 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porphyrins with a carbosilane dendrimer periphery as synthetic components for supramolecular self-assembly†
Zakariyya
Ishtaiwi
a,
Tobias
Rüffer
a,
Sami
Klaib
b,
Roy
Buschbeck
a,
Bernhard
Walfort
a and
Heinrich
Lang
*a
aTechnische Universität Chemnitz, Faculty of Natural Sciences, Institute of Chemistry, Inorganic Chemistry, D-09107 Chemnitz, Germany. E-mail: heinrich.lang@chemie.tu-chemnitz.de; Fax: +49-(0)371-531-21219; Tel: +49-(0)371-531-21210
bTafila Technical University, Faculty of Science, Department of Chemistry and Chemical Technology, Tafila, Jordan
First published on 7th March 2014
Abstract
The preparation of the shape-persistent carbosilane-functionalized porphyrins H2TPP(4-SiRR′Me)4, Zn(II)-TPP(4-SiRR′Me)4 (R = R′ = Me, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, CH2CH2CH2OH; R = Me, R′ = CH2CHCH2, CH2CH2CH2OH; TPP = tetraphenyl porphyrin), H2TPP(4-Si(C6H4-1,4-SiRR′Me)3)4, and Zn(II)-TPP(4-Si(C6H4-1,4-SiRR′Me)3)4 (R = R′ = Me, CH2CHCH2; R = Me, R′ = CH2CHCH2) using the Lindsey condensation methodology is described. For a series of five samples their structures in the solid state were determined by single crystal X-ray structure analysis. The appropriate 0th and 1st generation porphyrin-based 1,4-phenylene carbosilanes form 2D and 3D supramolecular network structures, primarily controlled by either π–π interactions (between pyrrole units and neighboring phenylene rings) or directional molecular hydrogen recognition and zinc–oxygen bond formation in the appropriate hydroxyl-functionalized molecules. UV-Vis spectroscopic studies were carried out in order to analyze the effect of the dendritic branches on the optical properties of the porphyrin ring.
CH2, CH2CH2CH2OH; R = Me, R′ = CH2CHCH2, CH2CH2CH2OH; TPP = tetraphenyl porphyrin), H2TPP(4-Si(C6H4-1,4-SiRR′Me)3)4, and Zn(II)-TPP(4-Si(C6H4-1,4-SiRR′Me)3)4 (R = R′ = Me, CH2CHCH2; R = Me, R′ = CH2CHCH2) using the Lindsey condensation methodology is described. For a series of five samples their structures in the solid state were determined by single crystal X-ray structure analysis. The appropriate 0th and 1st generation porphyrin-based 1,4-phenylene carbosilanes form 2D and 3D supramolecular network structures, primarily controlled by either π–π interactions (between pyrrole units and neighboring phenylene rings) or directional molecular hydrogen recognition and zinc–oxygen bond formation in the appropriate hydroxyl-functionalized molecules. UV-Vis spectroscopic studies were carried out in order to analyze the effect of the dendritic branches on the optical properties of the porphyrin ring.
Introduction
Highly branched macromolecular architectures as well as self-assembly processes have become very popular and represent fascinating research areas both in natural sciences and engineering.1 In this respect, dendrimers and metallo-dendrimers, repetitive branched molecules of structural perfection, have attracted considerable attention as nanoscale molecular materials due to their novel properties.2 Dendrimers possess a three-dimensional and well-designed highly symmetric spherical arrangement with flexible structures employing isotropic assembling processes.3 In the past, also snowflake-shaped dendrimers containing rigid backbones within the dendrimer side chains were prepared.4 Such systems were used, for example, as mediators in electron-transfer and energy-transfer processes,5 as dendritic boxes6 or as drug carrier systems.7Porphyrins can successfully be used as core molecules, as branching units, or, for example, in the stepwise synthesis of cross-shaped covalent assemblies.8 The micro-environments set-up by such molecules can be used among others to tune and control both optical and electrochemical properties of the appropriate porphyrin building block.8,9 Out of this, porphyrins are very useful molecules to probe and hence to characterize dendritic local environments. Recently, metallo-porphyrins tailored at a dendrimer core have been developed as synthetic models to mimic naturally occurring systems including light-harvesting and electron transfer processes (i.e., chlorophyll),8b,10 molecular oxygen storage and transport phenomena (i.e., hemoglobin) as well as oxidation enzymes (i.e., cytochrome c).11,12 In such systems, the porphyrin building blocks have site isolation effects imposed by the dendritic shell. This makes it possible to utilize such species in diverse applications including homogeneous catalysis,13 drug delivery14 and singlet oxygen generation.11a,14b,15 In addition, these compounds can be applied as non-linear optical,16 and light-emitting17 materials, as molecular sensors18 or photo-active systems which can be considered as artificial antennae devoted to solar energy conversion.19
Recently, we got interested in the synthesis of SiCH2CHCH2- and Si(CH2)3OH-functionalized tetraphenyl porphyrins and to use them as supramolecular tectons in the formation of ordered network arrays, since this family of compounds provides a relatively unexplored class of molecules due to their large size, ease of preparation and, for example, excellent coordination ability. Out of these reasons, we here report the Lindsey condensation methodology for the preparation of novel 0th and 1st generation 1,4-phenylene-based carbosilane dendrimer-functionalized porphyrins and zinc(II)-porphyrins. The single-crystal X-ray structure determination of five samples is reported as well showing different interporphyrin interactions.
Results and discussion
Synthesis
For the preparation of the carbosilane dendrimer-based porphyrins 3a–c (Scheme 1) the synthetic methodology developed by Lindsey was used.20 In this respect, the carbosilane aldehydes H(O)C-1-C6H4-4-SiRR′Me (2a, R = R′ = Me; 2b, R = Me, R′ = CH2CHCH2; 2c, R = R′ = CH2CHCH2), accessible by the consecutive treatment of 1-Br–C6H4-4-SiRR′Me (1a, R = R′ = Me; 1b, R = Me, R′ = CH2CHCH2; 1c, R = R′ = CH2CHCH2) with tBuLi and dimethylformamide, were reacted with pyrrole in the presence of catalytic amounts of [BF3·OEt2] followed by addition of 2,3-dichloro-5,6-dicyanobenzoquinone (= DDQ) in CH2Cl2 solutions at ambient temperature (Scheme 1, Experimental section). The appropriate porphyrins H2TPP(4-SiRR′Me)4 (3a, R = R′ = Me; 3b, R = Me, R′ = CH2CHCH2; 3c, R = R′ = CH2CHCH2; TPP = tetraphenyl porphyrin) were isolated as dark red solids with yields of between 25 and 30% (see the Experimental section). The corresponding zinc(II)-porphyrins Zn-TPP(4-SiRR′Me)4 (4a, R = Me, R′ = CH2CHCH2; 4b, R = R′ = CH2CHCH2) were obtained in virtually quantitative yields upon treatment of 3b or 3c with zinc(II) acetate in a mixture of CHCl3–MeOH (ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) (Scheme 1, see the Experimental section).
:1, v/v) (Scheme 1, see the Experimental section).
 | ||
| Scheme 1 Synthesis of 2–6.21–24 | ||
Porphyrins 3b and 3c, respectively, with their terminal SiCH2CHCH2 units could successfully be converted to the corresponding Si-propanolic-functionalized porphyrins 5a and 5b by a consecutive hydroboration–oxidation procedure (Scheme 1, see the Experimental section). Hydroboration of the appropriate end-grafted allyl groups with [BH3·dms] (dms = dimethyl sulfoxide) in thf gave the corresponding BH2-functionalized systems, which on addition of hydrogen peroxide were oxidized to the respective alcohols H2TPP(4-SiRR′′Me)4 (5a, R = Me, R′′ = CH2CH2CH2OH; 5b, R = R′′ = CH2CH2CH2OH) (Scheme 1). These porphyrins produce, when reacted with the transition metal salt [Zn(OAc)2·2H2O], the expected zinc(II) species Zn(II)-TPP(4-SiRR′′Me)4 (6a, R = Me, R′′ = CH2CH2CH2OH; 6b, R = R′′ = CH2CH2CH2OH) in virtually quantitative yield (Scheme 1, see the Experimental section). Zinc porphyrins 6a and 6b dissolve in most common organic solvents.
The synthesis procedure used in the preparation of 3–6 (Scheme 1) could successfully be transferred to the synthesis of the 1st generation carbosilane-based porphyrins 9a–c and 10a–c (Scheme 2). The therefore necessary key aldehyde starting materials 1-H(O)C–C6H4-4-Si(C6H4-1,4-SiRR′Me)3 (8a, R = R′ = Me; 8b, R = Me, R′ = CH2CHCH2; 8c, R = R′ = CH2CHCH2) were obtained with a two-step synthesis procedure from 1-Br-C6H4-4-Si(C6H4-1,4-SiRR′Me)3 (7a, R = R′ = CH3; 7b, R = CH3, R′ = CH2CHCH2; 7c, R = R′ = CH2CHCH2).
 | ||
| Scheme 2 Synthesis of 8–10.18a,24 | ||
After appropriate work-up, aldehydes 8a–c were obtained in excellent yield, and porphyrins 3, 5 and 9 in yields between 25 and 40%, while the formation of the respective zinc(II)-porphyrins 4, 6 and 10 was quantitative (see the Experimental section). All carbosilane-functionalized (metallo)porphyrins are, as the aldehyde starting materials, dark red colored solids soluble in most common polar organic solvents. They are air- and moisture-stable with decomposition or melting points between 200 and 350 °C (see the Experimental section). Aldehydes 8 and porphyrins 3–6 and 9–10 were characterized by elemental analysis, IR, UV-Vis and NMR spectroscopy (1H, 13C{1H}, 29Si{1H}) (see the Experimental section). ESI-TOF mass spectrometric measurements were additionally carried out with selected metal-free and zinc(II) metallated samples (2c, 3b,c, 4a,b, 5a,b, 6a,b and 8a,c). The identity of 3b,c, 4a, 6a and 9b in the solid state was confirmed by single X-ray diffraction studies (vide infra).
The IR spectra of the newly synthesized allyl carbosilane-based porphyrins (Schemes 1 and 2) show a characteristic νCC vibration at ca. 1630 cm−1 together with one or two typical absorptions in the range of 800–840 cm−1 for the Si–C stretching vibrations (see the Experimental section). The CH3 bending vibration of the SiMen entities (n = 1, 2, 3) is observed at ca. 1250 cm−1. These findings are in agreement with allyl-functionalized carbosilanes, i.e. Si(CH2CHCH2)4 (ref. 25). Further characteristic broad absorptions are observed at ca. 3315 and 3400 cm−1, which can be assigned to the NH as well as OH groups. The aldehyde functionalities present in 2 and 8 gave characteristic bands at 2732 and 2820 cm−1 for the CH and at 1705 cm−1 for the CO moieties. In addition, IR spectroscopy can be applied to monitor the progress of the hydroboration of 3 with [BH3·SMe2], since the νCC vibration of the allylic units in the respective starting compounds (ca. 1630 cm−1) disappears in the course of the reaction. After H2O2 treatment new bands for the terminal hydroxyl functionalities in 5a and 5b are found at ca. 3400 cm−1, which is typical for primary alcohols.26 Solely broad absorptions are observed in the IR spectra; hydrogen-bridge formation and hence formation of molecular networks are the most obvious.27
The 1H and 13C{1H} NMR spectra of all compounds are characterized by well-resolved resonance signals for the organic groups present (see the Experimental section). Most typical for aldehydes 2b,c and 8a–c is the resonance signal at 10.07 ppm. This functionality allows the monitoring of the progress of the appropriate porphyrin formation because this signal disappears during the course of the reaction. Further evidence for the successful formation of the porphyrins is the appearance of a singlet at 8.92 ppm, which can be assigned to the pyrrole hydrogen atoms.28 Also very distinctive is the resonance signal of the NH units at −2.8 ppm, while zincation leads to the disappearance of this signal and hence this unit can also be used to monitor the formation of the appropriate zinc porphyrins. Further indicative groups are the SiMe, Si(CH2)3Me, and SiCH2CHCH2 entities (see the Experimental section). Particularly the latter group is best suited to study the progress of the consecutive hydroboration–oxidation processes since new resonances for the Si(CH2)3OH building blocks are found (see the Experimental section). Similar to the IR spectra, the representative resonance signals for the SiCH2CHCH2 groups in 3b and 3c at ca. 2.0 (SiCH2), 5.0 (H2C) and 6.0 ppm (CH) (in CDCl3) disappear on hydroboration and after oxidation with H2O2 new signals can be found at 0.9 (SiCH2CH2CH2OH), 1.7 (SiCH2CH2CH2OH), 3.5 (SiCH2CH2CH2OH) and 4.5 ppm (SiCH2CH2CH2OH) for alcohols 5a and 5b (in dmso-d6), respectively. Similar observations were made in the 13C{1H} NMR spectroscopic studies (see the Experimental section).
Additionally, the 29Si{1H} spectra of the carbosilane-based porphyrins 3b,c, 4a,b, 9b,c and 10a,b and the aldehydes 2b,c and 8b,c were measured. For example, the 29Si{1H} NMR spectra of 9 and 10 (in CDCl3) show, as expected, two resonance signals at ca. −14.5 ppm and between −3.9 and −5.7 ppm, which can be assigned to the core and terminal silicon atoms (see the Experimental section).24 The values for the inner silicon atoms are in good agreement with tetraphenyl silane (−14.98 ppm).29
ESI-TOF mass spectrometric studies were carried out for all aldehyde derivatives and the 0th generation porphyrins. Compounds 2c, 3b,c and 5a show the protonated molecular ion peak [M + H]+, while for 4a,b [M]+ is characteristic. Compounds 5b, 6a,b and 8a,c could successfully be ionized by doping with KSCN and hence the ion [M + K]+ could be detected (see the Experimental section).
UV-Vis absorption spectra were additionally recorded for porphyrins 3b,c, 4a,b, 5a,b, 6a,b, 9a–c and 10a–c, in order to analyze the effect of the dendritic branches on the optical properties of the porphyrin ring. The spectra were measured in CH2Cl2 and thf as solvents (Table 1). The porphyrin core of 3b shows one Soret band at 420 nm in CH2Cl2 and four Q bands at 517, 552, 592 and 648 nm (Fig. 1a).30 Metallation of 3b with zinc(II) did not influence the position of the soret band (4a, 421 nm, in CH2Cl2) (Fig. 1b) but has a significant impact on the shape of the Q band pattern. Two characteristic bands at 549 and 588 nm are observed in CH2Cl2, which is typical for metallo-porphyrins.31Fig. 1 also shows the difference between the UV-Vis spectra of the corresponding 0th and 1st generation dendritic porphyrins. Conspicuous is that the transitions typical for the 1st generation dendritic porphyrins, for example, 9b, are nearly meeting the shape of the bands of the appropriate 0th generation systems, as, for example 3b, with just little enhancement of the band intensities and a very small red shift in the Soret band (Fig. 1a). Similar observations were made for zinc porphyrins 4a and 10b (Fig. 1b). The increasing shielding effect of growing dendrons around the porphyrin core as, for example, described by Aida and coworkers for an aryl ether scaffold30b,c does not appear in the case of our systems. The reason therefore is apparent when looking at the molecular structure of the 1st generation 9b (Fig. 10/11). Compared to aryl ether dendrons, the here reported aryl silyl dendrons (e.g.9b) are more rigid. A back-folding, as observed for the aryl ether dendrons, is impossible and that implies that the porphyrin core plane is even in the 1st generation type compounds easily accessible from above and below by solvents. A dendritic effect is therefore not observed by comparing the UV-Vis data of the 0th and 1st generation molecules. The data correlate well with reported literature spectra for silyl-functionalized porphyrins.32
 | ||
| Fig. 1 Absorption spectra of 3b (–) vs.9b (⋯) (a) and 4a (–) vs.10b (⋯) (b) in CH2Cl2. | ||
| Compound (solvent) | λ max (nm) (log ε) | Soret band (nm) (log ε) | Q bands (nm) (log ε) | |||
|---|---|---|---|---|---|---|
| 3b (CH2Cl2) | 420 (5.73) | 517 (4.35) | 552 (4.09) | 592 (3.87) | 648 (3.77) | |
| 3b (thf) | 419 (5.68) | 515 (4.28) | 550 (4.01) | 593 (3.73) | 648 (3.67) | |
| 3c (CH2Cl2) | 420 (5.78) | 517 (4.39) | 552 (4.14) | 592 (3.92) | 648 (3.85) | |
| 3c (thf) | 419 (5.72) | 515 (4.32) | 550 (4.05) | 593 (3.77) | 649 (3.70) | |
| 4a (CH2Cl2) | 402 (4.61) | 421 (5.75) | 549 (4.33) | 588 (3.67) | ||
| 4a (thf) | 405 (4.68) | 425 (5.83) | 558 (4.36) | 598 (3.98) | ||
| 4b (CH2Cl2) | 402 (4.71) | 421 (5.83) | 549 (4.42) | 588 (3.76) | ||
| 4b (thf) | 405 (4.67) | 426 (5.83) | 558 (4.36) | 597 (3.97) | ||
| 9a (CH2Cl2) | 421 (5.76) | 517 (4.38) | 552 (4.14) | 592 (3.91) | 648 (3.84) | |
| 9a (thf) | 420 (5.76) | 515 (4.32) | 550 (4.09) | 592 (3.80) | 648 (3.75) | |
| 9b (CH2Cl2) | 421 (5.80) | 517 (4.40) | 552 (4.16) | 592 (3.91) | 648 (3.83) | |
| 9b (thf) | 420 (5.74) | 515 (4.34) | 550 (4.10) | 592 (3.82) | 648 (3.76) | |
| 9c (CH2Cl2) | 421 (5.73) | 517 (4.34) | 552 (4.09) | 592 (3.85) | 648 (3.75) | |
| 9c (thf) | 420 (5.72) | 516 (4.32) | 550 (4.10) | 592 (3.81) | 649 (3.75) | |
| 10a (CH2Cl2) | 403 (4.79) | 422 (5.92) | 550 (4.52) | 588 (3.92) | ||
| 10a (thf) | 406 (4.69) | 427 (5.82) | 558 (4.38) | 598 (4.06) | ||
| 10b (CH2Cl2) | 403 (4.67) | 422 (5.81) | 550 (4.39) | 588 (3.78) | ||
| 10b (thf) | 406 (4.70) | 427 (5.82) | 559 (4.39) | 598 (4.07) | ||
| 10c (CH2Cl2) | 403 (4.70) | 422 (5.82) | 550 (4.44) | 588 (3.87) | ||
| 10c (thf) | 407 (472) | 427 (5.85) | 559 (4.40) | 598 (4.08) | ||
| 5a (CH2Cl2) | 420 (5.66) | 516 (4.24) | 552 (3.97) | 594 (3.69) | 648 (3.69) | |
| 5a (thf) | 419 (5.68) | 516 (4.28) | 550 (4.02) | 593 (3.73) | 650 (3.71) | |
| 5b (CH2Cl2) | 420 (5.66) | 517 (4.28) | 552 (4.05) | 594 (3.81) | 648 (3.78) | |
| 5b (thf) | 419 (5.62) | 517 (4.25) | 550 (4.03) | 594 (3.71) | 650 (6.77) | |
| 6a (CH2Cl2) | 403 (4.72) | 421 (5.84) | 550 (4.24) | 588 (3.81) | ||
| 6a (thf) | 405 (4.59) | 426 (5.69) | 558 (4.24) | 598 (3.91) | ||
| 6b (thf) | 426 (5.75) | 558 (4.31) | 598 (4.00) | |||
X-ray investigations
Single crystals of 3b (as 3b·1/4CH2Cl2), 3c, 4a and 9b (as 9b·3.5EtOH) were grown by slow diffusion of CH2Cl2 into EtOH solutions containing the respective compounds, while single crystals of 6a (as 6a·2thf) were obtained by layering a thf solution containing 6a with npentane at ambient temperature. The molecular structures of 3b,c, 4a and 6a are displayed in Fig. 2, 6, 3 and 8. In the case of 9b the asymmetric unit comprises two crystallographically independent centrosymmetric halves of 9b, denoted as 9bA and 9bB. Their molecular structures are depicted in Fig. 10 and 11. Crystal and intensity collection data of 3b·1/4CH2Cl2, 3c, 4a, 6a·2thf and 9b·3.5EtOH are summarized in Table S1,† while selected bond lengths and angles are given in Tables 2 and 3, respectively. | ||
| Fig. 2 ORTEP diagram (50% ellipsoid probability) of the molecular structure of 3b. All C-bonded hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to calculated interplanar angles between terminal C6H4 groups and the central C20N4H2 core. | ||
| 3b·1/4CH2Cl2 | 3c | 4a | 6a·2thf | 9b·3.5EtOH | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Symmetry code: −x, −y + 2, −z + 1. b Symmetry code: x − 1, y, z. c Symmetry code: −x + 2, −y, −z/−x + 1, −y, −z + 1. | |||||||||||||||
| C1–C2 | 1.399(6) | C11–C12 | 1.414(5) | C1–C10a | 1.448(4) | C1–C20 | 1.411(6) | C10–C11 | 1.405(6) | C2–C3 | 1.408(6) | C12–C13 | 1.398(6) | C9–C49c | 1.397(7)/1.407(6) |
| C2–C3 | 1.448(5) | C12–C13 | 1.435(5) | C1–C2 | 1.466(4) | C1–C2 | 1.424(6) | C11–C12 | 1.422(6) | C3–C4 | 1.446(6) | C13–C14 | 1.449(6) | C8–C9 | 1.433(6)/1.441(6) |
| C3–C4 | 1.361(5) | C13–C14 | 1.359(5) | C2–C3 | 1.406(4) | C2–C3 | 1.351(6) | C12–C13 | 1.339(6) | C4–C5 | 1.346(6) | C14–C15 | 1.350(7) | C7–C8 | 1.353(7)/1.367(6) |
| C4–C5 | 1.436(5) | C14–C15 | 1.464(5) | C3–C4 | 1.497(4) | C3–C4 | 1.438(6) | C13–C14 | 1.448(6) | C5–C6 | 1.440(6) | C15–C16 | 1.442(6) | C6–C7 | 1.427(6)/1.420(6) |
| N1–C2 | 1.368(4) | N3–C12 | 1.369(4) | N1–C1 | 1.442(3) | N1–C1 | 1.368(5) | N3–C11 | 1.366(5) | N2–C3 | 1.373(5) | N4–C13 | 1.371(5) | N2–C9 | 1.379(6)/1.377(6) |
| N1–C5 | 1.376(4) | N3–C15 | 1.374(5) | N1–C4 | 1.413(3) | N1–C4 | 1.366(5) | N3–C14 | 1.369(5) | N2–C6 | 1.370(5) | N4–C16 | 1.369(5) | N2–C6 | 1.378(6)/1.377(6) |
| C5–C6 | 1.395(5) | C15–C16 | 1.396(5) | C4–C5 | 1.460(4) | C4–C5 | 1.395(6) | C14–C15 | 1.391(6) | C6–C7 | 1.406(6) | C16–C17 | 1.409(6) | C5–C6 | 1.405(6)/1.410(6) |
| C6–C7 | 1.413(6) | C16–C17 | 1.411(5) | C5–C6 | 1.467(4) | C5–C6 | 1.408(6) | C15–C16 | 1.406(6) | C7–C8 | 1.398(6) | C17–C18 | 1.405(6) | C4–C5 | 1.414(6)/1.413(6) |
| C7–C8 | 1.454(5) | C17–C18 | 1.447(5) | C6–C7 | 1.517(4) | C6–C7 | 1.413(6) | C16–C17 | 1.435(6) | C8–C9 | 1.447(6) | C18–C19 | 1.445(6) | C3–C4 | 1.457(7)/1.468(6) |
| C8–C9 | 1.357(6) | C18–C19 | 1.350(5) | C7–C8 | 1.391(4) | C7–C8 | 1.320(6) | C17–C18 | 1.343(6) | C9–C10 | 1.345(6) | C19–C20 | 1.345(6) | C2–C3 | 1.350(7)/1.350(7) |
| C9–C10 | 1.435(5) | C19–C20 | 1.457(5) | C8–C9 | 1.492(4) | C8–C9 | 1.438(6) | C18–C19 | 1.453(6) | C10–C11 | 1.450(6) | C20–C1 | 1.445(6) | C1–C2 | 1.449(7)/1.455(7) |
| N2–C7 | 1.385(5) | N4–C17 | 1.390(5) | N2–C6 | 1.407(3) | N2–C6 | 1.367(5) | N4–C16 | 1.377(5) | N3–C8 | 1.381(5) | N1–C18 | 1.375(5) | N1–C4 | 1.369(6)/1.372(6) |
| N2–C10 | 1.384(5) | N4–C20 | 1.371(4) | N2–C9 | 1.440(3) | N2–C9 | 1.365(5) | N4–C19 | 1.370(5) | N3–C11 | 1.371(5) | N1–C1 | 1.368(5) | N1–C1 | 1.375(6)/1.379(6) |
| C10–C11 | 1.414(5) | C20–C1 | 1.427(5) | C9–C10 | 1.447(3) | C9–C10 | 1.397(6) | C19–C20 | 1.392(6) | C11–C12 | 1.404(6) | C1–C2 | 1.405(6) | C1–C49 | 1.415(6)/1.411(6) |
| C1–C21 | 1.502(5) | C11–C33 | 1.505(5) | C5–C11 | 1.540(3) | C5–C21 | 1.506(6) | C15–C43 | 1.505(6) | C2–C21 | 1.502(6) | C12–C43 | 1.498(6) | C5–C10 | 1.496(6)/1.489(6) |
| C6–C27 | 1.520(5) | C16–C39 | 1.503(5) | C10–C17 | 1.579(3) | C10–C32 | 1.511(6) | C20–C54 | 1.481(6) | C7–C32 | 1.504(5) | C17–C54 | 1.528(4) | C49–C40 | 1.493(7)/1.493(7) |
| N1⋯N3 | 4.160(5) | N2⋯N4 | 4.113(5) | N1⋯N1a | 4.325(5) | N1⋯N3 | 4.080(5) | N2⋯N4 | 4.059(5) | N1⋯N3 | 4.100(5) | N2⋯N4 | 4.108(5) | N1⋯N1c | 4.102(7)/4.095(7) |
| N2⋯N2a | 4.228(5) | Zn1–N1 | 2.040(3) | Zn1–N3 | 2.039(4) | Zn1–N1 | 2.077(3) | Zn1–N3 | 2.061(3) | N2⋯N2c | 4.154(7)/4.144(7) | ||||
| Zn1–N2 | 2.041(4) | Zn1–N4 | 2.019(3) | Zn1–N2 | 2.063(3) | Zn1–N4 | 2.071(3) | ||||||||
| Zn1–O4b | 2.155(3) | ||||||||||||||
| 3b·1/4CH2Cl2 | 3c | 4a | 6a·2thf | 9b·3.5EtOH | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ∑b | ∑b | ∑b | ∑b | ∑b | ||||||||||
| a Angles including atom O4 are not included. b ∑ = the sum of angles around 5,10,15,20-anellated atoms. c Symmetry code: −x, −y + 2, −z + 1. d Symmetry code: −x + 2, −y, −z/−x + 1, −y, −z + 1. | ||||||||||||||
| C2–C1–C20 | 125.0(4) | 359.9(6) | C4–C5–C6 | 126.5(2) | 360.0(4) | C1–C20–C19 | 124.0(4) | 360.0(7) | C1–C2–C3 | 125.0(4) | 360.0(6) | C4–C5–C6 | 124.5(4)/124.5(4) | 360.0(7) |
| C2–C1–C21 | 118.7(4) | C4–C5–C11 | 116.4(2) | C1–C20–C54 | 118.0(4) | C1–C2–C21 | 119.0(3) | C4–C5–C10 | 117.8(4)/118.0(4) | — | ||||
| C20–C1–C21 | 116.2(3) | C6–C5–C11 | 117.1(2) | C19–C20–C54 | 118.0(4) | C3–C2–C21 | 116.0(3) | C6–C5–C10 | 117.7(4)/117.5(4) | 360.0(7) | ||||
| C5–C6–C7 | 125.6(4) | 360.0(6) | C1–C10–C9c | 123.8(2) | 359.9(4) | C4–C5–C6 | 125.6(5) | 360.0(8) | C6–C7–C8 | 125.9(4) | 359.9(6) | C1–C49–C9d | 125.3(4)/125.2(4) | 359.9(7) |
| C5–C6–C27 | 117.0(3) | C1–C10–C17c | 116.9(2) | C4–C5–C21 | 116.4(4) | C6–C7–C32 | 116.6(3) | C1–C49–C50 | 118.0(4)/118.2(4) | — | ||||
| C7–C6–C27 | 117.4(3) | C9–C10–C17 | 119.2(2) | C6–C5–C21 | 118.0(4) | C8–C7–C32 | 117.4(3) | C9–C49–C50d | 116.6(4)/116.5(4) | 359.9(7) | ||||
| C10–C11–C12 | 125.4(4) | 360.0(6) | C9–C10–C11 | 125.4(5) | 360.0(8) | C11–C12–C13 | 124.4(4) | 359.9(7) | ||||||
| C10–C11–C33 | 116.7(3) | C9–C10–C32 | 116.8(4) | C11–C12–C43 | 117.8(4) | |||||||||
| C12–C11–C33 | 117.9(3) | C11–C10–C32 | 117.8(4) | C13–C12–C43 | 117.7(4) | |||||||||
| C15–C16–C17 | 124.6(4) | 360.0(6) | C14–C15–C16 | 124.4(5) | 360.0(8) | C16–C17–C18 | 125.9(4) | 359.9(7) | ||||||
| C15–C16–C39 | 117.9(3) | C14–C15–C43 | 117.2(4) | C16–C17–C54 | 116.1(4) | |||||||||
| C17–C16–C39 | 117.5(3) | C16–C15–C43 | 118.4(4) | C18–C17–C54 | 117.9(4) | |||||||||
| N1–Zn1–N3 | 177.9(3) | N1–Zn1–N3 | 164.6(1) | |||||||||||
| N2–Zn1–N4 | 178.6(3) | N2–Zn1–N4 | 167.0(1) | |||||||||||
| N1–Zn1–N2 | 90.4(2) | N1–Zn1–N2 | 88.6(1) | |||||||||||
| N1–Zn1–N4 | 89.3(2) | N1–Zn1–N4 | 89.6(1) | |||||||||||
| N2–Zn1–N3 | 90.0(2) | N2–Zn1–N3 | 89.9(1) | |||||||||||
| N3–Zn1–N4 | 90.3(2) | N3–Zn1–N4 | 88.4(1) | |||||||||||
As already mentioned for 9b, even 3c possesses in the solid state crystallographically imposed inversion symmetry, whereby the inversion centres are located in the middle of the atoms N2/N2A (3c, Fig. 6), N1/N1A (9bA, Fig. 10) and N4/N4A (9bB, Fig. 11). For all other crystallographically characterized porphyrins no crystallographically implied symmetry is observed, thus 3b·1/4CH2Cl2, 4a, and 6a·2thf are C1 symmetric in the solid state.
It should be emphasised that meso-tetraphenylporphyrins carrying at the para position any kind of Si-containing groups have been sparingly characterised by single crystal X-ray diffraction studies so far. The solid state structures of 5,10,15,20-tetrakis(4-pentamethyldisilanyl)phenyl)porphyrin32a and of 5,10,15,20-tetrakis[4-(diethoxymethylsilyl)phenyl]porphyrin32b are already described in the literature.
However, experimentally observed bond lengths and angles for the end-grafted carbosilane branches of all functionalized meso-tetraphenylporphyrins described here are in agreement with parameters typically found for phenylene-based carbosilanes.24,33
The molecular structures of H2TPP(4-SiMe2(CH2CHCH2))4 (3b) and its related zinc(II) species Zn(II)-TPP(4-SiMe2(CH2CHCH2))4 (4a) in the solid state are depicted in Fig. 2 and 3. Both porphyrins crystallize in the tetragonal space group P43 with similar dimensions of their respective unit cells (Table S1†). For 3b a partially occupied packing solvent molecule of CH2Cl2 is observed in the crystal structure, which should be found for 4a as well. However, no electron density peaks of 4a could be used for the refinement of an analogous packing solvent molecule. Despite this, 3b and 4a can be regarded as structurally isomorphic to each other.
 | ||
| Fig. 3 ORTEP diagram (50% ellipsoid probability) of the molecular structure of 4a. All hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to calculated interplanar angles between terminal C6H4 groups and the central C20N4Zn core. | ||
The asymmetric unit of both 3b and 4a possesses one crystallographically independent porphyrin molecule. Related bond lengths and angles of the C20N4 cores of 3b and 4a can be considered as identical to each other within standard deviations (Tables 2 and 3). Not surprisingly, the N⋯N distances of opposite nitrogen atoms of 3b are significantly longer, when compared with 4a (3B, N1⋯N3/N2⋯N4 = 4.160(5)/4.113(5) Å vs.4a, N1⋯N3/N2⋯N4 = 4.080(5)/4.059(5) Å), which nicely reflects the modification of the central C20N4 core upon complexation. The Zn(II) ion of 4a can be furthermore considered as being involved in an ideal quadratic planar ZnN4 coordination environment. Zn–N bond lengths of 4a cover a very narrow range (Zn1–N4 = 2.019(3) to Zn1–N2 = 2.041(4) Å) and N–Zn–N bond angles are very close to the ideal values of trans-/cis-ligated N donor atoms (trans: N1–Zn1–N3/N2–Zn1–N4 = 177.9(3)/178.6(3)°; cis: N1–Zn1–N4 = 89.3(2) to N1–Zn1–N2 = 90.4(2)°). Furthermore, the Zn1 atom is located practically in plane with respect to its N4 environment as it deviates by just 0.007(4) Å of the calculated mean plane of the atoms N1 to N4 (root-mean-square deviation from planarity (rmsd) = 0.030 Å, highest deviation from planarity (hdp) observed for N4 with 0.031(2) Å).
Porphyrins 3b and 4a are structurally isomorphic to each other (vide supra) and consequently their crystal structures are identical. For both porphyrins a 3D network structure is observed in the solid state of which a selected part has been illustrated in Fig. 4 (3b) and Fig. 5 (4a). Thereby it is observed that all four crystallographically different C6H4 rings are involved in T-shaped π–π contacts34 with the respective C20N4 cores (Fig. 4 and 5). Geometrical features of these π–π contacts of 3b and 4a (Fig. 4 and 5) are in good agreement with each other, when comparing both molecules.
 | ||
| Fig. 4 Above: Graphical illustration of a selected part of the 3D network formed by 3b due to intermolecular π–π interactions. Labels A–C refer to a 1st–3rd symmetry generated molecule of 3b. All hydrogen atoms and terminal substituents at the silicon atoms are omitted for clarity. Below: graphical illustration of the four different types of intermolecular π–π interactions between the aromatic C6H4 groups with respective parts of the porphyrin core. The sign ∢ refers to calculated interplanar angles between differently colored functionalities. Dotted lines indicate the shortest observed distances between the geometrical centroids of the C6H4 groups and the respective centroids/atoms of the porphyrin core with D1 = centroid of C21–C26, D3 = centroid of C27–C32, D4 = centroid of C33–C38, D6 = centroid of C39–C44, D2 = centroid of N1 and C2, D5 = centroid of C12–C14 and symmetry generated related atoms/fragments. | ||
 | ||
| Fig. 5 Above: graphical illustration of a selected part of the 3D network formed by 4a due to intermolecular π–π interactions. Labels A–C refer to a 1st to the 3rd symmetry generated molecule of 4a. All hydrogen atoms and terminal substituents at the silicon atoms are omitted for clarity. Below: graphical illustration of the four different types of intermolecular π–π interactions between the aromatic C6H4 groups with the respective parts of the porphyrin core. The sign ∢ refers to the calculated interplanar angles between differently colored functionalities. Dotted lines indicate the shortest observed distances between the geometrical centroids of the C6H4 groups and the respective centroids/atoms of the porphyrin core with D1 = centroid of C21–C26, D3 = centroid of C32–C37, D4 = centroid of C43–C48, D6 = centroid of C54–C59, D2 = centroid of N2 and C6, D5 = centroid of C16–C18, D7 = centroid of C12–C14 and symmetry generated related atoms/fragments. | ||
The molecular structure of H2TPP(4-SiMe(CH2CHCH2)2) (3c) in the solid state is depicted in Fig. 6. The replacement of one methyl group of the carbosilane SiMe2(CH2CHCH2) moiety in 3b by an allyl unit, as characteristic for 3c, induces considerable changes. In contrast to 3b, porphyrin 3c crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with crystallographically imposed inversion symmetry. Thus, the asymmetric unit of 3c comprises just half of the molecule, while the 2nd half is generated by an inversion center which is located at the crossing point of the atoms N1/N1A and N2/N2A (Fig. 3). Furthermore, it is astonishing to notice that related bond lengths of the C20N4 core of 3c, when compared with those of 3b and 4a and those of 6a and 9b as well, are significantly elongated (Table 2). As a wrongly determined space group may cause such deviations the structural refinement of 3c was checked with the utmost precision; however, there are no indications that the unit cell dimensions and space group of 3c are not accurate (Table S1†). For example, the N⋯N distances of opposite N atoms of 3c (N1⋯N1A/N2⋯N2A = 4.325(5)/4.228(5) Å) are by far the longest ones of the here described porphyrins (Table 2). A possible explanation of this unexpected observation for 3c might be drawn from the crystal structure of 3c, which is illustrated in Fig. 7. In contrast to the observation of 3D network structures for 3b and 4a (Fig. 4 and 6) which are induced by T-shaped π–π interactions, for 3c the formation of 2D layers has been noted. The 2D layers are formed along the crystallographic a- and b-axes, but not along the crystallographic c-axes as depicted in Fig. S1 and S2.† Moreover, only the C6H4 aromatic group comprising the atoms C11 to C16 and symmetry generated analogues is involved in T-shaped π–π interactions with the C20N4 core of adjacent molecules. Thereby, comparatively large centroid-to-centroid distances are observed (Fig. 7). To deduce that the different crystal structure of 3c is responsible for the observation of significantly different bond lengths of 3c, when compared to those of 3b and 4a, is certainly a more qualitative description. Hence, further work, e.g. quantum chemical calculations, is required to figure out the origin of this remarkable phenomenon.
with crystallographically imposed inversion symmetry. Thus, the asymmetric unit of 3c comprises just half of the molecule, while the 2nd half is generated by an inversion center which is located at the crossing point of the atoms N1/N1A and N2/N2A (Fig. 3). Furthermore, it is astonishing to notice that related bond lengths of the C20N4 core of 3c, when compared with those of 3b and 4a and those of 6a and 9b as well, are significantly elongated (Table 2). As a wrongly determined space group may cause such deviations the structural refinement of 3c was checked with the utmost precision; however, there are no indications that the unit cell dimensions and space group of 3c are not accurate (Table S1†). For example, the N⋯N distances of opposite N atoms of 3c (N1⋯N1A/N2⋯N2A = 4.325(5)/4.228(5) Å) are by far the longest ones of the here described porphyrins (Table 2). A possible explanation of this unexpected observation for 3c might be drawn from the crystal structure of 3c, which is illustrated in Fig. 7. In contrast to the observation of 3D network structures for 3b and 4a (Fig. 4 and 6) which are induced by T-shaped π–π interactions, for 3c the formation of 2D layers has been noted. The 2D layers are formed along the crystallographic a- and b-axes, but not along the crystallographic c-axes as depicted in Fig. S1 and S2.† Moreover, only the C6H4 aromatic group comprising the atoms C11 to C16 and symmetry generated analogues is involved in T-shaped π–π interactions with the C20N4 core of adjacent molecules. Thereby, comparatively large centroid-to-centroid distances are observed (Fig. 7). To deduce that the different crystal structure of 3c is responsible for the observation of significantly different bond lengths of 3c, when compared to those of 3b and 4a, is certainly a more qualitative description. Hence, further work, e.g. quantum chemical calculations, is required to figure out the origin of this remarkable phenomenon.
 | ||
| Fig. 6 ORTEP diagram (25% ellipsoid probability) of the molecular structure of 3c. All C-bonded hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to the calculated interplanar angles between terminal C6H4 groups and the central C20N4H2 core. Symmetry code for A: −x, −y + 2, −z + 1. | ||
 | ||
| Fig. 7 Above: graphical illustration of a selected part of one 2D layer formed by 3c due to intermolecular π–π interactions. Labels ′, ′′, * and # refer to symmetry generated atoms of crystallographically independent molecules of the asymmetric unit of 3c, label A to symmetry generated atoms of the asymmetric unit of 3c and labels B–E to symmetry generated atoms of A labelled atoms. All C-bonded hydrogen atoms and terminal substituents at the silicon atoms are omitted for clarity. Below: graphical illustration of the intermolecular π–π interactions between the aromatic C6H4 groups with the respective parts of the porphyrin core. The sign ∢ refers to the calculated interplanar angles between differently coloured functionalities. Dotted lines indicate the shortest observed distances between the geometrical centroids of the C6H4 groups and the respective centroids/atoms of the porphyrin core with D1 = centroid of C11–C16 and D2 = centroid of C6–C8, respectively, and symmetry generated related atoms/fragments. | ||
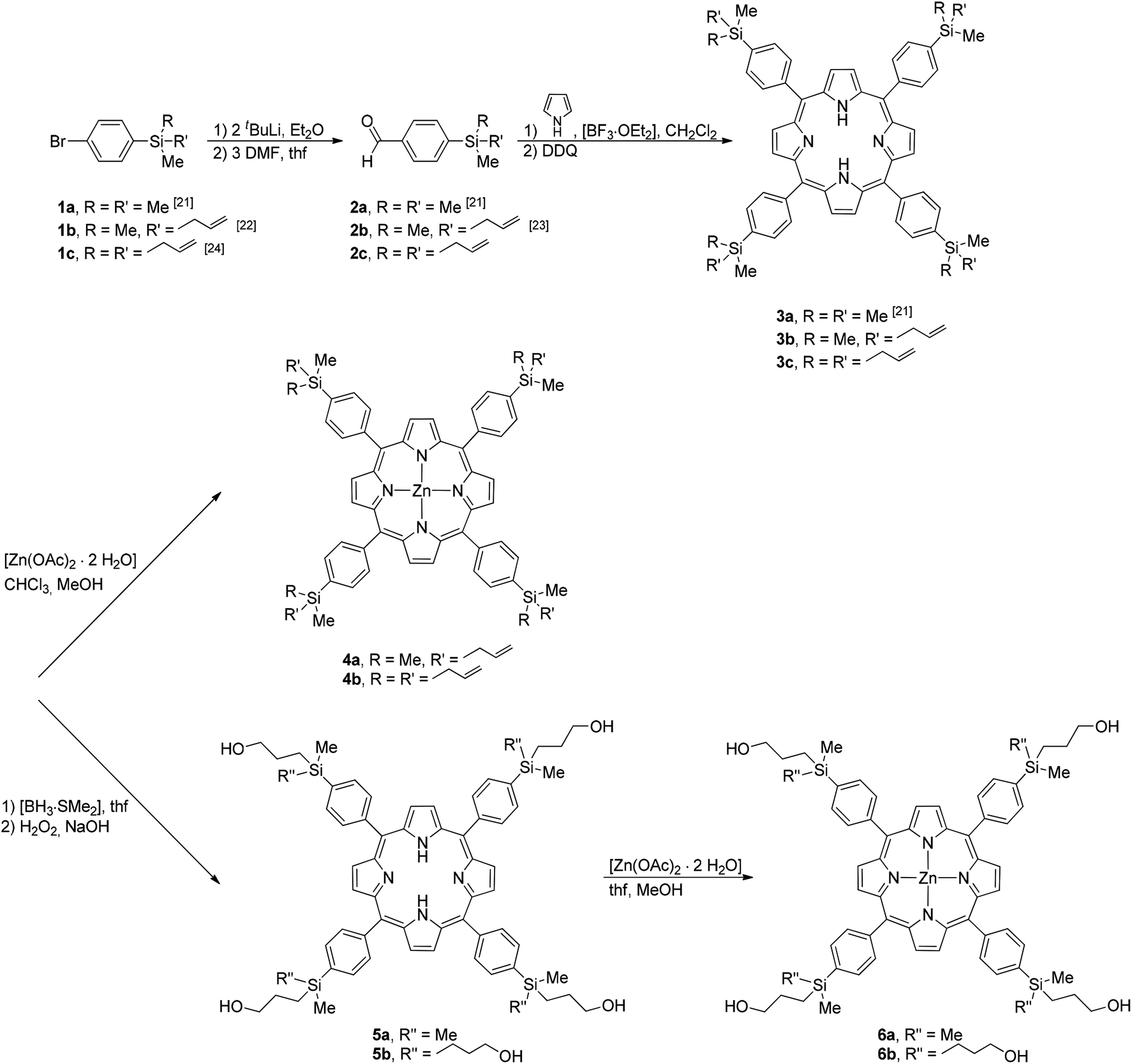
The molecular structure of Zn(II)-TPP(4-SiMe2((CH2)3OH))4 (6a) is depicted in Fig. 8. Porphyrin 6a crystallizes in the triclinic system P with one molecule of 6a in the asymmetric unit cell. As indicated before, bond lengths and angles of the central C20N4 core of 6a compare well with those of 3b, 4a and 9b (Tables 2 and 3). Due to formation of an intermolecular O(H)–Zn bond (Zn1–O1 = 2.155(3) Å), the Zn(II) ion of 6a is involved in an approximate square-based pyramidal ZnN4O coordination environment. Not surprisingly, as a consequence of the intermolecular O(H)–Zn bond formation the Zn1 atom of 6a is significantly moved out of the basal plane into the direction of the coordinated O donor atom. Thus, the Zn1 atom is located 0.255(2) Å above the calculated mean plane of the atoms N1 to N4 (rmsd = 0.022 Å, hdp observed for N3 with 0.022(2) Å).
 | ||
| Fig. 8 ORTEP diagram (50% ellipsoid probability) of the molecular structure of 6a. All C-bonded hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to the calculated interplanar angles between terminal C6H4 groups and the central C20N4Zn core. | ||
The exchange of the terminal allyl groups of 4a by 3-propyloxy functionalities, as present in 6a, resulted in a completely different packing mode. There are no π–π interactions of any kind observed for 6a in the solid state. Instead, the crystal structure is exclusively governed by reciprocal formation of intermolecular O(H)–Zn contacts along the crystallographic a-axes together with formation of intermolecular O(H)⋯O hydrogen bonds along the crystallographic b-axes. Fig. S3 and Table S2† show bond lengths and angles for the characteristic intermolecular hydrogen bonds. A part of the thus formed 2D layers is furthermore graphically illustrated in Fig. 9. It is surprising to note that the formation of 1D chains, as a part of the 2D network structure, due to mutual intermolecular O(H)–Zn contacts as observed for 6a, has not been observed so far for any kind of O-functionalised metalloporphyrins possessing 3d transition metal ions. Related metalloporphyrins functionalised by any kind of O-donor atoms, with the oxygen atoms belonging to alcohol, ether, carbonic acid and/or carbonyl functionalities, form either dimers,35 trimers36 or polymeric 2D37 and 3D38 networks, respectively.
 | ||
| Fig. 9 Graphical representation of a part of one of the 2D layers formed by 6a in the solid state due to formation of intermolecular O–H⋯O hydrogen bonds and O(H)–Zn bonds. All C-bonded hydrogen atoms and packing solvent molecules are omitted for clarity. Of disordered atoms only one atomic position is displayed. Labels A–E refer to a 1st–5th symmetry generated molecule of 6a. | ||
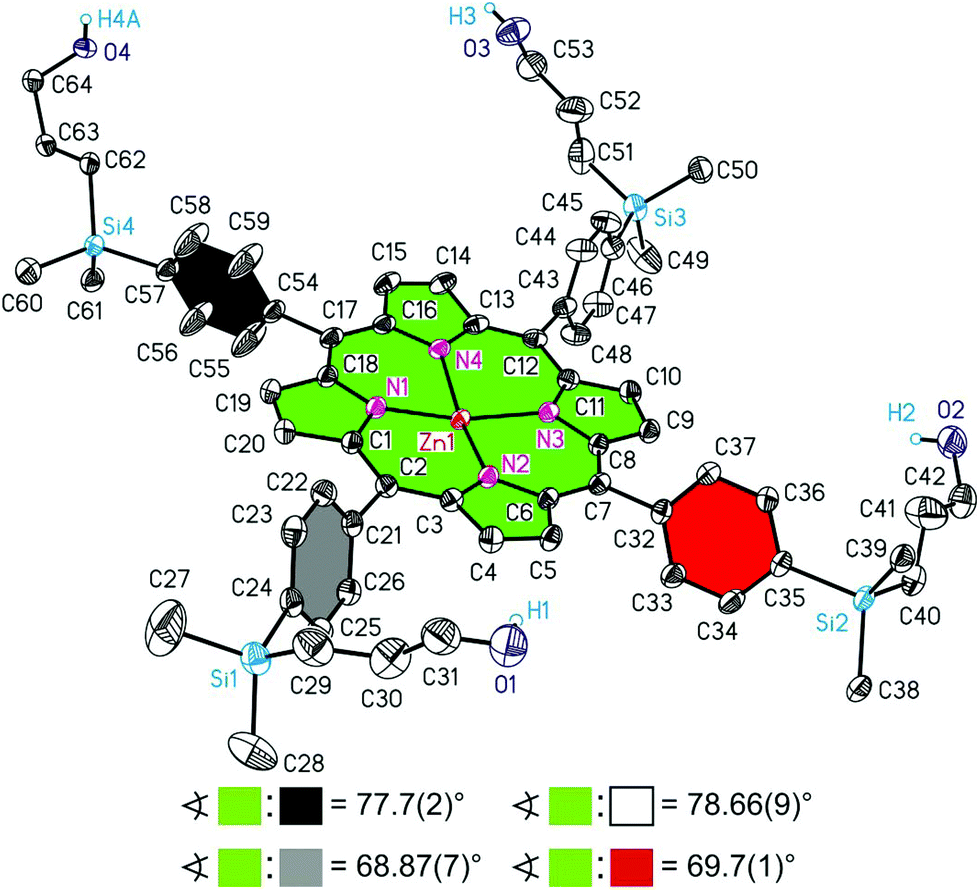
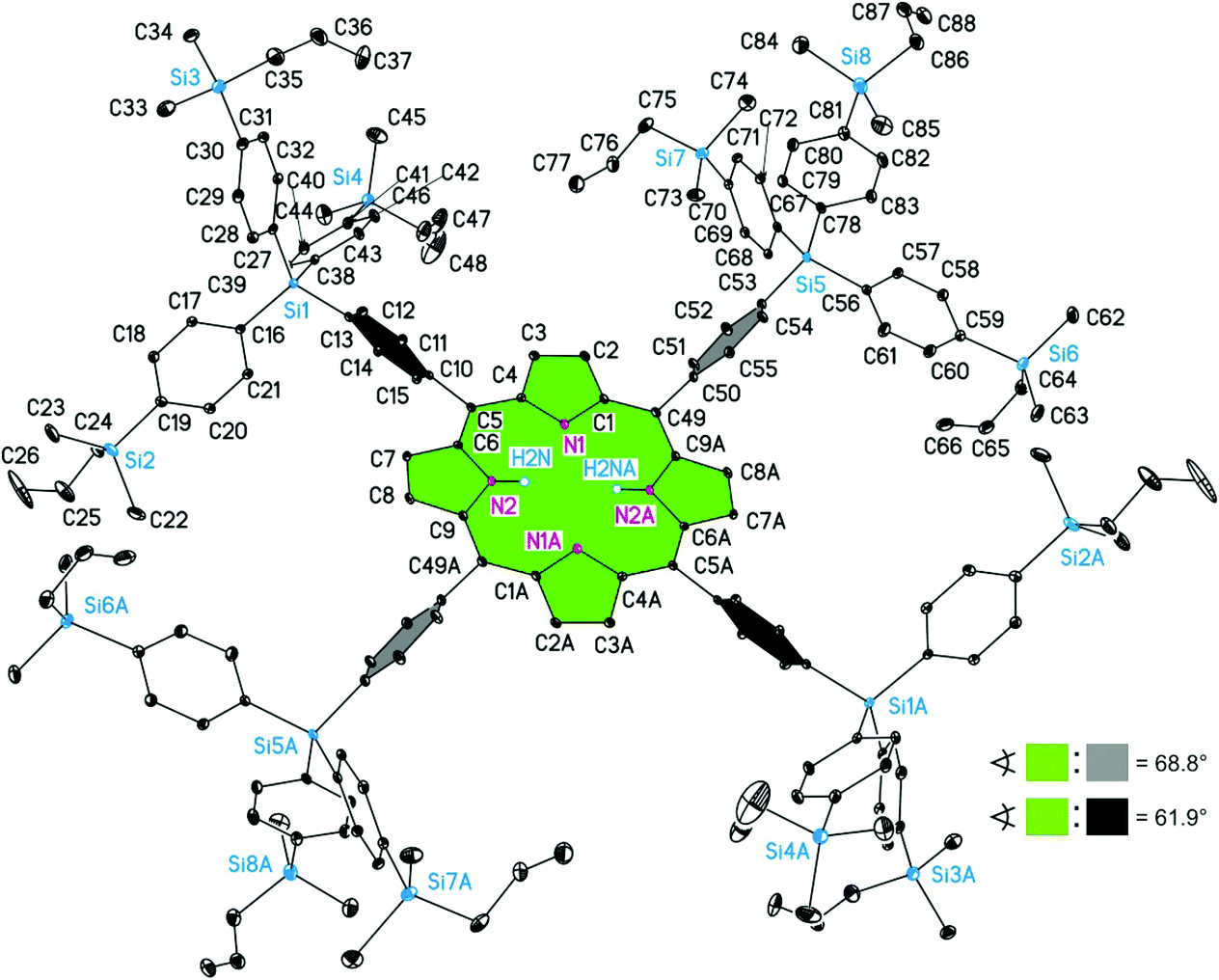
Porphyrin H2TPP(4-Si(C6H4-4-Si(CH2CHCH2)Me2)3)4 (9b) crystallises in the triclinic space group P. The asymmetric unit contains half of two crystallographically independent molecules of 9b, denoted as 9bA and 9bB. Both 9bA and 9bB possess in the solid state crystallographically imposed inversion symmetry, whereby the molecular structures of both individual molecules are depicted in Fig. 10 and 11. The bond distances and angles of the C20N4 cores of 9bA and 9bB do not only compare well with each other, they can be even considered as closely related to analogous data reported here for 3b, 4a and 6a but not 3c (see above and Tables 2 and 3).
 | ||
| Fig. 10 ORTEP diagram (20% ellipsoid probability) of the molecular structure of 9bA. All carbon-bonded hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to the calculated interplanar angles between the directly porphyrin bonded C6H4 groups and the central C20N4H2 core. Symmetry code for A: −x + 2, −y, −z. | ||
 | ||
| Fig. 11 ORTEP diagram (20% ellipsoid probability) of the molecular structure of 9bB. All carbon-bonded hydrogen atoms are omitted for clarity. Of disordered atoms only one atomic position is displayed. The sign ∢ refers to the calculated interplanar angles between the directly porphyrin bonded C6H4 groups and the central C20N4H2 core. Symmetry code for A: −x + 1, −y and for #:, −z + 1.40a | ||
Due to the presence of sixteen crystallographically independent C6H4 aromatic rings of both 9bA and 9bB, determination of the crystal structure of 9b with respect to possible π–π interactions is rather complicated.39 However, it was found that a 3D network is formed by 9b in the solid state due to T-shaped π–π interactions. This 3D network can be understood as being formed of 2D layers of molecules of 9bA and 9bB of which a part is illustrated in Fig. 12. Further descriptions of the individual interactions are given in Fig. 14 and S4–S6.† The 2D layers interact then with each other by means of T-shaped π–π interactions exclusively between molecules of either 9bA or 9bB (Fig. 13). The respective π–π interactions being responsible for the formation of the 2D layers and of the 3D network are then separately illustrated in Fig. 14. The inter-layer distance between 2D layers corresponds to 25.1408 Å (=b), (Fig. 13).
 | ||
| Fig. 12 Selected part of one 2D layer formed by 9b in the solid state due to T-shaped π–π interactions between molecules of 9bA and 9bB.40b | ||
 | ||
| Fig. 13 Graphical illustration of the mutual T-shaped π–π interactions between molecules of 9bA and 9bB, being responsible for the connection of the 2D layers of 9b to give a 3D network structure.40b | ||
 | ||
| Fig. 14 Graphical illustration of the respective T-shaped π–π interactions observed in the solid state structure of 9b being responsible for the formation of 2D layers (left and middle) and for the interaction of 2D layers to give a 3D network.40b | ||
Different types of non-planar distortions commonly observed for porphyrins have been already explicitly discussed.41 In the case of here structurally described porphyrins it can be determined that the central C20N4 porphyrin cores can be regarded as planar which can be concluded from, for example, the sum of angles around the 5,10,15,20-anellated atoms of the C20N4 cores (Table 3) and further data are given in Table S3 and Fig. S7,† including accompanying remarks.
In summary, the observation of the formation of 3D (3b, 4a and 9b) or 2D networks (3c) resulting from intermolecular T-shaped π–π interactions is not a specific feature of the here reported porphyrins. There are already two crystallographically described meso-tetraphenylporphyrins known bearing terminal Si-functionalities at the para-position of the phenyl rings, namely 5,10,15,20-tetrakis(4-pentamethyldisilanyl)phenyl)porphyrin32a and 5,10,15,20-tetrakis[4-(diethoxymethylsilyl)phenyl]porphyrin,32b which can be regarded as closely related to the here reported porphyrins. Bond lengths and angles of the C20N4 porphyrin cores of these two reported samples are in very good agreement with the corresponding data observed for 3b, 4a, 6a and 9b, respectively.
Especially for 5,10,15,20-tetrakis[4-(diethoxymethylsilyl)phenyl]porphyrin it is indicated that “no significant short contacts such as π-stacking” could be observed, which is attributed to the steric hindrance of the terminal silyl functionalities.32b It seems, however, likely, that only sandwich type π–π interactions have been ruled out.
In the discussion of the crystal structures of Zn(TPP) and Ag(TPP),42 which are isomorphous to H2TPP, T-shaped π–π interactions have been explicitly mentioned. Interplanar angles between interacting aromatic units are given, although no centroid-to-centroid distances and the final type of assemblies formed have been mentioned.42 In the case of a report on the crystallographic characterisation of Fe(TPP) as a toluene solvate, the formation of 1D chains, due to sandwich type π–π interactions, is observed.43 One can thus assume that for non-functionalised M(TPP) (M = 3d metal ion) type porphyrins, especially for those in which the central metal ion is not coordinated by one and/or two donor molecules, π–π interactions play a significant role in the crystal structures. Indeed, for Zn(TPP), being co-crystallized with different guest molecules, a review by Byrn et al.44 for over 200 different cases mentions explicitly the observation of intermolecular T-shaped π–π interactions, although no geometrical features or types of assemblies were given.
Conclusions
The preparation of a series of 0th and 1st generation carbosilane dendrimer-based porphyrins of types H2TPP(4-SiRR′Me)4, Zn(II)-TPP(4-SiRR′Me)4 (R = R′ = Me, CH2CHCH2, CH2CH2CH2OH; R = Me, R′ = CH2CHCH2, CH2CH2CH2OH; TPP = tetraphenyl porphyrin), H2TPP(4-Si(C6H4-1,4-SiRR′Me)3)4, and Zn(II)-TPP(4-Si(C6H4-1,4-SiRR′Me)3)4 (R = R′ = Me, CH2CHCH2; R = Me, R′ = CH2CHCH2) using the Lindsey condensation methodology is described. Functionalization of TPP with the carbosilane dendrons leads to a slight bathochromic shift of the Soret and Q bands in the UV-Vis spectra, which is in agreement with the literature.30 The structures of five samples (3b,c, 4a, 6a, 9b) in the solid state have been determined by single X-ray structure determination. The supramolecular structures of the allyl 0th generation species 3b,c, 4a and the 1st generation carbosilane-containing porphyrin 9b are primarily controlled by π–π interactions, while in the hydroxyl-functionalized porphyrin 6a the network formation is exclusively set by zinc–oxygen coordination and hydrogen bonding intermolecular interactions. Conspicuously, porphyrin 3b and its analogous zinc-metallated system 4a possess an identical 3D supramolecular structure as both compounds can be regarded as isomorphic in the solid state. On the other hand, the eight allyl groups in 3c, instead of the four allyl units present in 3b, modify the 3D network into a 2D network which might be responsible for the observation of significantly larger bond lengths of its C20N4 core in comparison with the related bond lengths of the other here described porphyrins.
For meso-tetraphenylporphyrins bearing any kind of substituent at the phenyl rings it seems less likely that intermolecular sandwich type π–π interactions can be observed, although T-shaped π–π interactions might be found. For such cases we do not find, to the best of our knowledge, precise comments for crystallographically described representatives. We assume, however, that especially T-shaped π–π interactions should be observed, as reported here for 3b, 3c, 4a and 9b, at least in such cases where the central metal ions are not coordinated by additional donor solvents or the two N(H)-protons of the central porphyrin rings are not involved in hydrogen bridges with protic donor solvents.
Experimental section
General data
All reactions were carried out under an atmosphere of nitrogen using standard Schlenk techniques. Tetrahydrofuran, toluene and npentane were purified by distillation from sodium–benzophenone ketyl; CH2Cl2 and CHCl3 were purified by distillation from calcium hydride. Diethylamine and diisopropylamine were distilled from KOH; absolute MeOH was obtained by distillation from magnesium.Instrumentation
Infrared spectra were recorded with a Perkin Elmer FT-IR spectrometer Spectrum 1000 (ṽ in cm−1). UV-Vis spectra were measured with a Thermo Electron Corporation Genesys 6 spectrometer. NMR spectra were recorded with a Bruker Avance 250 spectrometer (1H NMR at 250.12 MHz and 13C{1H} NMR at 62.86 MHz) in the Fourier transform mode. Chemical shifts are reported in δ units (parts per million) downfield from tetramethylsilane (δ = 0.00 ppm) with the solvent as the reference signal (CDCl3: 1H NMR, δ = 7.26; 13C{1H} NMR, δ = 77.16; DMSO-d6: 1H NMR, δ = 2.54; 13C{1H} NMR, δ = 40.45; thf-d8: 1H NMR, δ = 1.72; 13C{1H} NMR, δ = 24.45).45 The atom numbering is depicted in the ESI (Fig. S8†). The assignment of 13C{1H} and 29Si{1H} NMR spectroscopic signals is mainly based on 13C-DEPT-135 spectra and 2D-correlation spectra (gradient with sensitivity-enhanced heteronuclear multiple quantum correlation (gs-HMQC) for carbon and silicon and gradient with sensitivity-enhanced heteronuclear multiple bond correlation (gs-HMBC) for carbon). Please note that the resonance signal for the pyrrole-α-C unit could only be detected for the zinc complexes under the measurement conditions applied. ESI-TOF mass spectra were recorded using a Mariner biospectrometry workstation 4.0 (Applied Biosystems). Microanalyses were performed with a Thermo FlashAE 1112 instrument. Melting points of pure samples were measured with Gallenkamp MFB 595 010 equipment.Reagents
1a, 2a, 3a,211b,221c, 7a,c,242b23 and 7b, 8b, 9b18a were prepared according to published procedures. All other chemicals were purchased from commercial suppliers and were used as received.4-Diallylmethylsilylbenzaldehyde (2c)
t BuLi (1.7 M, 34.7 mmol, 20.4 mL, npentane) was added drop-wise to a Et2O (75 mL) solution containing 1c (17.37 mmol, 4.88 g) at −78 °C. After 1 h of stirring at this temperature the resulting solution was drop-wise transferred via a cannula to dimethyl formamide (52.12 mmol, 3.81 g, 4.04 mL) in thf (50 mL) at 0 °C and the obtained reaction mixture was kept at this temperature for 15 min. Afterwards, it was warmed to ambient temperature, stirring was continued for 2 h, and then it was quenched with aqueous HCl (3 N, 80 mL). The obtained residue was extracted with Et2O (100 mL) and the organic layer was washed with water (2 × 60 mL), saturated NaHCO3 (60 mL) and brine (60 mL) and was then dried over MgSO4. Afterwards, all volatiles were removed in oil-pump vacuum. The crude product was purified by column chromatography (a) nhexane, (b) 20 vol% CH2Cl2–nhexane to afford 2c as colorless oil (14.86 mmol, 3.42 g, 86% based on 1c).IR (film): ṽ = 3074, 3024, 2992, 2966, 2910, 2882, 2824 (w, CHO), 2732 (w, CHO), 1704 (s, CO), 1630 (w, CC), 1254 (w, CH3 bending), 808 (s, SiC). 1H NMR (CDCl3): δ = 10.02 (s, 1 H, CHO), 7.84 (dt, JHH = 8.1 Hz, JHH = 1.6 Hz, 2 H, H3), 7.68 (dt, JHH = 8.1 Hz, 1.6 Hz, 2 H, H4), 5.74 (ddt, JHH = 17.6 Hz, JHH = 9.5 Hz, JHH = 8.1 Hz, 2 H, H8), 4.89 (ddt, JHH = 17.6 Hz, JHH = 2.0 Hz, JHH = 1.1 Hz, 2 H, cis-H9), 4.88 (ddt, JHH = 9.5 Hz, JHH = 2.0 Hz, JHH = 1.1 Hz, 2 H, trans-H9), 1.84 (dt, JHH = 8.1 Hz, JHH = 1.1 Hz, 4 H, H7), 0.34 (s, 3 H, H6). 13C{1H} NMR (CDCl3): δ = 192.4 (1 C1), 145.5 (1 C5), 136.7 (1 C2), 134.4 (2 C4), 133.4 (2 C8), 128.5 (2 C3), 114.4 (2 C9), 21.2 (2 C7), −6.1 (1 C6). 29Si{1H} NMR (CDCl3): δ = −5.0. ESI-TOF: m/z = 231.10 [M + H]+ (calcd 231.12). Anal. calcd for C14H18OSi (230.38): C, 72.99; H, 7.88. Found: C, 72.54; H, 7.51.
meso-Tetrakis(4-allyldimethylsilylphenyl)porphyrin (3b)
To aldehyde 2b (6.00 mmol, 1.224 g) and pyrrole (6.00 mmol, 0.403 g, 0.416 mL) dissolved in CH2Cl2 (600 mL), [BF3·OEt2] (0.60 mmol, 0.085 g, 0.076 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and then 2,3-dichloro-5,6-dicyanobenzoquinone (4.50 mmol, 1.224 g) was added in a single portion and stirring was continued for 1 h. The reaction mixture was washed with saturated NaHCO3 (100 mL) and then with water (3 × 100 mL). The organic layer was filtered through a pad of silica gel and afterwards all volatiles were removed in oil-pump vacuum. The obtained solid material was purified by column chromatography (a) nhexane and (b) 20 vol% CH2Cl2–nhexane. Porphyrin 3b was obtained as a dark red solid (0.44 mmol, 0.44 g, 29% based on 2b).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3315 (s, NH), 3069, 3056, 3011, 2994, 2911, 2880, 1627 (w, CC), 1251 (m, CH3 bending), 834, 818, 804 (s, SiC). 1H NMR (CDCl3): δ = 8.87 (s, 8 H, H1), 8.23 (brd, JHH = 7.9 Hz, 8 H, H5), 7.90 (brd, JHH = 7.9 Hz, 8 H, H6), 6.01 (ddt, JHH = 17.0 Hz, JHH = 10.1 Hz, JHH = 8.1 Hz, 4 H, H10), 5.04 (ddt, JHH = 17.0 Hz, JHH = 2.4 Hz, JHH = 1.1 Hz, 4 H, cis-H11), 5.01 (ddt, JHH = 10.1 Hz, JHH = 2.4 Hz, JHH = 1.1 Hz, 3 H, trans-H11), 2.02 (dt, JHH = 8.1 Hz, JHH = 1.1 Hz, 16 H, H9), 0.54 (s, 24 H, H8), −2.74 (brs, 2 H, NH). 13C{1H} NMR (CDCl3): δ = 142.8 (4 C7), 137.8 (4 C4), 134.7 (4 C10), 134.1 (8 C6), 132.0 (8 C5), 131.2 (br, 8 C1), 120.1 (4 C3), 113.6 (4 C11), 24.0 (4 C9), −3.2 (8 C8). 29Si{1H} NMR (CDCl3): δ = −4.1. ESI-TOF: m/z = 1007.49 [M + H]+ (calcd 1007.48). Anal. calcd for C64H70N4Si4 (1007.61): C, 76.29; H, 7.00; N, 5.56. Found: C, 76.03; H, 7.26; N, 5.30.
meso-Tetrakis(4-diallylmethylsilylphenyl)porphyrin (3c)
To aldehyde 2c (10.00 mmol, 2.301 g) and pyrrole (10.00 mmol, 0.671 g, 0.69 mL) dissolved in CH2Cl2 (1000 mL) [BF3·OEt2] (1.00 mmol, 0.142 g, 0.13 mL) was added in a single portion. The reaction solution was stirred for 2 h and then 2,3-dichloro-5,6-dicyanobenzoquinone (7.50 mmol, 1.703 g) was added in a single portion and stirring was continued for 1 h. The reaction mixture was washed with saturated NaHCO3 (100 mL) and then with water (3 × 100 mL). The organic layer was filtered through a pad of silica gel and afterwards all volatiles were removed in oil-pump vacuum. The crude product was purified by column chromatography (a) nhexane and (b) 20 vol% CH2Cl2–nhexane. Porphyrin 3c was obtained as a dark red solid (0.69 mmol, 0.76 g, 27% based on 2c).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3314 (s, NH), 3070, 3006, 2994, 2966, 2912, 2880, 1628 (m, CC), 1250 (m, CH3 bending), 800 (s, SiC). 1H NMR (CDCl3): δ = 8.90 (s, 8 H, H1), 8.26 (brd, JHH = 7.9 Hz, 8 H, H5), 7.92 (brd, JHH = 7.9 Hz, 8 H, H6), 6.04 (ddt, JHH = 17.0 Hz, JHH = 10.1 Hz, JHH = 8.1 Hz, 8 H, H10), 5.10 (ddt, JHH = 17.0 Hz, JHH = 2.0 Hz, JHH = 0.9 Hz, 8 H, cis-H11), 5.06 (ddt, JHH = 10.1 Hz, JHH = 2.0 Hz, JHH = 0.9 Hz, 8 H, trans-H11), 2.10 (dt, JHH = 8.0 Hz, JHH = 0.9 Hz, 32 H, H9), 0.58 (s, 24 H, H8), −2.69 (brs, 2 H, NH). 13C{1H} NMR (CDCl3): δ = 142.9 (4 C7), 136.1 (4 C4), 134.3 (8 C10), 134.1 (8 C6), 132.3 (8 C5), 131.1 (br, 8 C1), 120.1 (4 C3), 114.1 (8 C11), 21.8 (8 C9), −5.6 (4 C8). 29Si{1H} NMR (CDCl3): δ = −5.2. ESI-TOF: m/z = 1111.50 [M + H]+ (calcd 1111.54). Anal. calcd for C72H78N4Si4 (1111.53): C, 77.78; H, 7.07; N, 5.04. Found: C, 77.68; H, 6.59; N, 4.80.
meso-Tetrakis(4-allyldimethylsilylphenyl)porphyrinato zinc (4a)
To 3b (0.050 mmol, 0.0504 g) dissolved in CHCl3 (15 mL), a MeOH solution (10 mL) containing [Zn(OAc)2·2H2O] (0.100 mmol, 0.0219 g) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and was then washed with saturated NaHCO3 (15 mL) followed by washing with water (2 × 15 mL). The organic layer was dried over Na2SO4. All volatiles were removed in oil-pump vacuum to afford 4a as a dark red solid (0.495 mmol, 0.053 g, 99% based on 3b).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3055, 3011, 2989, 2911, 2954, 2908, 2878, 1627 (w, CC), 125 (m, CH3 bending), 836, 820, 811, 796 (s, SiC). 1H NMR (CDCl3): δ = 9.00 (s, 8 H, H1), 8.25 (brd, JHH = 7.9 Hz, 8 H, H5), 7.90 (brd, JHH = 7.9 Hz, 8 H, H6), 6.02 (ddt, JHH = 17.0 Hz, JHH = 10.1 Hz, JHH = 8.1 Hz, 4 H, H10), 5.05 (brd, JHH = 17.0 Hz, 4 H, cis-H11), 5.02 (brd, JHH = 10.1 Hz, 4 H, trans-H11), 2.03 (brd, JHH = 8.1 Hz, 8 H, H9), 0.56 (s, 24 H, H8). 13C{1H} NMR (CDCl3): δ = 150.2 (4 C2), 143.4 (4 C7), 137.5 (4 C4), 134.8 (4 C10), 133.9 (8 C6), 132.1 (8 C1), 131.8 (8 C5), 121.2 (4 C3), 113.6 (4 C11), 24.0 (4 C9), −3.2 (8 C8). 29Si{1H} NMR (CDCl3): δ = −4.1 (4 Si). ESI-TOF: m/z = 1070.44 [M]+ (calcd 1070.40). Anal. calcd for C64H70N4Si4Zn (1073.00): C, 71.64; H, 6.58; N, 5.22. Found: C, 71.44; H, 6.58; N, 4.98.
meso-Tetrakis[4-(diallylmethylsilyl)phenyl]porphyrinato zinc (4b)
To porphyrin 3c (0.050 mmol, 0.0556 g) dissolved in CHCl3 (15 mL), [Zn(OAc)2·2H2O] (0.100 mmol, 0.0219 g) dissolved in MeOH (10 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and afterwards it was washed with saturated NaHCO3 (15 mL) and then with water (2 × 15 mL). The organic layer was dried over Na2SO4 and all volatiles were removed in oil-pump vacuum to afford 4b as a dark red solid (0.490 mmol, 0.0576 g, 98% based on 3c).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3070, 3056, 3008, 2990, 2966, 2952, 2912, 2876, 1628 (m, CC), 1252 (m, SiCH3 bending), 820 (s, SiC), 798 (s, SiC). 1H NMR (CDCl3): δ = 8.98 (s, 8 H, H1), 8.24 (brd, JHH = 7.9 Hz, 8 H, H5), 7.89 (brd, JHH = 7.9 Hz, 8 H, H6), 6.01 (ddt, JHH = 17.0 Hz, JHH = 10.1 Hz, JHH = 8.1 Hz, 8 H, H10), 5.07 (ddt, JHH = 17.0 Hz, JHH = 2.0 Hz, JHH = 0.9 Hz, 4 H, cis-H11), 5.03 (ddt, JHH = 10.1 Hz, JHH = 2.0 Hz, JHH = 0.9 Hz, 4 H, trans-H11), 2.02 (dt, JHH = 8.1 Hz, JHH = 0.9 Hz, 16 H, H9), 0.55 (s, 12 H, H8). 13C{1H} NMR (CDCl3): δ = 150.1 (4 C2), 143.6 (4 C7), 135.8 (4 C4), 134.3 (8 C10), 133.9 (8 C6), 132.2 (8 C5), 132.0 (8 C1), 121.1 (4 C3), 114.1 (8 C11), 21.9 (8 C9), −5.6 (4 C8). 29Si{1H} NMR (CDCl3): δ = −5.2. ESI-TOF: m/z = 1174.48 [M]+ (calcd 1174.46). Anal. calcd for C72H78N4Si4Zn (1177.15): C, 73.46; H, 6.68; N, 4.76. Found: C, 73.24; H, 6.45; N, 4.68.
meso-Tetrakis[4-dimethyl(3-hydroxypropyl)silylphenyl]porphyrin (5a)
To [BH3·SMe2] (2.0 M, 2.00 mmol, 1.00 mL) dissolved in thf (5.0 mL), 3b (0.397 mmol, 0.400 g) in thf (30 mL) was drop-wise added over 10 min at 0 °C. The reaction solution was stirred for 2 h at this temperature and afterwards it was quenched with aqueous NaOH (3 M, 0.75 mL) and stirring was continued for 15 min. Afterwards, H2O2 (30%, 0.75 mL) was added in a single portion, stirring was continued at ambient temperature for 30 min and then the reaction mixture was extracted with saturated aqueous K2CO3 (20 mL) and with a Et2O–thf mixture (ratio 1:1, v/v; 50 mL). The organic layer was washed with brine and filtered through a silica gel column (thf, column size 1.0 × 10 cm). From the eluate, all volatiles were removed in oil-pump vacuum and the residue was washed with Et2O (3 × 15 mL) and was then dried in oil-pump vacuum to give 5a (0.332 mmol, 0.358 g, 83% based on 3b) as a dark red solid.
Mp = 350 °C (dec). IR (KBr disc): ṽ = 3397 (s, OH), 3314 (s, NH), 3055, 3011, 2994, 2926, 2864, 1251 (m, CH3 bending), 833, 803 (s, SiC). 1H NMR (dmso-d6): δ = 8.85 (s, 8 H, H1), 8.23 (d, JHH = 7.4 Hz, 8 H, H5), 7.96 (d, JHH = 7.3 Hz, 8 H, H6), 4.53 (t, JHH = 5.3 Hz, 4 H, OH), 3.51 (q, JHH = 6.2 Hz, 8 H, H11), 1.67 (m, 8 H, H10), 0.96 (m, 8 H, H9), 0.50 (s, 24 H, H8), −2.87 (br, 2 H, NH). 1H NMR (thf-d8): δ = 8.84 (s, 8 H, H1), 8.22 (d, JHH = 7.8 Hz, 8 H, H5), 7.95 (d, JHH = 7.8 Hz, 8 H, H6), 3.58 (t, JHH = 6.2 Hz, 8 H, H11), 1.64 (m, 8 H, H10), 1.03 (m, 8 H, H9), 0.52 (s, 24 H, H8). 13C{1H} NMR (thf-d8): δ = 143.6 (4 C7), 139.4 (4 C4), 134.8 (8 C6), 132.7 (8 C5), 131.7 (br, 8 C1), 120.9 (4 C3), 65.5 (4 C11), 28.5 (4 C10), 12.5 (4 C9), −2.8 (8 C8). 29Si{1H} NMR (dmso-d6): δ −2.0. 29Si{1H} NMR (thf-d8): δ = −4.0. ESI-TOF: m/z = 1079.60 [M + H]+ (calcd 1079.52). Anal. calcd for C64H78N4O4Si4·thf (1150.57): C, 70.91; H, 7.53; H, 4.86. Found: C, 70.63; H, 7.65; N, 4.98.
meso-Tetrakis[4-methyldi(3-hydroxypropyl)silylphenyl]porphyrin (5b)
To [BH3·SMe2] (2.0 M, 2.00 mmol, 1.00 mL) dissolved in thf (5.0 mL), porphyrin 3c (0.207 mmol, 0.230 g) in thf (30 mL) was added drop-wise over 10 min at 0 °C. After 2 h of stirring at this temperature aqueous NaOH (3 M, 0.75 mL) was added and stirring was continued for 15 min. Afterwards, H2O2 (30%, 0.75 mL) was added in a single portion and the appropriate reaction mixture was stirred at ambient temperature for 30 min. It was extracted with saturated aqueous K2CO3 (20 mL) and then with a Et2O–thf mixture (ratio 1:1, v/v; 50 mL). The organic layer was washed with brine and filtered through silica gel (thf, column size 1 × 10 cm). All volatiles were removed in oil-pump vacuum and the residue was washed with Et2O (3 × 15 mL) and then dried in oil-pump vacuum to give 5b as a dark red solid (0.150 mmol, 0.186 g, 72% based on 3c).
Mp = 350 °C (dec). IR (KBr disc): ṽ = 3393 (s, OH), 3314 (s, NH), 3055, 3011, 2994, 2927, 2863, 1251 (m, CH3 bending), 801 (s, SiC). 1H NMR (dmso-d6): δ = 8.87 (s, 8 H, H1), 8.26 (d, JHH = 7.5 Hz, 8 H, H5), 7.97 (d, JHH = 7.5 Hz, 8 H, H6), 4.56 (t, JHH = 5.3 Hz, 8 H, OH), 3.51 (q, JHH = 6.1 Hz, 16 H, H11), 1.67 (m, 16 H, H10), 1.00 (m, 16 H, H9), 0.51 (s, 12 H, H8), −2.86 (br, 2 H, NH). 1H NMR (thf-d8): δ = 8.84 (s, 8 H, H1), 8.22 (d, JHH = 7.6 Hz, 8 H, H5), 7.96 (d, JHH = 7.6 Hz, 8 H, H6), 3.58 (t, JHH = 6.4 Hz, 16 H, H11), 1.74 (m, 16 H, H10), 1.06 (m, 16 H, H9), 0.52 (s, 12 H, H8). 13C{1H} NMR (thf-d8): δ = 143.5 (4 C7), 138.6 (4 C4), 134.8 (8 C6), 133.1 (8 C5), 131.7 (br, 8 C1), 121.0 (4 C3), 65.6 (8 C11), 28.5 (8 C10), 10.9 (4 C9), −4.8 (8 C8). 29Si{1H} NMR (dmso-d6): δ = −0.8. 29Si{1H} NMR (thf-d8): δ = −0.8. ESI-TOF: m/z = 1293.70 [M + K]+ (calcd 1293.58). Anal. calcd for C72H94N4O8Si4·H2O (1273.90): C, 67.88; H, 7.60; N, 4.40. Found: C, 67.68; H, 7.65; N, 4.31.
meso-Tetrakis[4-dimethyl(3-hydroxypropyl)silylphenyl]porphyrinato zinc (6a)
To 5a (0.0926 mmol, 0.100 g) dissolved in thf (30 mL) a solution of [Zn(OAc)2·2H2O] (0.185 mmol, 0.0407 g) in MeOH (10 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and afterwards was concentrated in oil-pump vacuum to dryness. The residue was dissolved in thf (20 mL) and filtered through a pad of silica gel (thf) and then all volatiles were removed in oil-pump vacuum. The remaining solid was washed with a Et2O–npentane mixture (ratio 1:4 v/v,(30 mL)) and then dried in oil-pump vacuum to give the title compound as a dark red solid (0.0857 mmol, 0.0980 g, 93% based on 5a).
Mp = 350 °C (dec). IR (KBr disc): ṽ = 3398 (m, OH), 3048, 3004, 2945, 2927, 2863, 1246 (s, CH3 bending), 831, 798 (s, SiC). 1H NMR (dmso-d6): δ = 8.76 (s, 8 H, H1), 8.16 (d, JHH = 7.5 Hz, 8 H, H5), 7.89 (d, JHH = 7.5 Hz, 8 H, H6), 4.49 (t, JHH = 6.2 Hz, 4 H, OH), 3.46 (q, JHH = 6.3 Hz, 8 H, H11), 1.63 (m, 8 H, H10), 0.92 (m, 8 H, H9), 0.47 (s, 24 H, H8). 13C{1H} NMR (dmso-d6): δ = 150.1 (4 C2), 144.2 (4 C7), 138.6 (4 C4), 134.7 (8 C6), 132.6 (8 C5 and 8 C1), 121.2 (4 C3), 64.8 (4 C11), 28.2 (4 C10), 12.4 (4 C9), −1.8 (8 C8). 29Si{1H} NMR (dmso-d6): δ = −2.1. ESI-TOF: m/z = 1179.50 [M + K]+ (calcd 1179.39). Anal. calcd for C64H76N4O4Si4Zn (1143.04): C, 67.25; H, 6.70; N, 4.90. Found: C, 67.44; H, 7.12; N, 4.81.
meso-Tetrakis[4-methyldi(3-hydroxypropyl)silylphenyl]porphyrinato zinc (6b)
To porphyrin 5b (0.0805 mmol, 0.100 g) dissolved in thf (30 mL), [Zn(OAc)2·2H2O] (0.1611 mmol, 0.03536 g) in MeOH (10 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and afterwards all volatiles were removed in oil-pump vacuum. The obtained residue was dissolved in thf (50 mL) and filtered through a pad of silica gel (thf). All volatiles were removed in oil-pump vacuum. The remaining residue was washed with Et2O (30 mL) and then dried in oil-pump vacuum to give 6b as a red solid (0.068 mmol, 0.089 g, 85% based on 5b).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3403 (m, OH), 3055, 3004, 2923, 2863, 1250 (w, CH3 bending), 858, 796 (s, SiC). 1H NMR (dmso-d6): δ = 8.77 (s, 8 H, H1), 8.18 (d, JHH = 7.7 Hz, 8 H, H5), 7.90 (d, JHH = 7.7 Hz, 8 H, H6), 4.49 (t, JHH = 6.2 Hz, 8 H, OH), 3.46 (q, JHH = 6.3 Hz, 8 H, H11), 1.64 (m, 16 H, H10), 0.95 (m, 16 H, H9), 0.47 (s, 12 H, H8). 13C{1H} NMR (dmso-d6): δ = 150.2 (4 C2), 144.2 (4 C7), 137.8 (4 C4), 134.8 (8 C6), 132.9 (8 C5), 132.6 (br, 8 C1), 121.3 (4 C3), 64.9 (4 C11), 28.2 (4 C10), 10.8 (4 C9), −3.8 (8 C8). 29Si{1H} NMR (dmso-d6): δ = −0.9. ESI-TOF: m/z = 1355.62 [M + K]+ (calcd 1355.49). Anal. calcd for C72H92N4O8Si4Zn (1319.24): C, 65.33; H, 6.95; H, 4.29. Found: C, 64.89; H, 7.11; N, 4.07.
4-[Tris(4-trimethylsilylphenyl)silyl]benzaldehyde (8a)
The synthetic procedure described for the preparation of 2b (see earlier) was applied to synthesize 8a: tBuLi (1.7 M, 13.00 mmol, 7.70 mL, npentane), 7a (6.50 mmol, 4.10 g) in Et2O (70 mL), dimethyl formamide (19.50 mmol, 1.43 g, 1.51 mL) in thf (40 mL) and aqueous HCl (3 N, 40 mL). After appropriate work-up, 8a could be isolated as a colorless solid (5.93 mmol, 3.45 g, 91% based on 7a).Mp = 212 °C. IR (film): ṽ = 3048, 2996, 2953 (m), 2894, 2816 (w, CHO), 2722 (w, CHO), 1706 (s, CO), 1249 (w, CH3 bending), 1133, 838 (s, SiC). 1H NMR (CDCl3): δ = 10.06 (s, 1 H, CHO), 7.87 (brd, JHH = 8.2 Hz, 2 H, H3), 7.78 (brd, JHH = 8.2 Hz, 2 H, H4), 7.55 (s, 12 H, H7 and H8), 0.29 (s, 27 H, H10). 13C{1H} NMR (CDCl3): δ = 192.6 (1 C1), 143.2 (1 C2), 142.4 (3 C9), 136.9 (2 C4), 135.5 (6 C7), 133.5 (3 C6), 132.8 (6 C8), 128.6 (2 C3), 127.8 (1 C5), −1.2 (9 C10). 29Si{1H} NMR (CDCl3): δ = −14.7 (1 Si1), −3.9 (3 Si2). ESI-TOF: m/z = 619.24 [M + K]+ (calcd 619.21). Anal. calcd for C35H44OSi4 (581.05): C, 70.28; H, 7.63. Found: C, 70.19; H, 7.45.
4-[Tris-(4-diallylmethylsilylphenyl)silyl]benzaldehyde (8c)
The same procedure as described for preparing 2b was applied in the synthesis of 8c (see above): tBuLi (1.7 M, 6.00 mmol, 3.60 mL, npentane), 7c (3.00 mmol, 2.361 g) in Et2O (40 mL), dimethyl formamide (9.00 mmol, 0.658 g, 0.70 mL) in thf (20 mL) and aqueous HCl (3 N, 25 mL). After appropriate work-up, 8c was isolated as colorless oil (2.46 mmol, 1.81 g, 82% based on 7c).IR (film): ṽ = 3074, 3050, 2996, 2969, 2913, 2878, 2819 (w, CHO), 2725 (w, CHO), 1705 (s, CO), 1629 (w, CC), 1252 (w, CH3 bending), 1132, 821 (s, SiC), 802 (s, SiC). 1H NMR (CDCl3): δ = 10.07 (s, 1 H, CHO), 7.89 (brd, JHH = 8.1 Hz, 2 H, H3), 7.77 (brd, JHH = 8.1 Hz, 2 H, H4), 7.55 (brs, 12 H, H7 and H8), 5.80 (ddt, JHH = 16.9 Hz, JHH = 10.2 Hz, JHH = 8.0 Hz, 6 H, H12), 4.91 (ddt, JHH = 16.9 Hz, JHH = 2.1 Hz, JHH = 1.1 Hz, 6 H, cis-H13), 4.89 (ddt, JHH = 10.2 Hz, JHH = 2.1 Hz, JHH = 1.1 Hz, 6 H, trans-H13), 1.84 (brd, JHH = 8.0 Hz, 12 H, H11), 0.31 (s, 9 H, H10). 13C{1H} NMR (CDCl3): δ = 192.5 (1 C1), 142.8 (1 C2), 139.1 (3 C9), 137.0 (1 C5), 136.9 (2 C4), 135.4 (6 C8), 134.0 (6 C12), 133.9 (3 C6), 133.4 (6 C7), 128.6 (2 C3), 113.5 (6 C13), 23.5 (6 C11), −3.6 (3 C10). 29Si{1H} NMR (CDCl3): δ = −14.7 (1 Si1), −5.7 (3 Si2). ESI-TOF: m/z = 775.01 [M + K]+ (calcd 775.30). Anal. calcd for C46H56OSi4 (737.28): C, 74.94; H, 7.66. Found: C, 74.93; H, 7.77.
meso-Tetrakis{4-[tris(4-trimethylsilylphenyl)silyl]phenyl}porphyrin (9a)
To 8a (4.00 mmol, 2.324 g) and pyrrole (4.00 mmol, 0.268 g, 0.28 mL) dissolved in CH2Cl2 (400 mL), [BF3·OEt2] (0.400 mmol, 0.057 g, 0.05 mL) was added in a single portion. The reaction solution was stirred for 2 h and then 2,3-dichloro-5,6-dicyanobenzoquinone (3.00 mmol, 0.681 g) was added in a single portion and stirring was continued for 1 h. The reaction mixture was washed with saturated NaHCO3 (40 mL) and afterwards with water (3 × 40 mL). The organic layer was filtered through a pad of silica gel and then all volatiles were removed in oil-pump vacuum. The crude product was purified by column chromatography (a) nhexane and (b) 20 vol% CH2Cl2–nhexane. After all volatiles were removed in oil pump vacuum, porphyrin 9a was obtained as a dark red solid (0.256 mmol, 0.643 g, 26% based on 8a).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3315 (s, NH), 3047, 2995, 2952, 2932, 2894, 1249 (m, CH3 bending), 1133, 847 (s, SiC), 839 (s, SiC), 803. 1H NMR (CDCl3): δ = 8.94 (s, 8 H, H1), 8.27 (brd, JHH = 8.0 Hz, 8 H, H5), 8.02 (brd, JHH = 8.0 Hz, 8 H, H6), 7.85 (brd, JHH = 8.0 Hz, 24 H, H9), 7.70 (brd, JHH = 8.0 Hz, 24 H, H10), 0.36 (s, 108 H, H12), −2.72 (br, 2 H, NH). 13C{1H} NMR (CDCl3): δ = 143.3 (4 C4), 142.2 (12 C11), 135.8 (24 C10), 134.7 (8 C6), 134.6 (12 C8), 134.2 (8 C5), 133.5 (4 C7) 132.9 (24 C9), 120.1 (4 C3), −1.1 (36 C12). 29Si{1H} NMR (CDCl3): δ = −14.2 (4 Si1), −4.5 (12 Si2). Anal. calcd for C152H182N4Si16 (2514.47): C, 72.60; H, 7.30; N, 2.23. Found: C, 72.54; H, 6.93; N, 2.13.
meso-Tetrakis{4-[tris(4-diallylmethylsilylphenyl)silyl]phenyl}porphyrin (9c)
To 8c (1.81 mmol, 1.333 g) and pyrrole (1.81 mmol, 0.122 g, 0.13 mL) dissolved in CH2Cl2 (180 mL), [BF3·OEt2] (0.18 mmol, 0.026 g, 0.02 mL) was added in a single portion. The reaction solution was stirred for 2 h and then 2,3-dichloro-5,6-dicyanobenzoquinone (1.36 mmol, 0.308 g) was added and stirring was continued for 1 h. The reaction mixture was washed with saturated NaHCO3 (40 mL) and then with water (3 × 40 mL). The organic layer was filtered through a pad of silica gel and afterwards all volatiles were removed in oil-pump vacuum. The crude product was purified by column chromatography (a) nhexane and (b) 20 vol% CH2Cl2–nhexane. After removal of all volatiles in a vacuum, porphyrin 9c was isolated as a dark red solid (0.143 mmol, 0.452 g, 32% based on 8c).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3322 (S, NH), 3074, 3050, 2996, 2968, 2954, 2912, 2878, 1630 (m, CC), 1252 (m, CH3 bending), 1132, 822 (s, SiC), 802 (s, SiC). 1H NMR (CDCl3): δ = 8.94 (s, 8 H, H1), 8.28 (brd, JHH = 7.6 Hz, 8 H, H5), 7.99 (brd, JHH = 7.6 Hz, 8H, H6), 7.82 (brd, JHH = 7.5 Hz, 24 H, H9), 7.68 (brd, JHH = 7.5 Hz, 24 H, H10), 5.86 (ddt, JHH = 17.2 Hz, JHH = 9.6 Hz, JHH = 8.1 Hz, 24 H, H14), 4.95 (brd, JHH = 17.2 Hz, 24 H, cis-H15), 4.93 (brd, 9.6 Hz, 24 H, trans-H15), 1.90 (brd, 8.00 Hz, 48 H, H13), 0.37 (s, 36 H, H12), −2.75 (s, 2 H, NH). 13C{1H} NMR (CDCl3): δ = 143.4 (4 C4), 138.9 (12 C11), 135.7 (24 C10), 135.0 (12 C8), 134.8 (8 C6), 134.3 (8 C5), 134.2 (24 C14), 133.5 (24 C9), 133.2 (4 C7), 120.1 (4 C3), 114.0 (24 C15), 21.6 (24 C13), −5.8 (24 C12). 29Si{1H} NMR (CDCl3): δ = −14.2 (4 Si1), −5.7 (12 Si2). Anal. calcd for C200H230N4Si16 (3153.39): C, 76.52; H, 7.38, N, 1.78. Found: C, 76.66; H, 7.17; N, 1.23.
meso-Tetrakis{4-[tris(4-trimethylsilylphenyl)silyl]phenyl}porphyrinato zinc (10a)
To 9a (0.0139 mmol, 0.0350 g) in CHCl3 (5 mL), [Zn(OAc)2·2H2O] (0.0347 mmol, 0.0076 g) in MeOH (2.5 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and afterwards was concentrated in oil-pump vacuum to dryness. The residue was extracted with CHCl3 (10 mL) and then with water (5 mL). The organic layer was dried over MgSO4 and all volatiles were removed in oil-pump vacuum to afford 10a as a dark red solid (0.0135 mmol, 0.340 g, 97% based on 9a).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3046, 2994, 2952, 2932, 2893, 1249 (m, CH3 bending), 1132, 845 (s, SiC), 839 (s, SiC), 803. 1H NMR (CDCl3): δ = 9.02 (s, 8 H, H1), 8.25 (brd, JHH = 7.9 Hz, 8 H, H5), 7.99 (brd, JHH = 7.9 Hz, 8 H, H6), 7.83 (brd, JHH = 7.8 Hz, 24 H, H9), 7.68 (brd, JHH = 7.8 Hz, 24 H, H10), 0.34 (s, 108 H, H12). 13C{1H} NMR (CDCl3): δ = 150.1 (8 C2), 144.0 (4 C4), 142.1 (12 C11), 135.8 (24 C10), 134.7 (12 C8), 134.6 (8 C6), 134.1 (8 C5), 133.2 (4 C7), 132.9 (24 C9), 132.1 (8 C1), 121.1 (4 C3), −1.1 (36 C12). 29Si{1H} NMR (CDCl3): δ = −14.3 (4 Si1), −3.9 (12 Si2). Anal. calcd for C152H180N4Si16Zn·EtOH (2619.02): C, 70.49; H, 7.14; N, 2.14. Found: C, 70.51; H, 6.95; N, 1.86.
meso-Tetrakis{4-[tris(4-allyldimethylsilylphenyl)silyl]phenyl]porphyrinato zinc (10b)
To 9b (0.0123 mmol, 0.0350 g) dissolved in CHCl3 (5 mL), [Zn(OAc)2·2H2O] (0. 0308 mmol, 0.0067 g) in MeOH (2.5 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and then all volatiles were removed in oil-pump vacuum. The residue was extracted with CHCl3 (10 mL) and then with water (5 mL). The organic layer was dried over MgSO4 and all volatiles were removed in oil-pump vacuum to afford 10b as a dark red solid (0.0118 mmol, 0.336 g, 96% based on 9b).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3074, 3047, 2994, 2953, 2912, 1628 (m, CC), 1249 (m, CH3 bending), 1132, 835 (s, SiC), 799 (s, SiC). 1H NMR (CDCl3): δ = 9.05 (s, 8 H, H1), 8.29 (brd, JHH = 8.0 Hz, 8 H, H5), 7.97 (brd, JHH = 8.0 Hz, 8H, H6), 7.85 (brd, JHH = 8.0 Hz, 24 H, H9), 7.70 (brd, JHH = 8.0 Hz, 24 H, H10), 5.88 (ddt, JHH = 16.9 Hz, JHH = 10.2 Hz, JHH = 8.1 Hz, 24 H, H14), 4.95 (ddt, JHH = 16.9 Hz, JHH = 2.1 Hz, JHH = 1.1 Hz, cis-H15), 4.92 (ddt, JHH = 10.2 Hz, JHH = 2.1 Hz, JHH = 1.1 Hz, trans-H15), 1.86 (dt, JHH = 8.1 Hz, JHH = 1.1 Hz, 24 H, H13), 0.38 (s, 72 H, H12). 13C{1H} NMR (CDCl3): δ = 150.1 (8 C2), 144.0 (4 C4), 140.5 (12 C11), 135.8 (24 C10), 134.9 (12 C8), 134.6 (12 C14 and 8 C6), 134.1 (8 C5), 133.2 (24 C9), 133.0 (4 C7), 132.1 (8 C1), 121.1 (4 C3), 113.5 (12 C15), 23.6 (12 C13), −3.5 (24 C12). 29Si{1H} NMR (CDCl3): δ = −14.2 (4 Si1), −4.5 (12 Si2). Anal. calcd for C176H204N4Si16Zn·2EtOH (2982.26): C, 72.49; H, 7.30, N, 1.88. Found: C, 72.17; H, 7.10; N, 1.67.
meso-Tetrakis{4-[tris(4-diallylmethylsilylphenyl)silyl]phenyl}porphyrinato zinc (10c)
To 9c (0.0317 mmol, 0.100 g) in CHCl3 (15 mL), a solution of [Zn(OAc)2·2H2O] (0.0793 mmol, 0.017 g) in MeOH (5 mL) was added in a single portion. The reaction solution was stirred for 2 h at ambient temperature and afterwards all volatiles were removed in oil-pump vacuum. The residue was extracted with CHCl3 (20 mL) and then with water (10 mL). The organic layer was dried over MgSO4 and all volatiles were removed in oil-pump vacuum to afford 10c (0.0308 mmol, 0.099 g, 97% based on 9c).Mp = 350 °C (dec). IR (KBr disc): ṽ = 3074, 3050, 2994, 2968, 2912, 2878, 1628 (m, CC), 1252 (m, CH3 bending), 1132, 820 (s, SiC), 802 (s, SiC). 1H NMR (CDCl3): δ = 9.03 (s, 8 H, H1), 8.27 (brd, JHH = 8.0 Hz, 8 H, H5), 7.97 (brd, JHH = 8.0 Hz, 8H, H6), 7.82 (brd, JHH = 8.0 Hz, 24 H, H9), 7.67 (brd, JHH = 8.0 Hz, 24 H, H10), 5.85 (ddt, JHH = 16.9 Hz, JHH = 9.6 Hz, JHH = 8.1 Hz, 24 H, H14), 4.94 (ddt, JHH = 16.9 Hz, JHH = 2.1 Hz, JHH = 1.0 Hz, cis-H15), 4.91 (ddt, JHH = 9.9 Hz, JHH = 2.1 Hz, JHH = 1.0 Hz, trans-H15), 1.89 (brd, JHH = 8.1 Hz, 48 H, H13), 0.36 (s, 36 H, H12). 13C{1H} NMR (CDCl3): δ = 150.1 (8 C2), 144.1 (4C4), 138.8 (12 C11), 135.7 (24 C10), 135.1 (12 C8), 134.6 (8 C6), 134.2 (24 C14), 134.1 (8 C5), 133.5 (24 C9), 132.9 (4 C7), 132.1 (8 C1), 121.1 (4 C3), 114.0 (24 C15), 21.6 (24 C13), −5.8 (12 C12). 29Si{1H} NMR (CDCl3): δ = −14.2 (4 Si1), −5.7 (12 Si2). Anal. calcd for C200H228N4Si16Zn·2EtOH (3294.87): C, 74.36; H, 7.34; N, 1.70. Found: C, 74.21; H, 7.30; N, 1.78.
X-ray crystallography
Single crystal X-ray diffraction measurements of 3b,c, 4a and 9b were performed with a Bruker Smart 1k CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å), while for data collection of 6a an Oxford Gemini S diffractometer and Cu Kα radiation (λ = 1.54184 Å) was used. Table S1† summarizes selected crystal and structural refinement data of 3b,c, 4a, 6a and 9b. All structures were solved by direct methods using the SHELXS-97 and refined by full-matrix least-squares procedures on F2 using the SHELXL-97 as part of the software package SHELXTL.46 All non-hydrogen atoms were refined anisotropically. All C-, N- and O-bonded hydrogen atoms were refined using a riding model. Only in the case of 3c the positions of N-bonded hydrogen atoms could be taken from the difference Fourier map and were refined freely. For 3b the atoms C52–C54 have been refined disordered on three positions with occupation factors of 0.26, 0.20 and 0.54. Furthermore, the CH2 group of the partially occupied CH2Cl2 (occupation factor 1/4) packing solvent molecule has been refined to split occupancies of 0.94 and 0.06. In the case of 3c the atoms C23–C29 and C30–C36 have been refined disordered with split occupancies of 0.68/0.32 and 0.51/0.49, respectively. In the case of 4a the atoms C38–C42 and C49–C53 have been refined disordered with split occupancies of 0.36/0.64 and 0.72/0.28, respectively. For 6a one thf packing solvent molecule (O6, C70–C73) has been refined disordered with split occupancies of 0.47 and 0.53. Furthermore, atoms O1, O2 and O3 are disordered and have been refined to split occupancies of 0.54/0.46, 0.50/0.50 and 0.50/0.50, respectively. In the case of 9b the atoms C25/C26, C36/C37 and C126 have been disordered with split occupancies of 0.76/0.24, 0.57/0.43 and 0.52/0.48, respectively. Furthermore, the ethyl group (C11O, C12O) of one partially occupied EtOH molecule (occupation factor 1/2) has been refined to split occupancies of 0.32 and 0.68.Data have been deposited at the Cambridge Crystallographic Data Centre under the CCDC deposition numbers 976300 (3c), 976301 (3b), 976302 (4a), 976303 (6a) and 976304 (9a).
Acknowledgements
We gratefully acknowledge the generous financial support from the Deutsche Forschungsgemeinschaft (Research Training Group Accumol, Research Unit “Towards Molecular Spintronics” FOR1154) and the Fonds der Chemischen Industrie. We thank Dipl.-Phys. L. Smykalla and Prof. Dr M. Hietschold (Technische Universität Chemnitz, Faculty of Sciences, Institute of Physics) for providing us with a DFT calculated “saddle-shaped” deformed molecular structure of tetra(p-hydroxy-phenyl) porphyrin (Fig. S7†) and for helpful discussion concerning the definition of “saddle-shaped” deformed geometries of porphyrins.References
- (a) S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2012, 64, 102 CrossRef; (b) D. Astrúc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857 CrossRef PubMed; (c) A. Sekiguchi, V. Y. Lee and M. Nanjo, Coord. Chem. Rev., 2000, 210, 11 CrossRef CAS; (d) M. Fischer and F. Vögtle, Angew. Chem., 1999, 111, 934 ( Angew. Chem. Int. Ed. , 1999 , 38 , 884 ) CrossRef; (e) A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665 CrossRef CAS PubMed; (f) G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689 CrossRef CAS PubMed.
- (a) D. A. Tomalia, Soft Matter, 2010, 6, 456–474 RSC; (b) A.-M. Caminade, C.-O. Turrin, R. Laurent, A. Ouali and B. Delavaux-Nicot, Dendrimers: Towards Catalytic, Material and Biomedical Uses, John Wiley & Sons, Chichester, 2011 Search PubMed; (c) D. Türp, T.-T.-T. Nguyen, M. Baumgarten and K. Müllen, New J. Chem., 2012, 36, 282 RSC; (d) X. Qi, C. Xue, X. Huang, Y. Huang, X. Zhou, H. Li, D. Liu, F. Boey, Q. Yan, W. Huang, S. De Feyter, K. Müllen and H. Zhang, Adv. Funct. Mater., 2010, 20, 43 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352 RSC.
- (a) S. M. Grayson and J. M. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS PubMed; (b) T. Kawaguchi, K. L. Walker, C. L. Wilkins and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 2159 CrossRef CAS.
- (a) A. K. Diallo, J. Ruiz and D. Astrúc, Chem.–Eur. J., 2013, 19, 8913 CrossRef CAS PubMed; (b) D. Astrúc, Nat. Chem., 2012, 4, 255 CrossRef PubMed; (c) X.-N. Han, J.-M. Chen, Z.-T. Huang and Q.-Y. Zheng, Eur. J. Org. Chem., 2012, 6895 CrossRef CAS; (d) G. Bergamini, P. Ceroni, P. Fabbrizi and S. Cicchi, Chem. Commun., 2011, 47, 12780 RSC.
- K. Chiad, M. Grill, M. Baumgarten, M. Klapper and K. Müllen, Macromolecules, 2013, 46, 3554 CrossRef CAS.
- (a) J. Lim and E. E. Simanek, Adv. Drug Delivery Rev., 2012, 64, 826 CrossRef CAS PubMed; (b) A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171 CrossRef CAS PubMed; (c) L. G. Weaver, Y. Singh, G. Vamvounis, M. F. Wyatt, P. L. Burn and J. T. Blanchfield, Polym. Chem., 2014, 5, 1173 RSC.
- (a) W. S. Li and T. Aida, Chem. Rev., 2009, 109, 6047 CrossRef CAS PubMed; (b) A. Uetomo, M. Kozaki, S. Suzuki, K. Yamanaka, O. Ito and K. Okada, J. Am. Chem. Soc., 2011, 133, 13276 CrossRef CAS PubMed; (c) Y.-H. Jeong, H.-J. Yoon and W.-D. Jang, Polym. J., 2012, 44, 512 CrossRef CAS.
- G. Zaragoza-Galán, M. A. Fowler, J. Duhamel, R. Rein, N. Solladié and E. Rivera, Langmuir, 2012, 28, 11195 CrossRef PubMed.
- (a) M.-S. Choi, T. Yamazaki, I. Yamazaki and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 150 CrossRef CAS PubMed; (b) H. Imahori, J. Phys. Chem. B, 2004, 108, 6130 CrossRef CAS PubMed; (c) E. M. Harth, S. Hecht, B. Helms, E. E. Malmstrom, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 3926 CrossRef CAS PubMed.
- (a) M. A. Oar, J. M. Serin, W. R. Dichtel, J. M. J. Fréchet, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Mater., 2005, 17, 2267 CrossRef CAS; (b) A. Zingg, B. Felber, V. Gramlich, L. Fu, J. P. Collman and F. Diederich, Helv. Chim. Acta, 2002, 82, 333 CrossRef; (c) J. P. Collman, L. Fu, A. Zingg and F. Diederich, Chem. Commun., 1997, 193 RSC.
- P. Weyermann, J.-P. Gisselbrecht, C. Boudon, F. Diederich and M. Gross, Angew. Chem., Int. Ed., 1999, 38, 3215 CrossRef CAS.
- (a) P. Bhyrappa, J. K. Young, J. S. Moore and K. S. Suslick, J. Am. Chem. Soc., 1996, 118, 5708 CrossRef CAS; (b) P. Bhyrappa, J. K. Young, J. S. Moore and K. S. Suslick, J. Mol. Catal. A: Chem., 1996, 113, 109 CrossRef CAS.
- (a) E. R. Gillies and J. M. J. Fréchet, Drug Discovery Today, 2005, 10, 35 CrossRef CAS PubMed; (b) N. Nishiyama, H. R. Stapert, G.-D. Zhang, D. Takasu, D.-L. Jiang, T. Nagano, T. Aida and K. Kataoka, Bioconjugate Chem., 2003, 14, 58 CrossRef CAS PubMed; (c) G.-D. Zhang, A. Harada, N. Nishiyama, D.-L. Jiang, H. Koyama, T. Aida and K. Kataoka, J. Controlled Release, 2003, 93, 141 CrossRef CAS.
- W. R. Dichtel, J. M. Serin, C. Edder, J. M. J. Fréchet, M. Matuszewski, L.-S. Tan, T. Y. Ochulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2004, 126, 5380 CrossRef CAS PubMed.
- (a) E. G. Morales-Espinoza, K. E. Sanchez-Montes, E. Klimova, T. Klimova, I. V. Lijanova, J. L. Maldonado, G. Ramos-Ortíz, S. Hernández-Ortega and M. Martínez-García, Molecules, 2010, 15, 2564 CrossRef CAS PubMed; (b) B. P. Singh, R. Vijaya, S. J. Shetty, K. Kandasamy, P. N. Puntambekar and T. S. Srivastava, J. Porphyrins Phthalocyanines, 2000, 4, 659 CrossRef CAS.
- (a) S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097 CrossRef CAS PubMed; (b) P. L. Burn, S.-C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675 CrossRef CAS; (c) J. M. Lupton, I. D. W. Samuel, M. J. Frampton, R. Beavington and P. L. Burn, Adv. Funct. Mater., 2001, 11, 287 CrossRef CAS; (d) A. Adronov and J. M. J. Fréchet, Chem. Commun., 2000, 1701 RSC.
- (a) V. K. Gupta, A. K. Jain, Z. Ishtaiwi, H. Lang and G. Maheshwari, Talanta, 2007, 73, 803 CrossRef CAS PubMed; (b) V. K. Gupta, A. K. Jain, G. Maheshwari, H. Lang and Z. Ishtaiwi, Sens. Actuators, B, 2006, 117, 99 CrossRef CAS.
- (a) H. Imahori and T. Umeyama, J. Phys. Chem. C, 2009, 113, 9029 CrossRef CAS; (b) V. Balzani, A. Credi and M. Venturi, ChemSusChem., 2008, 1, 26 CrossRef CAS PubMed.
- (a) K. W. Pollak, E. M. Sanford and J. M. J. Fréchet, J. Mater. Chem., 1998, 3, 519 RSC; (b) J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS.
- B.-H. Ye and Y. Naruta, Tetrahedron, 2003, 59, 3593 CrossRef CAS.
- C. J. Murphy and H. W. Post, J. Org. Chem., 1962, 27, 1486 CrossRef CAS.
- (a) Y. Lee and R. B. Silverman, Org. Lett., 2000, 2, 303 CrossRef CAS PubMed; (b) Y. Lee and R. B. Silverman, Tetrahedron, 2001, 57, 5339 CrossRef CAS.
- Z. Ishtaiwi, T. Rüffer, A. Hildebrandt, F. F. Awwadi, H. Hahn, A. Abylaikhan, D. Taher, U. Siegert, B. Walfort and H. Lang, Eur. J. Inorg. Chem., 2013, 2368 CrossRef CAS.
- M. Fishwick and M. G. H. Wallbridge, J. Organomet. Chem., 1970, 25, 69 CrossRef CAS.
- G. Socrates, Infrared Characteristic Group Frequencies, John Wiley & Sons, 1980, p. 126 Search PubMed.
- A. Nikolić, B. Jović, S. Csanady and S. Petrović, J. Mol. Struct., 2007, 834–836, 249 CrossRef.
- R. J. Abraham, S. C. M. Fell and K. M. Smith, Org. Magn. Resonance, 1977, 9, 367 CrossRef CAS.
- K. C. K. Swamy, V. Chandrasekhar, J. J. Harland, J. M. Holmes, R. O. Day and R. R. Holmes, J. Am. Chem. Soc., 1990, 112, 2341 CrossRef CAS.
- (a) R.-H. Jin, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun., 1993, 1260 RSC; (b) Y. Tomoyose, D.-L. Jiang, R.-H. Jin, T. Aida, T. Yamashita, K. Horie, E. Yashima and Y. Okamoto, Macromolecules, 1996, 29, 5236 CrossRef CAS; (c) R. Sadamoto, N. Tomioko and T. Aida, J. Am. Chem. Soc., 1996, 118, 3978 CrossRef CAS; (d) D. Takasu, N. Tomioka, D.-L. Jiang, T. Aida, T. Kamachi and I. Okura, J. Inorg. Biochem., 1997, 67, 242 CrossRef CAS.
- K. W. Pollak, J. W. Leon, J. M. J. Fréchet, M. Maskus and H. D. Abruña, Chem. Mater., 1998, 10, 30 CrossRef CAS.
- (a) S. Kyushin, K. Yoshimura, K. Sato and H. Matsumoto, Chem. Lett., 2009, 38, 324 CrossRef CAS; (b) S. Ishida, M. Ito, H. Horiuchi, H. Hiratsuka, S. Shiraishi and S. Kyushin, Dalton Trans., 2010, 39, 9421 RSC.
- (a) P. C. VanDort and P. L. Fuchs, J. Am. Chem. Soc., 1994, 116, 5657 CrossRef CAS; (b) A. Barbero, Y. Blanco and F. J. Puliolo, J. Org. Chem., 2005, 70, 6876 CrossRef CAS PubMed; (c) Z. Ishtaiwi, T. Rüffer, R. Mothes, B. Walfort and H. Lang, J. Organomet. Chem., 2011, 696, 1409 CrossRef.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherril, J. Am. Chem. Soc., 2002, 124, 10887 CrossRef CAS PubMed.
- (a) T. Straaso, S. Kapishnikov, K. Kato, M. Takata, J. Als-Nielsen and L. Leiserowitz, Cryst. Growth Des., 2011, 11, 3342 CrossRef CAS; (b) D. S. Bohle, E. L. Dodd, A. J. Kosar, L. Sharma, P. W. Stephens, L. Suarez and D. Tazoo, Angew. Chem., Int. Ed., 2011, 50, 6151 CrossRef CAS PubMed; (c) K. Yamanishi, M. Miyazawa, T. Yairi, S. Sakai, N. Nishina, Y. Kobori, M. Kondo and F. Uchida, Angew. Chem., Int. Ed., 2011, 50, 6583 CrossRef CAS PubMed; (d) T. Amaya, T. Ueda and T. Hirao, Tetrahedron Lett., 2010, 51, 3376 CrossRef CAS; (e) M. J. Gunter, T. P. Jeynes and P. Turner, Eur. J. Org. Chem., 2004, 193 CrossRef CAS; (f) H. M. Lee, M. M. Olmstead, G. G. Gross and A. L. Balch, Cryst. Growth Des., 2003, 3, 691 CrossRef CAS; (g) S. Pagola, P. W. Stephens, D. S. Bohle, A. D. Kosov and S. K. Madsen, Nature, 2000, 404, 307 CrossRef CAS PubMed; (h) R. G. Khoury, L. Jaquinod, R. Paolesse and K. M. Smith, Tetrahedron, 1999, 55, 6713 CrossRef CAS; (i) M. O. Senge, M. Speck, A. Wiehe, H. Dieks, S. Aguirre and H. Kurreck, Photochem. Photobiol., 1999, 70, 206 CrossRef CAS; (j) H. M. Goff, E. T. Shimomura, Y. Ja Lee and W. R. Scheidt, Inorg. Chem., 1984, 23, 315 CrossRef CAS.
- (a) D. Bonifazi, G. Accorsi, N. Armaroli, F. Song, A. Palkar, L. Echegoyen, M. Scholl, P. Seiler, B. Jaum and F. Diederich, Helv. Chim. Acta, 2005, 88, 1839 CrossRef CAS; (b) J. Wojaczynski, L. Latos-Grazynski, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1997, 36, 4548 CrossRef CAS PubMed.
- P. Bhyrappa, G. Vaijayanthimala and B. Verghese, Tetrahedron Lett., 2002, 43, 6427 CrossRef CAS.
- Y. Diskin-Posner, S. Dahal and I. Goldberg, Chem. Commun., 2000, 585 RSC.
- It needs to be emphasized that for 3b, 3c, 4a and 9b, π–π interactions involving the terminal allylic side chains have been left unattended. The allyl groups have been at least partially refined disordered, if possible. Moreover, the respective carbon atoms do have comparatively large Uij values. Due to that it appears less trustworthy to assign for them π–π interactions correctly apart from attractive van der Waals forces.
-
(a) Labelling code: symmetry generated equivalent atoms of Si10, Si11–Si16/C101, C102–C188 are assigned as Si0#, Si1#–Si6#/C1# and C2#–C88#. Of symmetry generated carbon atoms only selected ones have been labelled. Atom C126 is disordered on two positions of which only one is displayed and labelled with C12A;
(b) The assignment of symmetry generated atoms is for 9b more complicated than in conventional cases, as four digits are allowed only when working with crystallographic software. For example, for 9bB the atomic labelling of atom Si16 is trivial, its symmetry generated equivalent by applying the crystallographically imposed inversion symmetry should be then labelled as Si16A. A second full molecule of 9bB, fully symmetry generated, should then have the atom labelling Si16B and Si16BA. Such a labelling code is not accessible. Therefore, the following restrictions were applied:
. - W. Jentzen, M. C. Simpson, J. D. Hobbs, X. Song, T. Ema, N. Y. Nelson, C. J. Medforth, K. M. Smith, M. Veyrat, M. Mazzanti, R. Ramasseul, J.-C. Marchon, T. Takeuchi, W. A. Goddard, III and J. A. Shelnutt, J. Am. Chem. Soc., 1995, 117, 11085 CrossRef CAS.
- W. R. Scheidt, J. U. Mondal, C. W. Eigenbrot, A. Adler, L. J. Radonovich and J. L. Hoard, Inorg. Chem., 1986, 25, 795 CrossRef CAS.
- C. Hu, B. C. Noll, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2007, 46, 619 CrossRef CAS PubMed.
- M. P. Byrn, C. J. Curtis, Y. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXTL Version 5.1, An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Siemens Analytical X-ray Instruments, Madison, WI, 1990 Search PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystal structure data. Fig. S1/S2 and Fig. S3 display the crystal structure of 3c and 6a, respectively, with respect to the orientation of the unit cell. Table S1 gives crystal and intensity collection data of 3b·1/4CH2Cl2, 3c, 4a, 6a·2thf and 9b·3.5EtOH. Fig. S4–S6 illustrate T-shaped π–π interactions in the crystal structure of 9b·3.5EtOH. Table S2 gives selected geometric features of intermolecular hydrogen bonds of 6a. Table S3 gives structural parameters of the porphyrin cores of 3b, 3c, 4a, 6a and 9b. Fig. S7 illustrates geometrical features of saddling distorted porphyrins. Fig. S8 shows the atom labelling for the NMR data. CCDC 976300–976304. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt53535e |
| This journal is © The Royal Society of Chemistry 2014 |