model Hamiltonian, H(6), based on a smaller hexanuclear cluster with an Fe3(μ3-O)3 core and three pendant iron centers (ESI, S3, Scheme S1, eqn (S1) and (S2)†), is tractable even when t2 ≠ 0.26 This model shares the same n + n core/outer topology as the parent Fe8 cluster and, in the limiting case that t2 = 0, the H(8) and H(6) Hamiltonians generate very similar eigenvalue patterns.26 A comparison of the magnetic functions (μeff and Mmol) reconstructed using both H(8) and H(6) confirms that they are indeed almost identical in the limiting case of t2 = 0 (ESI, S3, Fig. S3†). However, H(6) (and therefore, by extension, H(8)) provides a very poor fit to the temperature dependence of μeff over the range 2 < T < 300 K when t2 is set to 0 (black line in Fig. 2). Moreover, we were unable to identify any physically reasonable combination of J1–4 and t1 that could reproduce the sharp rise in μeff between 0 and 100 K, suggesting that the separation between the ground doublet state and excited states with higher multiplicity is being systematically overestimated. When t2 is increased (now by necessity using only H(6)), states with S > 1/2 are indeed stabilized, and values in the region of 1750 cm−1 provide a reasonable match to μeff over the entire temperature range (2 < T < 300 K). The data are therefore entirely consistent with our computed value of t2 = 1784 cm−1 (orange curve in Fig. 2). The influence of the dominant cubane–outer exchange interactions and the intrinsic site asymmetry on μeff is explored in Fig. S4 and S5 (ESI, S4†).
The Mmol(B) curves measured using either SQUID or pulsed high-field magnetometry (Fig. 3) offer a rather different perspective: the agreement between experiment and the curve generated using t2 = 1784 cm−1 is strikingly poor. In the low-field region sampled by both the pulsed high-field measurements and SQUID magnetometry (up to 7 T, see the expanded plot), Mmol is rather insensitive to the value of t2 in the range 0–1500 cm−1 because only the ground doublet state is appreciably populated under these conditions (the curves for values of 1250 cm−1 and smaller lie directly below the red curve at 1500 cm−1). Based on the low-field data alone, therefore, we can do no more than place an upper limit of ∼1500 cm−1 on t2 at 2 K. The high-field data, however, pinpoint the crossover field Bc, the point where the S = 3/2 state becomes the ground state, at ∼63 T, a value that is consistent only with t2 values approaching zero (where Bc = 60.6 T). Values of 1500 cm−1, 1000 cm−1 and even 500 cm−1 for t2, in contrast, generate much smaller crossover fields that are clearly inconsistent with the data. Indeed, no physically reasonable alternative combination of parameters with t2 substantially greater than zero can reproduce the experimental data (ESI, S4, Fig. S6, S7†). The fact that t2 appears to be quenched at very low temperatures is indicative of significant vibronic coupling, which generates a number of distinct minima on the adiabatic potential surfaces that serve to localize the itinerant electron on the cubane core. Note that the localizing influence of vibronic coupling is particularly strong when S is small. This assertion is consistent with the energy of the IVCT transition, which leads to an upper estimate of ∼6000 cm−1 for the reorganization energy, λ.28 Based on the magnetic data alone, we are unable to distinguish between the alternative possibilities that it is localized on a single center (t1 = t2 = 0) or pairwise delocalized (t1 = −1438 cm−1, t2 = 0): the predicted values of Bc are almost identical, 61.4 T and 61.1 T, respectively. The X-ray photoelectron spectroscopy data reported in ref. 28 are, however, more consistent with localization on a single center. The sharp increase in μeff in the low-temperature region suggests, however, that the barriers to delocalization are low.
Conclusions
In this paper, we have presented new magnetic data measured using both conventional SQUID and pulsed high-field magnetometry that reveal the nature of mixed valency in the FeIIFe7III cluster, [Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4]−. The isotropic exchange proves to be very similar to that in the parent all-ferric Fe8III cluster, with a dominant antiferromagnetic coupling between the cubane and outer iron centers mediated by a μ4-oxo ligand. However, electron transfer, both within the cubane core (t1) and between the cubane and outer iron centers (t2), is an important component of the spin Hamiltonian. In particular, the μeff(T) curve cannot be fitted adequately over the entire temperature range (2 < T < 300 K) if the value of t2 is smaller than 1750 cm−1. The picture that emerges from the molar magnetization data measured up to very high field at 0.4 K is, in contrast, rather different: the plateau in the region 5 < B < 60 T is indicative of a large zero-field separation between ground doublet and first excited quartet states that is inconsistent with any value of t2 substantially greater than zero. The striking discrepancy between values of t2 that afford reasonable agreement with the low- and high-temperature data indicates that the measurements are sampling fundamentally different electronic distributions. The quenching of t2 at low temperatures is consistent with the presence of significant vibronic coupling that localizes the itinerant electron on the cubane core.Acknowledgements
Financial support from the EPSRC-EP/G002789/1 (UK), National Science Foundation-CHE-0822600 (USA), the Ministry of Education and Science project 8436 (Russia) and the Operational Program Research and Development for Innovations CZ.1.05/2.1.00/03.0058 of the Ministry of Education, Youth and Sports (Czech Republic) is gratefully acknowledged. Part of this work was carried out at the National High Magnetic Field Laboratory, which is funded by NSF (Cooperative Agreement DMR 1157490), the State of Florida, and Department of Energy. RGR is grateful to the NHMFL for the granting of instrument time to projects PO1587 and PO1965.This article is published in celebration of the 50th anniversary of the opening of the Chemistry Department at the University of York.
J.E.M. and R.G.R. conceived the project, designed the experiments and wrote the paper; E.M.Z. and W.M.C.S. carried out the computational work; E.M.Z. also contributed to the writing; R.H. collected and modeled the SQUID data; R.M. and J.S. collected the pulsed magnetic field data; E.V.G. prepared the studied materials; J.K. measured the HFEPR spectra; Y.S., S.A.B. and Z.T. contributed to the discussion of concepts and edited the paper.
References
- G. Blondin and J.-J. Girerd, Chem. Rev., 1990, 90, 1359–1376 CrossRef CAS.
- S. A. Borshch, E. L. Bominaar, G. Blondin and J.-J. Girerd, J. Am. Chem. Soc., 1993, 115, 5155–5168 CrossRef CAS.
- J. J. Borrás-Almenar, E. Coronado, B. S. Tsukerblat and R. Georges, in Molecular magnetism: from molecular assemblies to the devices, NATO Adv. Stud. Inst. Ser. 321, Kluwer Academic Publishers, Dordrecht, 1996, pp. 105–139 Search PubMed.
- J. J. Borrás-Almenar, E. Coronado, S. M. Ostrovsky, A. V. Palii and B. S. Tsukerblat, Chem. Phys., 1999, 240, 149–161 CrossRef.
- X. Yang, H. Hu and Z. Chen, Int. J. Quantum Chem., 2005, 103, 190–197 CrossRef CAS.
- C. Zener, Phys. Rev., 1951, 81, 440–444 CrossRef CAS.
- C. Zener, Phys. Rev., 1951, 82, 403–405 CrossRef CAS.
- P. W. Anderson and H. Hasegawa, Phys. Rev., 1955, 100, 675–681 CrossRef CAS.
- J.-J. Girerd, J. Chem. Phys., 1983, 79, 1766–1775 CrossRef CAS.
- L. Noodleman and E. J. Baerends, J. Am. Chem. Soc., 1984, 106, 2316–2327 CrossRef CAS.
- S. A. Borshch, I. N. Kotov and I. B. Bersuker, Sov. J. Chem. Phys., 1985, 3, 1009–1016 Search PubMed.
- M. I. Belinskii, B. S. Tsukerblat and N. V. Gerbeleu, Sov. Phys. Solid State, 1983, 25, 497–498 Search PubMed.
- S. A. Borshch, I. N. Kotov and I. B. Bersuker, Chem. Phys. Lett., 1984, 111, 264–270 CrossRef CAS.
- B. Bechlars, D. M. D'Alessandro, D. M. Jenkins, A. T. Iavarone, S. D. Glover, C. P. Kubiak and J. R. Long, Nat. Chem., 2010, 2, 362–368 CrossRef CAS PubMed.
- A. Somcini, T. Mallah and L. F. Chibotaru, J. Am. Chem. Soc., 2010, 132, 8106–8114 CrossRef PubMed.
- D. Lee, J. L. DuBois, B. Pierce, B. Hedman, K. O. Hodgson, M. P. Hendrich and S. J. Lippard, Inorg. Chem., 2002, 41, 3172–3182 CrossRef CAS PubMed.
- V. Barone, A. Bencini, I. Ciofini, C. A. Daul and F. Totti, J. Am. Chem. Soc., 1998, 120, 8357–8365 CrossRef CAS.
- X.-Q. Ding, E. L. Bominaar, E. Bill, H. Winkler, A. X. Trautwein, S. Drüeke, P. Chaudhuri and K. Wieghardt, J. Chem. Phys., 1990, 92, 178–186 CrossRef CAS.
- V. Papaefthymiou, J.-J. Girerd, I. Moura, J. J. G. Moura and E. Münck, J. Am. Chem. Soc., 1987, 109, 4703–4710 CrossRef CAS.
- A. X. Trautwein, E. Bill, E. L. Bominaar and H. Winkler, Struct. Bonding, 1991, 78, 1–95 CrossRef CAS.
- J.-M. Mouesca and B. Lamotte, Coord. Chem. Rev., 1998, 178–180, 1573–1614 CrossRef CAS.
- M. Kröckel, M. Grodzicki, V. Papaefthymiou, A. X. Trautwein and A. Kostikas, J. Biol. Inorg. Chem., 1996, 1, 173–176 CrossRef.
- M. Kröckel, M. Grodzicki, V. Papaefthymiou, A. X. Trautwein and A. Kostikas, J. Biol. Inorg. Chem., 1996, 1, 173–176 CrossRef.
- E. L. Bominaar, S. A. Borshch and J.-J. Girerd, J. Am. Chem. Soc., 1994, 116, 5362–5312 CrossRef CAS.
- J. J. Borrás-Almenar, J. M. Clemente, E. Coronado, A. V. Palii, B. S. Tsukerblat and R. Georges, J. Chem. Phys., 1996, 105, 6892–6909 CrossRef.
- J. M. Clemente-Juan, J. J. Borrás-Almenar, E. Coronado, A. V. Palii and B. S. Tsukerblat, Inorg. Chem., 2009, 48, 4557–4568 CrossRef CAS PubMed.
- P. Baran, R. Boča, I. Chakraborty, J. Giapintzakis, R. Herchel, Q. Huang, J. E. McGrady, R. G. Raptis, Y. Sanakis and A. Simopoulos, Inorg. Chem., 2008, 47, 645–655 CrossRef CAS PubMed.
- I. Chakraborty, P. Baran, Y. Sanakis, A. Simopoulos, E. Fachini and R. G. Raptis, Inorg. Chem., 2008, 47, 11734–11737 CrossRef CAS PubMed.
- E. M. Zueva, W. M. C. Sameera, D. M. Piñero, I. Chakraborty, E. Devlin, P. Baran, K. Lebruskova, Y. Sanakis, J. E. McGrady and R. G. Raptis, Inorg. Chem., 2011, 50, 1021–1029 CrossRef CAS PubMed.
- C. Greaves, J. Solid State Chem., 1983, 49, 325–333 CrossRef CAS.
- M. F. Fleet, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 917–920 CrossRef.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, Science, 2007, 316, 1726–1729 CrossRef CAS PubMed.
- P. Turano, D. Lalli, I. C. Felli, E. C. Theil and I. Bertini, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 545–550 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef.
- L. Engelhardt and M. Luban, Dalton Trans., 2010, 39, 4687–4692 RSC.
- A. Matsuo, K. Kindo, H. Nojiri, L. Engelhardt, M. Luban, E. K. Brechin and I. A. Gass, Phys. Rev. B: Condens. Matter, 2009, 80, 092401 CrossRef.
- L. Engelhardt, I. A. Gass, C. J. Milios, E. K. Brechin, M. Murrie, R. Prozorov, M. Vannette and M. Luban, Phys. Rev. B: Condens. Matter, 2007, 76, 172406 CrossRef.
- K. L. Taft, C. D. Delfs, G. C. Papaefthymiou, S. Foner, D. Gatteschi and S. J. Lippard, J. Am. Chem. Soc., 1994, 116, 823–832 CrossRef CAS.
- C. Boilleau, N. Suaud, R. Bastardis, N. Guihéry and J. P. Malrieu, Theor. Chem. Acc., 2010, 126, 231–241 CrossRef CAS.
- M. Shoji, K. Koizumi, T. Taniguchi, Y. Kitagawa, S. Yamanaka, M. Okumura and K. Yamaguchi, Int. J. Quantum Chem., 2007, 107, 116–133 CrossRef CAS.
- P. A. Goddard, J. Singleton, P. Sengupta, R. D. McDonald, T. Lancaster, S. J. Blundell, F. L. Pratt, S. Cox, N. Harrison, J. L. Manson, H. I. Southerland and J. A. Schlueter, New J. Phys., 2008, 10, 083025 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- L. Noodleman and D. A. Case, Adv. Inorg. Chem., 1992, 38, 423–470 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: High-frequency, high-field EPR spectra of [Bu4N][Fe8(μ4-O)4(μ-4-Cl-pz)12Cl4] (S1), ab initio computation of the spin Hamiltonian parameters (S2), development of model spin Hamiltonians (S3), plots of μeff(T) and Mmol(B) (S4), sample magnetic moment versus pulsed magnetic field plots for two [Fe8]0 compounds (S5). See DOI: 10.1039/c4dt00020j |
| ‡ Present address: Department of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA. |
| This journal is © The Royal Society of Chemistry 2014 |