 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multinuclear 1H, 13C and 11B solid-state MAS NMR study of 16- and 18-electron organometallic ruthenium and osmium carborane complexes
Nicolas P. E.
Barry
a,
Thomas F.
Kemp
b,
Peter J.
Sadler
*a and
John V.
Hanna
*b
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: J.V.Hanna@warwick.ac.uk
First published on 20th February 2014
Abstract
The first 1H, 13C, 31P and 11B solid state MAS NMR studies of electron-deficient carborane-containing ruthenium and osmium complexes [Ru/Os(p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] are reported. The MAS NMR data from these 16-electron complexes are compared to those of free carborane-ligand and an 18-electron triphenylphosphine ruthenium adduct, and reveal clear spectral differences between 16- and 18-electron organometallic carborane systems in the solid state.
Introduction
The icosahedral structure of dicarba-closo-dodecaborane (boron- and carbon-cluster; C2B10H12) results in a slight negative polarization of the 10 hydrogen atoms.1–3 These clusters contain characteristic non-classical bonding interactions: the hexacoordination of the carbon and boron atoms results in electron-deficient bonding and spreading of the bonding power of a pair of electrons over more than two atoms.4 The aggregation of atoms in 3-centre-2-electron bonding compensates for this low electron density.5 The electron-deficient complexes [Ru/Os(p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] (2 and 3 in Fig. 1) were reported in 2000,6 and although the osmium complex has been characterised by X-ray crystallography, there is no corresponding crystal structure of the ruthenium analogue. Furthermore, such stable 16-electron complexes react with Lewis bases (e.g. aromatic amines and phosphines) to give 18-electron complexes, such as complex [Ru(p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)(triphenylphosphine)] (4, Fig. 1).6–12 Recently, we reported that in dichloromethane and chloroform solutions at ambient temperature, the blue 16-e complex 2 readily forms adducts with aromatic amines to give the corresponding yellow 18-electron adducts, and the thermal displacement of this equilibrium results in marked thermochromic properties.13 | ||
| Fig. 1 Ligand and 16- and 18-electron organometallic ruthenium and osmium dithiolato carborane complexes studied in this work. | ||
Here, we investigate the use of 1D 1H, 13C, 31P and 11B MAS, and 2D 11B multiple quantum MAS (MQMAS) NMR spectroscopy to characterise 16- and 18-electron dithiocarborane ruthenium and osmium arene complexes 2–4. This methodology is of particular interest for investigating structural and electronic effects in arene metal carborane complexes which are not readily crystallisable. Furthermore, this approach is particularly useful for avoiding the coordination of donor-solvents in solution so ensuring the integrity of the 16-electron complexes.
Results and discussion
Free ligand 1 was synthesised by reaction of dicarba-closo-dodecaborane with two mol equiv of n-butyl lithium (nBuLi) in dry THF under nitrogen and by addition of sulfur powder. After removal of the solvent, the compound was used without further purification. The 1H MAS NMR spectrum of 1 shows the proton resonance of the BH vertices at 5.0 ± 0.1 ppm, along with a broad signal at 2.3 ± 0.2 ppm (Table 1). Similarly, a 13C MAS NMR resonance assignable to the carborane carbon atoms was found at 94.7 ± 0.1 ppm in the spectrum of 1, similar to the shift in solution (Table 2), while three other signals were observed at 69.4 ± 0.1, 26.0 ± 0.1 and 15.1 ± 0.1 ppm. No attempt was made to assign the latter peaks, but they may belong to other sulphide species in the reaction mixture.| Compound | 1H | 13C | 11B | 31P | |||
|---|---|---|---|---|---|---|---|
| Carborane | Aromatic arene | CH and CH3 | PPh3 | ||||
| 1 | 5.0 ± 0.1 2.3 ± 0.2 | 94.7 ± 0.1 | — | — | — | 1.2 ± 1 −4.9 ± 1 −12.5 ± 1 | — |
| 2 | 5.9 ± 0.2 2.4 ± 0.3 1.5 ± 0.2 | 101.8 ± 0.1 | 95.9 ± 0.1, 90.0 ± 0.1 88.5 ± 0.1, 78.9 ± 0.1 75.0 ± 0.1, 71.2 ± 0.1 | 34.7 ± 0.1 27.0 ± 0.1 22.4 ± 0.1 21.7 ± 0.1 | — | −9.3 ± 0.5 | — |
| 3 | 6.3 ± 0.2 2.4 ± 0.3 1.5 ± 0.2 0.2 ± 0.2 | 98.7 ± 0.1 | 94.9 ± 0.1, 88.5 ± 0.1 84.6 ± 0.1, 82.9 ± 0.1 72.1 ± 0.1, 64.8 ± 0.1 | 35.2 ± 0.1 27.5 ± 0.1 23.0 ± 0.1 22.2 ± 0.1 | — | −10.3 ± 1 | — |
| 4 | 7.1 ± 0.3 5.2 ± 0.2 2.1 ± 0.2 1.0 ± 0.1 | 126.5 ± 0.1 | 111.1 ± 0.1, 103.1 ± 0.1 98.9 ± 0.1, 97.7 ± 0.1 94.4 ± 0.1, 91.3 ± 0.1 | 32.7 ± 0.1 31.1 ± 0.1 19.6 ± 0.1 17.3 ± 0.1 | 140.8 ± 0.1, 138.6 ± 0.1 135.2 ± 0.1, 133.0 ± 0.1 130.4 ± 0.1, 129.3 ± 0.1 | −7.9 ± 0.5 −11.9 ± 0.5 | 32.5 ± 0.1 |
| Complex | Carborane | Arene | CH and CH3 | PPh3 |
|---|---|---|---|---|
| 2 | 93.7 | 79.4, 81.3 93.88, 104.1 | 20.2, 23.1 31.9 | — |
| 3 | 95.9 | 72.5, 74.9 87.6, 97.5 | 20.7, 23.4 32.4 | — |
| 4 | 93.7 | 92.8, 95.5 104.2, 114.2 | 17.5, 23.1 30.8 | 128.2, 130.5 134.6, 135.2 |
The 16-electron complexes 2 and 3 were synthesised following a published procedure,6 involving the reaction of ligand 1 with the corresponding ruthenium or osmium dimer [Ru/Os(p-cym)Cl2]2 under a nitrogen atmosphere. After purification by column chromatography on silica gel, the two compounds were isolated as powders. Both 1H and 13C MAS NMR spectra of 2 and 3 are characterised by similar resonances. The 1H MAS NMR resonances of the two complexes are broad and unresolved, but display the same overall characteristics of a strongly dipolar-coupled system, while the corresponding 13C MAS NMR spectra are remarkably well resolved. The carbon signals of the carborane ligand are found at 101.8 ± 0.1 and 98.7 ± 0.1 ppm, for 2 and 3 respectively. The CH carbon signal of the para-cymene ligand is observed at 34.7 ± 0.1 and 35.2 ± 0.1 ppm for 2 and 3, respectively, while the methyl carbons of the methyl and isopropyl groups are found at 27.0 ± 0.1, 22.4 ± 0.1, 21.7 ± 0.1 ppm, respectively. As expected, six signals are observed for the aromatic carbons of the arene ligand. These data are summarised in Table 1 and shown in Fig. 2.
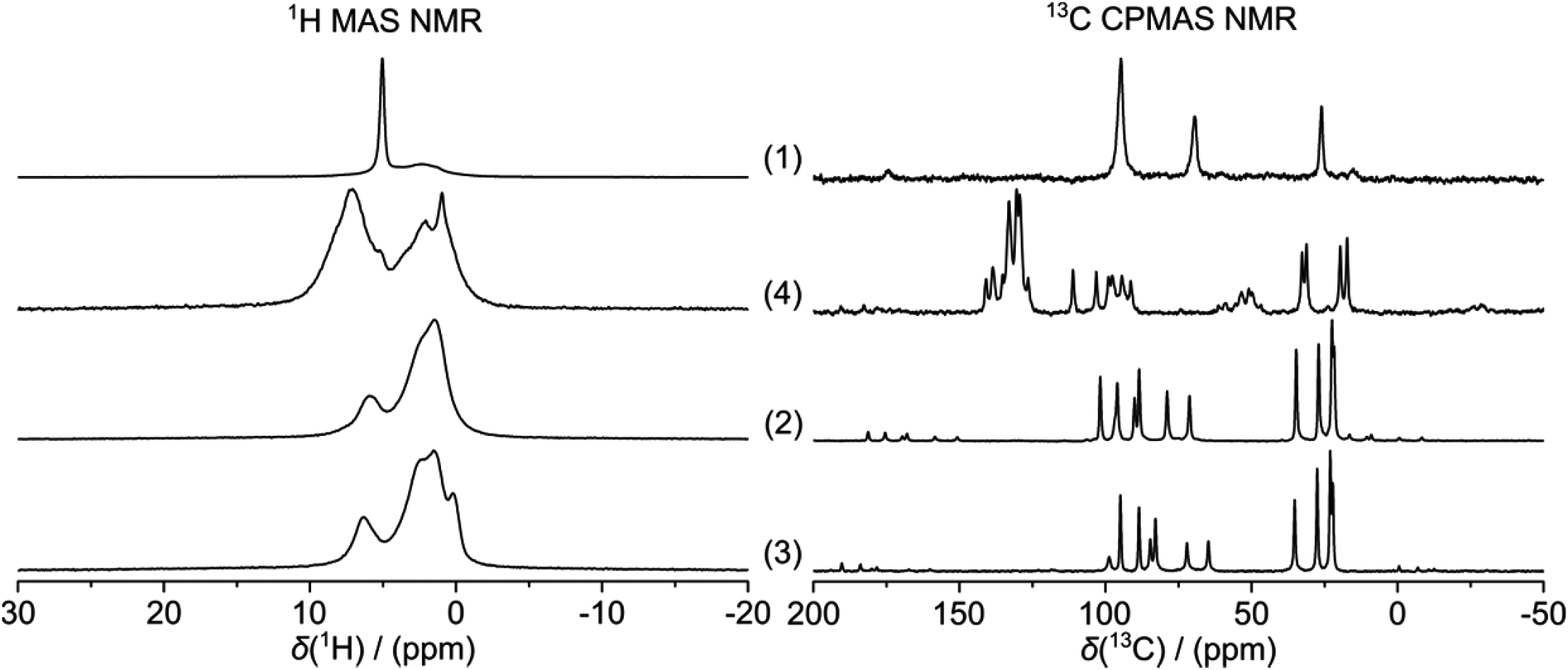 | ||
| Fig. 2 1H MAS and 13C CPMAS NMR spectra of free ligand 1, 18-electron complex 4 and 16-electron complexes 2 and 3. The 13C resonances between 150 and 200 ppm for 1–4 are assignable to spinning sidebands, as are bands between 50 and 60 ppm in the spectrum of complex 4. | ||
The 18-electron complex 4, synthesised by addition of triphenylphosphine to 2, according to literature,6 also exhibits a well-resolved 13C MAS NMR spectrum showing similar signals as 2 and 3, plus the 6 resonances associated with the triphenylphosphine ligand at 140.8 ± 0.1, 138.6 ± 0.1, 135.2 ± 0.1, 133.0 ± 0.1, 130.4 ± 0.1, 129.3 ± 0.1 ppm. Interestingly, the carborane 13C signal in 4 is shifted upfield to 126.5 ± 0.1 ppm in comparison to the corresponding resonances for 2 and 3 which are observed at ∼100 ppm. This shift may arise from the loss of pseudo-aromaticity between 16- and 18-electron species and elongation of the C–S bonds. X-ray crystal structures of the electron-deficient osmium-carborane complex 3 (CCDC 166814)14 and 18-electron ruthenium complex 4 (CCDC 136420)6 have been previously reported. This carborane 13C MAS NMR resonance might therefore be a useful probe for the 16- or 18-electron configurations of an arene metal carborane complex, and suggests that the complex 2, for which no X-ray crystal structure is available, is indeed an electron-deficient complex in the solid state. The chemical shifts of most of the 13C signals observed in CDCl3 solution (Table 2) are very similar to those observed in the solid state and also are in accord with previous SSNMR studies on arene ruthenium complexes (Table 3).15
| Ru/Os complex/carborane | Nuclei | Application | Ref. |
|---|---|---|---|
| Tp* = hydridotris(3,5-dimethylpyrazolyl)borate; THT = tetrahydrothiophene; Cy = cyclohexyl. | |||
| Half-sandwich ruthenium complexes | 13C | Analysis of mixtures of diastereomers | 15 |
| Tp*RuD(THT)2; Tp*RuD(D2)(THT); Tp*RuD(D2)2; Cp*RuD3(PPh3); RuD2(η2-D2)2(PCy3)2 | 2H; 10–300 K | 2H quadrupolar interaction studies and characterisation of 2H-metal bonding | 21 |
| trans-[Ru(D2)Cl(PPh2CH2CH2PPh2)2]PF6 | 2H; 5.4–320 K | Observation of coherent rotational tunnelling in a solid di-deuteride complex | 22 |
| Sc3RuC4; Sc3OsC4 | 13C, 45Sc; 298 K | Probing structure and bonding in carbides | 23 |
| Ru nanoparticles stabilised by hexadecylamine | 2H and 13C cross-polarisation | Direct evidence for the presence of hydride ligands coordinated to ruthenium nanoparticles | 24 |
| Silyl-carborane (1,7-C2B10H10) hybrid diethynylbenzene-silylene polymers | 11B, 13C, and 29Si; 298 K | Structural study | 25 |
| Alkali metal dodecahydro-closo-dodecaborates M2(B12X12), (X = H, Cl, Br, I) | 11B; 120–370 K | Molecular dynamics of [B12X12]2− | 26 |
Clear NMR spectroscopic differences between 16- and 18-electron complexes are also observed from the 1D 11B MAS and 2D 11B multiple quantum magic angle spinning (MQMAS) NMR data of complexes 2–4 (see Fig. 3). 2D MQMAS NMR experiments on half-integer quadrupolar nuclei often provide increased resolution as the narrower central transition resonances are distributed along a second dimension.16–18 The 2D 11B MQMAS spectrum of the free ligand 1 exhibits three well resolved resonances with apparent shifts in the 2–13 ppm range. Although four inequivalent B positions are expected from the schematic representation of 1 in Fig. 1, positions B8,10H and B9,12H are not resolved and the intensity ratio of the manifold of resonances is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2 instead of 2:2:4:2. Similarly, the 2D 11B MQMAS data from the 18-electron complex 4 also exhibit three resonances with positions B8,10H and B9,12H again not being resolved. These observations contrast markedly with the 1D 11B MAS and 2D 11B MQMAS NMR data from the 16-electron complexes 2 and 3 which show single broadened resonances at −9.3 ± 0.5 and −10.3 ± 1, respectively. The results for 2 and 3 suggest that significant motional averaging is occurring on the timescale of the NMR experiment, due to either full or restricted rotation of the carborane cage and its pendant metallocene linker, yielding an averaged spectrum represented by a single broadened resonance. This contrasts with the unaveraged/resolved 2D 11B MQMAS result from the 18-electron complex 4 which is unlikely to possess the same rotational freedom due to the bulky additional PPh3 group stabilizing this structure. The additional PPh3 ligand in complex 4 is evident from the 31P MAS NMR spectrum shown in Fig. 3 where one major resonance is observed at 32.5 ± 0.1 ppm; this 31P shift for PPh3 is located further downfield and deshielded in comparison to other transition metal PPh3 adducts involving Cu and Ag.19,20 This observation is also in agreement with corresponding 31P solution NMR measurements where a similar shift at 31.19 ppm is observed for complex 4 in CDCl3 solution.
:4:2 instead of 2:2:4:2. Similarly, the 2D 11B MQMAS data from the 18-electron complex 4 also exhibit three resonances with positions B8,10H and B9,12H again not being resolved. These observations contrast markedly with the 1D 11B MAS and 2D 11B MQMAS NMR data from the 16-electron complexes 2 and 3 which show single broadened resonances at −9.3 ± 0.5 and −10.3 ± 1, respectively. The results for 2 and 3 suggest that significant motional averaging is occurring on the timescale of the NMR experiment, due to either full or restricted rotation of the carborane cage and its pendant metallocene linker, yielding an averaged spectrum represented by a single broadened resonance. This contrasts with the unaveraged/resolved 2D 11B MQMAS result from the 18-electron complex 4 which is unlikely to possess the same rotational freedom due to the bulky additional PPh3 group stabilizing this structure. The additional PPh3 ligand in complex 4 is evident from the 31P MAS NMR spectrum shown in Fig. 3 where one major resonance is observed at 32.5 ± 0.1 ppm; this 31P shift for PPh3 is located further downfield and deshielded in comparison to other transition metal PPh3 adducts involving Cu and Ag.19,20 This observation is also in agreement with corresponding 31P solution NMR measurements where a similar shift at 31.19 ppm is observed for complex 4 in CDCl3 solution.
 | ||
| Fig. 3 1D 11B and 31P MAS NMR, and 2D 11B MQMAS NMR spectra of the free ligand 1, 18-electron complex 4 and 16-electron complexes 2, 3. | ||
Solid state MAS NMR spectroscopy has been only rarely employed for the study of the molecular structures of ruthenium and osmium complexes and carborane-containing systems. Some reported examples of MAS and static NMR studies for such compounds are listed in Table 3. This study demonstrates that MAS NMR spectroscopy is a potentially powerful tool for probing the electronic structures of arene metal carborane complexes in the solid state, and highlights some particular spectral characteristics, which can aid differentiation between 16- and 18-electron systems. Further work is needed to elucidate the nature of the dynamic motion which appears to be associated with the carborane cage and its pendant metallocene linker for the 16-electron complexes 2 and 3 (in comparison to their 18-electron counterpart 4), as initially observed through the 11B MAS NMR studies. Further 11B, 1H and 2D solid state NMR studies should help to clarify this phenomenon.
Conclusions
The interconversion of 16- and 18-electron organometallic arene complexes is of particular importance in the design of Ru catalysts, e.g. for azide–alkyne cycloaddition reactions,27 racemisation of aromatic and aliphatic secondary alcohols,28 and for transfer hydrogenation.29 The conversion from 16- to 18-electron carborane-containing organometallic complexes has also been shown to be a useful synthetic strategy for multimetallic clusters containing direct metal–metal bonds.30,31 Sixteen-electron complexes are often thought to be reactive intermediates in catalytic cycles but can be difficult to isolate and characterise. In solution solvent molecules may bind weakly to the vacant 6th coordination site. Equilibria can exist in solution between 16- and 18-electron species, for example between electron-deficient complex [Ru(p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] (2) and corresponding 18-electron amine and phosphine adducts (e.g.4). Here we have used multinuclear solid state MAS NMR to characterise the stable 16-electron complex [Ru(p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] (2) and its OsII analogue (3). The comparison of 1H, 13C and 11B solid-state NMR spectra of 16-electron ruthenium complex 2 with the 16-electron osmium complex 3 and 18-electron ruthenium complex 4, complexes which have been characterised by X-ray crystallography, provided a clear structural correlation between electron deficient complexes 2 and 3, and highlighted some differences between 16- and 18-electron complexes 2 and 4. The increased resolution afforded by the 2D 11B MQMAS NMR technique has highlighted possible dynamic processes affecting the carborane cage in the 16- and 18-electron structures. Further 11B, 1H and 2D solid state NMR studies may be fruitful for charactering such dynamics.Experimental section
Materials
The syntheses of the ligand [(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)Li2], 16-electron precursors [Ru(η6-p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] (2), [Os(η6-p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)] (3), and the 18-electron complex [Ru(η6-p-cym)(1,2-dicarba-closo-dodecaborane-1,2-dithiolate)(triphenylphosphine)] (4) were based on a previous report.6 All the complexes were purified by column chromatography on silica gel, using a mixture 2/1 hexane–dichloromethane. The purity of the complexes was assessed by 1H NMR spectroscopy in CDCl3 and was in accord with previous reports.Methods and instrumentation
All 13C MAS NMR data were acquired at 11.7 T using a Bruker Avance III spectrometer (13C Larmor frequency of 125.76 MHz). The 1H–13C cross-polarisation experiment was used in each case, with these measurements being performed using a Bruker 4 mm triple-channel HXY probe operating at a MAS frequency of 10 kHz. An initial 1H π/2 excitation pulse of 2.5 μs, a Hartmann–Hahn contact period of 1 ms and a recycle delay of 3 s were used throughout. All 13C isotropic chemical shifts were referenced to the primary reference of TMS via the secondary reference of solid alanine. The 31P MAS NMR data for complex 4 was acquired at 14.1 T using a Bruker Avance II+ spectrometer (31P Larmor frequency of 243.81 MHz) and a Bruker 4 mm triple channel HXY probe operating at a MAS frequency of 12 kHz. A single pulse experiment was used employing a 31P π/4 excitation pulse of 2.5 μs and a recycle delay of 30 s. 31P chemical shifts were referenced to the primary reference of 85% H3PO4via a secondary solid reference of (NH4)H2(PO4). The 1D 11B MAS NMR data were acquired at 14.1 T using Varian 600 spectrometer (11B Larmor frequency of 193.23 MHz) using a Varian 4 mm T3 probe operating at a MAS frequency of 12 kHz. A single pulse experiment was used employing a 11B π/2 ‘selective’ (solids) pulse of 3 μs and a recycle delay of 5 s. All apparent 11B chemical shifts are referenced to NaBH4 and do not represent isotropic values as they remain uncorrected for second order quadrupole effects. The corresponding 2D 11B MQMAS NMR data were acquired at 14.1 T using a Bruker Avance II+ spectrometer and a Bruker 3.2 mm triple-channel HXY probe operating at a MAS frequency of 20 kHz. A 3Q Z-filtered experiment was used with a recycle delay of 4 s. Finally, all 1H MAS NMR measurements were undertaken using a Bruker Avance II+ spectrometer (1H Larmor frequency of 600.1 MHz) and a Bruker 2.5 mm triple channel HXY probe operating at a MAS frequency of 30 kHz. A single pulse experiment was used employing a 1H π/4 excitation pulse of 2 μs and a recycle delay of 30 s. All 1H chemical shifts were referenced to the primary reference of TMS.Acknowledgements
We thank the Swiss National Science Foundation (grant no. PA00P2-145308 to NPEB), the ERC (grant no. 247450 to PJS), EPSRC (grant no. EP/F034210/1) and EC COST Action CM1105 for support. JVH thanks EPSRC and the University of Warwick for partial funding of the solid state NMR infrastructure at Warwick, and acknowledges additional support obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and partial funding by the European Regional Development Fund (ERDF).Notes and references
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- Z. J. Lesnikowski, New Opportunities in Boron Chemistry for Medical Applications, in Boron Sciences. New technologies and Applications, ed. N. S. Hosmane, CRC Press., Boca Raton, 2011, pp. 3–19 Search PubMed.
- Z. J. Lesnikowski, Collect. Czech. Chem. Commun., 2007, 72, 1646–1658 CrossRef CAS.
- S. Korbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef PubMed.
- Z. Xie, Acc. Chem. Res., 2003, 36, 1–9 CrossRef CAS PubMed.
- M. Herberhold, H. Yan and W. Milius, J. Organomet. Chem., 2000, 598, 142–149 CrossRef CAS.
- S. Liu, G.-L. Wang and G.-X. Jin, Dalton Trans., 2008, 425–432 RSC.
- Y.-F. Han, J.-S. Zhang, Y.-J. Lin, J. Dai and G.-X. Jin, J. Organomet. Chem., 2007, 692, 4545–4550 CrossRef CAS PubMed.
- Z.-J. Yao, G. Su and G.-X. Jin, Chem.–Eur. J., 2011, 17, 13298–13307 CrossRef CAS PubMed.
- X.-K. Huo, G. Su and G.-X. Jin, Dalton Trans., 2010, 39, 1954–1961 RSC.
- D.-H. Wu, C.-H. Wu, Y.-Z. Li, D.-D. Guo, X.-M. Wang and H. Yan, Dalton Trans., 2009, 285–290 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264–3279 RSC.
- N. P. E. Barry, R. J. Deeth, G. J. Clarkson, I. Prokes and P. J. Sadler, Dalton Trans., 2013, 42, 2580–2587 RSC.
- M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Chem.–Eur. J, 2002, 8, 388–395 CrossRef CAS.
- V. Ritleng, P. Bertani, M. Pfeffer, C. Sirlin and J. Hirschinger, Inorg. Chem., 2001, 40, 5117–5122 CrossRef CAS PubMed.
- A. Medek, J. S. Harwood and L. Frydman, J. Am. Chem. Soc., 1995, 117, 12779–12787 CrossRef CAS.
- J. V. Hanna, M. E. Smith and H. J. Whitfield, J. Am. Chem. Soc., 1996, 118, 5772–5777 CrossRef CAS.
- J. V. Hanna and M. E. Smith, Solid State Nucl. Magn. Reson., 2010, 38, 1–18 CrossRef CAS PubMed.
- G. A. Bowmaker, Effendy, J. V. Hanna, P. C. Healy, J. C. Reid, C. E. F. Rickard and A. H. White, J. Chem. Soc., Dalton Trans., 2000, 753–761 RSC.
- J. V. Hanna, M. E. Smith and S. N. Stuart, J. Phys. Chem., 1992, 96, 7560–7567 CrossRef CAS.
- B. Walaszek, A. Adamczyk, T. Pery, X. Yeping, T. Gutmann, N. d. S. Amadeu, S. Ulrich, H. Breitzke, H. M. Vieth, S. Sabo-Etienne, B. Chaudret, H.-H. Limbach and G. Buntkowsky, J. Am. Chem. Soc., 2008, 130, 17502–17508 CrossRef CAS PubMed.
- F. Wehrmann, T. P. Fong, R. H. Morris, H.-H. Limbach and G. Buntkowsky, Phys. Chem. Chem. Phys., 1999, 1, 4033–4041 RSC.
- L. Zhang, C. Fehse, H. Eckert, C. Vogt, R.-D. Hoffmann and R. Pöttgen, Solid State Sci., 2007, 9, 699–705 CrossRef CAS PubMed.
- T. Pery, K. Pelzer, G. Buntkowsky, K. Philippot, H.-H. Limbach and B. Chaudret, ChemPhysChem, 2005, 6, 605–607 CrossRef CAS PubMed.
- H. Kimura, K. Okita, M. Ichitani, T. Sugimoto, S. Kuroki and I. Ando, Chem. Mater., 2002, 15, 355–362 CrossRef.
- I. Tiritiris, T. Schleid and K. Müller, Appl. Magn. Reson., 2007, 32, 459–481 CrossRef CAS.
- M. Lamberti, G. C. Fortman, A. Poater, J. Broggi, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, Organometallics, 2012, 31, 756–767 CrossRef CAS.
- R. Karvembu, R. Prabhakaran and K. Natarajan, Coord. Chem. Rev., 2005, 249, 911–918 CrossRef CAS PubMed.
- R. Noyori and S. Hashigushi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- S. Liu, Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2007, 36, 1543–1560 RSC.
- X. Meng, F. Wang and G.-X. Jin, Coord. Chem. Rev., 2010, 254, 1260–1272 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |