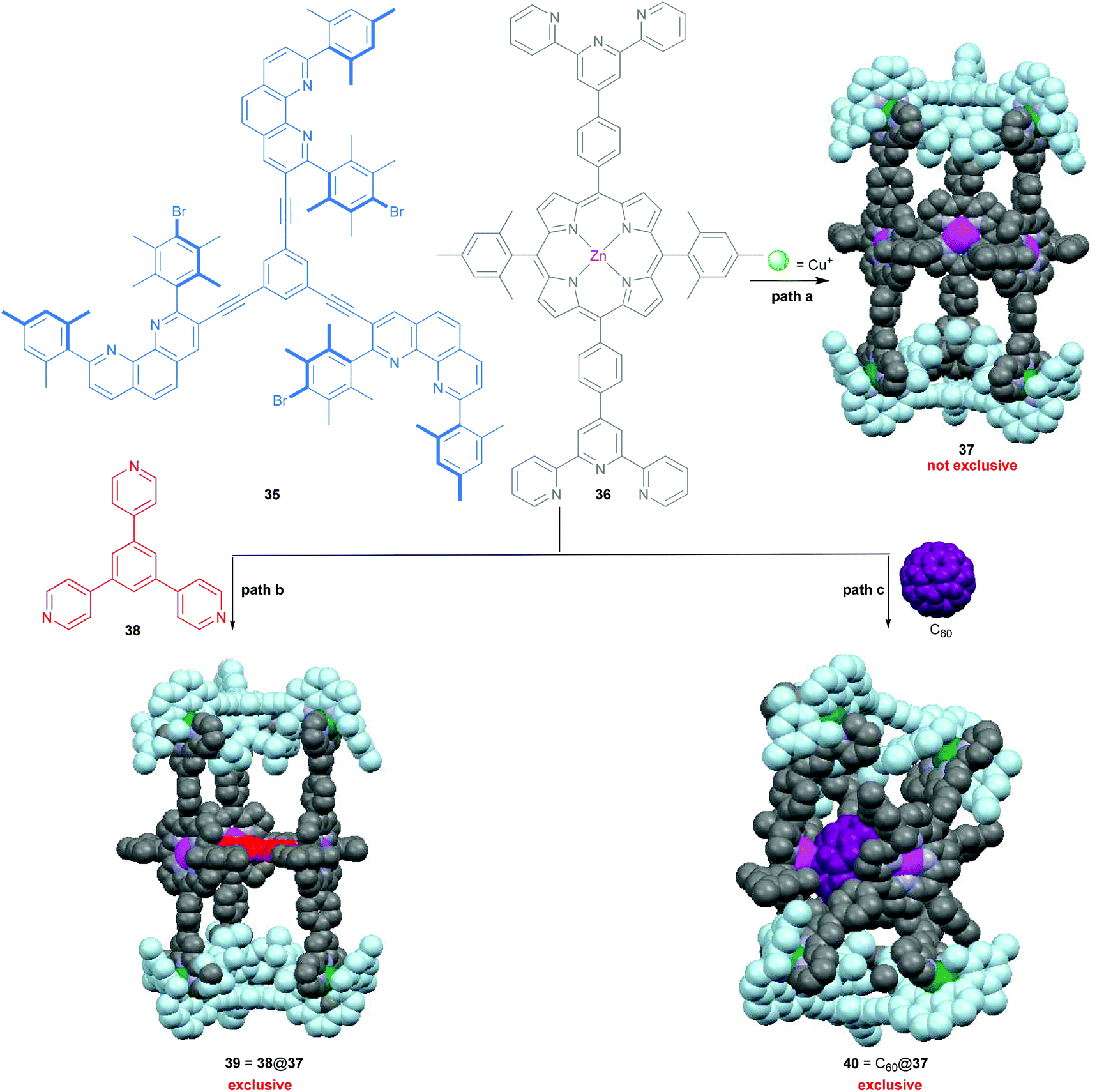DOI:
10.1039/C3DT53570C
(Perspective)
Dalton Trans., 2014,
43, 3815-3834
Dynamic heteroleptic metal-phenanthroline complexes: from structure to function†
Received
19th December 2013
, Accepted 13th January 2014
First published on 27th January 2014
Abstract
Dynamically heteroligated metal centres are auspicious platforms to access multicomponent supramolecular systems, the latter showing unique structures, amazing properties and even emergent functions. The great potential of heteroleptic complexes has materialised after the development of appropriate strategies that warrant quantitative formation in spite of the dynamic character. In this perspective, we discuss our endeavours at developing various heteroleptic self-assembly protocols based on sterically bulky 2,9-diarylphenanthrolines and our work toward self-sorted multicomponent architectures and assemblies with new and useful functions.
 Manik Lal Saha | Manik Lal Saha was born in 1986 in West Bengal, India. He received his B.Sc. (Honours) in Chemistry from University of Calcutta (India) in 2006. After finishing his M.Sc. from the Indian Institute of Technology Kanpur in 2008, he joined Prof. Schmittel's group at the University of Siegen (Germany). He is currently working towards his PhD on the synthesis of self-sorted multicomponent architectures. |
 Subhadip Neogi | Subhadip Neogi received his B.Sc. in Chemistry from University of Burdwan (India) and M.Sc. from Banaras Hindu University (India). He obtained his Ph.D. from the Indian Institute of Technology Kanpur in 2008. After completing Humboldt postdoctoral research in Germany with Prof. Michael Schmittel at the University of Siegen, he joined the Department of Chemistry, Indian Institute of Technology Kanpur as a postdoctoral research fellow. His research interests focus on multicomponent supramolecular assemblies and organic–inorganic hybrid porous materials. |
 Michael Schmittel | Michael Schmittel studied chemistry and French in Freiburg and Paris. After a postdoctoral stay in Rochester/USA and habilitation at the University of Freiburg, he joined the University of Würzburg as an associate professor in 1993 and since 1999 holds a Chair in Organic Chemistry at the University of Siegen. His broad interests, published in ca. 230 papers and one textbook, cover research areas as diverse as diradical cyclisations, metallosupramolecular coordination chemistry, lab-on-a-molecule chemosensors and, starting recently, (supra)molecular machines and nanoswitches. |
1. Introduction
Nature masterfully employs self-assembly and self-organisation in combination with self-sorting for constructing the elegant and intricate molecular machinery from which life is built on our planet.1 For instance, all cellular devices, such as ribosomes, mitochondria and many smaller multicomponent enzyme complexes, are made by arranging a multitude of prefabricated covalent building blocks in a proper spatial and functionally active arrangement through noncovalent interactions. As a result, heteroaggregates are at the centre of many specific biological functions, signalling cascades and feedback protocols. It is thus of no surprise that the development of approaches to intricate heteroaggregates in a holistic manner has turned into an attractive research area within abiological self-assembly.2 The latter has already experienced an ontogenetic growth, starting from magnificent nanoscale topologies3 to host–guest chemistry,4 artificial molecular machines,5 and smart materials.6 As a result, contemporary self-assembly has evolved as a broad multidisciplinary domain that provides a fertile ground for breathtaking research.7
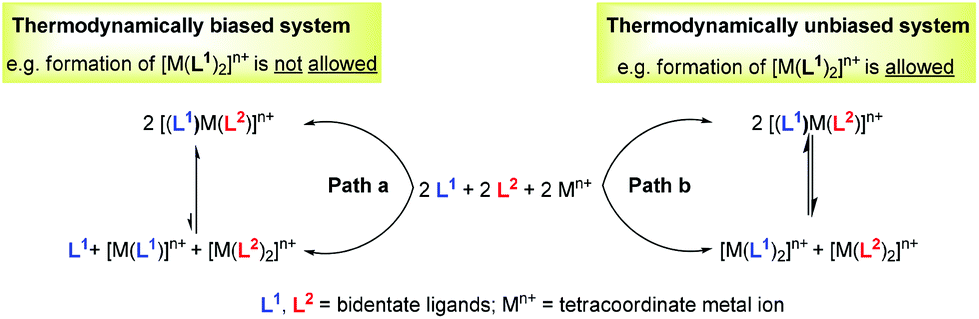
For example, over the last two decades, metallosupramolecular self-assembly8 has materialised as a promising research area for the synthesis of well-defined, discrete two- and three-dimensional nano-architectures. Despite the notable wealth of the known nano-architectures, nearly all systems comprise only two different components – a ligand and a metal ion – and thus are homoleptic in nature and suffer from a lack of diversity.9 To our understanding, heteroleptic architectures are less frequently targeted due to the lack of convincing strategies that enable the exclusive formation of dynamic mixed complexes and that require control over the concurrent binding of two or more different freely exchanging ligands at a single metal centre.10 For instance, even in the simplest possible case, i.e. starting with an equimolar mixture of two ligands and one metal ion (Fig. 1, path b), one would risk the parallel formation of two homoleptic complexes, [M(L1)2]n+ and [M(L2)2]n+, along with the desired mixed complex [(L1)M(L2)]n+. Clearly, in such set-ups, labile metal complexes allow very rapid ligand exchange reactions, thus counteracting the clean formation of a heteroleptic complex in solution.
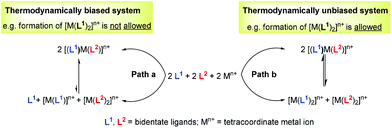 |
| | Fig. 1 Thermodynamic bias to control the heteroleptic complex [(L1)M(L2)]n+ formation over that leading to homoleptic complexes [M(L1)2]n+ and [M(L2)2]n+. | |
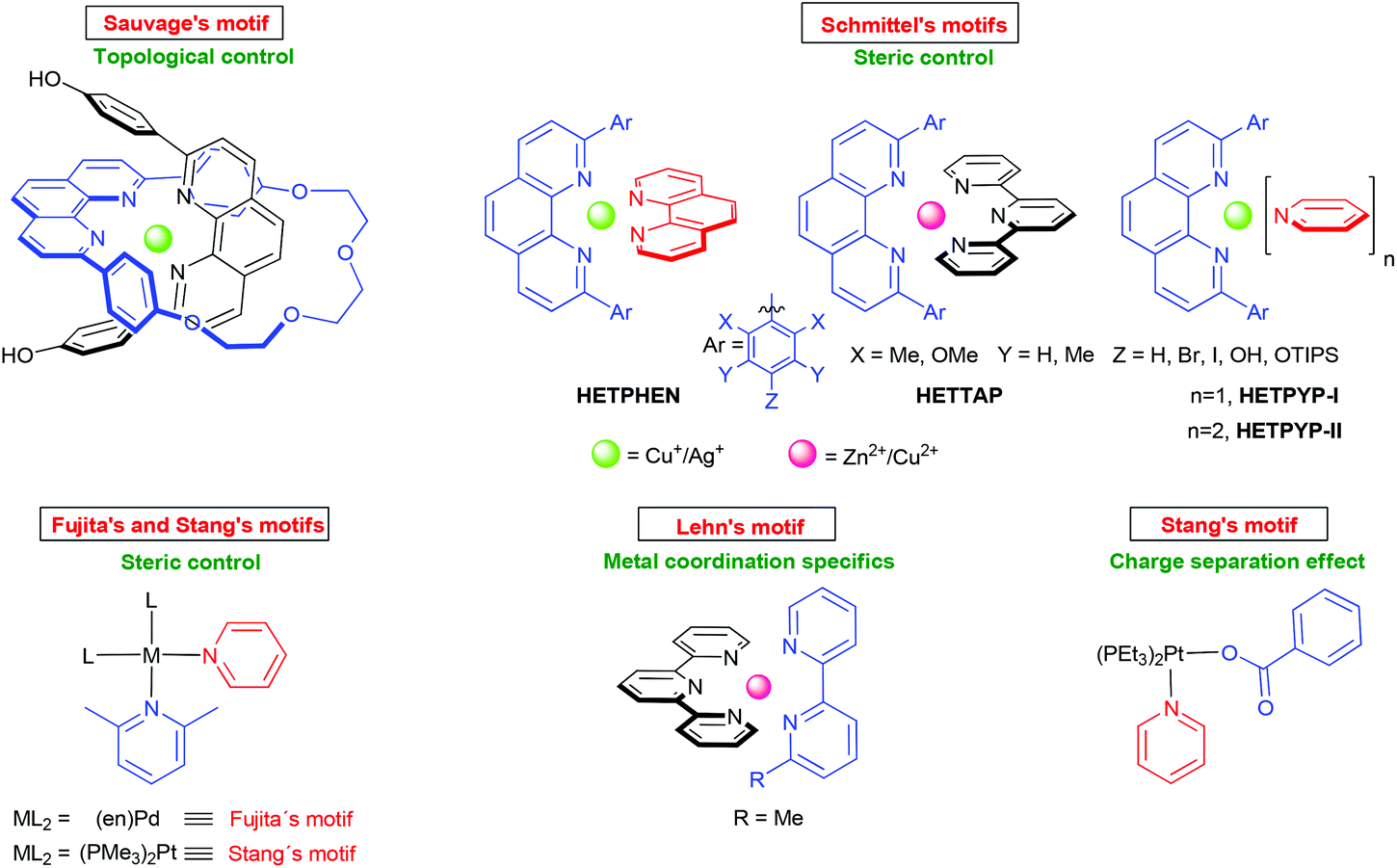
To drive any dynamic heteroaggregation to completion one needs to impose a sizeable thermodynamic bias in the mixed complex as compared to the homoleptic alternatives (Fig. 1, path a). In this respect, chemists often rely on maximum site occupancy, requiring optimal saturation of all donor (ligand) and acceptor (metal-ion) sites while matching all stereochemical requirements.10,11 In principle, if one is able to suppress formation of at least one of the homoleptic combinations, e.g. [M(L1)2]n+ (Fig. 1, path a), using the appropriate ligand design, metal coordination specifics and/or stoichiometry bias, then maximum site occupancy will drive quantitative formation of the mixed complex [(L1)M(L2)]n+ on enthalpic grounds. Otherwise, energy-rich and coordinatively frustrated entities, e.g. [M(L1)]n+ and L1, would form along with [M(L2)2]n+ and [(L1)M(L2)]n+. Over the years, Sauvage,12 Lehn,13 Fujita,14 Stang,15,16 Schmittel17 and others18,19 have used and refined the principle (Fig. 2) in the context of metal-coordination driven dynamic heteroleptic architectures.10 For example, Sauvage implemented topological constraints at one ligand preventing formation of its bishomoleptic assembly, while Schmittel precluded one of the bishomoleptic complexation motifs due to steric shielding. In other cases (Fig. 2), where both bishomoleptic complexes are equally possible, the hetero-selectivity is attained either by applying a well-balanced steric programming (Fujita and Stang), capitalising on the coordination specifics of penta-coordinating copper(II) ions (Lehn) or profiting on the charge separation effect (Stang).
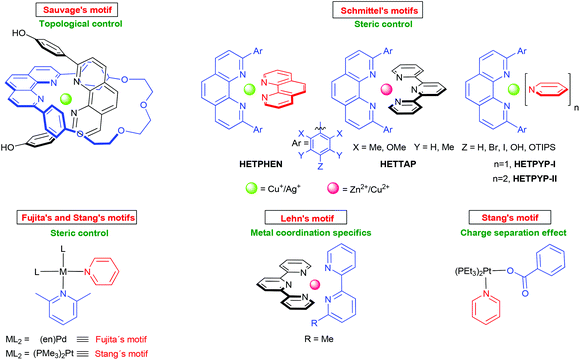 |
| | Fig. 2 Strategies to quantitatively prepare mixed metal complexes in solution. | |
Undoubtedly, considering the minute number of well-established heteroligation protocols (Fig. 2), the vision of setting up functional assemblies that replicate even partly some complex biological function remains quite far-fetched. To mimic biological machineries, however, the required multicomponent assemblies need to possess several orthogonal homo- and/or hetero-recognition motifs20 that precisely cooperate with each other.21 For this, one may apply the same strategy as that used by nature: amalgamate self-assembly and self-sorting22 protocols to integrate different subunits into the final architecture with precise spatial/positional and constitutional control. Hence, there has been, in recent years, a renewed quest for deeper understanding of the various concepts which are vital to multicomponent assembly such as orthogonality in coordination motifs,20completive and integrative self-sorting,22c cooperative and allosteric effects, etc.21
This perspective describes our efforts and advancement in understanding and exploring the concepts of dynamic heteroleptic complexation using 2,9-diarylphenanthrolines as the key building blocks (Fig. 2). In the subsequent sections, we will discuss the rational design and the ensuing synthesis of heteroleptic assemblies, thereafter the blending of optimised heteroleptic motifs into multicomponent self-sorted aggregates and finally architectures that offer new and useful functions.
2. Our heteroleptic toolkits and polynuclear complexes derived therefrom
In 1997, our studies on metal phenanthroline complexes revealed an interesting behaviour (Scheme 1a).23 An equimolar mixture of Cu+ and the two phenanthrolines 1, 2 at equilibrium did not afford a statistical mixture of all three possible complexes 3–5 (Scheme 1a). Instead, only the complexes 3 = [Cu(1)2]+ and 4 = [Cu(1)(2)]+ were furnished in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, suggesting that complex 3 with its four intramolecular π–π stacking interactions is thermodynamically favoured over the heteroleptic complex 4 (two π–π stacking interactions) and homoleptic complex 5 = [Cu(2)2]+ (zero π–π stacking interactions). The thermodynamics of this copper(I) complex equilibration is qualitatively reflected by the energy levels depicted in Scheme 1b. Clearly, to realise exclusive formation of the desired heteroleptic complex, one needs simply to increase the barrier for the [Cu(1)(2)]+ → [Cu(1)2]+ transformation. At this juncture, we reasoned that a sterically shielded 2,9-diarylphenanthroline (phenAr2) is a highly promising candidate (Scheme 1c), because its bishomoleptic copper complex should be impossible applying a proper balance of steric and electronic effects. Indeed, the combination of various [Cu(phenAr2)]+ motifs with a sterically unpretentious counterligand, used at correct stoichiometry, provided exclusively the heteroleptic complex that is driven by maximum site occupancy (vide supra). Over the years, we have refined this approach for various ligands and metal ions and used it for constructing a rich variety of dynamic oligonuclear structures. In the following, we will portray the different protocols and some interesting architectures derived therefrom.
:1, suggesting that complex 3 with its four intramolecular π–π stacking interactions is thermodynamically favoured over the heteroleptic complex 4 (two π–π stacking interactions) and homoleptic complex 5 = [Cu(2)2]+ (zero π–π stacking interactions). The thermodynamics of this copper(I) complex equilibration is qualitatively reflected by the energy levels depicted in Scheme 1b. Clearly, to realise exclusive formation of the desired heteroleptic complex, one needs simply to increase the barrier for the [Cu(1)(2)]+ → [Cu(1)2]+ transformation. At this juncture, we reasoned that a sterically shielded 2,9-diarylphenanthroline (phenAr2) is a highly promising candidate (Scheme 1c), because its bishomoleptic copper complex should be impossible applying a proper balance of steric and electronic effects. Indeed, the combination of various [Cu(phenAr2)]+ motifs with a sterically unpretentious counterligand, used at correct stoichiometry, provided exclusively the heteroleptic complex that is driven by maximum site occupancy (vide supra). Over the years, we have refined this approach for various ligands and metal ions and used it for constructing a rich variety of dynamic oligonuclear structures. In the following, we will portray the different protocols and some interesting architectures derived therefrom.
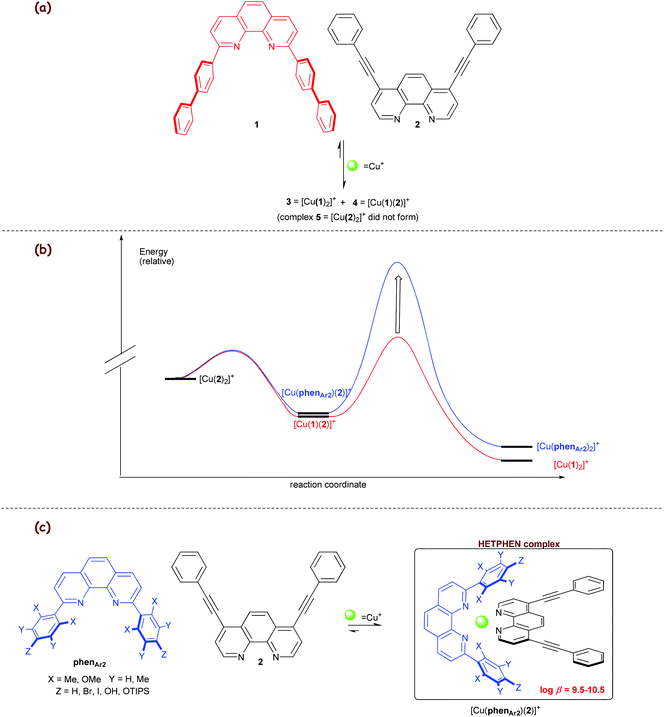 |
| | Scheme 1 (a) Influence of intracomplex π–π interactions on copper(I) complex equilibria. (b) Energy diagram depicting the complexation equilibria. (c) The HETPHEN concept.23 | |
2.1. HETPHEN concept
The HETPHEN complexation is a three-component assembly strategy developed for the selective formation of ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) eroleptic bis-
eroleptic bis-![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) anthroline metal complexes (Scheme 1c and Fig. 3).23 Over the years, the HETPHEN approach has proved its value in the preparation of heteroleptic mononuclear Cu+-, Ag+- and Zn2+-bisphenanthroline,24 mixed phenanthroline-2,2′-bipyridine (bipy) complexes,25 and a rich variety of supramolecular polynuclear structures derived therefrom, such as triangles,26 ring-in-ring assemblies,27 nanoscaffolds,28 tripledeckers,29 nanogrids,30 nanoracks,31 porphyrin stacks32 and nanobaskets.33 In addition, using a variant of the HETPHEN strategy, we recently developed an access to constitutionally dynamic heteroleptic [Cu(phenAr2)(iminopyridine)]+ complexes (log β ≈ 9–11)34,35 (Fig. 3) through four-component assembly. During those studies, we recognised that the much weaker binding picolinaldehydes, our versatile precursors to iminopyridines, themselves undergo heteroleptic complex formation to [Cu(phenAr2)(picolinaldehyde)]+ at log β ≈ 8–8.5.34c It is a big advantage of iminopyridine complexes that, due to their high dissociation–association dynamics, overall kinetic barriers in intricate complexation processes are lower than those of the sturdy phenanthroline ligands, an important feature to drive supramolecular exchange and repair processes (vide infra).
anthroline metal complexes (Scheme 1c and Fig. 3).23 Over the years, the HETPHEN approach has proved its value in the preparation of heteroleptic mononuclear Cu+-, Ag+- and Zn2+-bisphenanthroline,24 mixed phenanthroline-2,2′-bipyridine (bipy) complexes,25 and a rich variety of supramolecular polynuclear structures derived therefrom, such as triangles,26 ring-in-ring assemblies,27 nanoscaffolds,28 tripledeckers,29 nanogrids,30 nanoracks,31 porphyrin stacks32 and nanobaskets.33 In addition, using a variant of the HETPHEN strategy, we recently developed an access to constitutionally dynamic heteroleptic [Cu(phenAr2)(iminopyridine)]+ complexes (log β ≈ 9–11)34,35 (Fig. 3) through four-component assembly. During those studies, we recognised that the much weaker binding picolinaldehydes, our versatile precursors to iminopyridines, themselves undergo heteroleptic complex formation to [Cu(phenAr2)(picolinaldehyde)]+ at log β ≈ 8–8.5.34c It is a big advantage of iminopyridine complexes that, due to their high dissociation–association dynamics, overall kinetic barriers in intricate complexation processes are lower than those of the sturdy phenanthroline ligands, an important feature to drive supramolecular exchange and repair processes (vide infra).
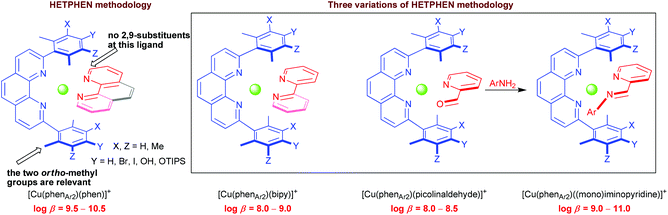 |
| | Fig. 3 Variations in the HETPHEN concept: heteroleptic [Cu(phenAr2)(bipy)]+,25 [Cu(phenAr2)(picolinaldehyde)]+ and [Cu(phenAr2)(iminopyridine)]+ motifs.34b | |
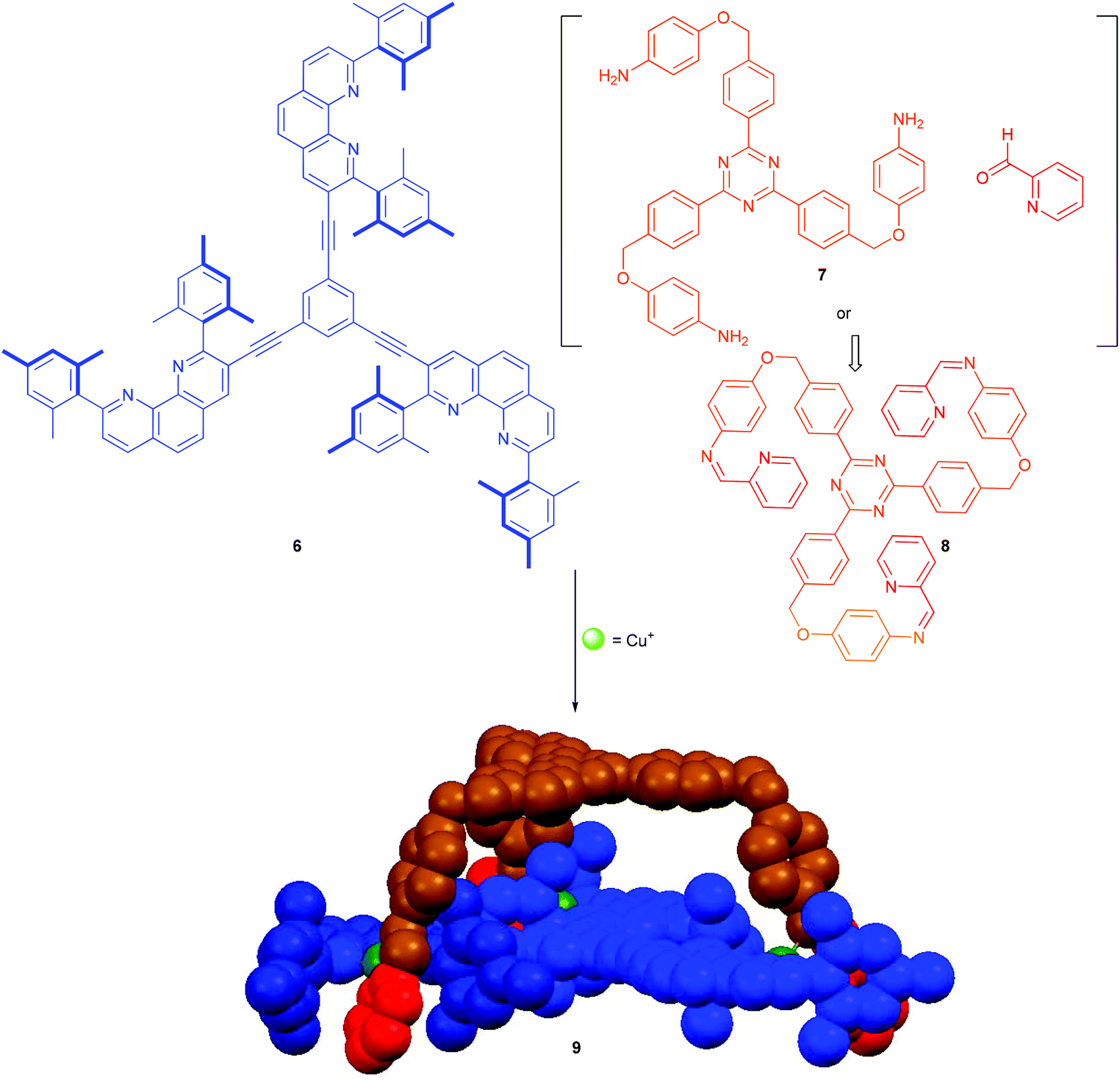
Based on the efficient repair in constitutionally dynamic iminopyridines and their complexes, we recently realised the formation of the diastereomerically pure heteroleptic metal–organic cage 9.34a Cage 9 was manufactured from the C3-symmetric building blocks 6 and 8. The latter may be used either in the prefabricated form or generated in situ from picolinaldehyde and trianiline 7 (Scheme 2).34a
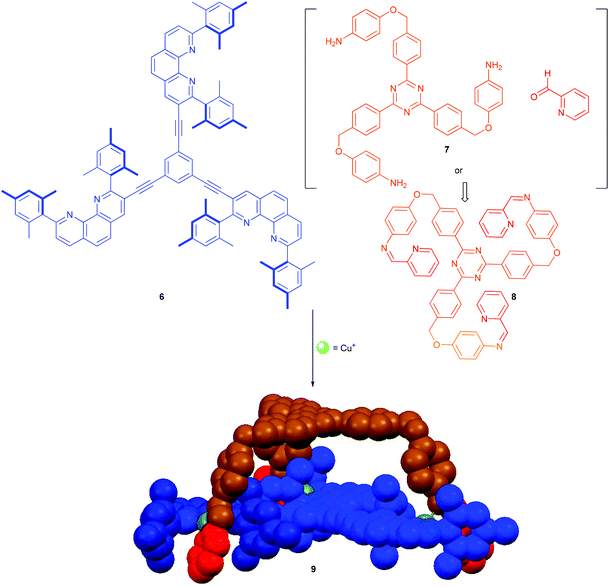 |
| | Scheme 2 Cage structure 9 prepared from the triple-arm tris-iminopyridine 8 and trisphenanthroline 6 by the formation of three [Cu(phenAr2)(iminopyridine)]+ linkages.34a | |
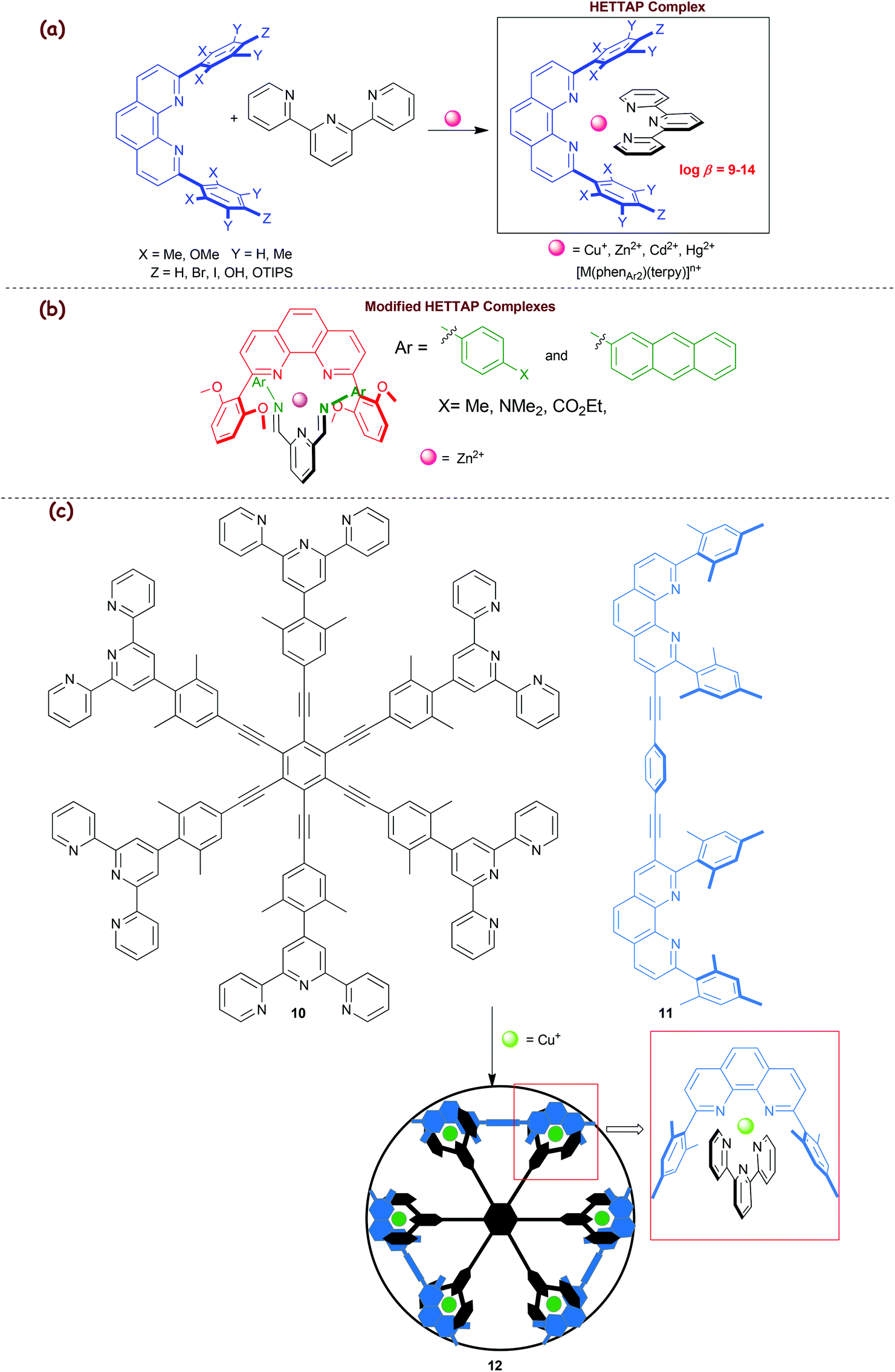
2.2. HETTAP concept
In 2005, we developed the HETTAP concept (eroleptic erpyridine ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) nd henanthroline complexation) that allows one to prepare heteroleptic complexes of the type [M(phenAr2)(terpy)]n+ with terpy representing a terpyridine ligand (Scheme 3a).36 Lately, this concept was extended to constitutionally dynamic heteroleptic [M(phenAr2)(diiminopyridine)]n+ complexes (Scheme 3b). 2,9-Bis(2,6-dimethoxyphenyl)-1,10-phenanthroline and Zn2+ ions turned out to be the most suitable combination to accomplish the heteroleptic complexation, as otherwise either diiminopyridines would form incompletely or the kinetically rather stable hexa-coordinated bis(diiminopyridine) zinc complexes would arise in sizable amounts.37
nd henanthroline complexation) that allows one to prepare heteroleptic complexes of the type [M(phenAr2)(terpy)]n+ with terpy representing a terpyridine ligand (Scheme 3a).36 Lately, this concept was extended to constitutionally dynamic heteroleptic [M(phenAr2)(diiminopyridine)]n+ complexes (Scheme 3b). 2,9-Bis(2,6-dimethoxyphenyl)-1,10-phenanthroline and Zn2+ ions turned out to be the most suitable combination to accomplish the heteroleptic complexation, as otherwise either diiminopyridines would form incompletely or the kinetically rather stable hexa-coordinated bis(diiminopyridine) zinc complexes would arise in sizable amounts.37
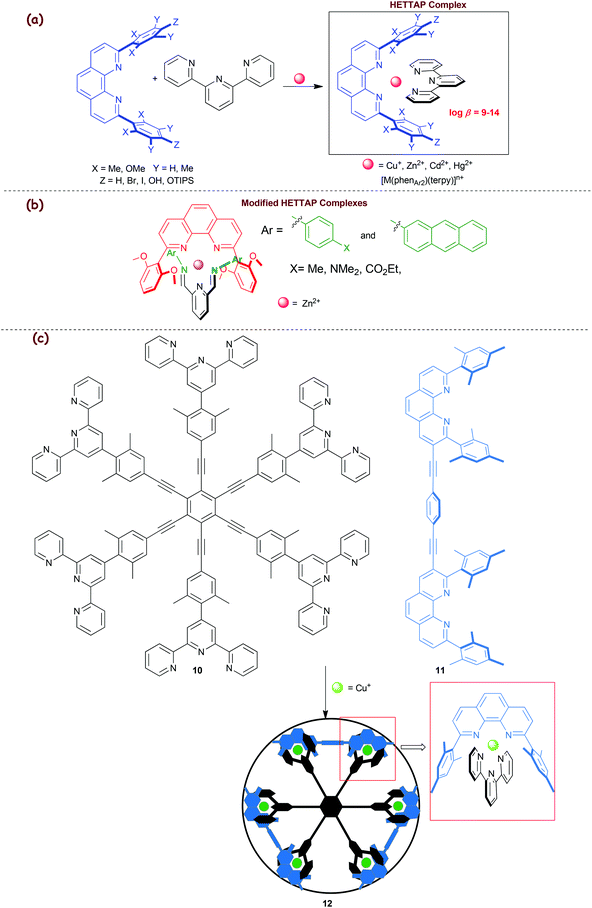 |
| | Scheme 3 (a) Illustration of the HETTAP approach. (b) Diiminopyridine-based HETTAP complexes. (c) Extension of the HETTAP approach toward the spoked-wheel architecture 12.39 | |
The high reliability of the HETTAP complexation process was demonstrated by preparing a number of supramolecular structures, such as nanoladders,36 supramolecular dumbbells,38 molecular-wheels,39 nano-prisms,40 isosceles triangles,41 tweezers,42etc. In 2008, we interrogated the approach toward the synthesis of the technomimetic spoked wheel 1239 that was prepared by the copper(I)-based self-assembly protocol (Scheme 3c). As depicted in Scheme 3c, the D6h-symmetric hexakis(terpyridine) 10 acts as a molecular spoke set in the structure of 12, while bisphenanthroline 11 acts as a rim. The MM+ computations on the structure suggested a diameter and a perimeter of 3.9 and 12.3 nm, respectively.
2.3. HETPYP concept
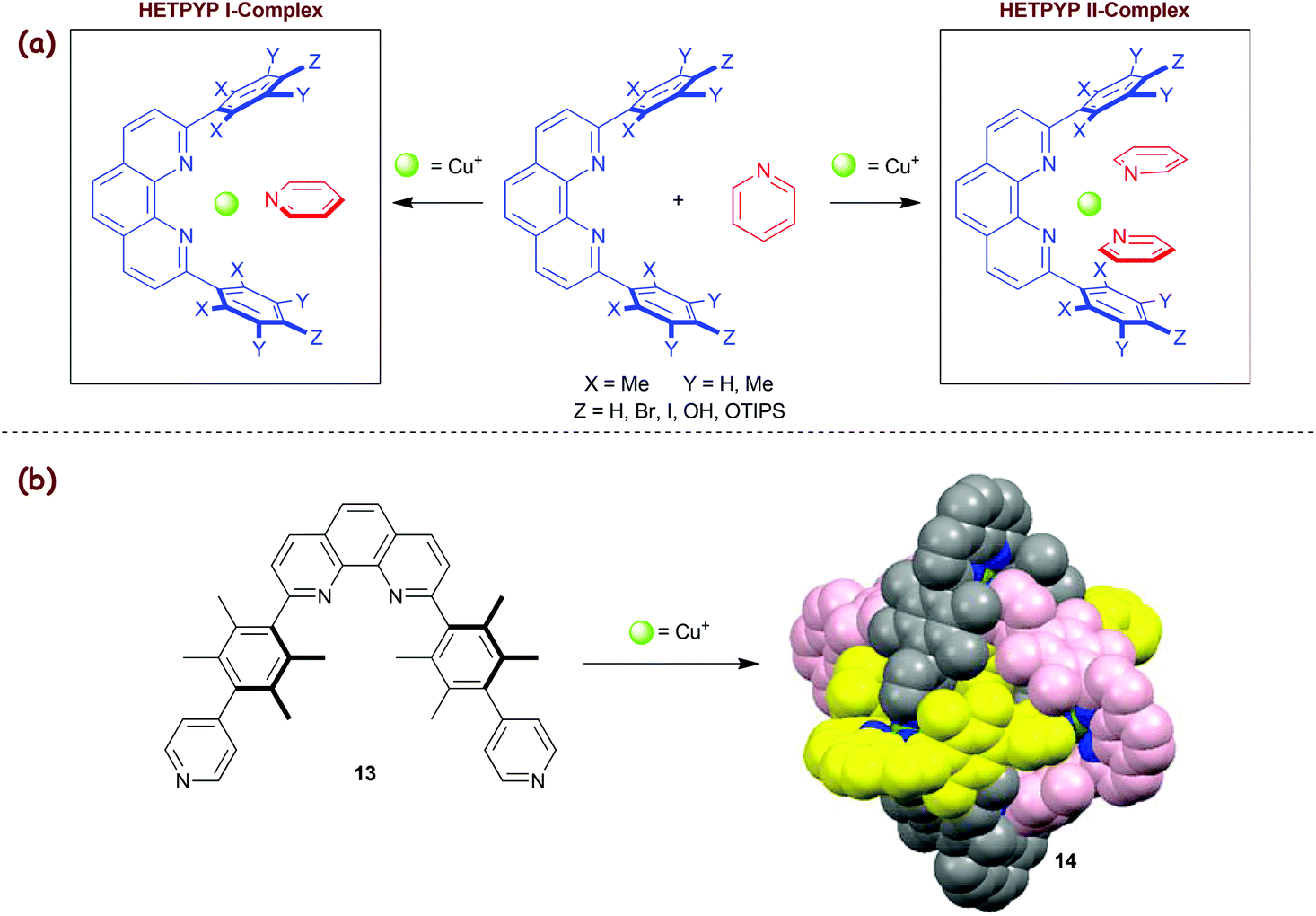
In 2009, we added the HETPYP-II concept43 (eroleptic ![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) ridine and henanthroline complexation) that allows attachment of two pyridine (py) ligands to a copper(I) centre yielding [Cu(phenAr2)(py)2]+ complexes. Lately, the concept has been extended to the HETPYP-I protocol (Scheme 4a),44,45 opening the way toward trigonal [Cu(phenAr2)(py)]+ units. As expected, HETPYP-I complexes (log β ≈ 8–8.5) are less stable than their HETPHEN and HETTAP analogues. Nevertheless, this complexation motif has proved its reliability in various supramolecules both in solution and in the solid state.45a For instance, various three-component racks, rectangles and trigonal prisms were recently prepared based on the HETPYP-I complexation.45b Similarly, the HETPYP-II motif was of use in several 2D and 3D-nanoarchitectures, again both in solution and in the solid state. For example, the aesthetically pleasing and topologically interesting Borromean cage14 was readily prepared from a 1:1 mixture of ligand 13 and Cu+ ions (Scheme 4b).43 While the formation of cage 14 is driven by maximum site occupancy and entropic constraints, an analogous cage was even synthesised without the HETPYP control.18b
ridine and henanthroline complexation) that allows attachment of two pyridine (py) ligands to a copper(I) centre yielding [Cu(phenAr2)(py)2]+ complexes. Lately, the concept has been extended to the HETPYP-I protocol (Scheme 4a),44,45 opening the way toward trigonal [Cu(phenAr2)(py)]+ units. As expected, HETPYP-I complexes (log β ≈ 8–8.5) are less stable than their HETPHEN and HETTAP analogues. Nevertheless, this complexation motif has proved its reliability in various supramolecules both in solution and in the solid state.45a For instance, various three-component racks, rectangles and trigonal prisms were recently prepared based on the HETPYP-I complexation.45b Similarly, the HETPYP-II motif was of use in several 2D and 3D-nanoarchitectures, again both in solution and in the solid state. For example, the aesthetically pleasing and topologically interesting Borromean cage14 was readily prepared from a 1:1 mixture of ligand 13 and Cu+ ions (Scheme 4b).43 While the formation of cage 14 is driven by maximum site occupancy and entropic constraints, an analogous cage was even synthesised without the HETPYP control.18b
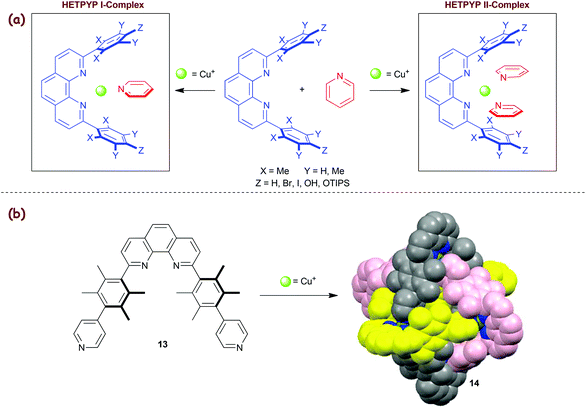 |
| | Scheme 4 (a) Illustration of HETPYP-I and II approaches. (b) Synthesis of the HETPYP-II based Borromean cage 14.43 | |
3. Multicomponent self-sorted nano-assemblies
Highly symmetrical architectures from multiple identical building blocks have shown their great utility in various examples of host–guest chemistry.4,46,47 To reach a higher degree of multifacetedness, though, multiple different components as well as multiple interactions are needed, because the latter may open the venue to functional aggregates with each component adding new functions (vide infra).20 In recent years, thus a good part of our efforts has been devoted to elaborate new multicomponent self-sorting protocols with a particular focus on completive processes (Fig. 4).22c In general, x-fold completive self-sorting describes a process in which x self-assembled entities form from a library containing n constituents by making quantitative use of all members.48 With respect to our systems, we desired to amalgamate two or more dynamic heteroleptic binding motifs in such a way that they remain orthogonal20 despite the fact that all motifs can easily undergo ligand and/or metal exchange reactions. Clearly, the required constitutional selectivity itself is not implemented in our heteroleptic toolkits (vide supra), as they are programmed at preventing the formation of homoleptic complexes, but at the onset do not provide the means of differentiating between various alternative heteroleptic combinations (Fig. 4). To address the latter issue, we probed x-fold completive self-sorting in a library of mononuclear cornerstones (Fig. 4, step I) until we were able to identify heteroleptic complexes that formed without interference. These motifs were later merged in multiligand building blocks for integrative (= 1-fold completive) self-sorting49 thus precluding any undesired side products (Fig. 4, step II). Undoubtedly, even in such an approach each individual component needs to be carefully instructed in such a way that sophisticated recognition patterns identify correct combinations while rejecting wrong arrangements.
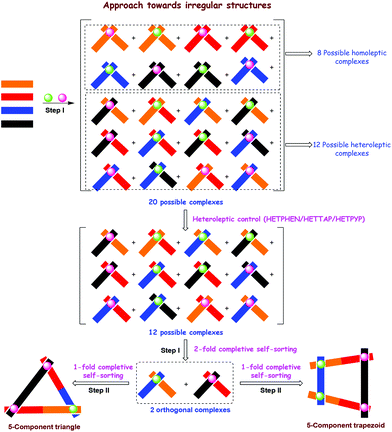 |
| | Fig. 4 Cartoon representation of 2-fold completive and 1-fold completive self-sorting processes. | |
Our methodology allows one to selectively fabricate geometrically and compositionally irregular supramolecules (Fig. 4) as a single species from a pool of communicative ligands and metal ions.20 Clearly, in the exemplarily shown triangle and trapezoid the diversity and complexity are largely increased in contrast to those of our previous oligonuclear three-component structures (chapter 2), because the latter are only based on one type of heteroleptic algorithm, e.g. either HETPHEN, HETTAP or HETPYP. Based on this strategy we are able to prepare metallocycles that utilise five to seven different components in their framework (vide infra, Schemes 5–7). However, to our appreciation, the approach as outlined in Fig. 4 holds promise for even larger dynamic structures encompassing more than seven components in the near future, quite a significant advancement in a field long dominated by two- or three-component structures.22b In the following, we will portray some of our recent examples by classifying them along an increasing number of heteroleptic algorithms.
3.1. Amalgamation of two heteroleptic motifs
Merging HETPHEN and N→zinc(II) porphyrin complexation.
In 2006, by demonstrating orthogonality between the HETPHEN and the well-known N→zinc(II) porphyrin coordination, we accomplished our first example of a 2-fold completive self-sorting.28b,32,50 The 2-fold self-sorting algorithm was highly useful to quantitatively prepare dynamic three-component heterometallic rectangles,28b four-component discrete porphyrin stacks such as 15 (Fig. 5),32 and a fullerene-porphyrin-Cu(phen)2-ferrocene tetrad.50 Notably, all structures are accessible by mixing the different components in any sequence as long as the stoichiometry is obeyed.
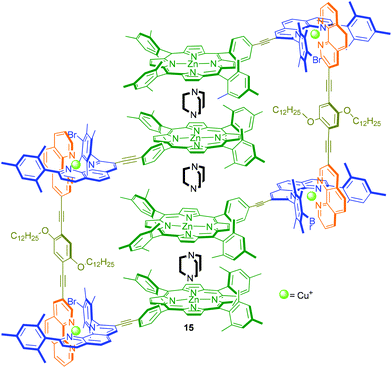 |
| | Fig. 5 Four-component porphyrins stack 15.32 | |
Merging HETPHEN and HETTAP complexation.
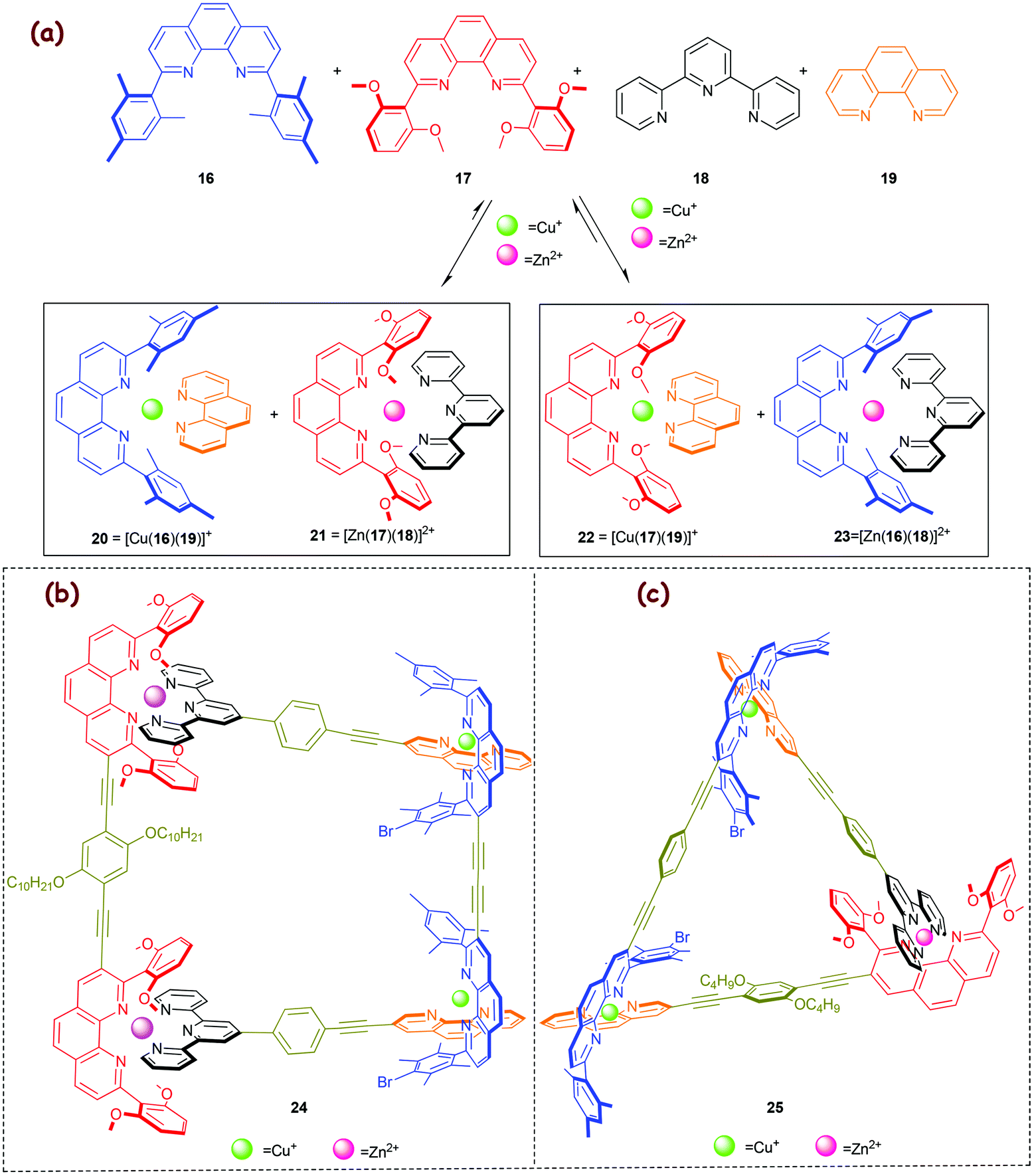
Lately, in 2009, we developed a 2-fold completive self-sorting22c process furnishing the HETPHEN and HETTAP complexes 20 and 21 in an interference-free manner (Scheme 5a).51 Considering the stability of other possible HETPHEN and HETTAP complexes, one expects at least the formation of four complexes, i.e., 20–23, from an equimolar mixture of ligands 16–19, Zn2+ and Cu+ ions. To our understanding, the observed selectivity toward 20 and 21 is guided by the correct stoichiometry, precise tuning of steric and electronic effects, π–π interactions, and metal-ion coordination specifics. With reference to copious reported complexes, we52 and others have stated that a Cu+ ion as a rule will be tetrahedrally coordinated by four donor atoms only – even if the surrounding ligands provide more than four. In contrast, Zn2+ has the ability to vary and expand its coordination number while preferring an octahedral geometry.37 Based on this fact, we reasoned that the additional Zn⋯OMe ion–dipole interaction(s) present in 21 would provide a suitable pseudo-octahedral geometry to the Zn2+ ions thus enthalpically (log β ≈ 14) supporting its formation while disfavouring the alternative HETTAP complex 23 (log β ≈ 12). At the accurate stoichiometry, the Zn2+ ions will make full use of ligands 17 & 18 to afford 21 and thus drive the formation of 20via maximum site occupancy and HETPHEN control. Interestingly, in a control experiment an equimolar mixture of ligands 16, 17, 19 and Cu+ ions did not contain the needed information to bias formation of 20 over that of 22, but instead produced a jumble of 20, 22 and free ligands 16, 17. To our appreciation, complex 21 thus behaves as a molecular chaperone that guides the 2-fold completive self-sorting process (Scheme 5a).
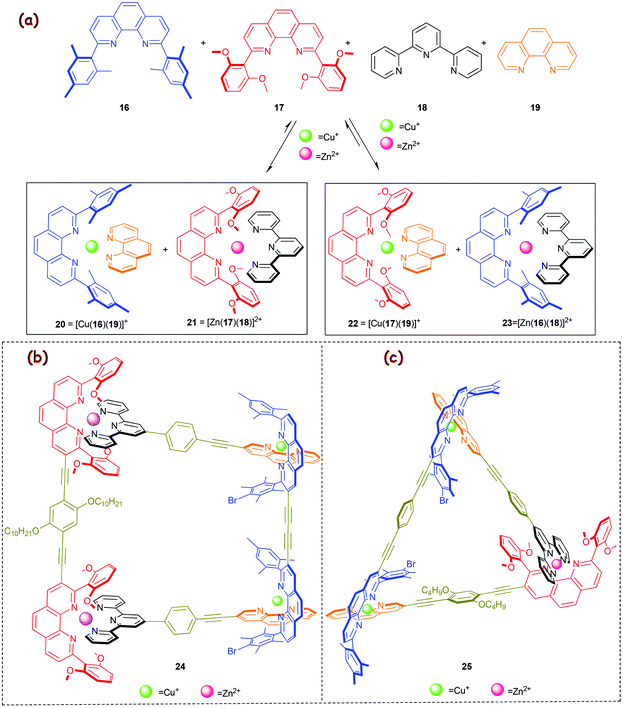 |
| | Scheme 5 (a) Merging HETPHEN and HETTAP approaches in a 2-fold completive self-sorting process.51 Five-component supramolecular (b) trapezoid 2451 and (c) triangle 2553 fabricated from 1-fold completive self-sorting. | |
The above 2-fold completive self-sorting protocol was first challenged in the fabrication of the five-component isosceles trapezoid 24 (Scheme 5b).51 Later, the same motifs were applied to construct the five-component triangle 25 (Scheme 5c).53,54
Merging HETPHEN and HETPYP-I complexation.
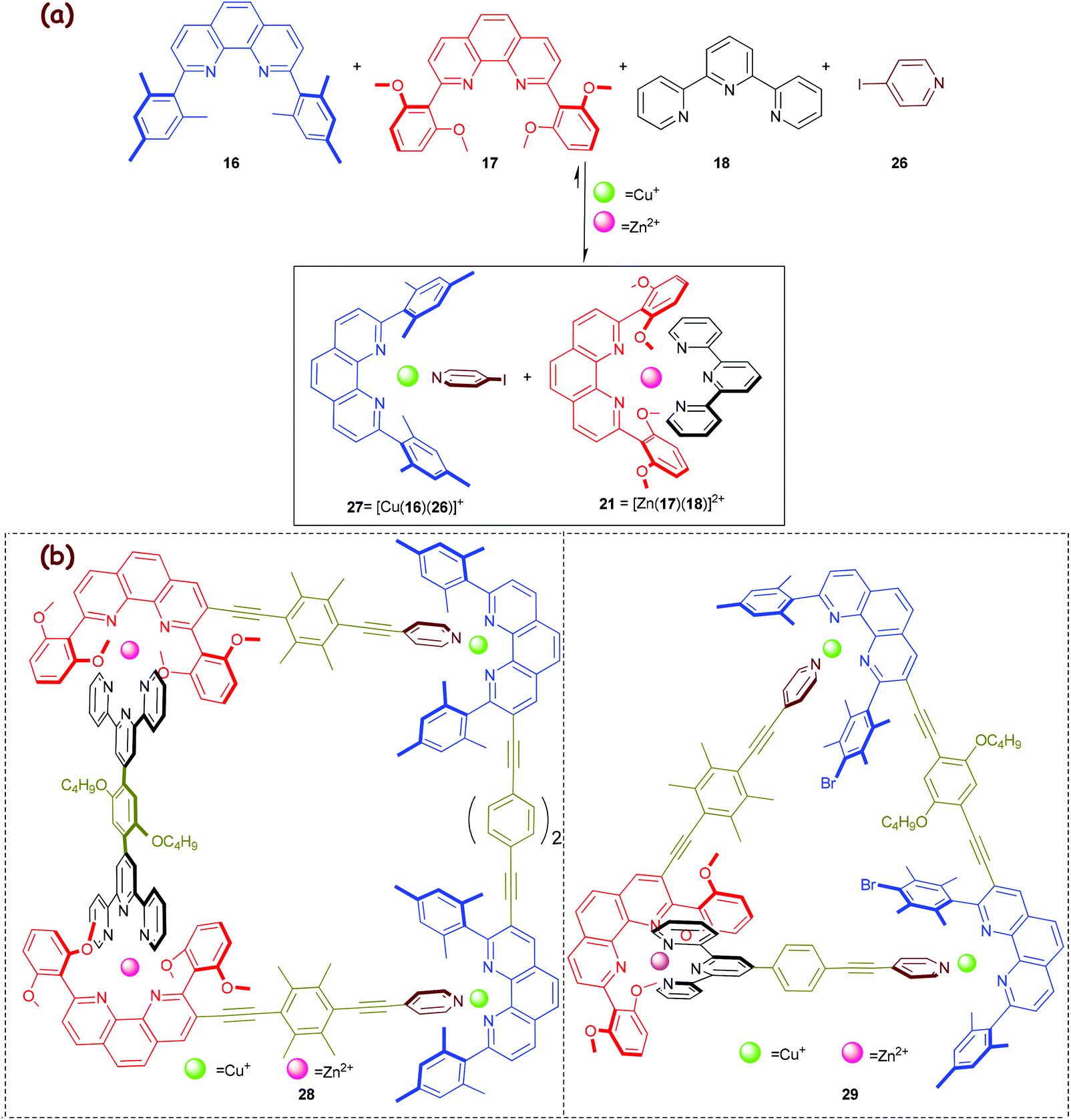
In 2013, we capitalised on the aforementioned molecular chaperone21 (Scheme 5) to direct a much more demanding self-sorting scenario toward the valence frustrated tri-coordinated HETPYP-I copper(I) complex 27 = [Cu(16)(26)](PF6) (log β ≈ 8) that is depicted in Scheme 6.55 To our delight, the higher stability of complex 21 (Δlog β ≈ 2) compared to that of the alternative complex 23 (Scheme 5a) is sufficient to drive the much weaker HETPYP-I complex 27 based on thermodynamic preferences.56 In a follow-up 1-fold completive self-sorting we efficaciously used both orthogonal motifs for the clean preparation of the five-component trapezoid 28 and triangle 29, with the weakly binding HETPYP-I motif contributing to 2 out of 4 (in 28) or 3 (in 29) cornerstones (Scheme 6). A comparison of trapezoids 24 (Scheme 5) and 28 (Scheme 6) reveals that the use of the Cs-symmetric HETPYP-I complex units in 28 avoids the formation of diastereomeric structures, thus greatly facilitating its characterisation via1H NMR.
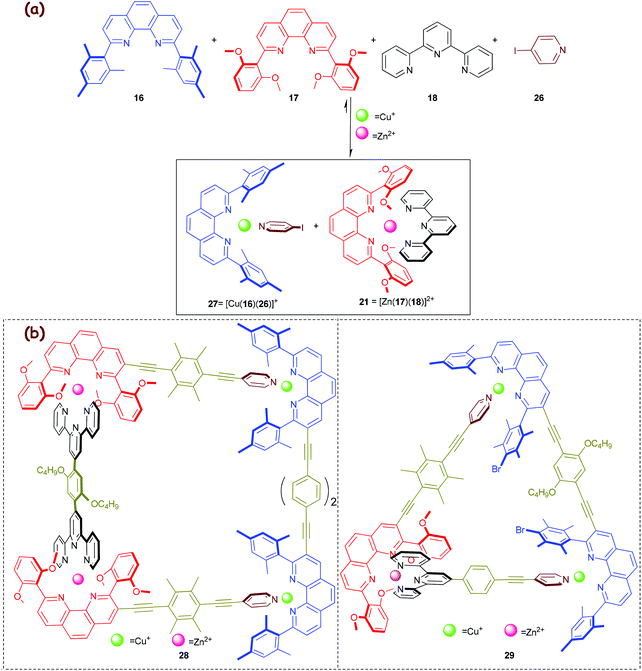 |
| | Scheme 6 (a) Exclusive formation of the weakly linked HETPYP-I complex 27 and the strongly associated HETTAP complex 21 from a 6-component library. (b) Chemical structures of the five-component supramolecular isosceles trapezoid 28 and triangle 29.55 | |
3.2. Merging HETPHEN, HETTAP and N→zinc(II) porphyrin complexation
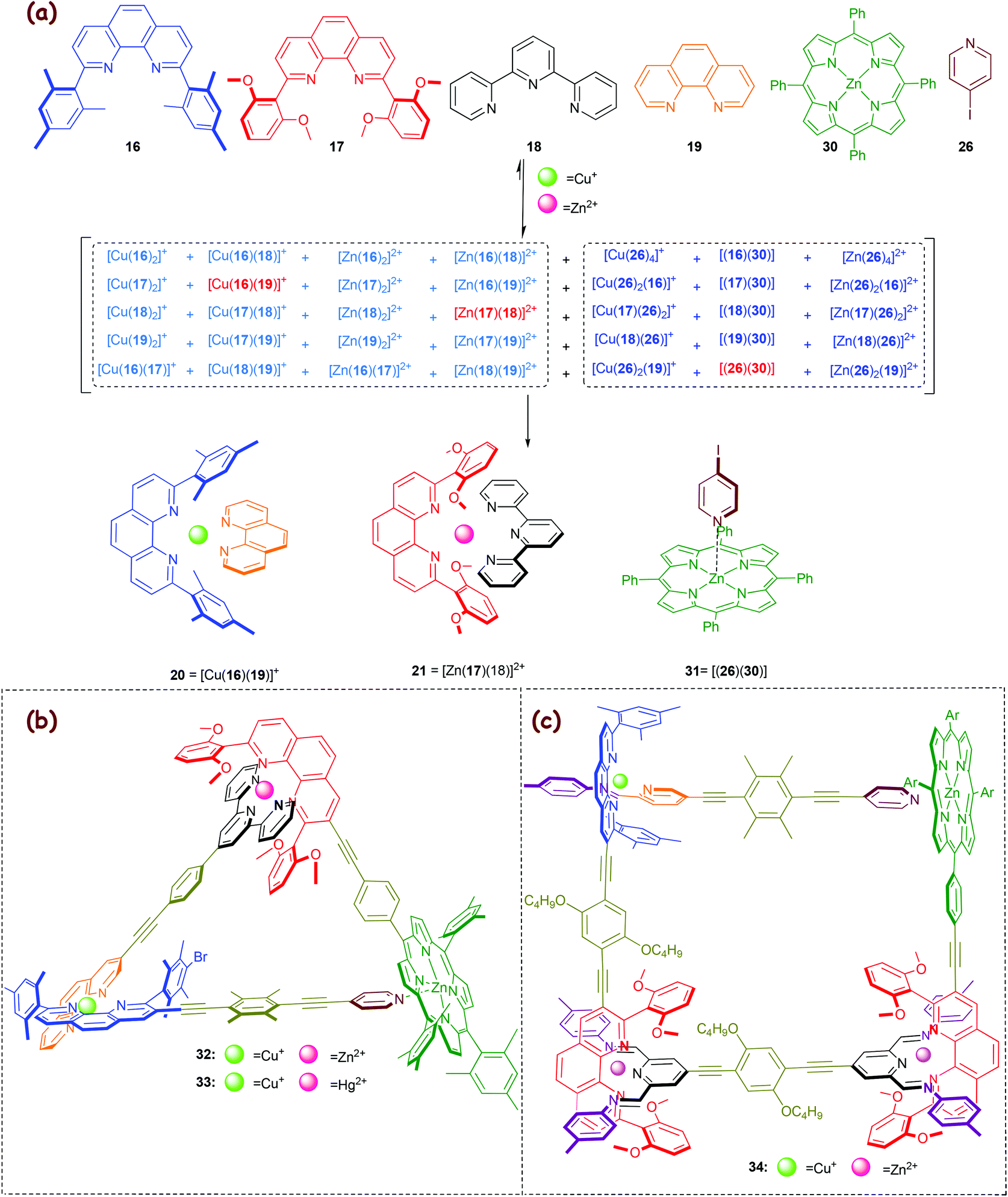
In 2010, a new eight-component library (Scheme 7a)57 was elaborated from the earlier six-component library (Scheme 5a), by juxtaposing the pyridine→zinc porphyrin coordination (log K ≈ 3.0–3.7) aside of the HETPHEN and HETTAP motifs. Orthogonality of the pyridine→zinc porphyrin motif 31 = [(26)(30)] with our heteroleptic complexes is readily implemented due to the optimal saturation of all ligands and metal ions in complexes 20, 21 and 31. Moreover, it is supported by the inability of the bi- or tridentate ligands 16–19 to interact with zinc porphyrin 30 due to high steric shielding. Noticeably, in the present library (Scheme 7a) only three of 35 possible complexes do form (3-fold completive self-sorting), while from our original library (Scheme 5) two complexes emerged out of twenty different combinations. Because detrimental cross-talk in general rapidly increases with an augmenting number of components,20 a more sophisticated molecular programming is required for any larger self-sorted library.49
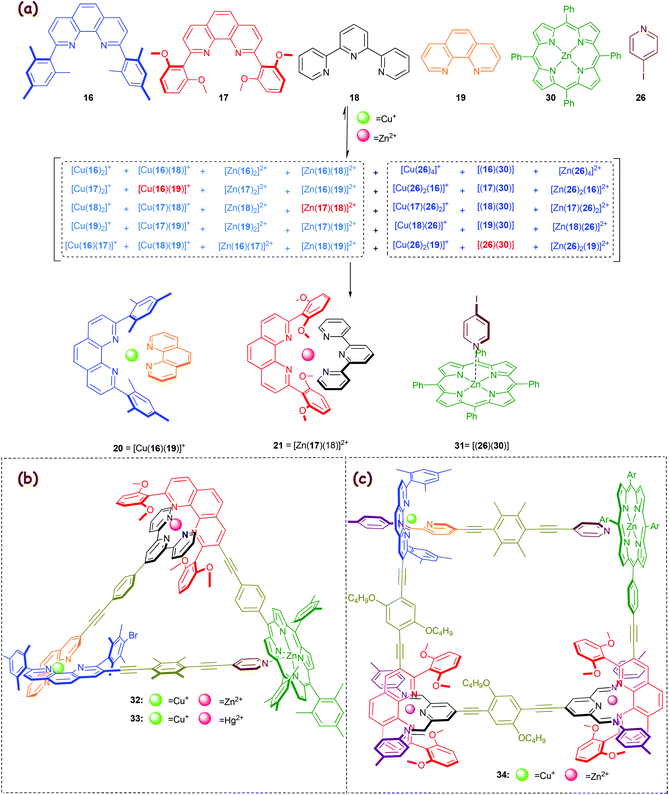 |
| | Scheme 7 (a) 3-Fold completive self-sorting in an eight-component library. (b) Chemical structure of the scalene triangles 32–3357 and (c) the scalene quadrilateral 34.37 | |
We exploited the three orthogonal complexes as three cornerstones in the scalene triangle 32 (Scheme 7b). Because Hg2+ ions form stronger HETTAP complexes than Zn2+ (Δlog β ≈ 2),36 addition of one equivalent of Hg2+ to 32 caused a metal displacement furnishing 33. To the best of our knowledge, 33 is the first trisheterometallic supramolecular scalene triangle with three different self-assembled corners.57
Very recently, we used the insight on constitutionally dynamic heteroleptic [M(phenAr2)(iminopyridine)]n+ complexes (Fig. 3 and Scheme 3b) in a 3-fold completive self-sorting as described in Scheme 7a for the fabrication of the scalene quadrilateral 34 that encompasses seven distinct components in its framework (Scheme 7c).37 To the best of our knowledge the quadrilateral represents the state-of-art in self-sorting of supramolecular entities, as the degree of self-sorting is much higher than that in any other case reported so far.
Summing up section 3, clearly the amalgamation of multiple similar and archetypically different interactions has been successful to access multicomponent structures of much higher complexity and diversity. Considering the present level of self-sorting, the scene is set for devising novel functional properties that emerge from spatial and electronic specifics in multicomponent aggregates.
4. Functions and applications
Implementation of emergent functions in multicomponent assemblies is currently one of the major challenges. Because any emergence arises only when all components find themselves in the proper spatiotemporal arrangement, it may not be directly predicted by considering the subcomponents in isolation.58,59 To this end, our heteroleptic toolkits with their high level of constitutional control in combination with self-sorting (vide supra) offers new chances for the rational design of multicomponent assemblies with tunable properties, as guided by compositional, constitutional and conformational changes. In recent years, our research has thus shifted toward functional nano-assemblies in various areas, as diverse as supramolecular splicing,60 metal ions sensing,38 nanoswitches,61 molecular electronics,62 host–guest chemistry,63 dynamic polymers,34cetc. In addition, our heteroleptic toolkits have served other groups64–67 to build nanorotors and solar cells among others. In the ensuing section, we will portray some of our recent systems that have already exhibited promising potential for future applications.
4.1. Guest adaptive frameworks
Over the past two decades, 3D metallosupramolecular hollow complexes of well-defined shapes and sizes have received frequent attention in the context of fascinating host–guest chemistry, as evidenced by impressive examples by Stang,7 Fujita,46 Raymond,46 Nitschke,4 Clever68 and many others.59 Recent highlights even include modulation of guest reactivity, cavity-controlled catalysis, gas storage, separation, etc. Quite often, such complex structures provide an adaptable cavity size that allows encapsulation of different guests in a regulated manner. Along this line, we described in 2008 how added guests regulate the shape of a supramolecular three-component assembly (Scheme 8) by providing either the nanoprism 39 = 38@37 or the constricted nanoframework 40 = C60@37.69 Explicitly, the desired nanoprism 37 = [Cu6(35)2(36)3]6+ is not exclusively assembled from its precursors without the strong supramolecular assistance from either 38 or C60, most likely due to the thermodynamically competitive formation of some small oligomeric aggregates. Clearly, the strong binding and good spatial match between the three pyridine nitrogens of 38 and the three zinc porphyrin units in 37 is the key to convert the dynamic mixture into a single species 39. The role of C60 is more remarkable as it forces 37 to adapt a smaller cavity size in order to maximise the porphyrin–fullerene interactions, affording an accordion-type host–guest system that wraps around C60, as demonstrated in Scheme 8.
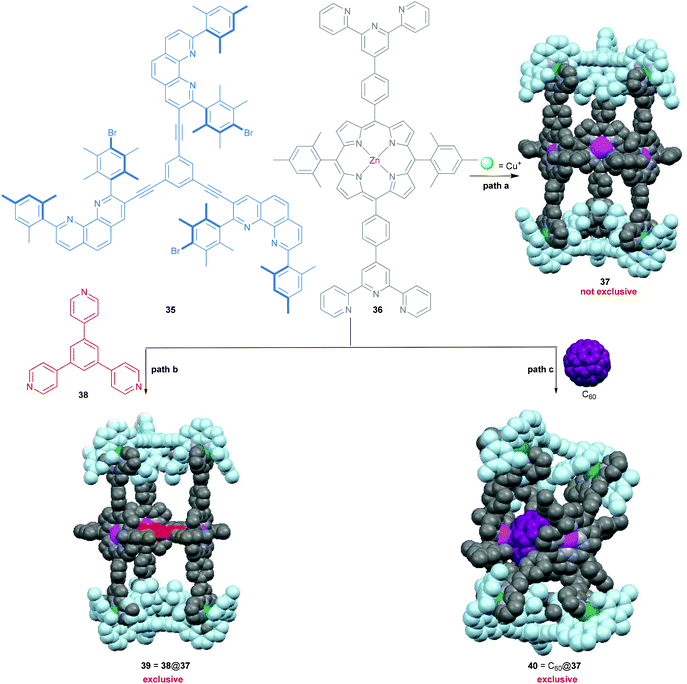 |
| | Scheme 8 Synthesis of the shape-adaptive nanoprism 37 = [Cu6(35)2(36)3]6+.69 | |
Lately, using the [Cu(phenAr2)(picolinaldehyde)]+ complexation motif, we fabricated a heteroleptic 3D metallosupramolecular tetragonal prism that behaves as a flexible and guest-adaptive host encapsulating either C60 or coronene.63
4.2. Metal exchange and fluorescence signalling
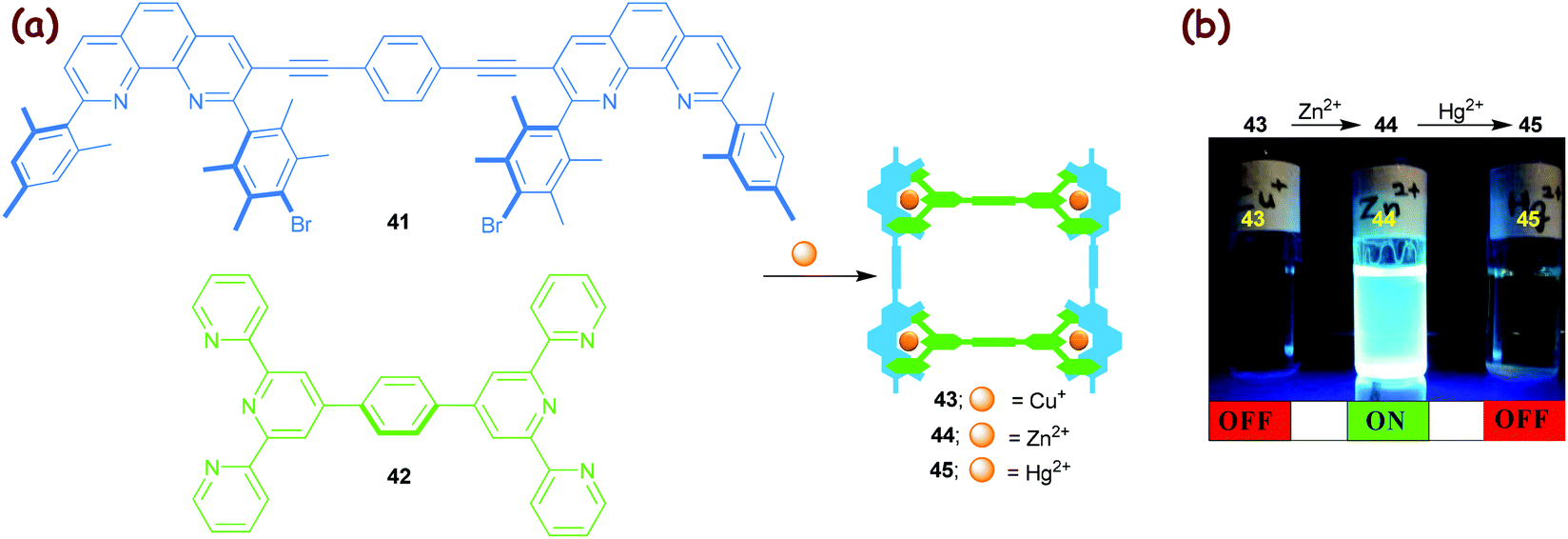
The diverse luminescence properties of polynuclear d10 metal complexes have found ample application.70 Our efforts in this area focused on a Cu+→Zn2+→Hg2+ transmetalation71 in a HETTAP nanoladder assembly that was readily recognised by the naked eye, as zinc(II) ladders have strong emissions in contrast to their copper(I) and mercury(II) analogues (Fig. 6).36 The protocol is easily understood considering the association constants that vary substantially for HETTAP complexes along the series: log β ≈ 9.3 (Cu+), 12.4 (Zn2+) and 14.7 (Hg2+). As a result, the transmetalation (Cu+→Zn2+→Hg2+) resulted in an OFF→ON→OFF emission response.
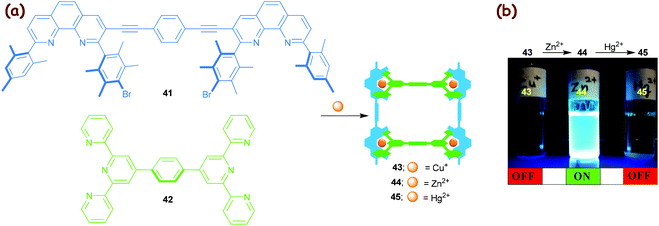 |
| | Fig. 6 (a) Synthesis of the three-component HETTAP ladders 43–45. (b) A two-step transmetalation OFF→ON→OFF fluorescence cascade. The zinc(II) ladder 44 shows a strong emission at λem = 463 nm (λex = 389 nm).36 The artwork in (b) is reprinted from ref. 36 with the permission of the American Chemical Society (Copyright 2005). | |
Capitalising on the above insight, we realised chemosensing with the zinc(II) based HETTAP clip 47 (Scheme 9),38 containing an oligoether unit at the periphery for metal cation recognition. Indeed, within the small series of cations (Na+, K+, Mg2+, Ca2+, Pb2+ and Ba2+), the strongest luminescence increase was found upon addition of Pb2+ ions (50% enhancement with respect to 47). In contrast, Hg2+ behaves ambivalently, as the luminescence strongly depends on its relative amount. Upon substoichiometric addition, the luminescence intensity of clip 47 was first increased (∼20% to 30%) but then became constant upon addition of just 1 equiv. of Hg2+. Further addition of Hg2+ efficiently quenched the luminescence. To our understanding, the increase of the luminescence may be assigned to an association of the metal ions with the oligoether tether of 47 (Scheme 9b). A second mechanism operates for Hg2+ at higher concentration. The decrease in luminescence is due to a Zn2+→Hg2+ exchange process in 47, analogous to that in Fig. 6b, which leads to quenching. The clip 47 thus exhibits a highly selective response toward Hg2+ due to supramolecular effects.
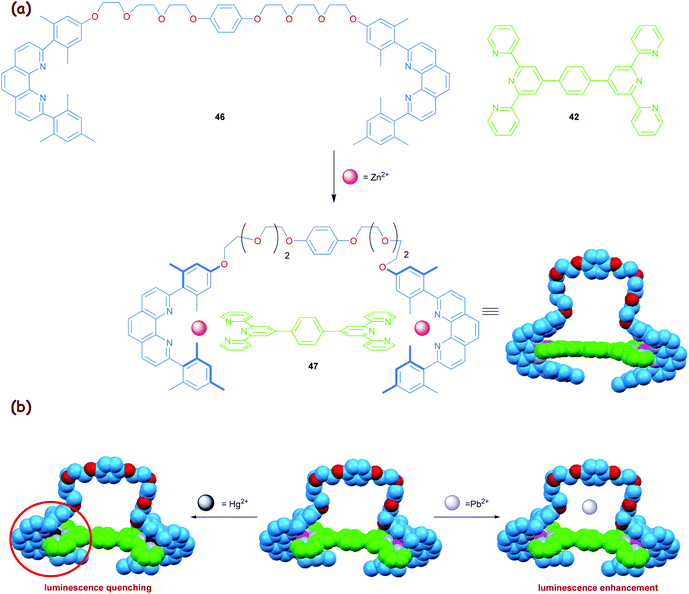 |
| | Scheme 9 (a) Synthesis of clip 47 and (b) its metal ions sensing by a luminescence probe.38 | |
4.3. Supramolecular fusion
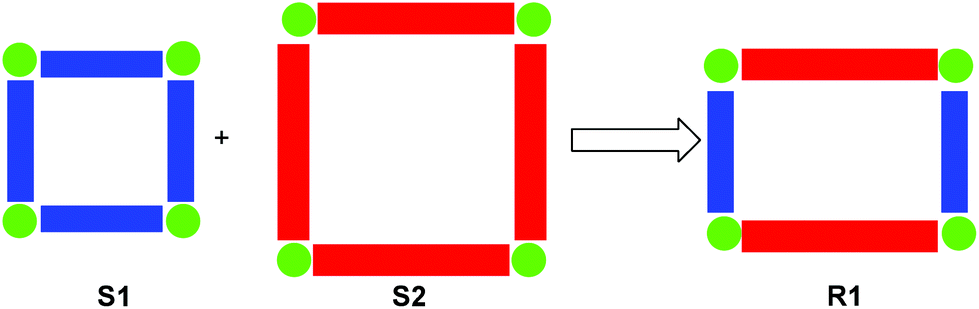
Stimuli-triggered supramolecular rearrangements have attracted continued attention due to their critical importance in both biological systems72 and toward the preparation of sophisticated molecular machines and switches73 (vide infra). At present, the current generation of switchable assemblies mainly involves two-state self-assembled aggregates, where the connectivity patterns are altered by switching molecular binding properties through chemical, photochemical or electrochemical inputs. In this modus operandi, self-assemblies are quite often either disassembled upon stimulation (e.g., pseudorotaxanes and tetrahedra) or only toggled within the same architectural manifold.74 In contrast, Nature elegantly uses gene shuffling, i.e. combination of dissimilar genes, to produce new emergent genes.75 To our understanding, as the original supramolecules already represent some chemical information, the implementation of an analogous supramolecular transformation may result in an evolution toward higher information content. For example, consider a simple case where two square architectures are fused into a rectangle, i.e.S1 + S2 → R1 (Fig. 7). The precursor squares are fully defined by the length of one side, while the description of the rectangle requires two pieces of information, i.e. two different lengths. Clearly, in such processes the diversity and complexity will be increased by enlarging the number of different components.
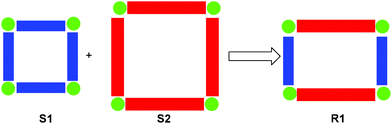 |
| | Fig. 7 Cartoon representation of a supramolecular fusion. | |
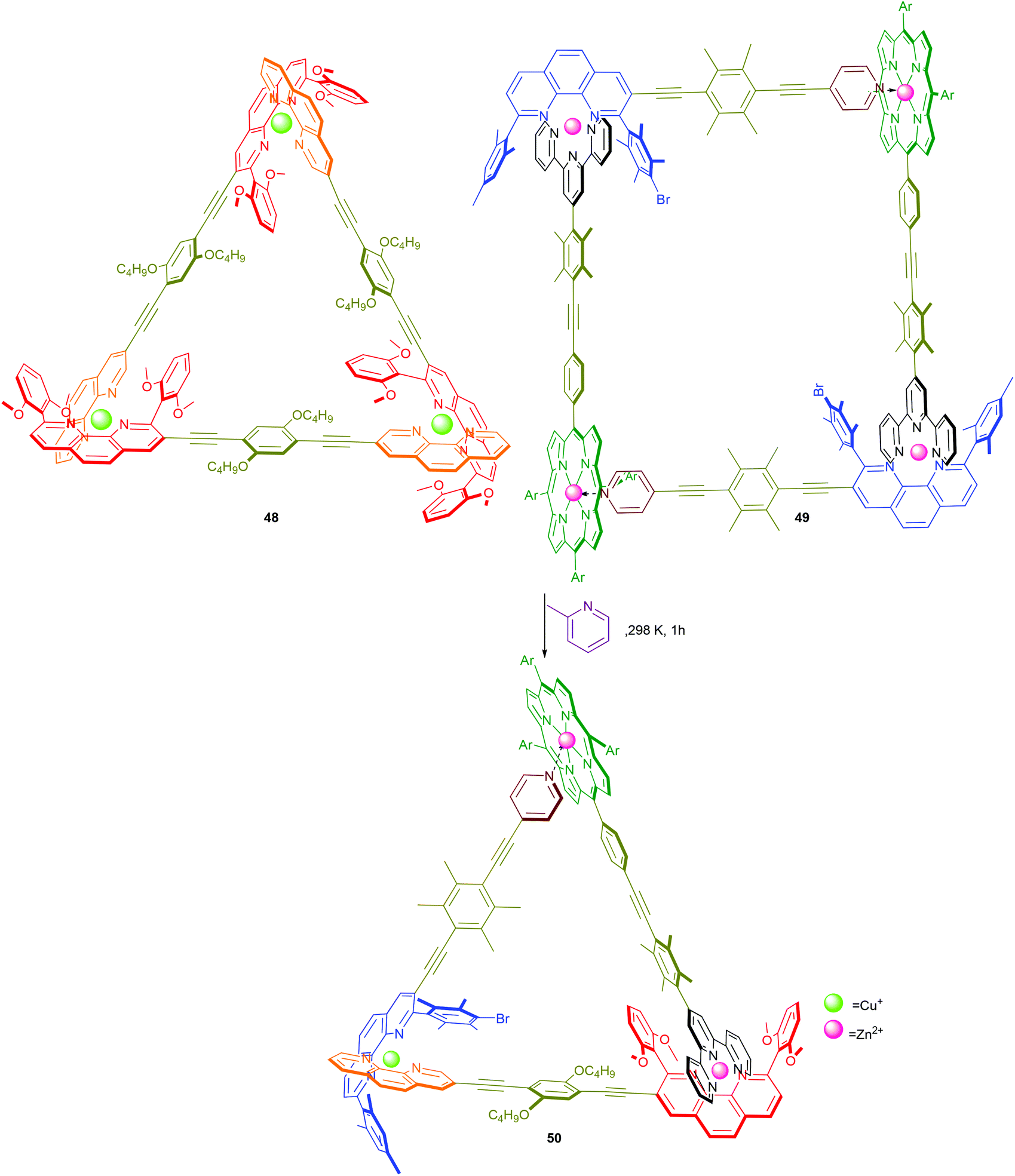
To this end, initial proof-of-concept experiments in our laboratories established the fabrication of the 5-component scalene triangle 50 by a spontaneous or catalytic supramolecular fusion of two distinct and dynamic supramolecules, namely equilateral triangle 48 and rectangle 49 (Scheme 10).60 Because the metal corners are thermodynamically less favoured in both 48 and 49 (Scheme 7), upon mixing the components re-shuffle to form the more favourable assembly 50. Whilst the spontaneous process takes 15 h at room temperature, the transformation is completed within 1 h in the presence of catalytic amounts of 2-methylpyridine. To our evaluation, the latter transformation comprises several distinct chemical events, including (i) favoured pair-selection due to a high fidelity 3-fold completive self-sorting (Scheme 7); (ii) error-correction under thermodynamic control; and (iii) acceleration of a supramolecular fusion reaction via labilisation of the metal–ligand bonds.
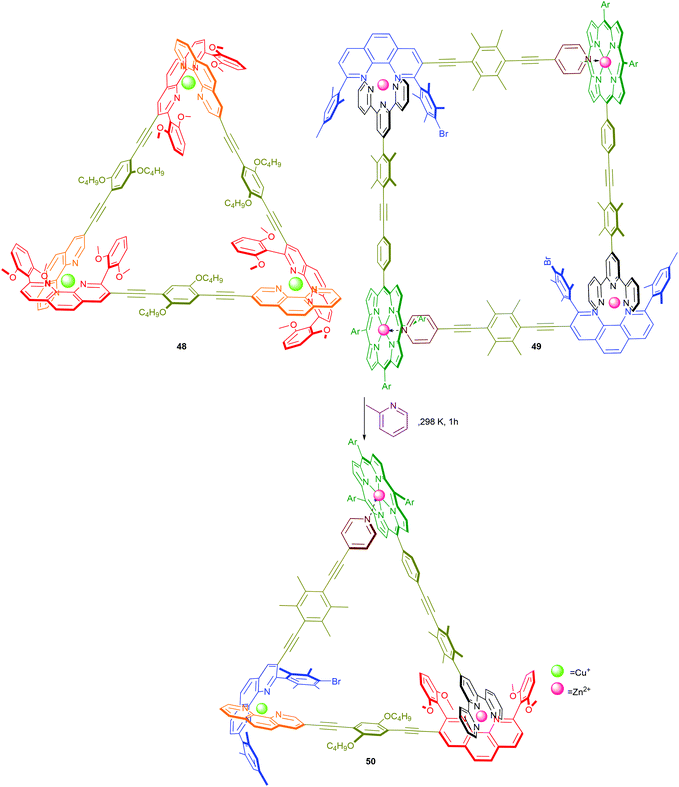 |
| | Scheme 10 Catalytic fusion of 48 and 49 to 50.60 | |
4.4. Multiple-stranded dynamers
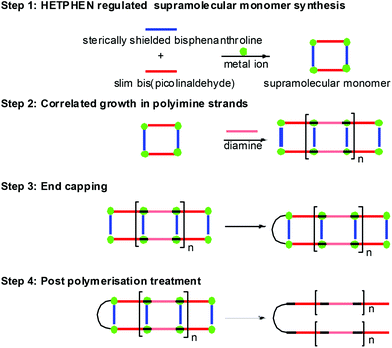
With the growing interest in functional supramolecular materials, the art of dynamer synthesis6 has become an escalating field of research in the last two decades. Consequently, many effective synthetic approaches have been developed that provide easy access to single-stranded dynamers.76 In contrast, multiple-stranded dynamers are hitherto extremely rare because of preparative problems as well as serious characterisation difficulties. With the aim of developing a ready-to-use synthetic methodology for multiple-stranded dynamers we recently developed the Template Directed Constitutional Dynamic Polymerisation (TD-CDP) approach34c (Fig. 8) based on constitutional dynamic chemistry and heteroleptic aggregation (Fig. 3). In Fig. 8, the full process is depicted in a pictorial manner for a difunctional supramolecular template.
 |
| | Fig. 8 Cartoon representation of the TD-CDP approach for a difunctional supramolecular template. | |
To illustrate the conceptual idea in a schematic manner, consider HETPHEN-based supramolecular aggregates (Step 1) that may act as sophisticated “monomeric units” in the TD-CDP. With their reactive aldehyde terminals they are perfectly suited for constitutionally dynamic polyimine formation in the presence of suitable diamines (step 2). Most importantly, the polymerisation step intrinsically bears the promise for effective control of the parallel growth of two, three or even more polymer strands, as all strands are expected to grow at the same pace. The metal phenanthrolines have a triple role. They act as a supramolecular template, catalyse the reversible iminopyridine formation and finally bind them as ligands thus setting up two, three or more parallely growing polymer strands. For example, elongation at any arbitrary strand, e.g. ‘strand one’, will preorganise the template such that the second and following strands will profit from cooperative effects in their elongation step. Hence, due to the dynamic heteroleptic coordination at the terminus of the growing polymer, a template with n phenanthroline sites will support parallel growth of n polymer strands in high fidelity. Finally, to evaluate the correlated growth of strands in multi-stranded polymers, end capping at one end of the multi-stranded dynamer (step 3) followed by a reductive post-polymerisation treatment of the resulting dynamic polymer (step 4) was required to kinetically lock and liberate the polymer from the supramolecular scaffold.
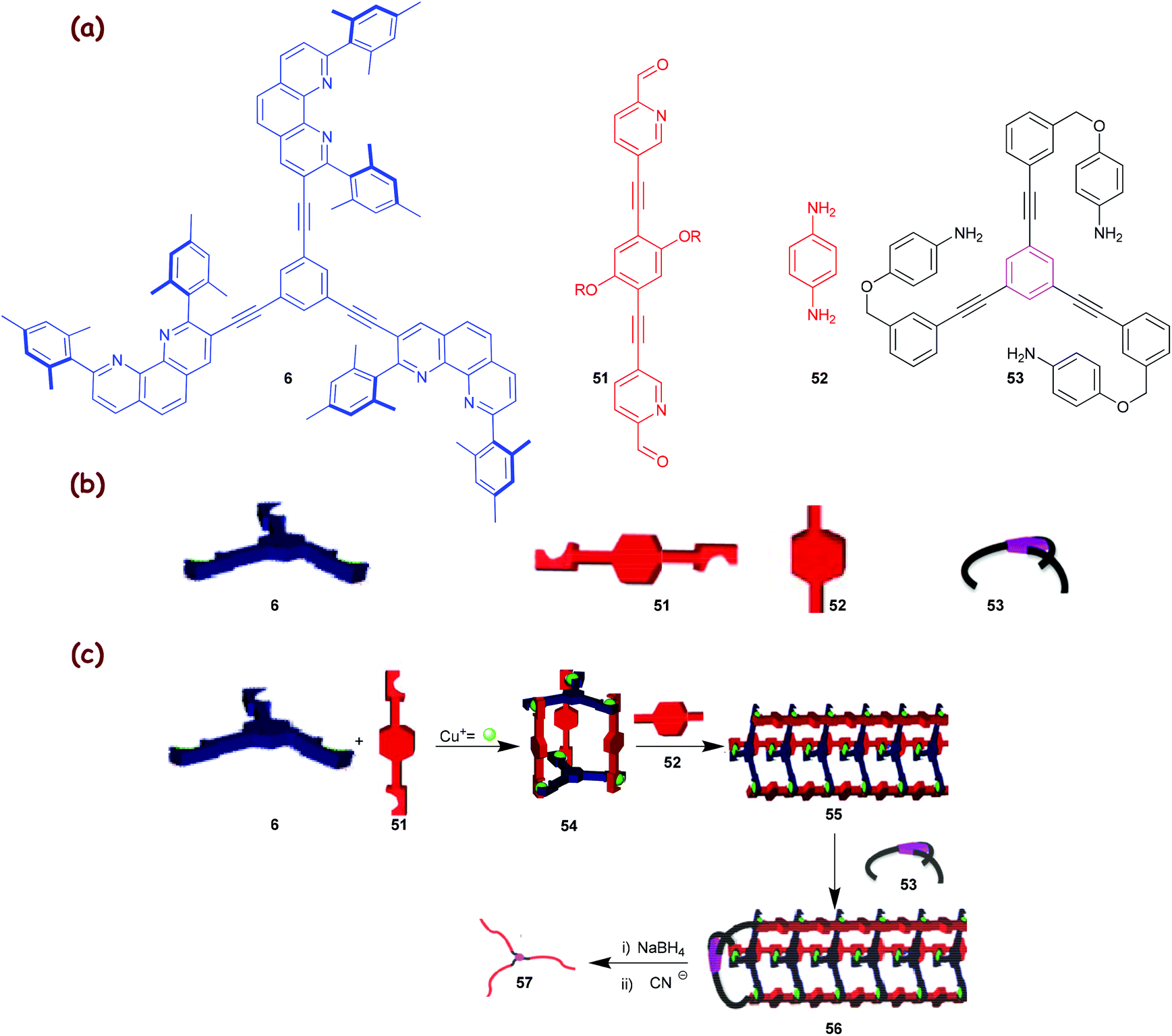
The principle, laid out in Fig. 8, opens many options for the preparation of compounds and materials with uncommon topology, such as supramolecular nanoscale cocoons, ribbons and filaments or polymers with a high constitutional fidelity. The methodology should also allow the preparation of bicyclic, polycyclic and star polymers with identical lengths of all strands, with the prospect that these yet unknown polymeric materials may convey interesting properties. In a recent communication, we demonstrated the usefulness of our concept developing dynamer 55 (Scheme 11) with three strands and its transformation into the covalent star polymer 57 that has arms of similar length (∼72%) as demonstrated by AFM.34c
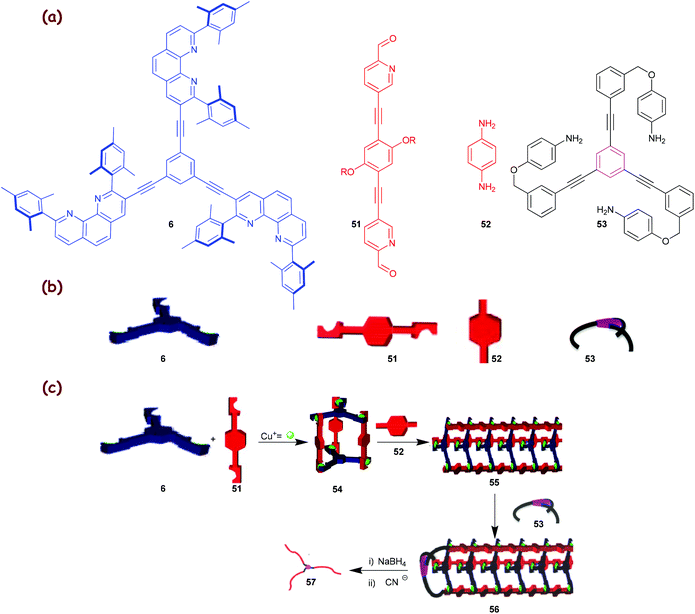 |
| | Scheme 11 (a) Chemical structures of ligands 6 and 51–53; (b) their cartoon representations; (c) the TD-CDP approach with a trigonal nanocage 54.34c The artwork in Scheme 11 is reprinted from ref. 34c with the permission of the American Chemical Society (Copyright 2012). | |
4.5. Novel nanoswitches
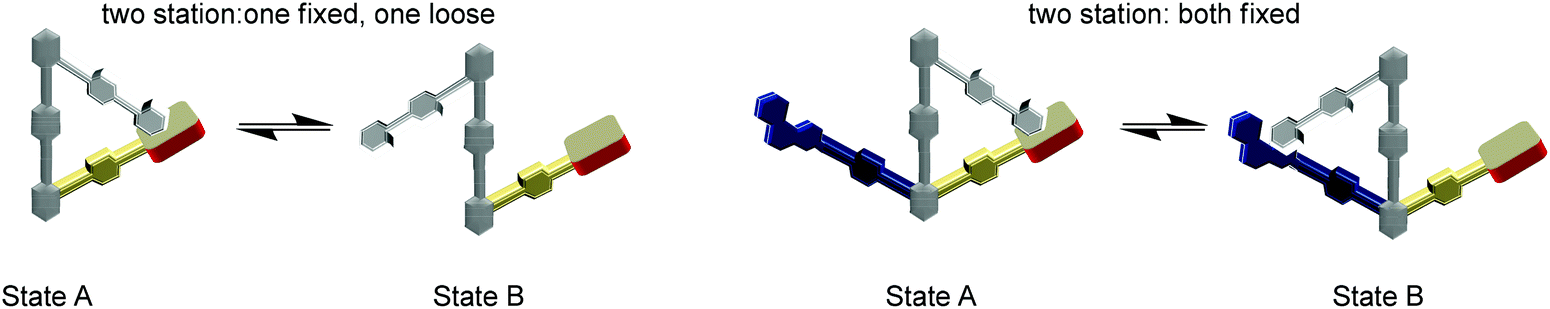
There is no life without molecular switching, as it plays a crucial role in almost all aspects of our existence.77 For example, many important biological machines (F1-ATPase, kinesin, RNA polymerase, helicases, etc.) rely on immense structural changes as a basis for their action. The structural changes actually provide the required electronic and steric alterations in the active site that empower the desired action. Thus, all machines must receive clear ON/OFF signals from the signalling network of the organism to control their action.78 Over the years, the modus operandi of the biological machinery has been serving as a constant source of inspiration for the design of prototypical artificial switches and machines, with the goal to mimic at least some part of the biological complexity. Our endeavours in this direction have led to two-stage rotary nanoswitches with a triangular architecture (Fig. 9) that can be reversibly and repeatedly toggled to control a chemical reaction in an ON and OFF manner.61
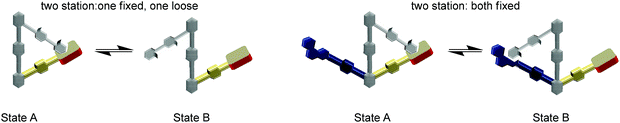 |
| | Fig. 9 Development of new rotary nanoswitches. | |
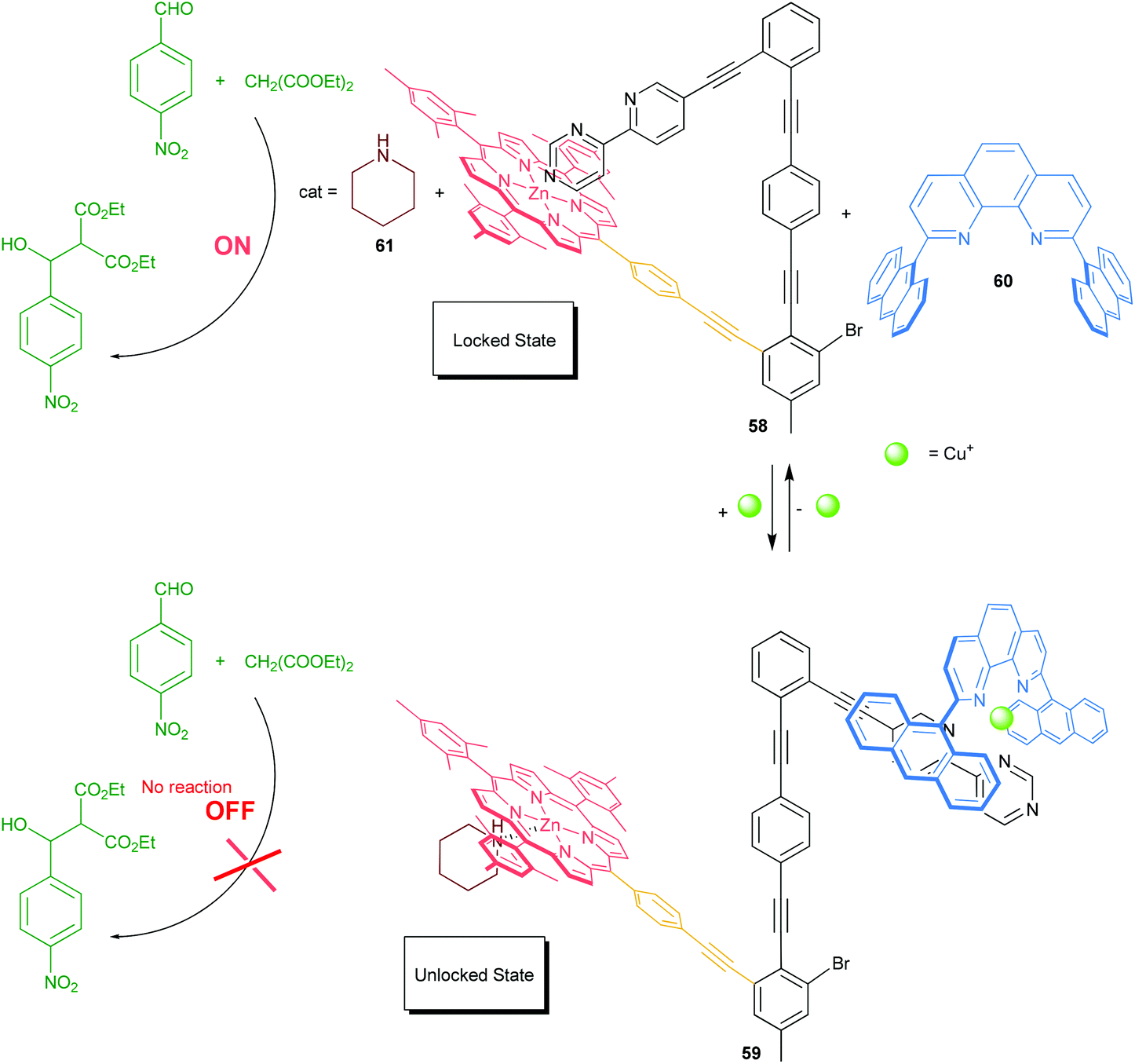
In 2012, we reported on the chemically triggered self-locking nanoswitch 58 and its reversible locking and unlocking, which mimics in part the working principle of CaMKII (Scheme 12).61a To serve the purpose, we implemented a sophisticated azabipyridine unit into the nanomechanical arm that may engage in the formation of either a pyridine→zinc(II) porphyrin motif (locked state) or HETPHEN-like [Cu(phenanthroline)(bipyridine)]+ motif (unlocked state). Originally, the nanoswitch 58 is locked at the azabipyridine→zinc(II) porphyrin linkage, but switching is possible by addition or removal of [Cu(60)]+ ions. The switching of 58 was exploited to reversibly toggle ON–OFF an organocatalytic Knoevenagel addition. As depicted in Scheme 12, in the ON state, the strong intramolecular association at the pyridine→zinc(II) porphyrin linkage prevents piperidine (61) from binding to the zinc(II)-porphyrin unit of the switch. Addition of [Cu(60)]+ ions as an input signal generates the heteroleptic complex 59 = [Cu(58)(60)(61)]+ liberating the arm from the zinc porphyrin station in 58. The liberated zinc porphyrin binds strongly to piperidine (61) preventing its function as an organocatalyst. Lately, we refined the approach by merging the shielded phenanthroline unit into the switch. The modified switch acts as a photocatalyst for the isomerisation of diazastilbene.61b
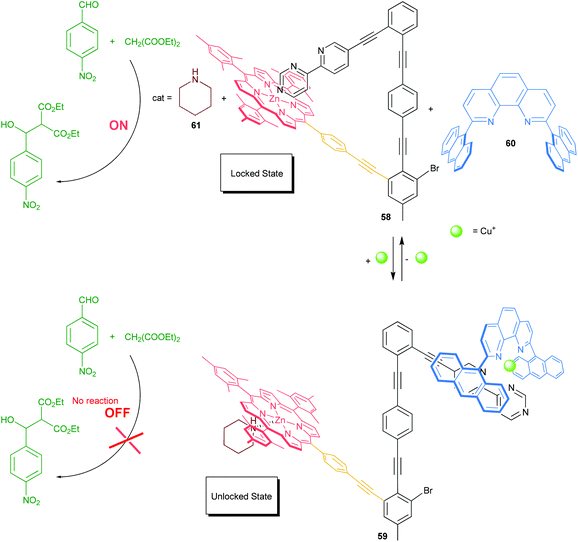 |
| | Scheme 12 Organocatalysis toggled ON/OFF with nanoswitch 58.61a | |
4.6. Novel supramolecular nanorotors
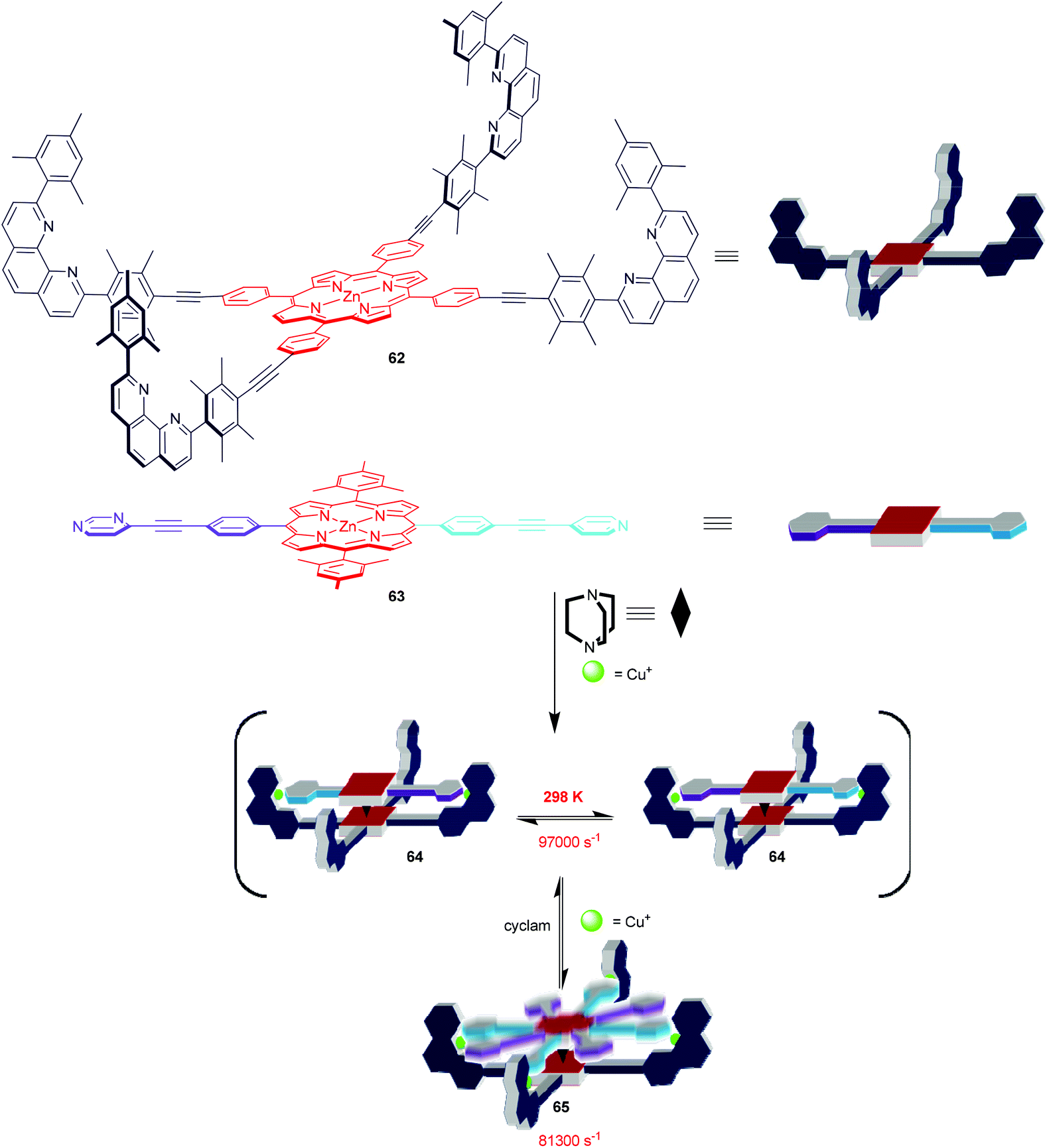
The rational design and synthesis of multicomponent supramolecular machines constitute a fascinating challenge in the field of nanoscience.5 Indeed, a huge majority of reports cover covalent machines, while fully dynamic and artificial supramolecular devices, like Stoddart's supramolecular valves,79a Aida's nanomechanical tweezers79b or Shionoya's double-ball bearing complexes,79c constitute rare examples in a wide terra incognita. Adding to the latter field, we recently elaborated a family of four-component supramolecular nanorotors that undergo fast (≥78000 s−1) stochastic rotary oscillations at 298 K (Scheme 13) but that are virtually stopped (≤18 s−1) at 198 K.80 Remarkably, both kinetic and thermodynamic data suggest the spinning motion in the rotors to take place solely by an intrasupramolecular pathway (>99.9%) despite operating across a dynamic hinge. For instance, the nanorotor 64 (Scheme 13) was designed based on two orthogonal coordination motifs, i.e. a HETPYP-I type and a DABCO→zinc(II) porphyrin complexation. Reacting the two different zinc(II) porphyrins 62 & 63, DABCO as a dynamic hinge and copper(I) ions in a ratio of 1:1:1:2 furnished quantitatively the self-assembled nanorotor 64 with platform 62 representing the stator and 63 the rotator. To our understanding, the two HETPYP-I units in 64 actually ensure and guide the formation of the DABCO-bis(porphyrin) hetero-sandwich. In nanorotor 64, the rotator 63 is spinning across the dynamic axle in 180° steps, while upon feeding the unloaded phenanthroline stations with Cu+ ions thus generating nanorotor 65, the oscillating rotation will proceed via mixed 90°/180° steps. A kinetic analysis reveals that depending on the mode of rotation the speed of the spinning motion is different and that speed regulation is thus possible through a reversible 64 ⇌ 65 nanorotor transformation.
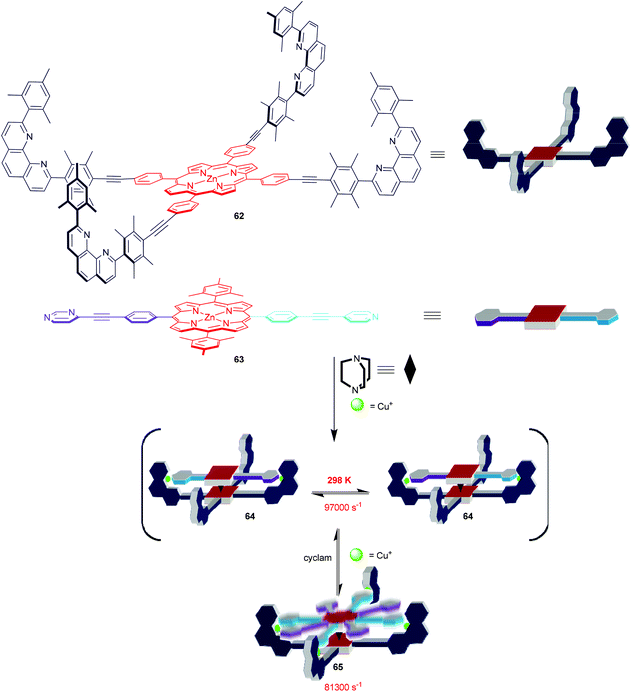 |
| | Scheme 13 Self-assembly of 4-component nanorotors 64 and 65.80 | |
5. Conclusion
Ever since the birth of supramolecular chemistry, achieving superior structural features as well as efficacious functions analogous to those in natural systems has been an immense challenge for chemists. To this end, our heteroleptic self-assembly protocols together with self-sorting represent a powerful tool for constructing demanding multicomponent supramolecules. Based on these newly available construction tools, we have elaborated in recent years a wide range of diverse multicomponent assemblies that exhibit interesting properties for various fields of application, such as smart materials, sophisticated nanomachines, switchable catalysis, etc. Efforts to extend our coordination tools even further to prepare aggregates with more than 7 components will be the focus of our laboratory in the years to come.
Acknowledgements
We are indebted to the Deutsche Forschungsgemeinschaft (Schm 647/15-1 and 19-1), the Alexander von Humboldt foundation and the University of Siegen for financial support over many years. Moreover, we are most grateful to all coworkers and collaborators who contributed with great ideas, dedicated work and many publications for the advancement of our research in this field.
References
-
(a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89 CrossRef;
(b) J.-M. Lehn, Science, 1993, 260, 1762 CAS;
(c) D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229 CrossRef CAS.
- P. J. Stang, J. Am. Chem. Soc., 2012, 134, 11829 CrossRef CAS PubMed.
-
(a) S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1 CrossRef CAS PubMed;
(b) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476 RSC.
-
(a) M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656 RSC;
(b) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC.
-
(a) J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814 CrossRef CAS;
(b) T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171 CrossRef CAS PubMed;
(c) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed;
(d) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC.
- M. Schmittel and K. Mahata, Angew. Chem., Int. Ed., 2008, 47, 5284 CrossRef CAS PubMed.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555 RSC.
-
(a) R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef;
(b) J.-M. Lehn and A. V. Eliseev, Science, 2001, 291, 2331 CrossRef CAS PubMed.
-
(a) J.-P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1985, 107, 6108 CrossRef CAS;
(b) J.-F. Nierengarten, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375 CrossRef CAS;
(c) J. Frey, C. Tock, J.-P. Collin, V. Heitz, J.-P. Sauvage and K. Rissanen, J. Am. Chem. Soc., 2008, 130, 11013 CrossRef CAS PubMed.
-
(a) B. Hasenknopf, J.-M. Lehn, G. Baum and D. Fenske, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1397 CrossRef CAS PubMed;
(b) V. C. M. Smith and J.-M. Lehn, Chem. Commun., 1996, 2733 RSC.
- M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383 CrossRef CAS.
- L. Zhao, B. H. Northrop, Y.-R. Zheng, H.-B. Yang, H. J. Lee, Y. M. Lee, J. Y. Park, K.-W. Chi and P. J. Stang, J. Org. Chem., 2008, 73, 6580 CrossRef CAS PubMed.
-
(a) K.-W. Chi, C. Addicott, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 16569 CrossRef CAS PubMed;
(b) Y.-R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873 CrossRef CAS PubMed.
- M. Schmittel and V. Kalsani, Top. Curr. Chem., 2005, 245, 1 CAS.
-
(a) S. Ghosh, D. R. Turner, S. R. Batten and P. S. Mukherjee, Dalton Trans., 2007, 1869 RSC;
(b) O. V. Dolomanov, A. J. Blake, N. R. Champness, M. Schröder and C. Wilson, Chem. Commun., 2003, 682 RSC.
-
(a) D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6 CrossRef CAS PubMed;
(b) Q. Zhang, J. Ding, Y. Cheng, L. Wang, Z. Xie, X. Jing and F. Wang, Adv. Funct. Mater., 2007, 17, 2983 CrossRef CAS.
- M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860 RSC.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
-
(a) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed;
(b) K. Osowska and O. Š. Miljanić, Synlett, 2011, 1643 CAS;
(c) M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651 RSC.
- M. Schmittel and A. Ganz, Chem. Commun., 1997, 999 RSC.
- M. Schmittel, A. Ganz, D. Fenske and M. Herderich, J. Chem. Soc., Dalton Trans., 2000, 353 RSC.
-
(a) M. Schmittel, A. Ganz, W. A. Schenk and M. Hagel, Z. Naturforsch., B: Chem. Sci., 1999, 559 CAS;
(b) S. De, S. Pramanik and M. Schmittel, Dalton Trans., 2013, 42, 15391 RSC.
- M. Schmittel and K. Mahata, Chem. Commun., 2008, 2550 RSC.
- M. Schmittel, A. Ganz and D. Fenske, Org. Lett., 2002, 4, 2289 CrossRef CAS PubMed.
-
(a) M. Schmittel, H. Ammon, V. Kalsani, A. Wiegrefe and C. Michel, Chem. Commun., 2002, 2566 RSC;
(b) R. S. K. Kishore, V. Kalsani and M. Schmittel, Chem. Commun., 2006, 3690 RSC.
- M. Schmittel, V. Kalsani, D. Fenske and A. Wiegrefe, Chem. Commun., 2004, 490 RSC.
-
(a) M. Schmittel, V. Kalsani and L. Kienle, Chem. Commun., 2004, 1534 RSC;
(b) M. Schmittel, V. Kalsani and J. W. Bats, Inorg. Chem., 2005, 44, 4115 CrossRef CAS PubMed.
- V. Kalsani, H. Bodenstedt, D. Fenske and M. Schmittel, Eur. J. Inorg. Chem., 2005, 1841 CrossRef CAS.
- R. S. K. Kishore, T. Paululat and M. Schmittel, Chem.–Eur. J., 2006, 12, 8136 CrossRef CAS PubMed.
- V. Kalsani, H. Ammon, F. Jäckel, J. P. Rabe and M. Schmittel, Chem.–Eur. J., 2004, 10, 5481 CrossRef CAS PubMed.
-
(a) J. Fan, J. W. Bats and M. Schmittel, Inorg.
Chem., 2009, 48, 6338 CAS;
(b) M. Schmittel, M. L. Saha and J. Fan, Org. Lett., 2011, 13, 3916 CrossRef CAS PubMed;
(c) J. Fan, M. L. Saha, B. Song, H. Schönherr and M. Schmittel, J. Am. Chem. Soc., 2012, 134, 150 CrossRef CAS PubMed.
- Depending on the substituent in the 4-position of anilines, the association constant Kass of iminopyridines towards [Cu(phenAr2)]+ in CH2Cl2 varies from 104 to 106 M–1.
- M. Schmittel, V. Kalsani, R. S. K. Kishore, H. Cölfen and J. W. Bats, J. Am. Chem. Soc., 2005, 127, 11544 CrossRef CAS PubMed.
- M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743 CrossRef CAS PubMed.
- M. Schmittel, V. Kalsani, P. Mal and J. W. Bats, Inorg. Chem., 2006, 45, 6370 CrossRef CAS PubMed.
- M. Schmittel and P. Mal, Chem. Commun., 2008, 960 RSC.
- M. Schmittel and B. He, Chem. Commun., 2008, 4723 RSC.
- M. Schmittel and K. Mahata, Inorg. Chem., 2009, 48, 822 CrossRef CAS PubMed.
- M. Schmittel and S. K. Samanta, J. Org. Chem., 2010, 75, 5911 CrossRef CAS PubMed.
- M. Schmittel, B. He, J. Fan, J. W. Bats, M. Engeser, M. Schlosser and H.-J. Deiseroth, Inorg. Chem., 2009, 48, 8192 CrossRef CAS PubMed.
- S. K. Samanta, D. Samanta, J. W. Bats and M. Schmittel, J. Org. Chem., 2011, 76, 7466 CrossRef CAS PubMed.
-
(a) S. Neogi, G. Schnakenburg, Y. Lorenz, M. Engeser and M. Schmittel, Inorg. Chem., 2012, 51, 10832 CrossRef CAS PubMed;
(b) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975 CrossRef CAS PubMed.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351 CrossRef PubMed.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703 RSC.
- For a detailed classification of completive self-sorting systems see ref. 22c.
-
(a) W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852 CrossRef CAS PubMed;
(b) W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425 CrossRef CAS PubMed.
- M. Schmittel, R. S. K. Kishore and J. W. Bats, Org. Biomol. Chem., 2007, 5, 78 CAS.
- K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544 CrossRef CAS PubMed.
- M. L. Saha, K. Mahata, D. Samanta, V. Kalsani, J. Fan, J. W. Bats and M. Schmittel, Dalton Trans., 2013, 42, 12840 RSC.
- M. Schmittel and K. Mahata, Chem. Commun., 2010, 46, 4163 RSC.
- K. Mahata and M. Schmittel, Beilstein J. Org. Chem., 2011, 7, 1555 CrossRef CAS PubMed.
- M. L. Saha, J. W. Bats and M. Schmittel, Org. Biomol. Chem., 2013, 11, 5592 CAS.
- In the 1H-NMR characterisation of the library (Scheme 6a) the authors found some additional signals (≤8%) that represent the alternative heteroleptic complexes [Zn(16)(18)](OTf)2 and [Cu(17)(26)](PF6).
- K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933 CrossRef CAS PubMed.
- J. R. Nitschke, Nature, 2009, 462, 736 CrossRef CAS PubMed.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC.
- M. L. Saha, S. Pramanik and M. Schmittel, Chem. Commun., 2012, 48, 9459 RSC.
-
(a) M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832 CrossRef CAS PubMed;
(b) M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730 RSC.
- M. Schmittel, P. Mal and A. de los Rios, Chem. Commun., 2010, 46, 2031 RSC.
- S. K. Samanta and M. Schmittel, Org. Biomol. Chem., 2013, 11, 3108 CAS.
-
(a) K. Nomoto, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2009, 131, 3830 CrossRef CAS PubMed;
(b) S. Kume, K. Nomoto, T. Kusamoto and H. Nishihara, J. Am. Chem. Soc., 2009, 131, 14198 CrossRef CAS PubMed;
(c) S. Kume and H. Nishihara, Chem. Commun., 2011, 47, 415 RSC;
(d) M. Nishikawa, K. Nomoto, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2012, 134, 10543 CrossRef CAS PubMed;
(e) M. Nishikawa, S. Kume and H. Nishihara, Phys. Chem. Chem. Phys., 2013, 15, 10549 RSC.
-
(a) Y. Pellegrin, M. Sandroni, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11309 CrossRef CAS PubMed;
(b) M. Sandroni, M. Kayanuma, A. Planchat, N. Szuwarski, E. Blart, Y. Pellegrin, C. Daniel, M. Boujtita and F. Odobel, Dalton Trans., 2013, 42, 10818 RSC.
-
(a) F. Sozmen, B. S. Oksal, O. A. Bozdemir, O. Buyukcakir and E. U. Akkaya, Org. Lett., 2012, 14, 5286 CrossRef CAS PubMed;
(b) M. G. Fraser, H. van der Salm, S. A. Cameron, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2013, 52, 2980 CrossRef CAS PubMed.
- Y. Fang, T. Murase, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 613 CrossRef CAS PubMed.
-
(a) M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906 RSC;
(b) S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476 CrossRef CAS PubMed;
(c) S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem.–Eur. J., 2013, 19, 2114 CrossRef CAS PubMed.
- M. Schmittel, B. He and P. Mal, Org. Lett., 2008, 10, 2513 CrossRef CAS PubMed.
-
(a) V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323 RSC;
(b) S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741 RSC;
(c) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- M. E. Carnes, M. S. Collins and D. W. Johnson, Chem. Soc. Rev., 2014 10.1039/c3cs60349k.
- A. Aderem, Cell, 2005, 121, 511 CrossRef CAS PubMed.
-
(a)
V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed;
(b)
B. L. Feringa and W. R. Browne, Molecular Switches 1 + 2, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- K. Parimal, E. H. Witlicki and A. H. Flood, Angew. Chem., Int. Ed., 2010, 49, 4628 CrossRef CAS PubMed.
-
(a) G. C. Conant and A. Wagner, Genome Biol., 2005, 6, R50 CrossRef PubMed;
(b) S. Pasek, J.-L. Risler and P. Brézellec, Bioinformatics, 2006, 22, 1418 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS PubMed.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377 CrossRef CAS PubMed.
- U. Lüning, Angew. Chem., Int. Ed., 2012, 51, 8163 CrossRef PubMed.
-
(a) R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370 CrossRef CAS PubMed;
(b) H. Kai, S. Nara, K. Kinbara and T. Aida, J. Am. Chem. Soc., 2008, 130, 6725 CrossRef CAS PubMed;
(c) S. Hiraoka, Y. Hisanaga, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2010, 49, 1669 CrossRef CAS PubMed.
- S. K. Samanta and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 18794 CrossRef CAS PubMed.
Footnote |
| † The present review is dedicated to Prof. em. Dr H. D. Lutz (Siegen) on the occasion of his 80th birthday. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence