 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anti-Markovnikov oxidation and hydration of terminal olefins
Jiayi
Guo
a and
Peili
Teo
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, S(117543), Singapore
bInstitute of Chemical & Engineering Sciences, 1 Pesek Road, Jurong Island, S(627833), Singapore. E-mail: peiliteo@nus.edu.sg; Fax: +(65)-67791691; Tel: +(65)-65161377
First published on 30th January 2014
Abstract
Efficient syntheses of aldehydes and primary alcohols from terminal olefins are critical to the chemical laboratory and industry. Heavy emphasis has been placed on regioselective olefin functionalization in recent decades. This perspective mainly focuses on advances in the Wacker-type oxidation and tandem hydration of terminal olefins with anti-Markovnikov selectivity.
 Jiayi Guo | Jiayi Guo received her Ph.D in 2013 from Nanyang Technological University, Singapore. Her thesis focused on the synthesis and characterization of bis-phosphorus stabilized metal complexes. In April 2013, she joined the group of Assistant Professor Peili Teo at the National University of Singapore, as a postdoctoral fellow. Her current research is focused on olefin functionalization via efficient and selective catalysis systems. |
 Peili Teo | Peili Teo received her Ph.D in 2008 from the National University of Singapore under the supervision of Professor T. S. Andy Hor as an A*STAR Graduate Scholar. Her thesis focused on coordination assemblies of d8 and d10 metals with N,O-mixed-donor ligands. Peili then went on to work as a A*STAR postdoctoral research fellow with Professor Robert H. Grubbs at California Institute of Technology where she worked on Z-selective olefin metathesis catalysts and anti-Markovnikov olefi oxidation and hydration. Returning to Singapore in 2012, Peili began her independent research career as a scientist at the Institute of Chemical & Engineering Sciences, A*STAR, and as an assistant Professor at the National University of Singapore. |
1. Introduction
Current methods for aldehyde production from carbon–carbon multiple bonds are hydroformylation (eqn (1)) and alkyne hydration. However, hydroformylation produces the homologous aldehyde and anti-Markovnikov alkyne hydration often employs catalysts that are non-trivial to prepare.1–3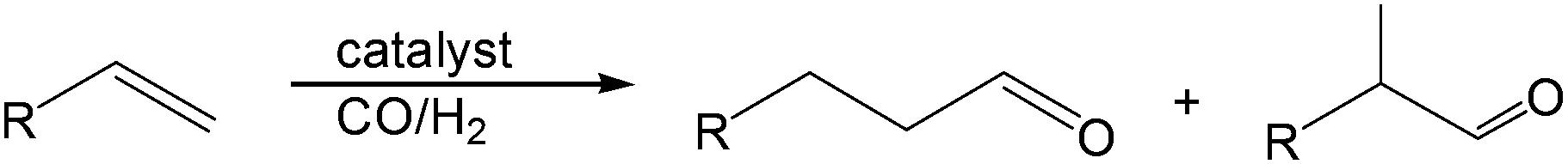 | (1) |
Another popular protocol is via alcohol oxidation but the formation of primary alcohols can itself be a very difficult process.4,5 The PdII-catalyzed oxidation of olefins to carbonyl compounds, usually known as the Wacker oxidation, is one of the most well-known Pd-mediated reactions and has been so extensively adopted that terminal olefins are often viewed as masked ketones.6 The process involves the coordination of olefin to PdII followed by reaction of the η2-Pd-alkene complex with water. This reaction obeys Markovnikov's rule in the majority of cases (Scheme 1).4 Nevertheless, aldehydes are sometimes produced in specific cases.7–10
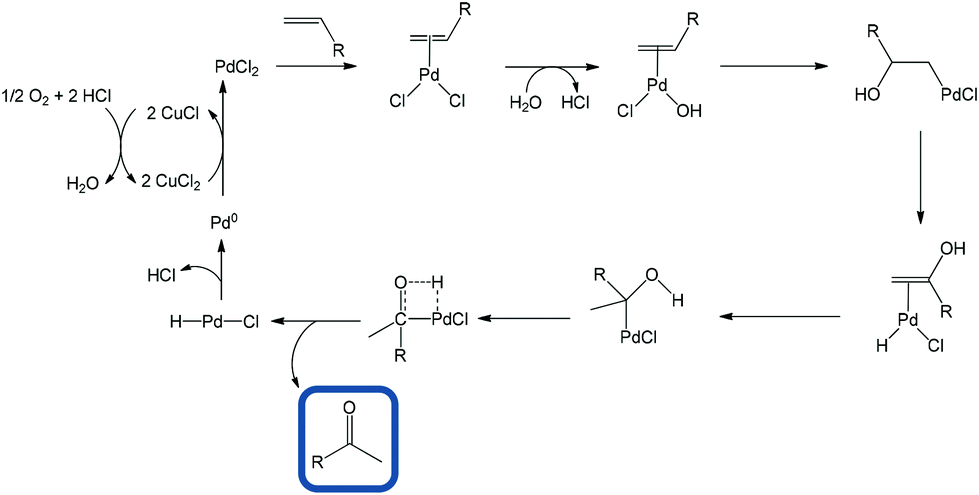 | ||
| Scheme 1 General mechanism of Wacker oxidation. | ||
Anti-Markovnikov hydration of olefins has been listed as one of the top ten challenges of catalysis since 1993.11a Direct addition of water across a carbon–carbon double bond is an important industrial process for alcohol production. Linear alcohols are commonly used in the flavoring, perfumery, lubricant and cosmetic industries. For example, 1-dodecanol, commonly known as lauryl alcohol, is commonly used as a surfactant or emulsifier in detergents and personal care products.11b However, in accordance with Markovnikov's rule,4 the primary alcohols are difficult to obtain. Hydroboration/oxidation is currently a popular indirect method to afford hydration products with anti-Markovnikov selectivity (eqn (2)).4 This process requires stoichiometric borane reagents that are expensive and the boron waste generated is difficult to be recycled. Moreover, the usage of peroxides in the oxidation step of hydroboration poses safety issues when the process is adapted for large scale production. As a result, hydroboration has not been adopted in the industrial production of alcohols from olefins yet. Hydroformylation/reduction is another two-step method that can produce primary alcohols from olefins albeit through a homologation process (eqn (3)).12,13 Thus, there is a compelling need to develop more efficient catalysis systems for anti-Markovnikov hydration of olefins.
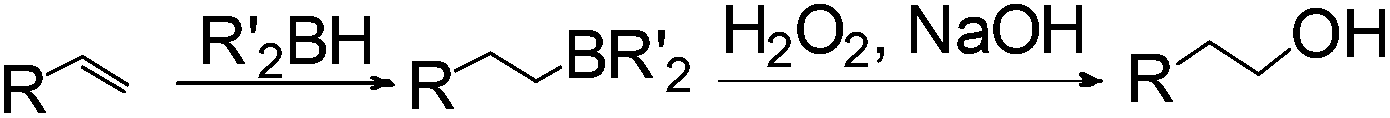 | (2) |
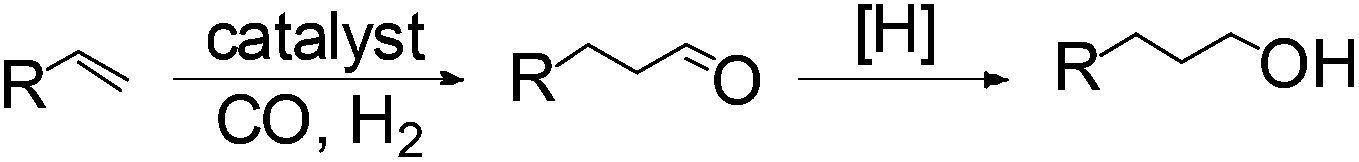 | (3) |
It is encouraging to note that some progress has been made recently from investigations on Wacker-type oxidation for aldehyde production. By controlling the regioselectivity of the oxidation step, anti-Markovnikov hydration of olefins can be realized through the subsequent reduction process in a tandem catalytic system. Despite this not being the most ideal method in the direct hydration of an olefin, it is still a major advancement in the development of selective catalysts for the anti-Markovnikov hydration of olefins.
Much progress in aldehyde-selective Wacker oxidation and anti-Markovnikov olefin hydration has been made since the last review on the former in 2007 by Muzart.6 In this perspective, we illustrate the most significant contributions to anti-Markovnikov olefin oxidation in recent years, especially in areas of Wacker-like processes for olefin oxidation and tandem hydration.
2. Aldehyde-selective Wacker-type reactions of terminal olefins
In 2006, an anti-Markovnikov Wacker reaction of styrene-related substrates was reported by Spencer et al.10 Reversal of the usual regioselectivity occurred stoichiometrically in the absence of reoxidants (eqn (4)) or catalytically, when heteropolyacid (HPA) was used as the terminal oxidant.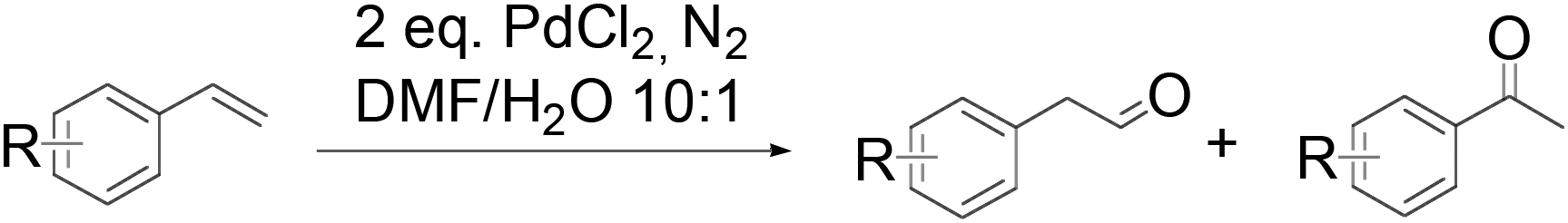 | (4) |
The substrate scope of this optimised condition was studied for a large range of styrenes from electron-deficient to electron-rich (Table 1). The usage of a heteropolyacid (HPA) containing molybdenum and vanadium H4[PMo11VO40] was reported to successfully achieve 75% yield of aldehyde in the presence of 12 mol% of PdCl2 (eqn (5)). With HPA, the process is catalytic but only styrene and methylstyrene can be oxidized to the corresponding aldehyde in reasonable yields.
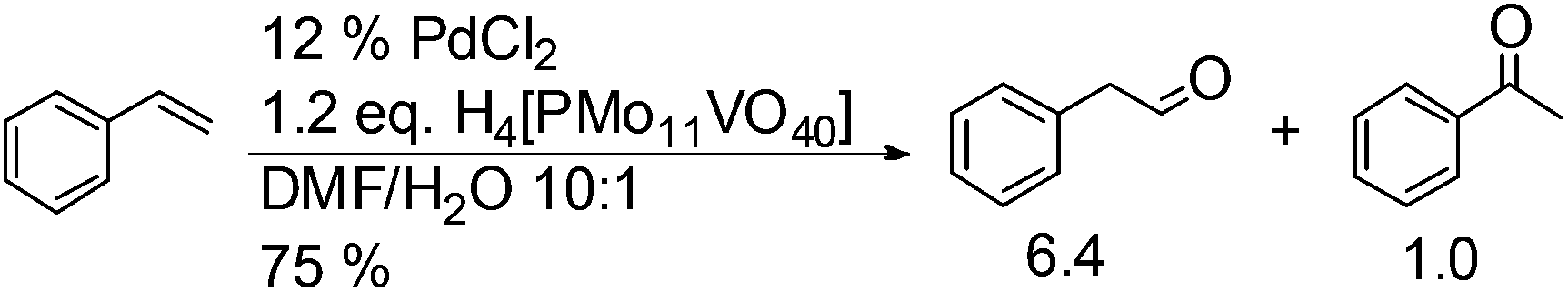 | (5) |
| R group | Ratio of aldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ketone :ketone |
Yield % |
|---|---|---|
| 4-OMe | 10:1 |
63 |
| H | 11:1 |
71 |
| 2-Me | >25:1 |
89 |
| 2,6-Di-Me | 2:1 |
60 |
| Naphthyl | 13:1 |
38 |
| 4-Me | 7:1 |
61 |
| 4-Cl | >25:1 |
76 |
| 4-CF3 | >25:1 |
69 |
| 4-tBu | 9:1 |
82 |
| 3-Me | 5:1 |
50 |
| 3-Cl | >25:1 |
80 |
| 3-NO2 | 16:1 |
54 |
| 3,5-di-tBu | 6:1 |
— |
| 2-OMe | 17:1 |
95 |
| 2-F | >25:1 |
78 |
| 2-Br | >25:1 |
— |
| 2,4-Di-Me | >25:1 |
82 |
The proposed mechanism for aldehyde formation from styrene-related substrates was also disclosed in this work (Scheme 2). The η4 nature of styrene acts as a pseudo-diene to form the η3-palladium intermediate followed by nucleophilic attack. This η4-interaction between the palladium and substrate apparently contributes to the anti-Markovnikov behaviour.
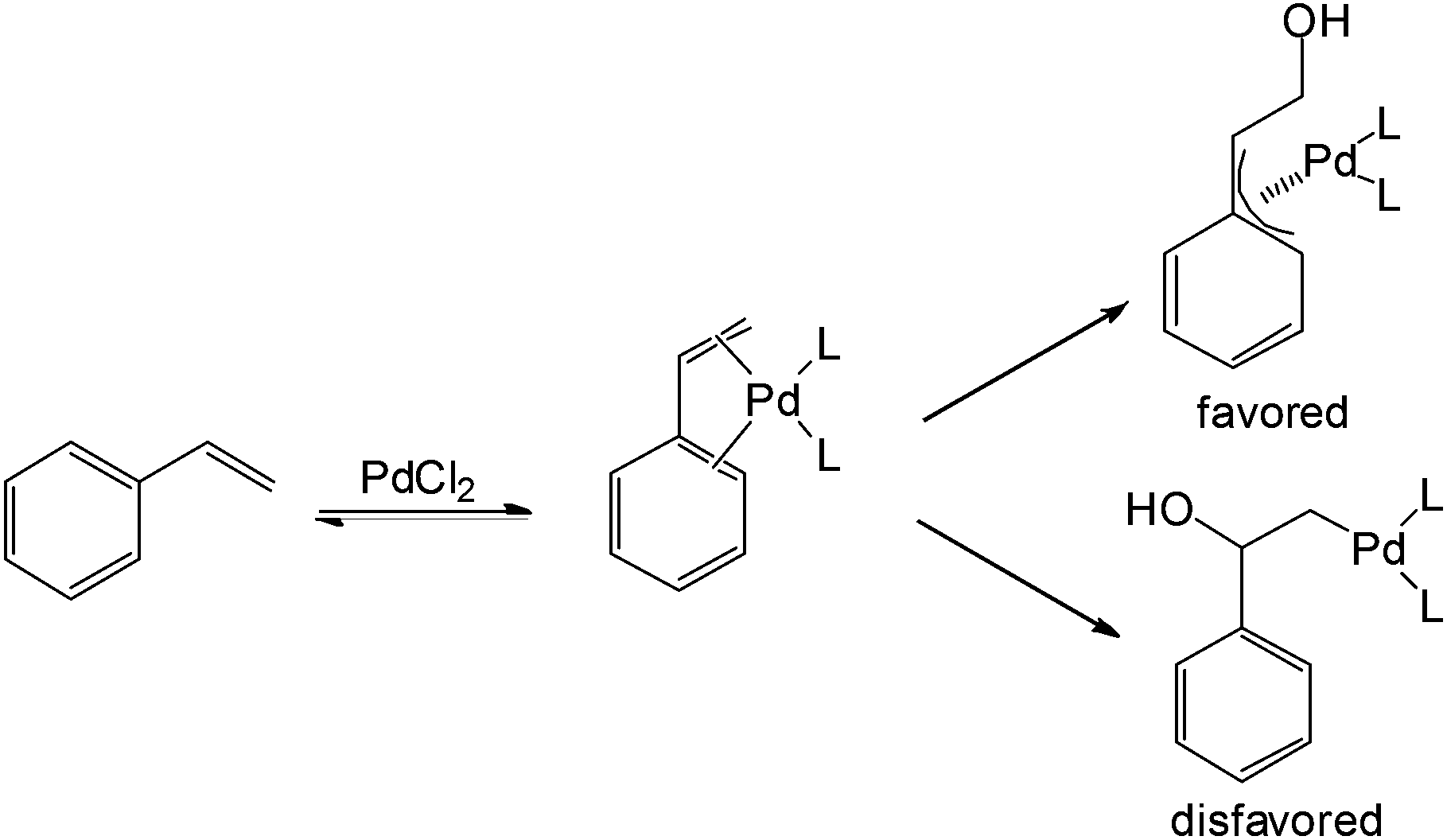 | ||
| Scheme 2 Proposed mechanism for anti-Markovnikov regioselectivity. | ||
Although moderate aldehyde selectivity was obtained in Spencer's work, it required either stoichiometric heteropolyacid or stoichiometric palladium. Also, a limited substrate scope is compatible with this system. With linear olefins, Markovnikov selectivity was obtained.
In 2007, the synthesis of aldehydes with moderate yields via Pd-catalyzed oxidation from functionalized olefins were summarized by Muzart.6 In his review, Wacker-type reactions involving successive intermediate steps of hydroxy-, alkoxy- and acetoxy-palladation were described. Most processes were restricted to activated olefins, that is, olefins containing a chelating group located at an appropriate position, such as a heteroatom or unsaturation, in order to result in the regioselectivity.
When the aldehyde-selective Wacker oxidation was employed towards terminal olefins of group-protected allylic amines, a new catalytic methodology for the synthesis of β3-amino acids was reported by Feringa et al. in 2009.8 Phthalimide proved to be the optimal protecting group resulting in good conversion in all the methods A, B and C, investigated (Table 2).
|
|
||
|---|---|---|
| Cat. | Conversion % | Ratio aldehyde:ketone |
| A | 100 | 96:4 |
| B | 100 | >99:1 |
| C | 80 | 94:6 |
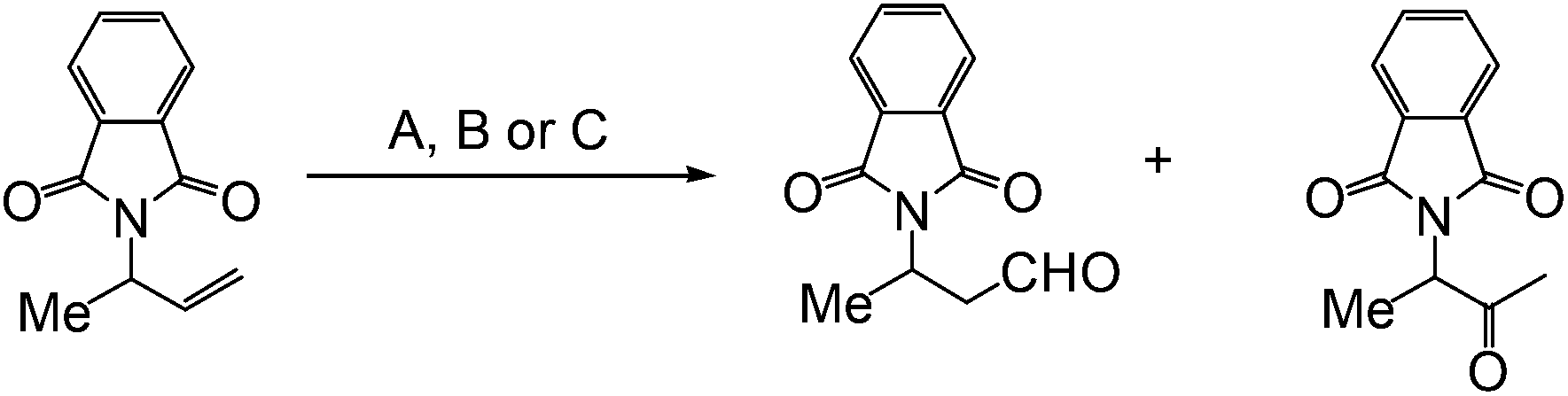
The substrate scope was also studied using methods A and B that gave full conversions (Table 3). It is noteworthy that substrates with long alkyl chains could be oxidized with excellent yields and selectivities (R = C5H11). The high anti-Markovnikov selectivity was proposed to have resulted from coordination of the protecting group to the palladium species.
|
|
|||
|---|---|---|---|
| Substrate | Cat. | Ratio aldehyde:ketone |
Yield % |
| R = CH3 | B | >99:1 |
94 |
| R = C5H11 | B | >99:1 |
91 |
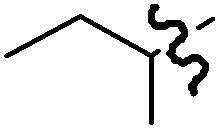
|
A | >99:1 |
93 |
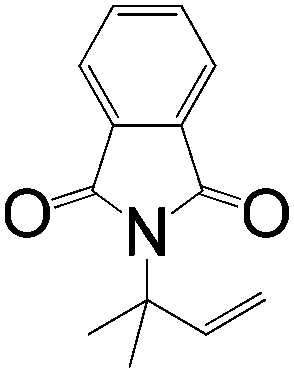
|
A | 94:6 |
74 |
| R = Bn | B | >99:1 |
94 |
| R = BnOCH2 | B | >99:1 |
93 |
| R = Ph | B | >99:1 |
95 |
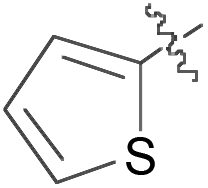
|
B | >99:1 |
77 |
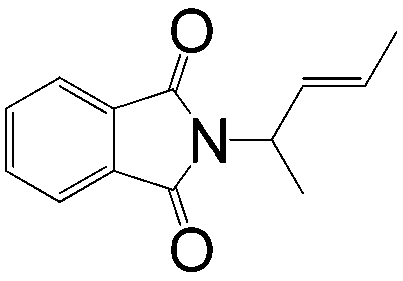
|
A | >1:99 |
89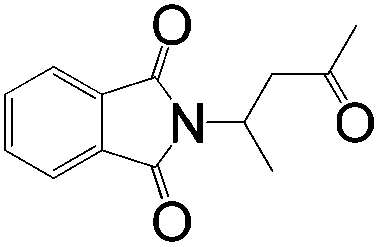 |
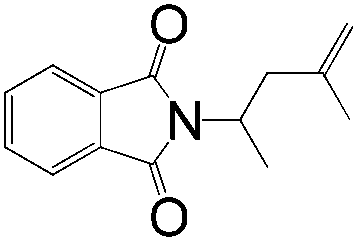
|
A | — | 0 |
| B | — | 0 | |
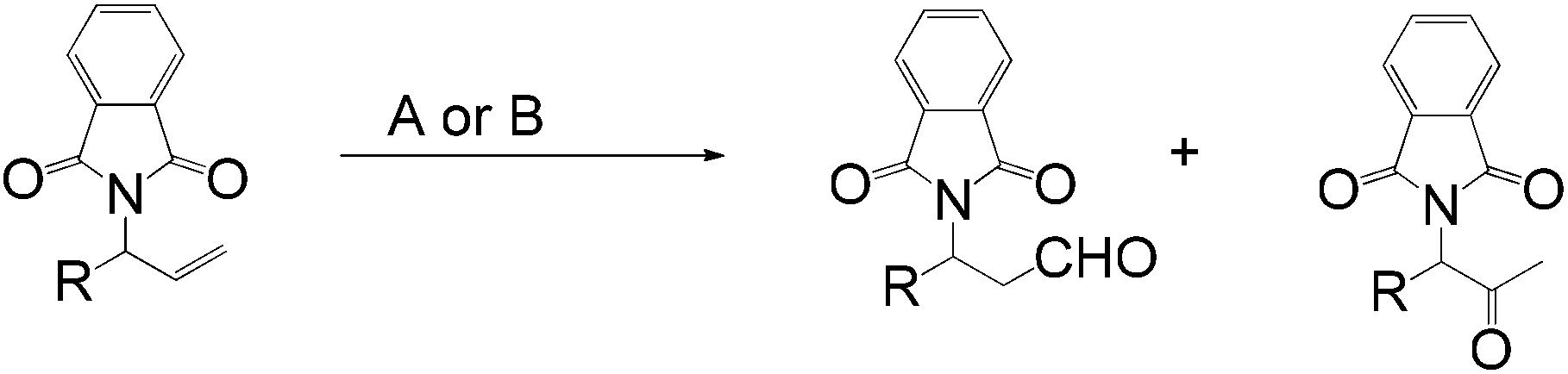
This new synthetic route to amino aldehydes provides an alternative method to prepare amino acids or amino alcohols via consecutive oxidation, reduction and deprotonation procedures (eqn (6)).14–16
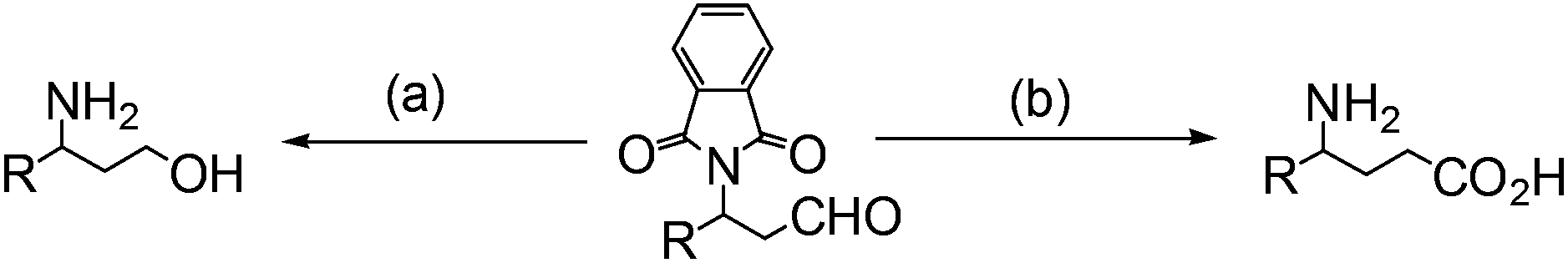 | (6) |
(a) (i) NaBH4, MeOH; R = Me 95%; (ii) H2NH2, EtOH, Δ; R = Me 90%; (b) (i) 0.5% Mn-tmtacn (1,4,7-trimethyl-1,4,7-triazacyclononane), Cl3CO2H, H2O2, MeCN; R = Me 87%; (ii) H2NH2, EtOH, Δ; R = Me 100%.
The palladium-catalyzed anti-Markovnikov oxidation of allylic esters was also achieved by Feringa et al.17 In this system, excellent aldehyde selectivity could be obtained with catalyst loadings as low as 0.5 mol% Pd(II), in tBuOH solvent under ambient conditions (Table 4). p-Benzoquinone was employed as the oxidant. Most aliphatic allylic esters resulted in high aldehyde selectivity. The phenyl- and benzyl substrates were oxidized with slightly lower yields.
|
|
|||
|---|---|---|---|
| R | Conv. | A/M | Yield % |
| H | Full | 11:1 |
78 |
| CH3 | Full | 7:1 |
71 |
| C2H5 | Full | 20:1 |
79 |
| C5H11 | Full | 20:1 |
73 |
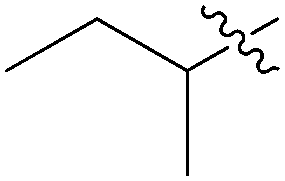
|
95% | 20:1 |
45 |
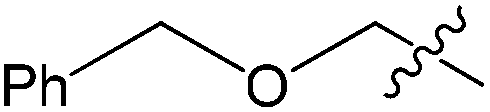
|
Full | 20:1 |
71 |
| Ph | Full | 20:1 |
52 |
| Bn | 95% | 20:1 |
64 |
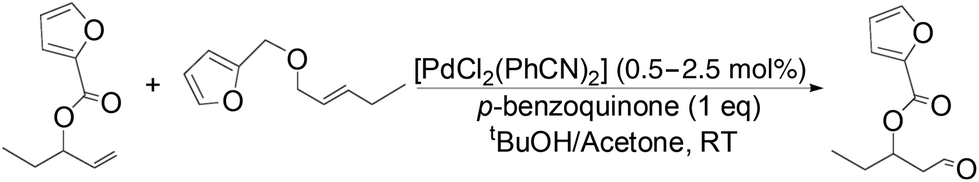
Owing to rapid isomerization between allylic esters under the reaction conditions, a major advantage of this system is that only the anti-Markovnikov product is afforded in the oxidation even when a mixture of branched and linear allylic esters was used (Scheme 3).
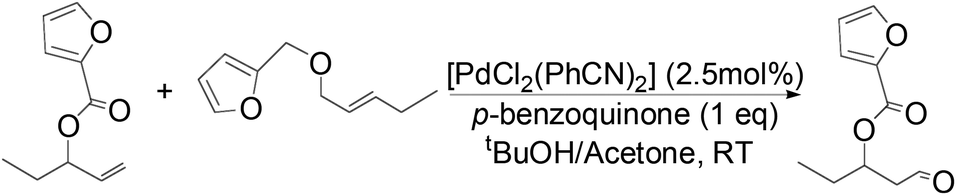 | ||
| Scheme 3 Oxidation of a mixture of linear and branched allylic esters. | ||
This anti-Markovnikov oxidation of allylic esters provides a facile route to the synthesis of protected β-hydroxy aldehyde with low Pd catalyst loadings from either the branched or linear allylic esters, or even a mixture of both.
PdII-catalyzed oxidation has also been adopted in allylic C–H acetoxylation. In 2010, Stahl et al. demonstrated a 4,5-diazafluorenone-ligated palladium (Fig. 1) catalytic strategy for the conversion of terminal alkenes to linear allylic acetoxylation products under 1 atm of O2, eliminating the requirement for the commonly-used oxidant 1,4-benzoquinone (BQ).18
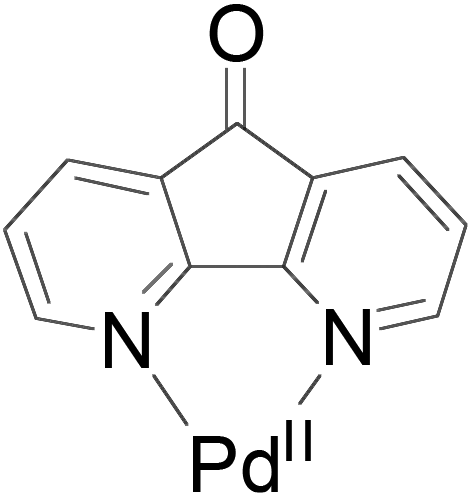 | ||
| Fig. 1 4,5-Diazafluorenone-ligated palladium catalyst. | ||
In this work, three possible roles of BQ in the catalytic cycle were reported (Scheme 4): (1) enhancement of nucleophilic attack from acetate on the π-allyl-PdII species (step II);19 (2) displacement of the allylic acetate product in the Pd0 species upon C–O bond formation (step III);20 and (3) regeneration of Pd0 to PdII (step IV).21 It is envisioned that geometric properties of the diazafluorenone ligand such as the bite angle, may destabilize PdII and facilitate C–O elimination from the π-allyl-PdII intermediate (step II), thus enabling the BQ-free catalytic turnover.
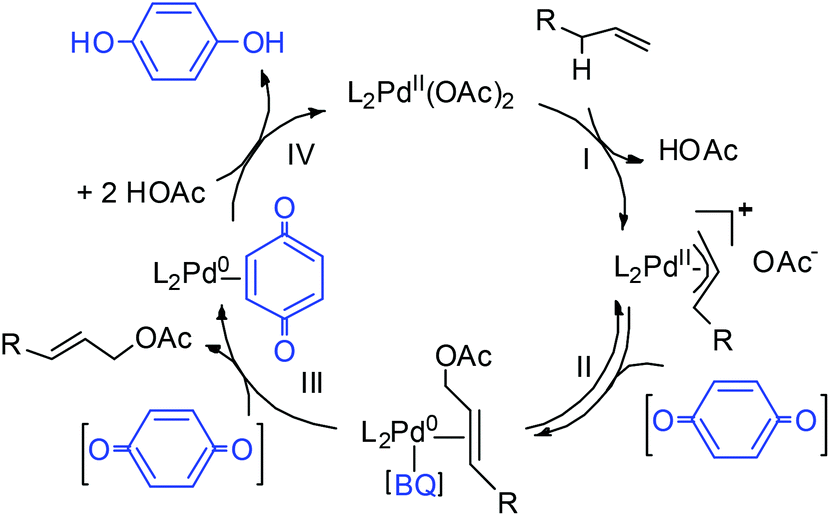 | ||
| Scheme 4 Proposed mechanism for Pd-catalyzed allylic acetoxylation. | ||
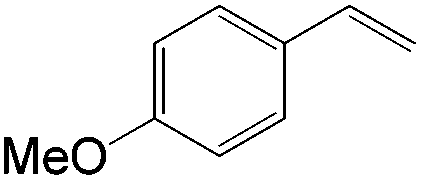
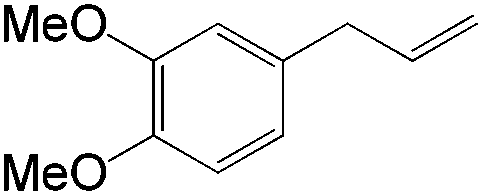
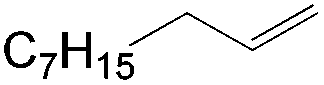
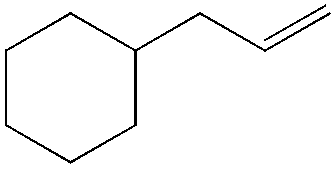
This PdII-catalyzed allylic acetoxylation was investigated with a number of alkenes under aerobic conditions (Table 5). Nearly all the substrates examined resulted in good yields of the linear acetoxylation products. Furthermore, anti-Markovnikov hydration of α-olefins was achieved using an O2/H2-coupled process based on this PdII-catalyzed allylic acetoxylation in a straightforward one-pot reaction (Scheme 5).
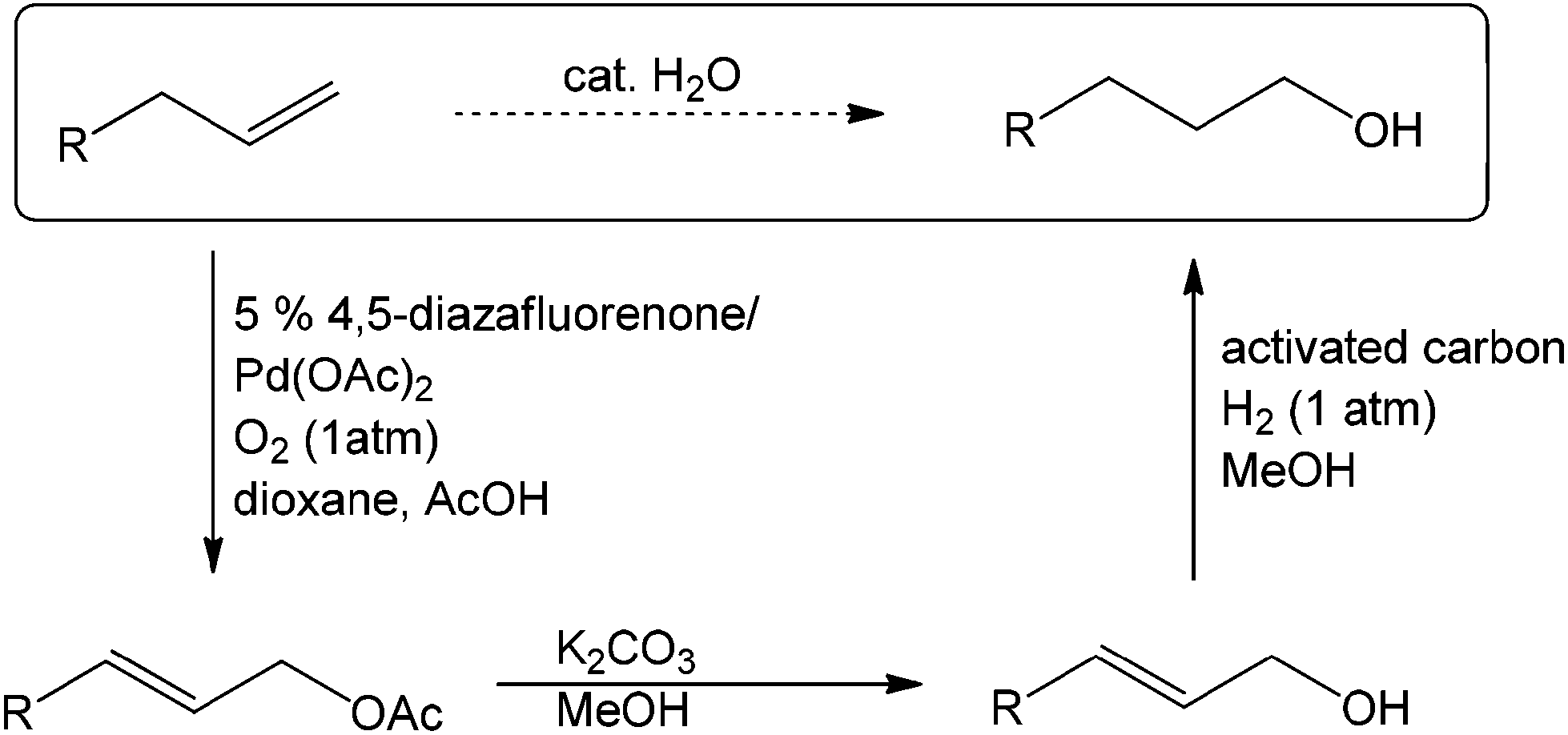 | ||
| Scheme 5 Net anti-Markovnikov hydration of terminal alkenes via a one-pot, three-step sequence. | ||
|
|
|||
|---|---|---|---|
| Substrate | Time (h) |
E:Z |
Yield % |
|
24 | 17:1 |
81 |
|
24 | 14:1 |
79 |
|
24 | 6:1 |
68 |
|
48 | 10:1 |
76 |
|
24 | 19:1 |
70 |
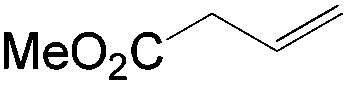
|
24 | 21:1 |
84 |
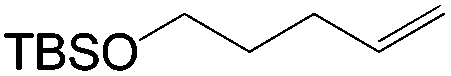
|
48 | 6:1 |
52 |
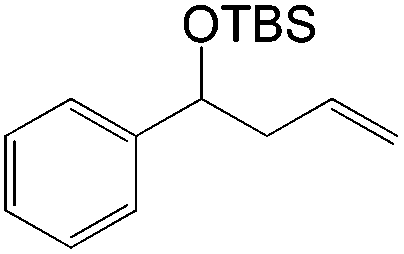
|
48 | 29:1 |
76 |
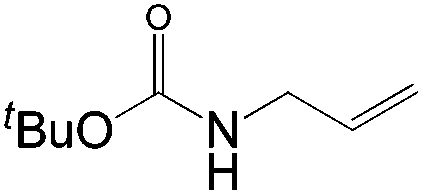
|
24 | 7:1 |
76 |
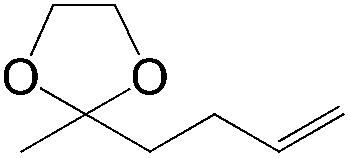
|
48 | 36:1 |
71 |
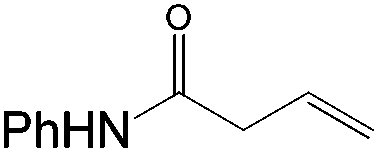
|
24 | 16:1 |
74 |
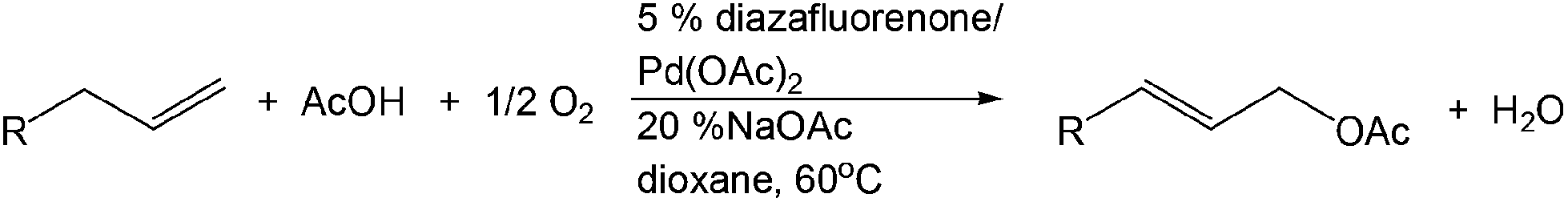
This aerobic ligand-based strategy serves as an inspiration for future work on the role of ancillary ligands in PdII-catalyzed oxidation and accordingly, omission of the use of undesirable oxidants.
Lin et al. reported another example of aerobic Wacker-type intramolecular aldol cyclization.22 Two frameworks of 2-naphthols and benzofurans were generated in acceptable yields. This aerobic PdII-catalyzed oxidation is an attractive method for the bioactive synthesis of natural products (Scheme 6).
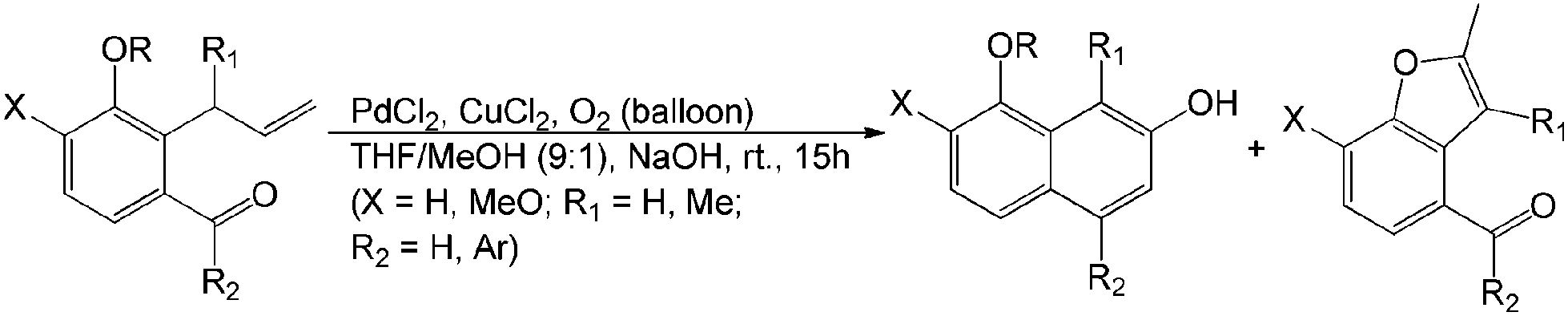 | ||
| Scheme 6 One-pot aerobic Wacker-type synthetic route toward 2-naphthols and benzofurans. | ||
The steric demand of nucleophiles is considered as another potential factor to influence the regioselectivity. In 2012, Kataoka et al. reported an anti-Markovnikov Pd-catalyzed oxidation mediated by the steric bulkiness of pinacol.23 This process enables the synthesis of terminal acetals from vinylarenes, allyl aryl ethers, and 1,5-dienes in the presence of 10 mol% PdCl2(MeCN)2 and 2 equiv. benzoquinone as the oxidant (Table 6). The terminal acetals can undergo acid-catalysed hydrolysis to result in the corresponding aldehydes.
|
|
|
|---|---|
| Substrate | Total yield of A and B (%) |
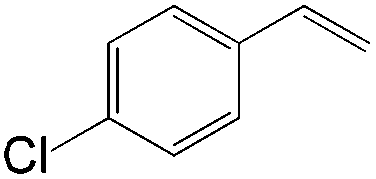
|
73 |
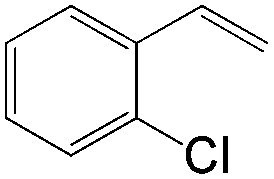
|
79 |
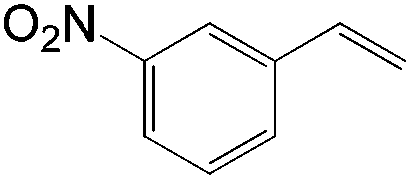
|
76 |
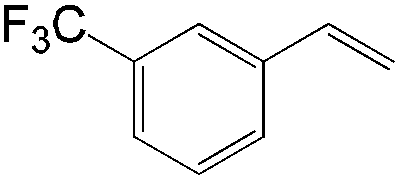
|
83 |
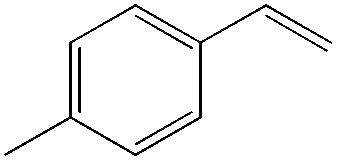
|
64 |
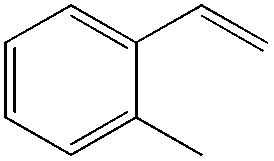
|
69 |
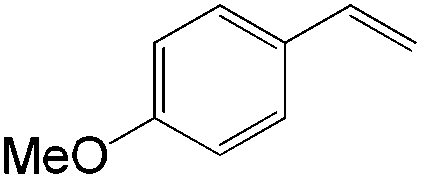
|
39 |
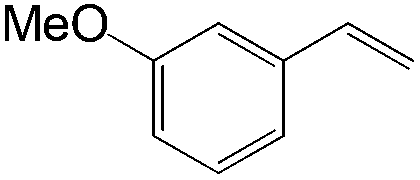
|
71 |
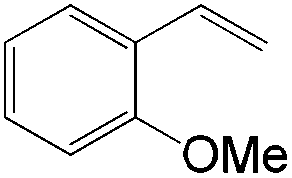
|
61 |
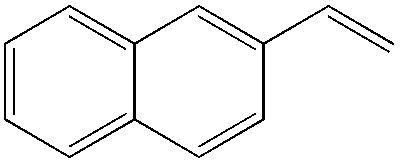
|
50 |
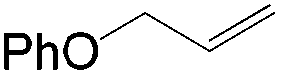
|
75 |
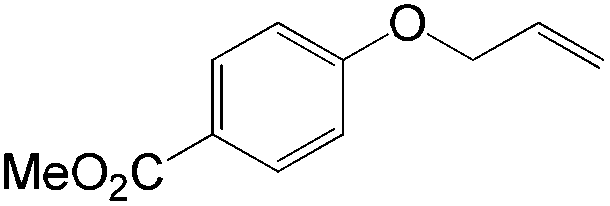
|
84 |
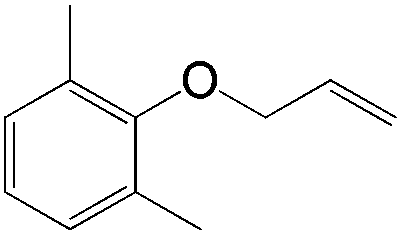
|
68 |

|
48 |
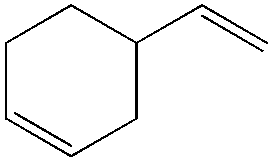
|
44 |

The proposed mechanism for this acetalization is shown in Scheme 7. Firstly η4-coordination occurred between styrene and the PdII species. The nucleophilic attack of the pinacol in an anti-Markovnikov manner then afforded the benzyl intermediate. Attack on the internal carbon would be unfavourable due to the steric repulsion between the phenyl group and pinacol. The final acetal product was formed via β-hydride elimination followed by cyclization. The formation of a benzyl intermediate in this process is considered to be the main factor leading to anti-Markovnikov selectivity.
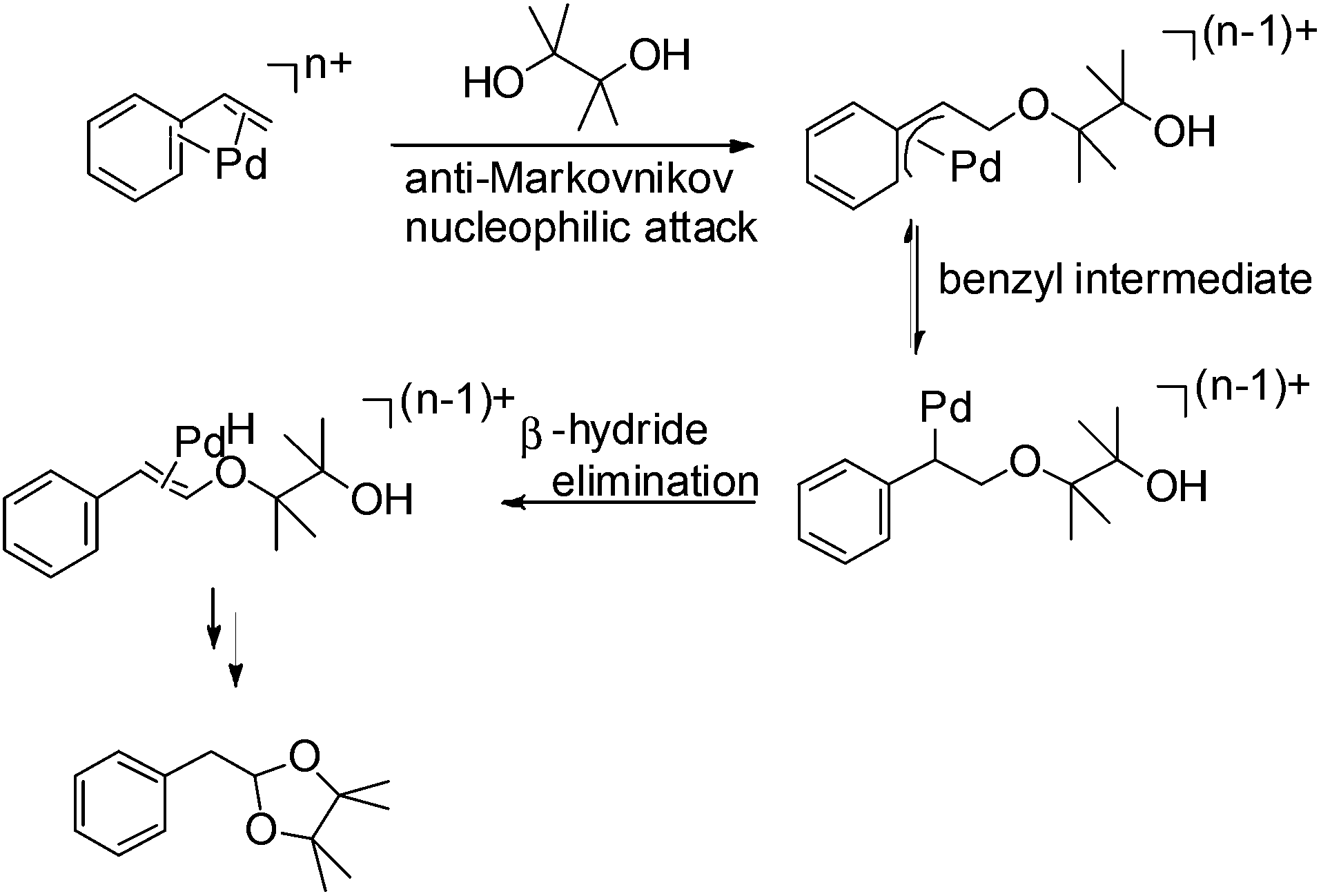 | ||
| Scheme 7 The proposed reaction mechanism for acetalization. | ||
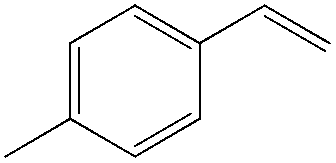
Although regioselectivity control by means of a bulky solvent is effective in Kataoka's system, the acetalization method is still an indirect way of obtaining the aldehyde. Recently, a facile PdII-catalyzed process to convert olefins directly into aldehydes was achieved by Grubbs et al.24 By combining PdCl2(MeCN)2, 1,4-benzoquinone (BQ) and t-BuOH in the presence of stoichiometric amounts of water, both high aldehyde selectivity and yields could be obtained with vinyl arenes (Table 7).
|
|
||
|---|---|---|
| Substrate | Aldehyde yield | Selectivity |
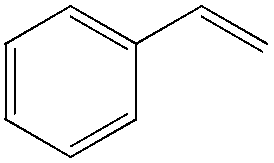
|
83 | 98 |
|
81 | 97 |
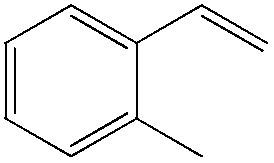
|
90 | 96 |
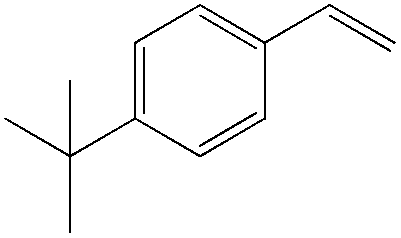
|
42 | 98 |
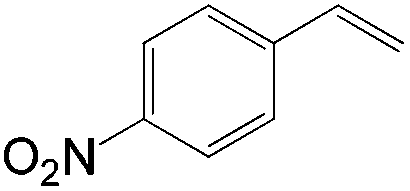
|
96 | 99 |
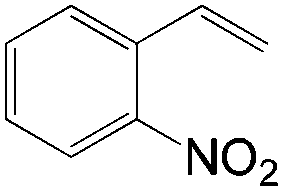
|
96 | >99 |
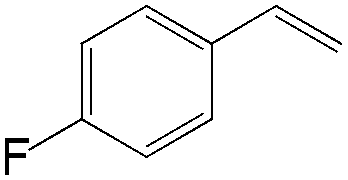
|
74 | 99 |
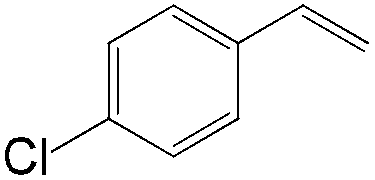
|
92 | 99 |
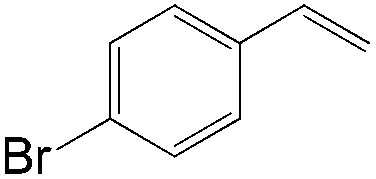
|
90 | 99 |
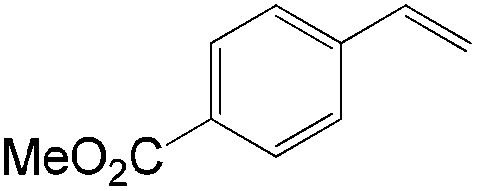
|
59 | 96 |
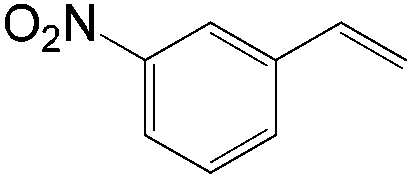
|
72 | 99 |
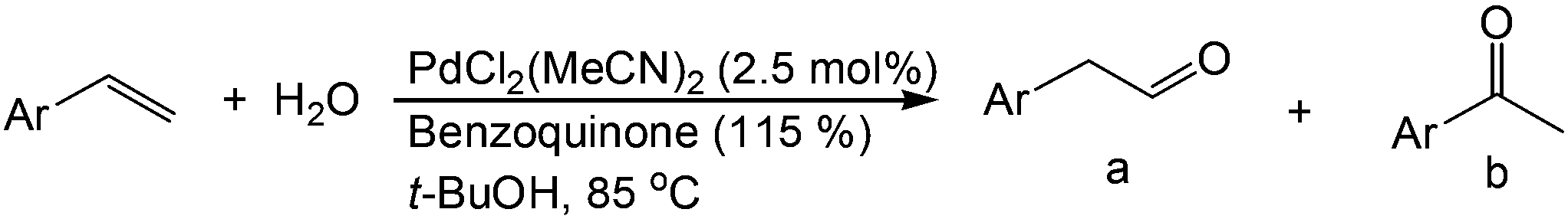
Similar to the solvent effect of Kataoka’s acetalization,23 the use of t-BuOH critically influences the regioselectivity in Grubbs's system. Due to the bulkiness of t-BuOH, the linear vinyl ether is preferred, which constitutes the key factor for high anti-Markovnikov selectivity. During the process, acid is generated in the presence of water, upon which the vinyl ether is converted to aldehyde via acid-catalyzed hydrolysis (Fig. 2).
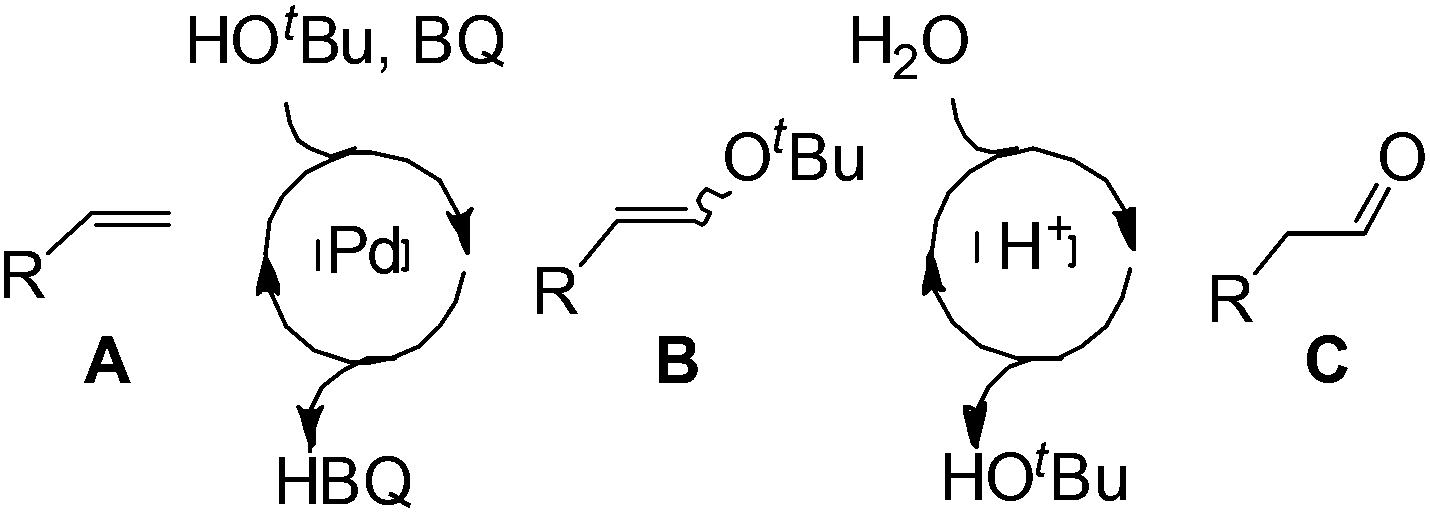 | ||
| Fig. 2 Proposed strategy for olefin oxidation. | ||
This highly aldehyde-selective system shows a major improvement to current methods for Wacker oxidation. The by-product 1,4-hydroquinone (HBQ) in this reaction could be easily converted to 1,4-benzoquinone (BQ) via aerobic oxidation.25,26 Moreover, its simplicity leads to more practical applications.
More recently, Grubbs et al. also reported the aldehyde-selective Wacker-type oxidation of unbiased olefins using a combination of catalytic tert-butyl nitrite with PdCl2(PhCN)2 and CuCl2 catalysts in the presence of oxygen.27 High anti-Markovnikov selectivity without reliance on substrate control was achieved (Table 8). In this system, AgNO2 plays a key role in the significant improvement of selectivity and yield. MeNO2 was found to be an important co-solvent. Good functional-group tolerance was observed with the optimized conditions.
|
|
||
|---|---|---|
| Substrate | Yield of aldehyde | Sel. |
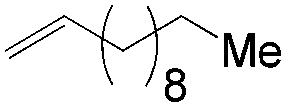
|
63 | 79 |
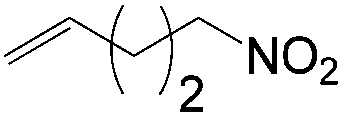
|
61 | 79 |
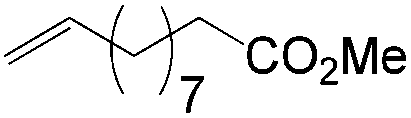
|
70 | 89 |
| 59 | 79 | |
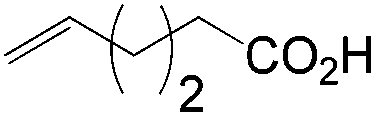
|
51 | 67 |
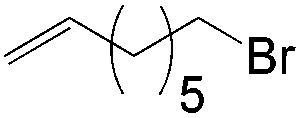
|
65 | 82 |
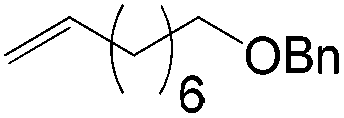
|
59 | 81 |
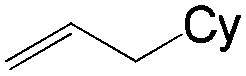
|
60 | 80 |
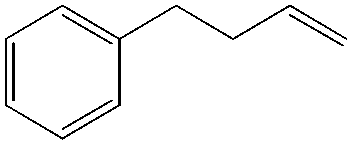
|
69 | 89 |
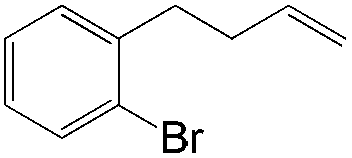
|
64 | 90 |
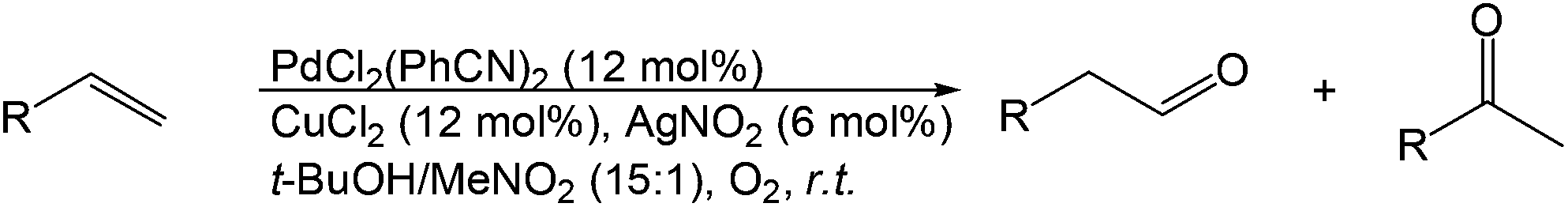
An 18O-labeling experiment was carried out and it was observed that the oxygen atom was derived from the nitrite salt where the 18O was incorporated into the aldehyde in an 81% yield. Thus the mechanistic feature of this reaction was proposed to be a metal-mediated radical-type addition to terminal olefins by NO2 species (Scheme 8).
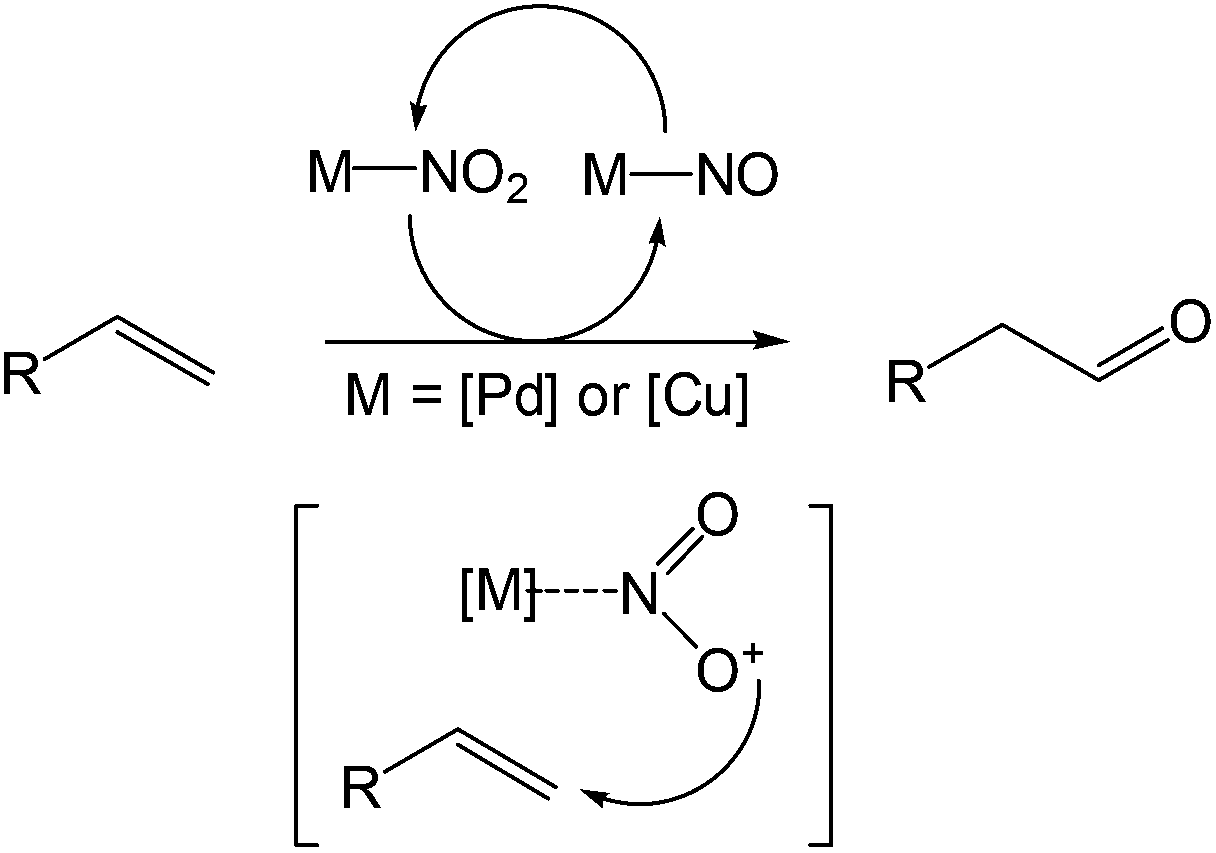 | ||
| Scheme 8 18O-labeling experiment and radical model. | ||
The success of this system provides a crucial lead for further development of catalytic anti-Markovnikov oxidation systems using unbiased aliphatic substrates. Later on, the authors further extended the study to the ketone synthesis from the more challenging internal olefins by Wacker-type oxidation.28
In fact, substrate control is an inherent challenge for Wacker-type processes. Sigman and Werner made efforts to develop a useful catalyst-controlled oxidation method which is not affected by functional groups on olefins or steric effects of solvents.29 The study revealed a TBHP (tert-butylhydroperoxide)-mediated oxidation arising from a Pd-quinox (quinoline oxazoline) catalyst formed in situ. It is believed that the electron-poor quinoline module of the ligand facilitates olefin coordination (Scheme 9). Markovnikov selectivity is achieved here.
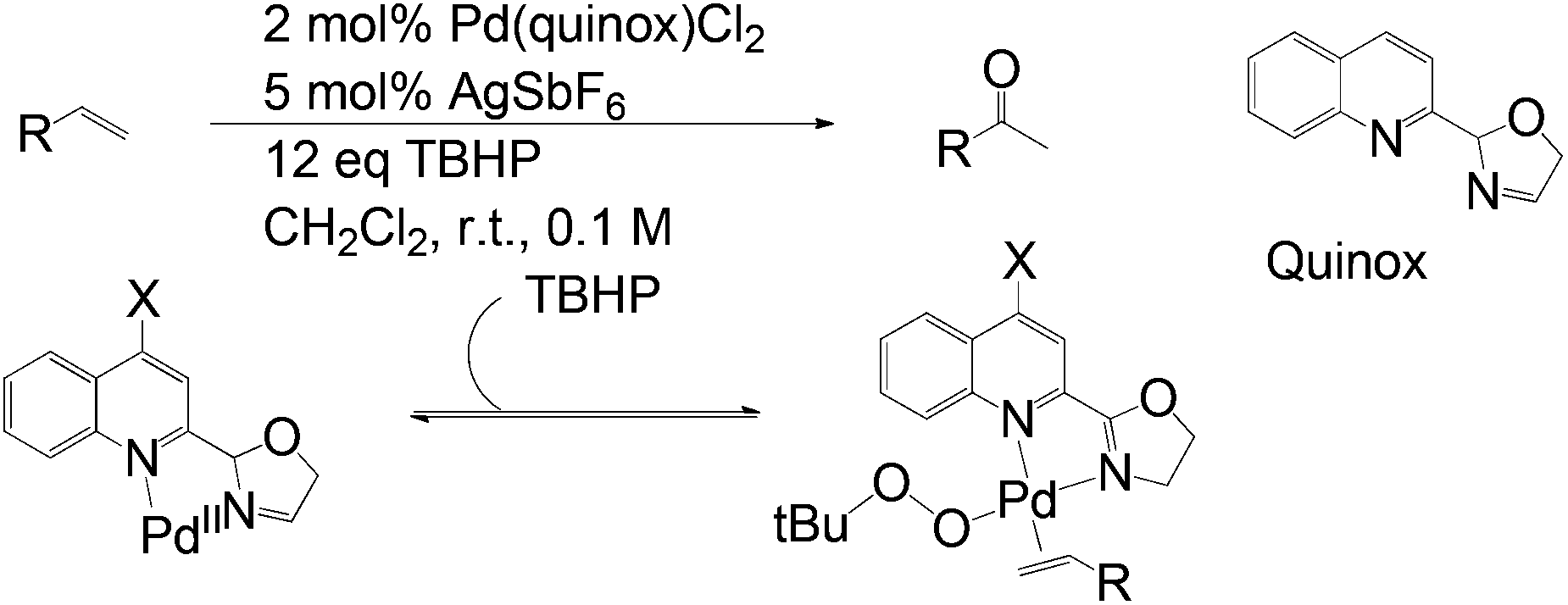 | ||
| Scheme 9 TBHP-mediated Wacker oxidation using the Pd-quinox catalyst. | ||
The use of an electronically asymmetric ligand in this Wacker oxidation has been shown to critically affect the environment of the metal center. Thus ligand design is crucial to achieve catalyst control for Wacker-type process and provides insight to realizing more challenging anti-Markovnikov functionalization.
Apart from the widely known Wacker-type methods for synthesizing aldehydes from olefins, Che et al. reported a ruthenium-porphyrin-catalyzed aldehyde synthesis in 2008 (Table 9)30. It was the first example of an aerobic epoxidation–isomerization (E–I) reaction (Scheme 10), forming aldehydes in up to 93% yields, using the ruthenium porphyrin catalyst [RuVI(tmp)Cl2] (Fig. 3).
|
|
||
|---|---|---|
| Substrate | Time (h) | Yield (%) |
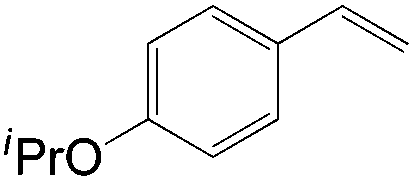
|
4.5 | 92 |
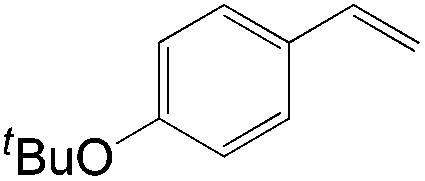
|
7 | 87 |
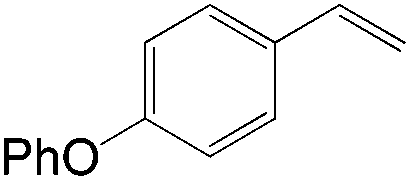
|
5 | 87 |
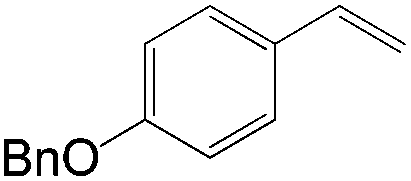
|
4 | 89 |
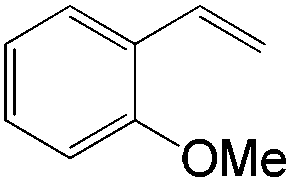
|
7 | 93 |
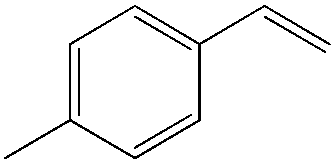
|
5 | 84 |
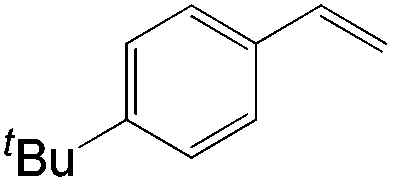
|
5 | 81 |
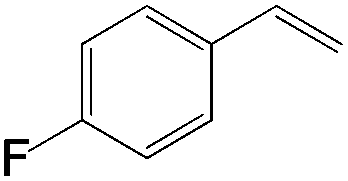
|
7 | 71 |
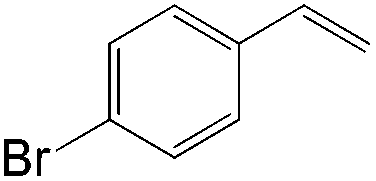
|
7 | 69 |
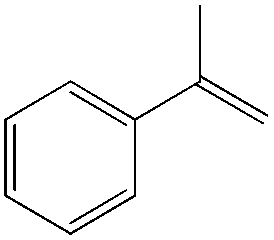
|
7 | 81 |
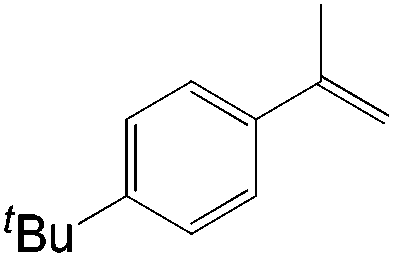
|
7 | 71 |
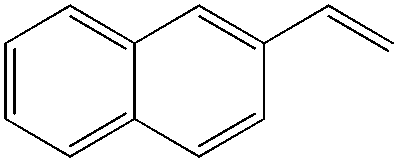
|
5 | 73 |
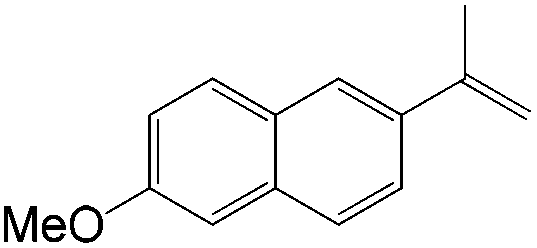
|
7 | 74 |
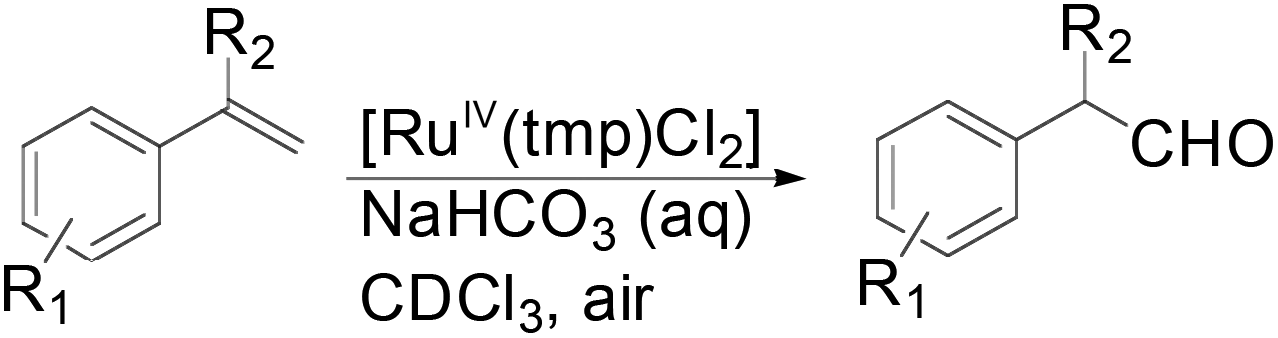
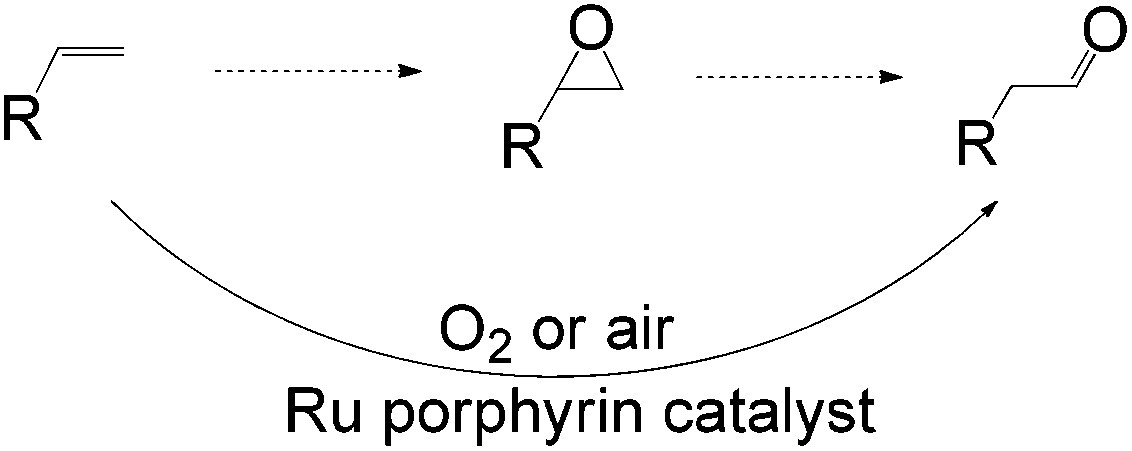 | ||
| Scheme 10 Ruthenium porphyrin-catalyzed oxidation of 1-alkenes to aldehyde (E–I reaction). | ||
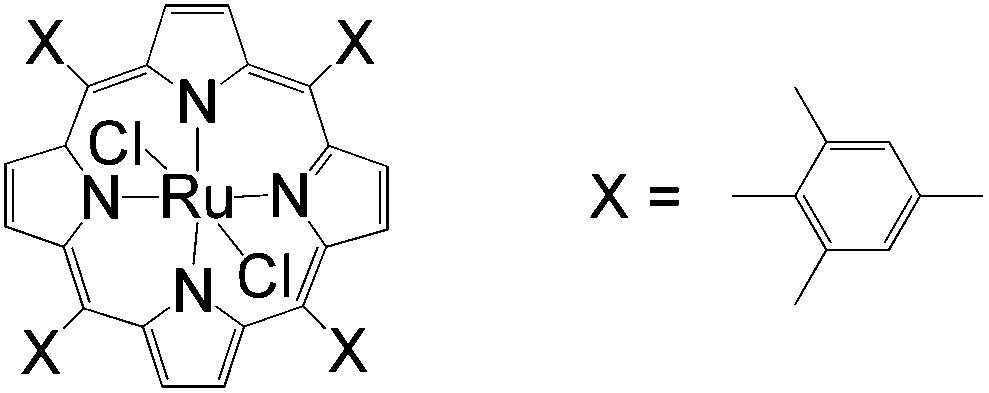 | ||
| Fig. 3 Ruthenium porphyrin catalyst [RuVI(tmp)Cl2]. | ||
Lahiri et al. reported an iron catalyzed aldehyde-selective oxidation of terminal olefins using Fe(BF4)2·6H2O with the commercially available ligand dipic (pyridine-2,6-dicarboxylic acid) and PhIO as an oxidant in CHCl3 at room temperature (Scheme 11).31 This catalytic system functions well with the inexpensive and simple iron catalyst under mild conditions.
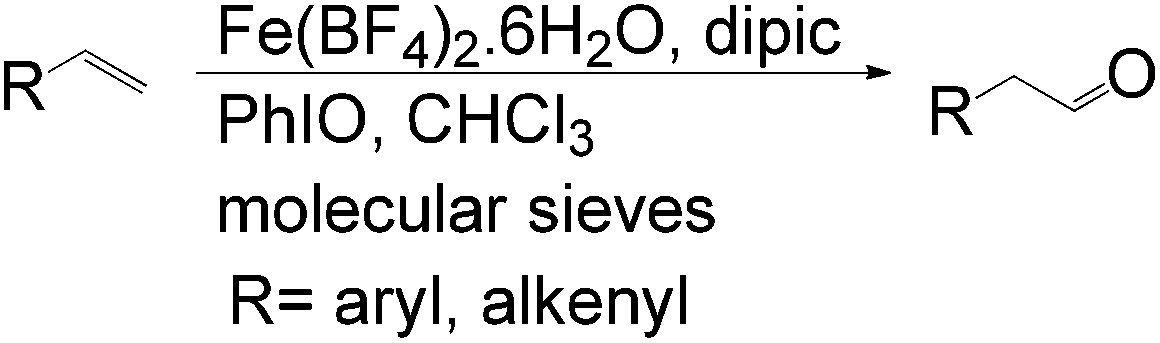 | ||
| Scheme 11 Fe-catalyzed oxidation of terminal olefins. | ||
In summary, Che's ruthenium- and Lahiri's iron-catalyzed systems provide unique and efficient methods for aldehyde synthesis from terminal olefins, which significantly complements the conventional Wacker-type oxidation.
3. Tandem anti-Markovnikov hydration from terminal olefins
Anti-Markovnikov hydration of terminal olefins remains a long-standing challenge to organic chemists.11 Studies on direct and efficient catalytic processes have attracted much attention in the last few decades. In 1986, Trogler and Jensen reported the first example of Pt-catalyzed regioselective hydration of non-activated terminal alkenes to primary alcohols.32 The direct transformation of 1-hexene to 1-hexanol is catalyzed by a species formed from the reaction between trans-PtHCl(PMe3)2 and NaOH in a water and 1-hexene mixture, containing a phase-transfer catalyst, NEt3(CH2Ph)Cl. Hydration of 1-dodecene to 1-dodecanol by the same procedure occurs at 100 °C.The proposed mechanism involves the formation of a hydrido-Pt(II)-hydroxo complex, followed by protonation to form an aqua complex and subsequent coordination of the olefin to platinum. The hydroxide anion then attacks the coordinated olefin to form a Pt-alkyl complex which then undergoes trans–cis isomerization resulting in the alkyl and hydride moiety being cis to one another. The complex then undergoes C–H reductive elimination to yield the primary alcohol (Scheme 12).
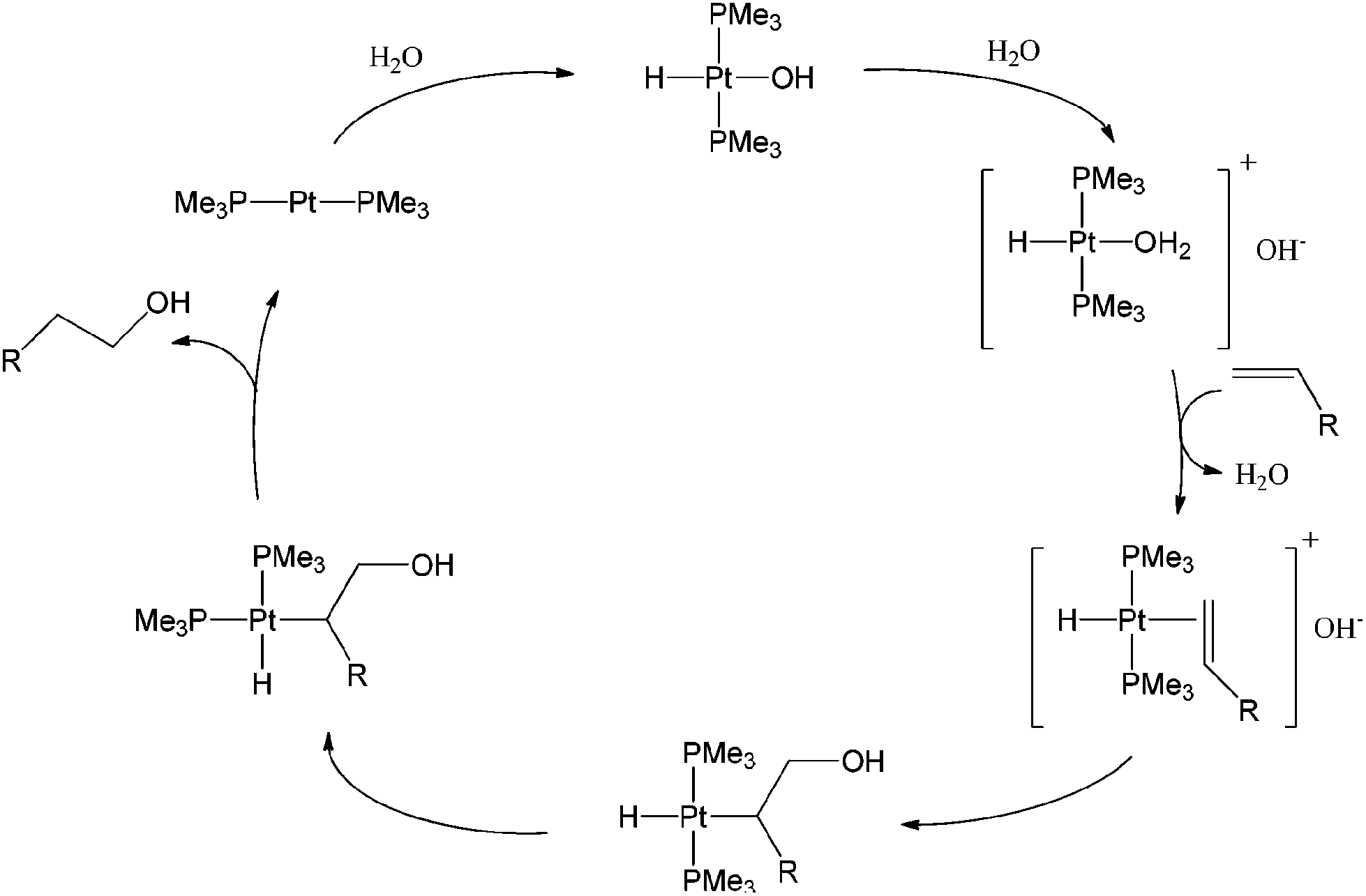 | ||
| Scheme 12 Proposed catalytic cycle for the hydration of terminal olefins to primary alcohols. | ||
Despite the elegance of Trogler's method, the system was subsequently claimed to be irreproducible by Ramprasad et al.33 It was also speculated by Grushin et al.34 that the observed reactivity in Trogler's system may be due to some impurity present in the reaction mixture.
In common with Grushin, Parkins et al.35 carried out tests on the platinum catalysts used in Trogler's process. In consideration of the unusual method adopted by Trogler for the preparation of the catalyst precursor trans-[(Me3P)2PtHCl], Parkins et al. tested the catalytic activity of dinuclear platinum complexes [(dppe)Pt(μ-H)2-PtH(dppe)]BF4,36 [(Me3P)2Pt(μ-H)2PtH(PMe3)2]Cl and [(Et3P)2Pt(μ-H)2PtH(PEt3)2]PF6,37 with sodium hydroxide, water and 1-octene in either THF or methanol (Fig. 4). Although these conditions corresponded most closely to that reported by Trogler, no 1-octanol was detected.
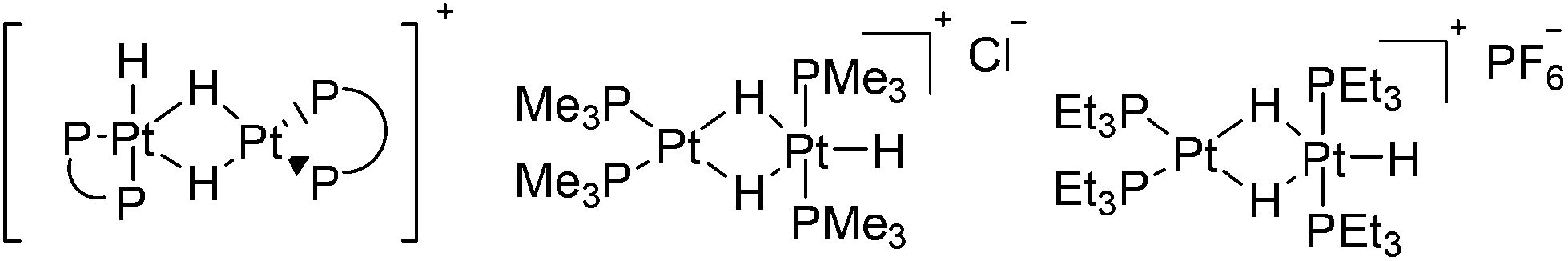 | ||
| Fig. 4 Dinuclear platinum complexes employed by Parkins and Richard for octene hydration. | ||
Furthermore, during the investigation of hydration of 1-octene using [Ph(PPh3)Pt(μ-H)(μ-PPh2)Pt(PPh3)2]BF438 (Fig. 5), an impurity identified as oct-1-ene-3-hydroperoxide, was observed in the acidic methanolic solution of 1-octene. The impurity underwent a Hock rearrangement and 1-methoxy-1-vinyloxyhexane was formed as shown in Scheme 13.39
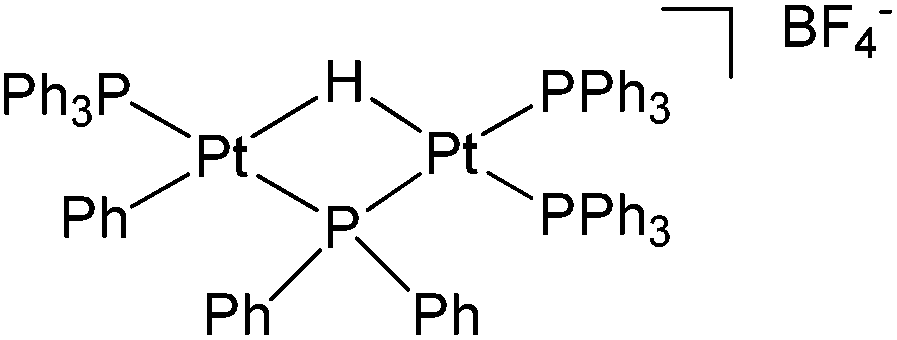 | ||
| Fig. 5 Platinum complex [Ph(PPh3)Pt(μ-H)(PPh2)Pt(PPh3)2]BF4. | ||
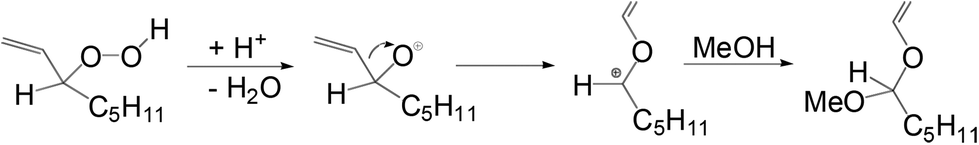 | ||
| Scheme 13 Reaction mechanism showing the Hock rearrangement of oct-1-ene-3-hydroperoxide in acidic methanol. | ||
It was suspected that Trogler's catalytic system32 might have contained some trinuclear species. The trinuclear platinum cluster [(dppe)3Pt3H3]+40 (Fig. 6) was also tested by Parkins et al. for hydration activity.
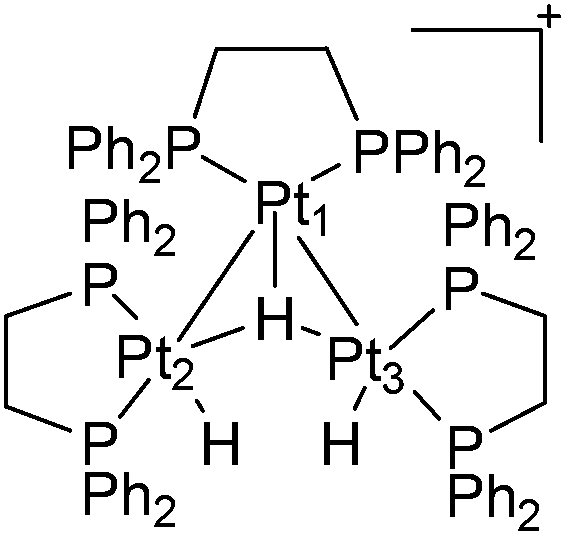 | ||
| Fig. 6 Parkin's trinuclear platinum cluster. | ||
1-Octanol was still not detected using this trinuclear platinum cluster as the hydration catalyst. However, the anti-Markovnikov product 1-methoxyoctane was observed in the product mixture. This suggests that a reaction between the hydroperoxide impurities in 1-octene and the trinuclear platinum cluster occurred, leading to the formation of 1-methoxyoctane as the product instead. The proposed mechanism for 1-methoxyoctane production from 1-octene is shown in Scheme 14.
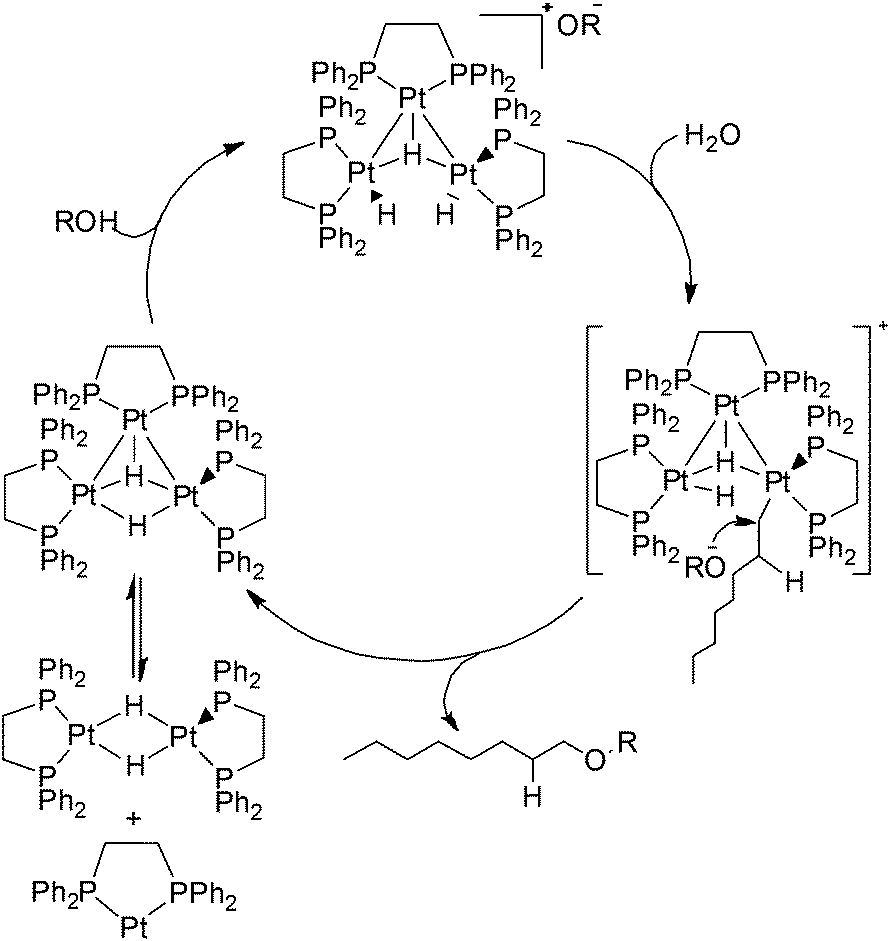 | ||
| Scheme 14 Proposed mechanism for the formation of 1-methoxy-octane using trinuclear platinum catalyst. | ||
Due to the presence of varying amounts of hydroperoxide impurities in treated or untreated 1-octene, Parkins et al.’s results were somewhat erratic. However, their studies shed some light on the problems of the platinum-catalyzed anti-Markovnikov hydration of terminal olefins initially reported by Trogler.32
In 2003, Toste et al. disclosed a base-catalyzed method for hydration and hydroalkoxylation of activated olefins using trialkylphosphine.41 Catalytic amounts of trimethylphosphine proved to be very active in catalyzing the hydration or hydroalkoxylation reaction, even in the absence of metals in their system. A variety of α,β-unsaturated substrates were tolerated under the reported conditions and only the highly conjugated 4-phenyl-3-buten-2-one was unreactive (Table 10).
|
|
||||
|---|---|---|---|---|
| EWG | R1 | ROH | Time (h) | Yield % |
| COEt | Me | H2O | 20 | 77 |
| MeOH | 24 | 85 | ||
| 16 | 63 | |||
| COMe | H | MeOH | 1 | 56 |
| Me2CHOH | 1 | 83 | ||
| PhOH | 16 | 59 | ||
| Ph | MeOH | 72 | 0 | |
| CO2Me | Me | MeOH | 36 | 71 |
| CN | H | MeOH | 4 | 79 |
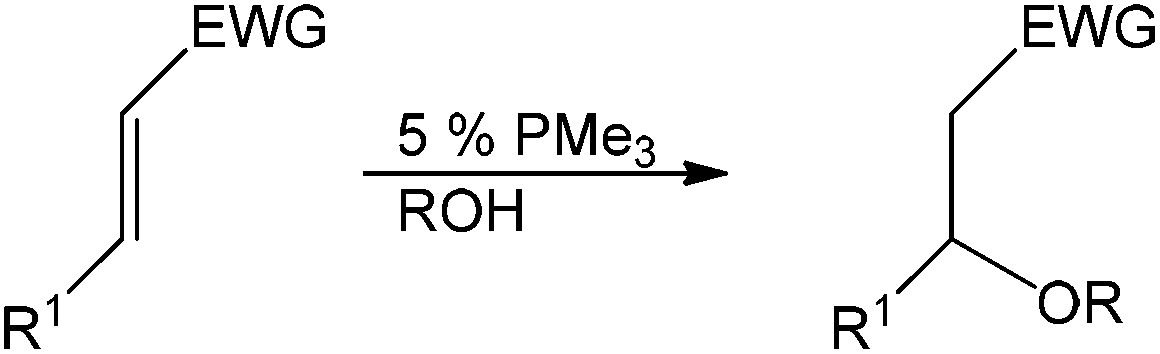
The proposed catalytic cycle is shown in Scheme 15. The phosphonium enolate is formed by nucleophilic attack from the phosphine to the unsaturated carbonyl compound, which spontaneously generates the alcohol by deprotonation. The alkoxide of the phosphonium ion pair then undergoes subsequent addition to give an enolate ion pair.42 Protonation of the enolate leads to the formation of the β-alkoxy carbonyl and regeneration of the alkoxide with an ion pair to continue the cycle. This nucleophilic phosphine catalytic system can be used in the absence of additional transition metals, strong acids or bases but is limited to the α β-unsaturated olefin class of substrates.
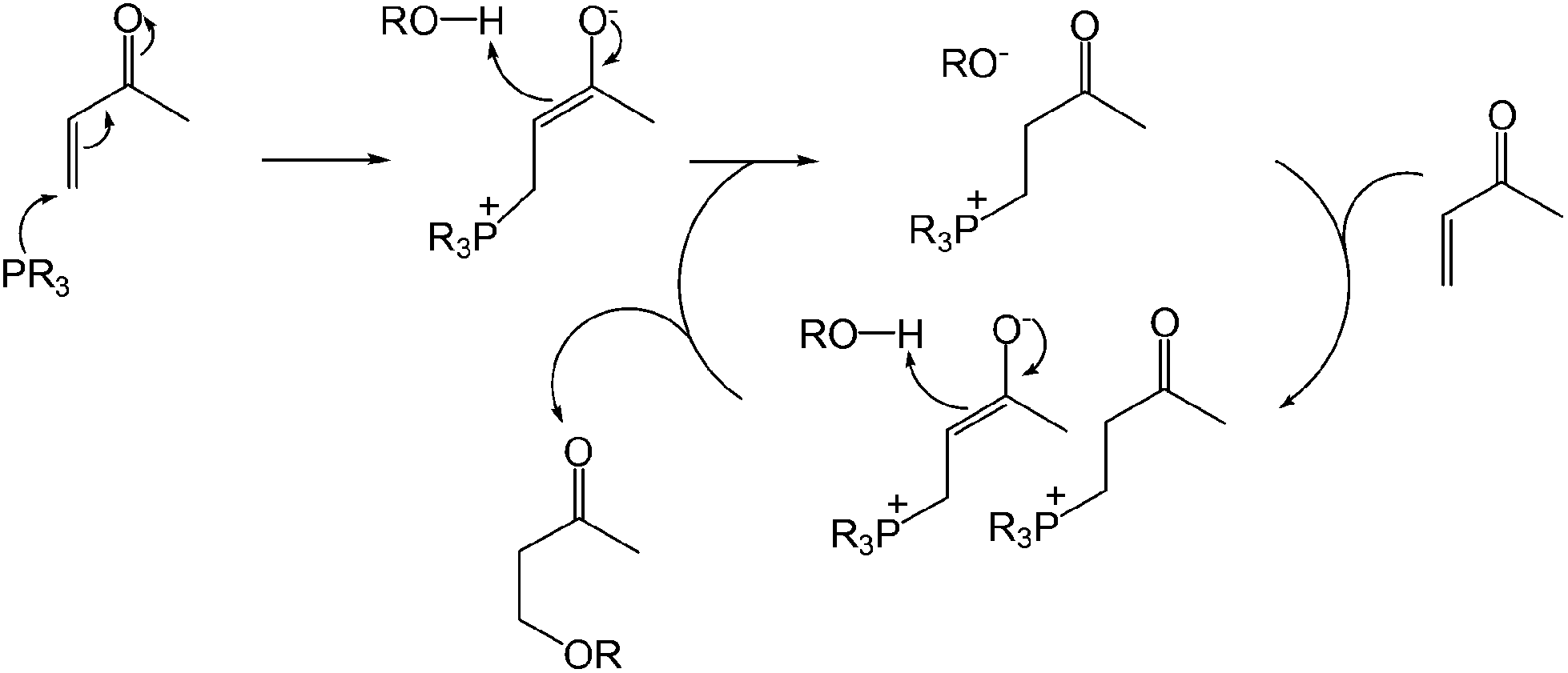 | ||
| Scheme 15 Proposed catalytic cycle. | ||
Recently, a tandem catalyst system for the synthesis of primary alcohols from non-activated terminal olefins was reported by Grubbs et al.25 This strategy is based on PdII-catalyzed oxidation, acid-catalyzed hydrolysis and Ru-catalyzed reduction cycles (Scheme 16). This triple-relay system relies on three criteria, firstly, the oxidation step must be aldehyde-selective; secondly, the oxidation cycle must be compatible with the reduction cycle and thirdly, the migration of the hydride from Pd to Ru must be facile.
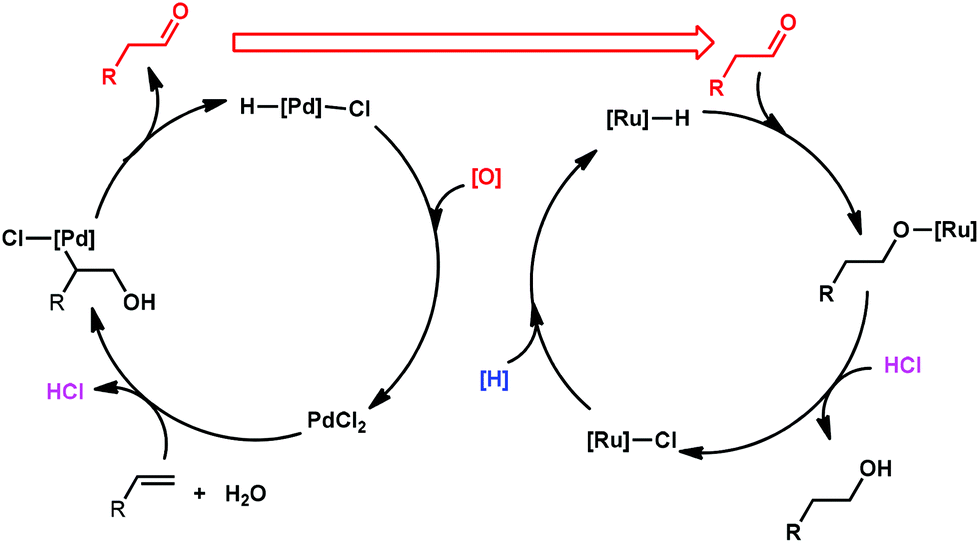 | ||
| Scheme 16 Proposed cooperative catalytic system for alcohol synthesis from olefins and water. | ||
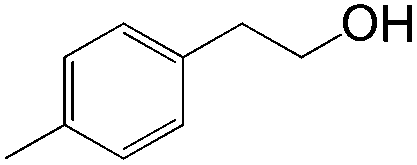
In this catalytic system, t-BuOH was chosen as the solvent for PdII-catalyzed oxidation to result in aldehyde selectivity.43 A transfer hydrogenation catalyst, Shvo's complex,44 was employed in the reduction step and i-PrOH was employed as the sacrificial hydrogen source.45 Primary alcohols were obtained under the optimized conditions with high selectivity. A wide range of functional groups was tolerated by the system. Despite the difficulty in functionalizing the aliphatic olefin, it is encouraging to see that even the aliphatic olefin is able to provide hydration products, albeit at reverse selectivity (Table 11).
|
|
|||
|---|---|---|---|
| Substrate | Product | Yield % | Selectivity |
|
|
84 | ≥20:1 |
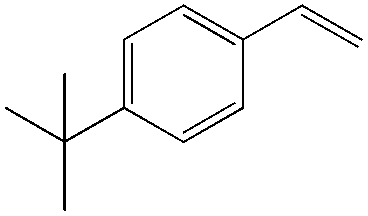
|
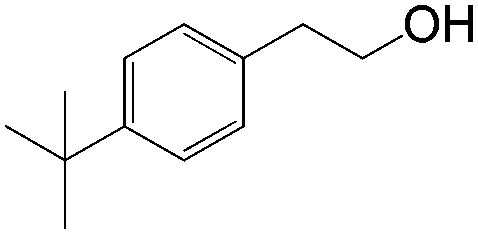
|
42 | ≥20:1 |
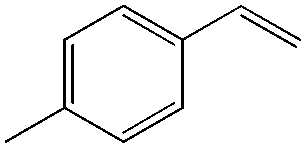
|
|
61 | ≥20:1 |
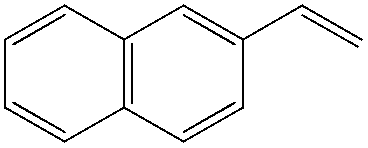
|
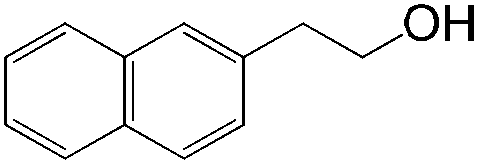
|
60 | ≥20:1 |
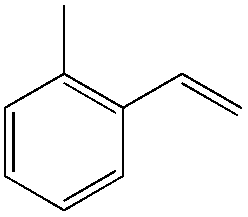
|
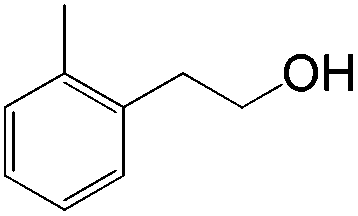
|
72 | ≥20:1 |
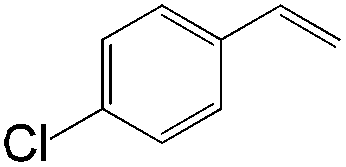
|
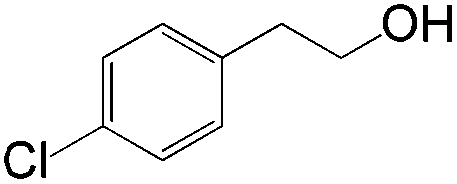
|
75 | ≥20:1 |
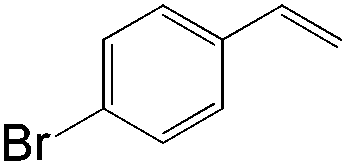
|
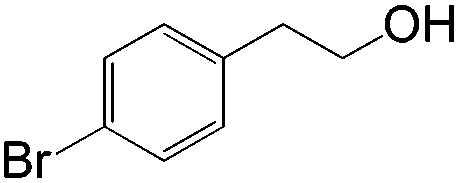
|
72 | ≥20:1 |
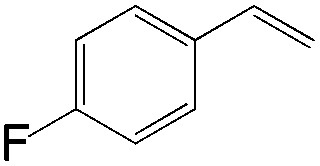
|
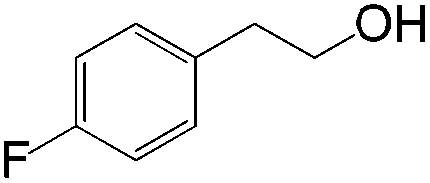
|
63 | ≥20:1 |
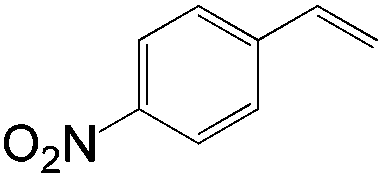
|
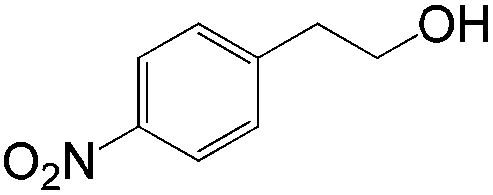
|
83 | ≥20:1 |
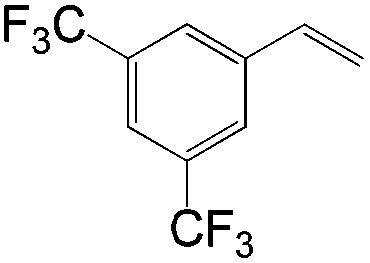
|
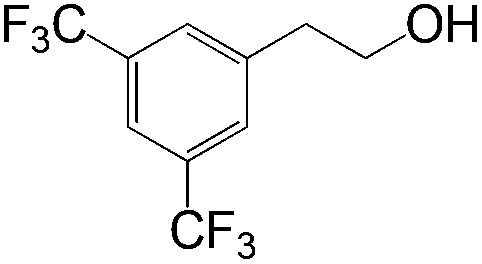
|
74 | ≥20:1 |
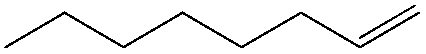
|
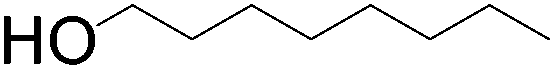
|
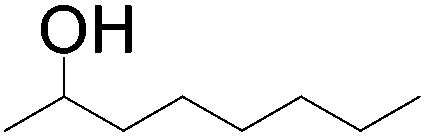
56 (1°OH:2°OH = 1:1.4) |
|
|
54 (1°OH:2°OH = 1:1.9) |
||
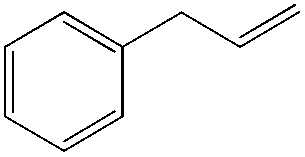
|
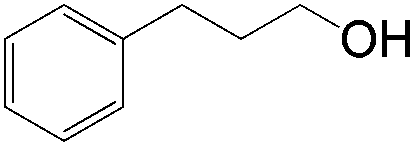
|
12 (1°OH:2°OH = 1:2.1) |
|
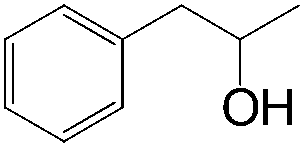
|
|||
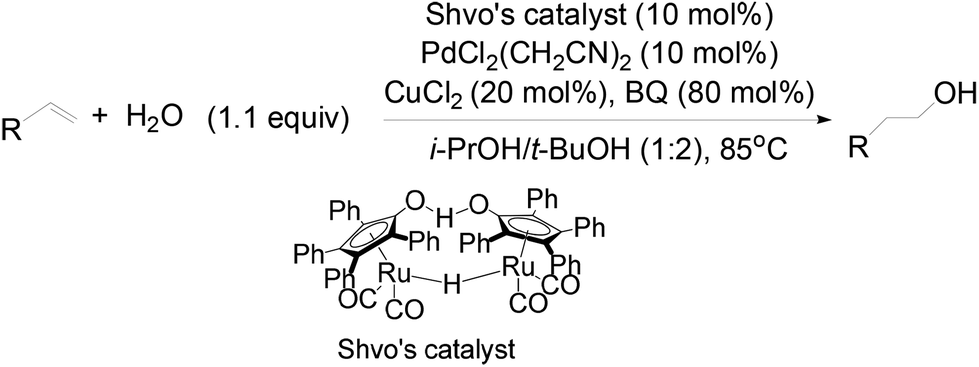
The role of each reactant was studied by a series of control experiments with styrene as the substrate (Table 12).25 The absence of Pd catalyst shut down the oxidation process totally and only afforded the over-reduction product, ethylbenzene, in a 26% yield. Without Shvo's catalyst, it is observed that no alcohol products were formed and the aldehyde was the major product. CuCl2 originally serving as a co-oxidant, appeared to be critical in slowing down the over-reduction process. In the absence of CuCl2, substantial quantities of ethylbenzene were formed. The exclusive formation of aldehyde in the absence of i-PrOH illustrated its role as the reductant. The critical role of 1,4-Benzoquinone (BQ)46 in the reactivity and selectivity of this catalytic system was shown in the lower yield and regioselectivity of the primary alcohol obtained, when the reaction was carried out without BQ. No oxygenated product was formed under anhydrous conditions by removal of water from the reaction using 4 Å molecular sieves.
| Change | Conversion (%) | Yield (%) | Byproducts (%) | |||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| None | 89 | 77 | 1.3 | 0.7 | 1.5 | 1.4 |
| No PdCl2(CH2CN)2 | 34 | 0 | 26 | 0 | 0 | 0 |
| No Shvo's catalyst | 80 | 0 | 0.2 | 0 | 42 | 0 |
| No CuCl2 | >99 | 48 | 32 | 5.9 | 0.5 | 1.4 |
| No BQ | 58 | 0 | 0.9 | 0 | 0 | 0 |
| No i-PrOH | 88 | 0 | 0.5 | 0 | 57 | 0 |
| No t-BuOH | 48 | 18 | 2.0 | Trace | Trace | Trace |
| No t-BuOH, but 28 equiv. H2O | 75 | 64 | 2.0 | 4.9 | 0.96 | 9.9 |
| Replace H2O with 4 Å molecular sieves | >99 | 0 | 57 | 0 | 0 | 0 |
The mechanism of this triple-relay catalyst system is proposed as three tandem reaction cycles as shown in Scheme 17. In the presence of t-BuOH, the olefin undergoes PdII-catalyzed oxidation to form a linear t-butyl vinyl ether, which is preferred due to the steric bulk of the t-butyl group. It is noteworthy that this t-BuOH nucleophilic attack plays a key role in the high anti-Markovnikov selectivity.6,43b Subsequently, the generation of acids (HCl and hydroquinone) during the oxidation process converts the vinyl ether to an aldehyde in the presence of water through acid-catalyzed hydrolysis. Finally, the primary alcohol is formed via Ru-catalyzed transfer-hydrogenation of the aldehyde.
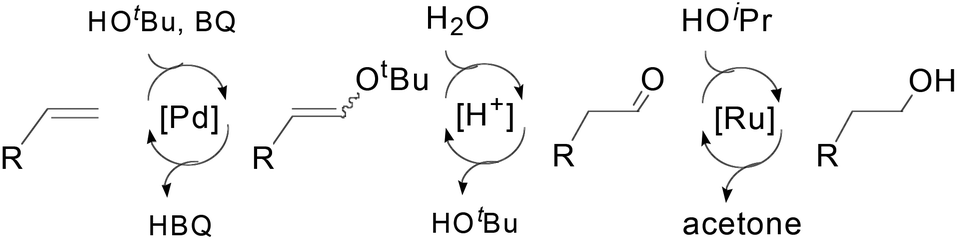 | ||
| Scheme 17 Anti-Markovnikov olefin hydration by a triple-relay catalysis system. | ||
With the high catalyst loadings and use of stoichiometric BQ, this approach is far from perfect. Nevertheless, it is still highly encouraging that promising results have been obtained with non-activated olefins such as styrenes and 1-octene using this efficient tandem catalytic system. This process is an attractive strategy to realize complex structural transformation that leads to potential industrial applications, as highlighted by Hintermann more recently.47
Along with this Wacker oxidation/hydrogenation approach, tandem hydroformylation/hydrogenation simultaneously attracted investigation albeit the homologation effect in the process. Nozaki et al. reported a one-pot regioselective olefin hydroformylation/hydrogenation using Rh/Ru (Shvo's catalyst) dual catalyst system.48 The success of this system was attributed to the orthogonal relationship between each catalyst and that the Shvo's catalyst is relatively inert in the hydroformylation step. Furthermore, it was found that the regioselective hydroformylation/hydrogenation of 1-decene could be mediated by just Shvo's catalyst or cyclopentadienone-ligated Ru tricarbonyl complexes alone (Scheme 18).
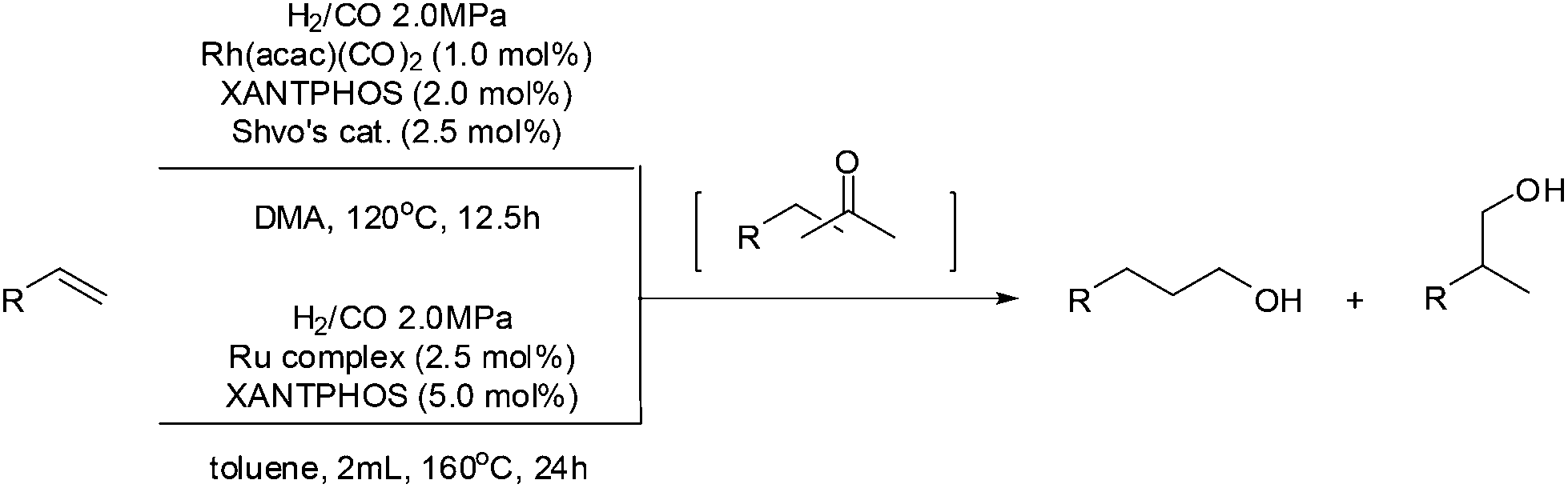 | ||
| Scheme 18 Tandem hydroformylation/hydrogenation. | ||
This system reported by Nozaki et al. offers an alternative pathway to the tandem synthesis of linear alcohols. It was reported that Shvo's catalyst was more active than the conventional Ru hydrogenation catalyst due to its robustness under CO pressure, which is insightful for selective hydrogenation catalyst design for more attractive industrial applications.
Other tandem catalytic sequences of branched olefins were also carried out for alcohol formation. One sustainable process that can be tailored to the production of plasticizer alcohols was reported by Harvey et al.49 The approach begins with oxidation of 2-ethyl-1-hexene to a diol followed by a dehydration and subsequent hydrogenation to yield an anti-Markovnikov product (Scheme 19).
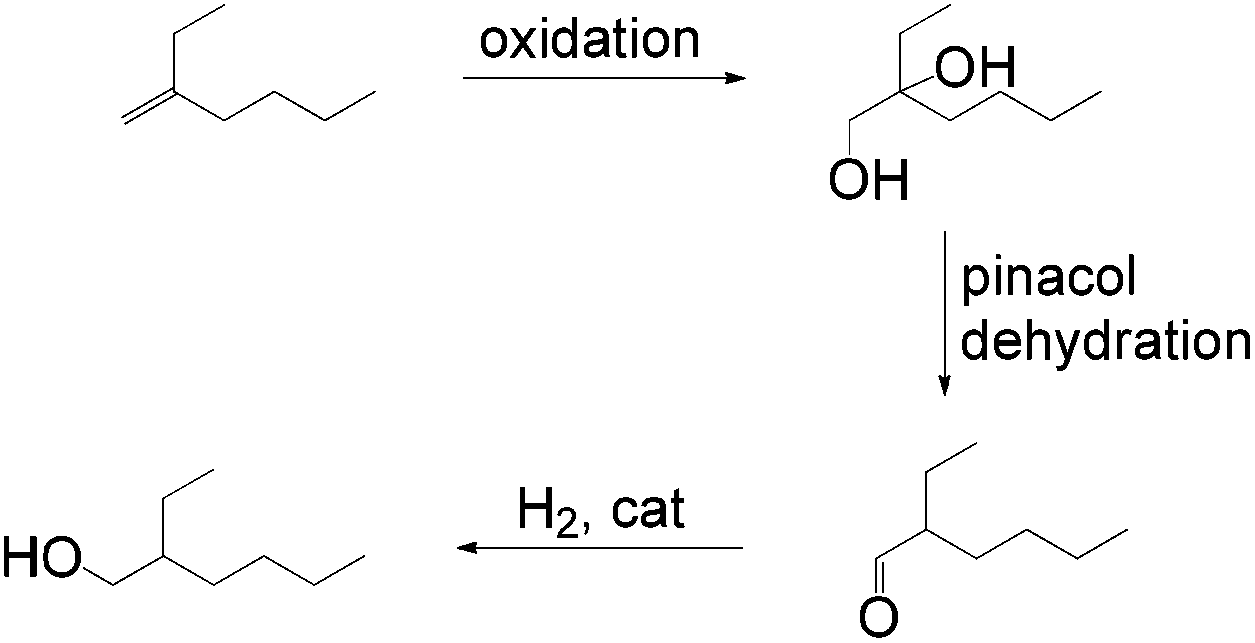 | ||
| Scheme 19 Oxidation/dehydration/hydrogenation route to an anti-Markovnikov product. | ||
This process generally complements the oxidation/reduction tandem strategy for alcohol formation especially from branched olefins. However, the system is rather complicated and harsh conditions involving the usage of peracetic acid for oxidation are required.
The tandem catalytic strategy was further extended to the regioselective hydration of alkynes that retain many of the characteristics of alkenes. Herzon and Li reported a Ru-catalyzed anti-Markovnikov hydration system from alkynes (Scheme 20).50 This strategy provides yet another route to linear alcohol formation from unsaturated aliphatic compounds.
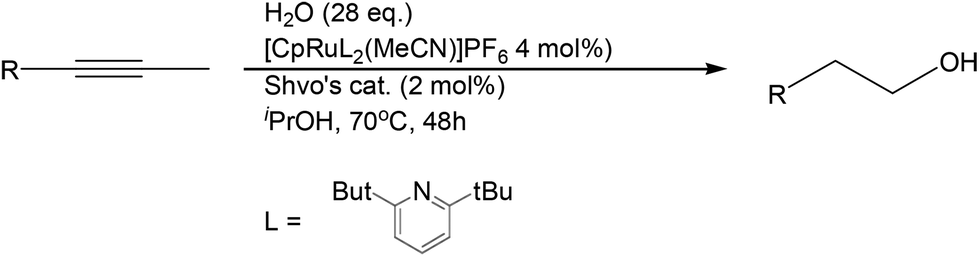 | ||
| Scheme 20 Anti-Markovnikov reductive hydration of alkynes. | ||
4. Perspectives
As described above, the PdII-catalyzed Wacker-type reaction has attracted much interest in its application toward anti-Markovnikov oxidation of olefins. Apart from the high aldehyde selectivity achieved with specific olefins bearing directing groups,6,8 much effort has been focused on the modification of each factor, such as terminal oxidant,10,17 solvent6,23,24 and additive in the oxidation process to facilitate its high regioselectivity with non-activated olefins. Other methods such as ruthenium-porphyrin-catalyzed30 and iron-catalyzed aldehyde formation31 provide motivation for the development of new catalytic pathways as well.Anti-Markovnikov hydration is a long-standing problem facing organic chemists.11 Solving this challenge would be highly rewarding for both the scientific and industrial communities. Although the first example of Pt-catalyzed hydration of aliphatic olefins32 had reproducibility issues, it nonetheless shed some light on the topic of anti-Markovnikov olefin hydration. From then on, efforts were directed towards this main goal. Grubbs's report25 on tandem catalysts for anti-Markovnikov olefin hydration has certainly contributed significantly to this area. The multi-relay catalytic approach via regioselectivity control in PdII-catalyzed oxidation coupled with a Ru-catalyzed transfer hydrogenation process provides an indirect solution to this challenging problem. Subsequently, studies on tandem catalytic strategies48–50 for linear alcohol formation from olefins and even alkynes are increasingly being reported.
The anti-Markovnikov functionalization of terminal olefins is still a tough challenge facing organic and organometallic chemists. This is especially true for non-activated aliphatic olefins. Solving this long-standing challenge of catalysis is the current focus of our research group. Much progress has been achieved and work on this area is ongoing.
Acknowledgements
This contribution was financially supported by the National University of Singapore and A*STAR through generous research grants (R143-000-523-133 and R143-000-535-305). J. Guo is grateful to A*STAR SERC for a postdoctoral grant (PSF R143-000-535-305).Notes and references
- D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232 CrossRef CAS PubMed.
- M. Tokunaga and Y. Wakatsuki, Angew. Chem., Int. Ed., 1998, 37, 2867 CrossRef CAS.
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS.
- M. B. Smith and J. March, March's advanced organic chemistry, John Wiley and Sons, New York, 2001 Search PubMed.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed.
- J. Muzart, Tetrahedron, 2007, 63, 7505 CrossRef CAS.
- T. T. Wenzel, J. Chem. Soc., Chem. Commun., 1993, 862 RSC.
- B. Weiner, A. Baeza, T. Jerphagnon and B. L. Feringa, J. Am. Chem. Soc., 2009, 131, 9473 CrossRef CAS PubMed.
- B. W. Michel, J. R. McCombs, A. Winkler and M. S. Sigman, Angew. Chem., Int. Ed., 2010, 49, 7312 CrossRef CAS PubMed.
- J. A. Wright, M. J. Gaunt and J. B. Spencer, Chem.–Eur. J., 2006, 12, 949 CrossRef CAS PubMed.
- (a) J. Haggin, Chem. Eng. News, 1993, 71, 23 CrossRef; (b) K. Noweck and W. Grafahrend, Fatty Alcohols, in Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Lurgi AG, Germany, 2006 Search PubMed.
- P. Eilbracht, L. Bärfacker, C. Buss, C. Hollman, B. E. Kitsoz-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329 CrossRef CAS PubMed.
- K. Takahashi, M. Yamashita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., Int. Ed., 2010, 49, 4488 CrossRef CAS PubMed.
- J. W. De Boer, J. Brinksma, W. R. Browne, P. L. Alsters, R. Hage and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 7990 CrossRef CAS PubMed.
- M. Tiecco, L. Testaferri, A. Temperini, R. Terlizzi, L. Bagnoli, F. Marini and C. Santi, Tetrahedron Lett., 2007, 48, 4343 CrossRef CAS.
- D. L. G. Clive, J. Wang and M. Yu, Tetrahedron Lett., 2005, 46, 2853 CrossRef CAS.
- J. Dong, M. F. Mastral, P. L. Alsters, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2013, 52, 5561 CrossRef CAS PubMed.
- A. N. Campbell, P. B. White, L. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15119 Search PubMed.
- (a) J. E. Bäckvall and A. Gogoll, Tetrahedron Lett., 1988, 29, 2243 CrossRef; (b) K. J. Szabó, Organometallics, 1998, 17, 1677 CrossRef.
- B. L. Lin, J. A. Labinger and J. E. Bercaw, Can. J. Chem., 2009, 87, 264–271 CrossRef CAS.
- H. Grennberg, A. Gogoll and J. E. Bäckvall, Organometallics, 1993, 12, 1790 CrossRef CAS.
- M.-Y. Chang, C.-K. Chan and S.-Y. Lin, Tetrahedron, 2013, 69, 1532 CrossRef CAS.
- M. Yamamoto, S. Nakaoka, Y. Ura and Y. Kataoka, Chem. Commun., 2012, 48, 1165 RSC.
- P. Teo, Z. K. Wickens, G. Dong and R. H. Grubbs, Org. Lett., 2012, 14, 3237 CrossRef CAS PubMed.
- G. Dong, P. Teo, Z. K. Wickens and R. H. Grubbs, Science, 2011, 333, 1609 CrossRef CAS PubMed.
- S. Kim, D. Kim and J. Parka, Adv. Synth. Catal., 2009, 351, 2573 CrossRef CAS.
- (a) Z. K. Wickens, B. Morandi and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 11257 CrossRef CAS PubMed; (b) Z. K. Wickens, K. Skakuj, B. Morandi and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 890 CrossRef CAS PubMed.
- B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 2944 CrossRef CAS PubMed.
- M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874 CrossRef CAS PubMed.
- G. Jiang, J. Chen, H.-Y. Thu, J.-S. Huang, N. Zhu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 6638 CrossRef CAS PubMed.
- A. D. Chowdhury, R. Ray and G. K. Lahiri, Chem. Commun., 2012, 48, 5497 RSC.
- (a) C. M. Jensen and W. C. Trogler, Science, 1986, 233, 1069 CAS; (b) C. M. Jensen and W. C. Trogler, J. Am. Chem. Soc., 1986, 108, 723 CrossRef CAS.
- (a) D. Ramprasad, H. J. Yue and J. A. Marsella, Inorg. Chem., 1988, 27, 3151 CrossRef CAS; (b) W. C. Trogler, J. Chem. Educ., 1988, 65, 294 CrossRef CAS.
- V. V. Grushin, I. S. Akhrem and M. E. Volpin, J. Organomet. Chem., 1989, 371, 403 CrossRef CAS.
- C. J. Richard and A. W. Parkins, New J. Chem., 2008, 32, 151 RSC.
- G. Minghetti, A. Albinati, A. L. Bandini and G. Banditelli, Angew. Chem., Int. Ed. Engl., 1985, 24, 120 CrossRef.
- (a) R. S. Paonessa and W. C. Trogler, Inorg. Chem., 1983, 22, 1038 CrossRef CAS; (b) S. Chaloupka and L. M. Venanzi, Inorg. Synth., 1990, 27, 30 CrossRef CAS.
- A. W. Parkins, C. J. Richard and J. W. Steed, Inorg. Chim. Acta, 2005, 358, 2827 CrossRef CAS.
- H. Hock and H. Kropf, Angew. Chem., 1957, 69, 313 CrossRef CAS.
- G. Minghetti, A. L. Bandini, G. Banditelli and F. Bonati, J. Organomet. Chem., 1981, 214, C50 CrossRef CAS.
- I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 8696 CrossRef CAS PubMed.
- A. A. Allen, R. Duffner and F. Kurzer, Tetrahedron, 1978, 34, 1247 CrossRef CAS.
- (a) T. T. Wenzel, J. Chem. Soc., Chem. Commun., 1993, 862 RSC; (b) B. L. Feringa, J. Chem. Soc., Chem. Commun., 1986, 909 RSC; (c) T. Ogura, R. Kamimura, A. Shiga and T. Hosokawa, Bull. Chem. Soc. Jpn., 2005, 78, 1555 CrossRef CAS.
- (a) Y. Shvo and D. Czarkie, J. Organomet. Chem., 1986, 315, C25 CrossRef CAS; (b) K. Itami, A. Palmgren, A. Thorarensen and J.-E. Backvall, J. Org. Chem., 1998, 63, 6466 CrossRef CAS.
- M. J. Krische and Y. Sun, eds. Acc. Chem. Res, 2007, 40, 1237 Search PubMed.
- T.-K. Yang and C.-Y. Shen, in Encyclopedia of Reagents for Organic Synthesis, ed. L. Paquette, Wiley, New York, 2009, 2ndedn, pp. 544 Search PubMed.
- L. Hintermann, ChemCatChem, 2012, 4, 321 CrossRef CAS.
- K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746 CrossRef CAS PubMed.
- B. G. Harvey, H. A. Meylemans and R. L. Quintana, Green Chem., 2012, 14, 2450 RSC.
- L. Li and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 17376 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |