 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variable photon energy photoelectron spectroscopy of tris-cyclopentadienyl lanthanides
Marcello
Coreno
a,
Monica
de Simone
b,
Jennifer C.
Green
*c,
Nikolas
Kaltsoyannis
d,
Rosemary
Coates
d,
Charlene
Hunston
d,
Naima
Narband
d and
Andrea
Sella
d
aCNR-IMIP, Montelibretti, Rome I-00016, Italy
bCNR-IOM, Laboratorio TASC, 34149 Trieste, Italy
cDepartment of Chemistry, Oxford University, South Parks Road, Oxford OX1 3QR, UK. E-mail: jennifer.green@chem.ox.ac.uk
dDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 3rd February 2014
Abstract
The gas phase photoelectron (PE) spectra of LnCp3 (Cp = η-C5H5; Ln = Pr, Nd, Sm), measured with a wide range of photon energy, are reported. Resonances observed in the photon energy regions of 4d to 4f excitation enable identification of ion states resulting from 4f ionization. For all three compounds molecular ion states characteristic of both 4fn and 4fn−1 configurations are observed (Pr, n = 2; Nd, n = 3; Sm, n = 6). The molecular ion ground states have a hole in the uppermost ligand orbital of a′ symmetry and are reached by either ligand or f electron ionization. The results are discussed in the context of the previously reported spectra of the Ce, Yb and Lu analogues. For YbCp3 f orbital/ligand interaction is proposed in the molecular ground state and for CeCp3+ in the molecular ion ground state. For PrCp3 and NdCp3 final state effects are proposed as the origin of the dual configuration structure in their PE spectra. When the contributing orbitals are close in energy the 4f/a′ interaction can give rise to significant covalent bonding even in the absence of effective overlap.
For d block transition metal complexes, gas phase photoelectron (PE) spectroscopy has had considerable success in revealing their electronic structure. For closed shell molecules in the valence region, there is normally a close correspondence between the photoelectron bands and the orbital structure predicted by density functional theory (DFT).1,2 Lanthanide complexes are not so straightforward. The f-electrons are energetically part of the valence region, and for some elements are active in redox chemistry, but spatially are part of the core, the f-orbitals lacking significant overlap with ligand orbitals. Core ionization may result in significant perturbation of a molecule's electronic structure, which manifests itself in satellite structure accompanying the primary core PE bands, often referred to as shake-up or shake-down bands. These bands are assigned to many electron processes in which a valence excitation or transition accompanies the core ionization.
Ionization of the 4f electrons of lanthanides gives very characteristic final state structure that depends on the electronic ground state adopted by the 4fn configuration. The relative intensities of the ion states arising from ionization of the 4f electrons have been predicted using a fractional parentage description of the ground state molecule,3 and these predictions are on the whole consistent with data obtained from solid state photoemission studies.4 Although consideration of spin orbit coupling leads to modification of the relative intensity predictions,5 the pattern of the bands associated with a particular 4fn−1 configuration are characteristic and may be assigned by consideration of the electronic spectrum of the Ln3+ ion of the preceding element, i.e. the lanthanide with an atomic number of one less and a 4fn−1 configuration.6
The cross sections of 4f electrons differ substantially from those of the higher lying ligand-based electrons, which are predominantly of p character.7 The 4f cross sections have a delayed maximum in the photon energy region between 75 and 150 eV depending on the particular lanthanide. By contrast, the cross sections of s and p bands have maxima close to threshold, hence employing incident photon energies away from p based maxima yet near to 4f maxima will enable unambiguous characterization of the final state structure. The 4f bands also exhibit cross section maxima in the region of the 4d to 4f resonant excitation.2,8,9 Such excitation opens another channel for photoionization, since the excitation can be followed by super Coster–Kronig (SCK) decay in which the core hole is refilled and an f electron is ejected. Thus, in a PE experiment, with careful selection of photon energies, as is enabled by synchrotron radiation, ion states accessible by f ionization may be distinguished from those accessible by ligand ionization.
PE studies of lanthanides and their compounds in the solid state normally show f ionization bands superimposed on the valence bands. In some cases, particularly intermetallic compounds and those with counterions of low electronegativity, f ionization bands corresponding to two different f configurations are found. Thus, if the compound has a formal oxidation state implying a 4fn configuration, both 4fn−1 and 4fn band patterns are observed. Such observations have been interpreted in a number of ways. In some cases the initial state is implicated and a mixture of oxidation states is proposed for the compound.10 In other cases the 4fn states are presumed to result from ionization of an f electron being accompanied by valence band to 4f electron transfer i.e. a final state effect.11,12
Gas phase PE studies on lanthanide complexes are less common than those of solid compounds. This is in part due to their low volatility but also because discharge sources, common in house, are of insufficient energy to be able to identify unambiguously 4f ionization bands. The availability of an angle resolved photoelectron spectrometer (ARPES) on the gas phase photoemission beam line of the Elettra synchrotron13 has enabled a programme of study of the tris-cyclopentadienyl lanthanides, LnCp3 (Cp = η-C5H5). We have reported so far on the PE spectra of CeCp3,14,15 YbCp3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and LuCp3.6 The PE spectrum of LuCp3 shows two ion states, 2F7/2 and 2F5/2 associated with a 4f13 configuration for the molecular ion as might be anticipated on ionizing the closed 4f14 shell of Lu3+. The PE spectrum of YbCp3 shows structure corresponding to both 4f12 and 4f13 ion configurations. Consequently it was proposed that YbCp3 has a mixed configuration ground state that can be represented by the superposition of an ionic configuration Yb(III):4f13(Cp3) and a charge-transfer configuration Yb(II):4f14(Cp3)−1. Subsequent confirmation was obtained by the study of the magnetism, epr spectrum and analysis of the electronic spectrum of YbCp3, which were all in agreement in weighting the charge transfer configuration in the range 12–17%.16 In addition, the hole in the ligand set in the charge transfer configuration was found to be in an orbital of a′ symmetry (C3h molecular symmetry) which interacts with one of the real parts of the 4f±3 orbitals. In the case of CeCp3 the PE spectrum also indicated two ion state configurations, but in this case it is the molecular ion that has a mixed configuration ground state, Ce(IV):4f0(Cp3) mixed with Ce(III):4f1(Cp3)−1. The molecular ion ground state is accessible by direct photoionization from the Ce(III):4f1(Cp3) molecular configuration by virtue of the former term.14 Calculations using a CASSCF/CASPT2 approach confirmed the presence of configuration interaction in the ground state of the CeCp3+ cation.15
6 and LuCp3.6 The PE spectrum of LuCp3 shows two ion states, 2F7/2 and 2F5/2 associated with a 4f13 configuration for the molecular ion as might be anticipated on ionizing the closed 4f14 shell of Lu3+. The PE spectrum of YbCp3 shows structure corresponding to both 4f12 and 4f13 ion configurations. Consequently it was proposed that YbCp3 has a mixed configuration ground state that can be represented by the superposition of an ionic configuration Yb(III):4f13(Cp3) and a charge-transfer configuration Yb(II):4f14(Cp3)−1. Subsequent confirmation was obtained by the study of the magnetism, epr spectrum and analysis of the electronic spectrum of YbCp3, which were all in agreement in weighting the charge transfer configuration in the range 12–17%.16 In addition, the hole in the ligand set in the charge transfer configuration was found to be in an orbital of a′ symmetry (C3h molecular symmetry) which interacts with one of the real parts of the 4f±3 orbitals. In the case of CeCp3 the PE spectrum also indicated two ion state configurations, but in this case it is the molecular ion that has a mixed configuration ground state, Ce(IV):4f0(Cp3) mixed with Ce(III):4f1(Cp3)−1. The molecular ion ground state is accessible by direct photoionization from the Ce(III):4f1(Cp3) molecular configuration by virtue of the former term.14 Calculations using a CASSCF/CASPT2 approach confirmed the presence of configuration interaction in the ground state of the CeCp3+ cation.15
Here we present the variable photon energy PE spectra of PrCp3, NdCp3 and SmCp3 and compare the results with those reported previously for Ce, Yb and Lu.
Experimental
The tris-cyclopentadienyl lanthanides of praseodymium, neodymium and samarium were prepared by the method of Birmingham and Wilkinson and sublimed at 200 °C/10−2 mmHg to remove coordinated THF.17 The resulting product was sealed in a glass ampoule and subsequently transferred to a copper container under nitrogen, which was inserted into an oven attached to the angle resolved photoelectron spectrometer.Measurements were carried out at the GAs PHase photoemission (GAPH) beam line of the ELETTRA storage ring in Trieste, Italy.13 An undulator source provides high-intensity synchrotron radiation in the photon energy range 20–900 eV. The highly polarized light is dispersed by a Variable Angle Spherical Grating Monochromator that is equipped with five interchangeable gratings, to cover the energy range 13 to 900 eV, and fixed entrance and exit slits, with a photon energy resolution ΔE/E ≤ 10−4.
The samples were held in an oven close to the ionization region and evaporated through a small hole by holding the temperature of the oven at around 161 °C.
The PE measurements on NdCp3 were carried out using the ARPES chamber. The ARPES chamber was equipped with a 50 mm mean radius electron energy analyser (VSW Ltd), mounted at the magic angle. Pass energies of 10 eV were used for valence PE spectra at low photon energies and 15 eV at higher photon energies. The PE spectra of PrCp3 and SmCp3 were analyzed by a VG220i hemispherical electron energy analyzer with a mean radius of 150 mm equipped with six channel electron multipliers. This electron analyser was mounted in the plane defined by the (linearly polarized) electric vector of the light and the photon propagation direction at an angle of 54.7° with respect to the electric vector of the light. In this geometry the axis of the analyzer is set at the pseudo magic angle, and so measurements should be insensitive to the PE asymmetry β parameter. Electron energy resolution is linear along all the kinetic energy scale and equal to 2% of pass band for both analyzers. PE spectra were acquired at the magic angle or pseudo magic angle depending on the analyzer used and normalised using the signal from a calibrated photodiode (IRD, Inc.).18 Pass energies of 5 eV were used for valence PE spectra. PE spectra of the valence region from 6–20 eV of binding energy were measured at the pseudo magic angle for a series of photon energies ranging from 24–180 eV. The binding energy scale was calibrated according to the Ar photoelectron lines.
Results
A selection of the PE spectra of LnCp3 (Ln = Pr, Nd, Sm) measured with both low and higher photon energies are shown in Fig. 1–3.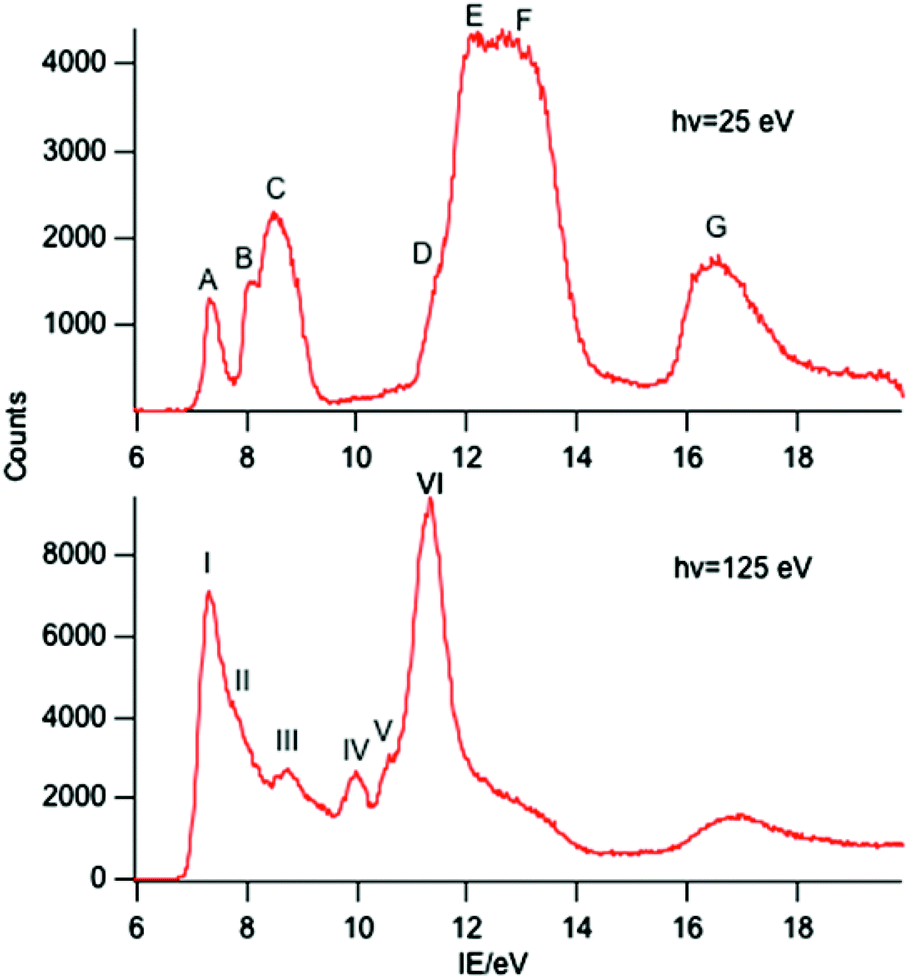 | ||
| Fig. 1 PE spectra of PrCp3 with photon energies of 25 and 125 eV. | ||
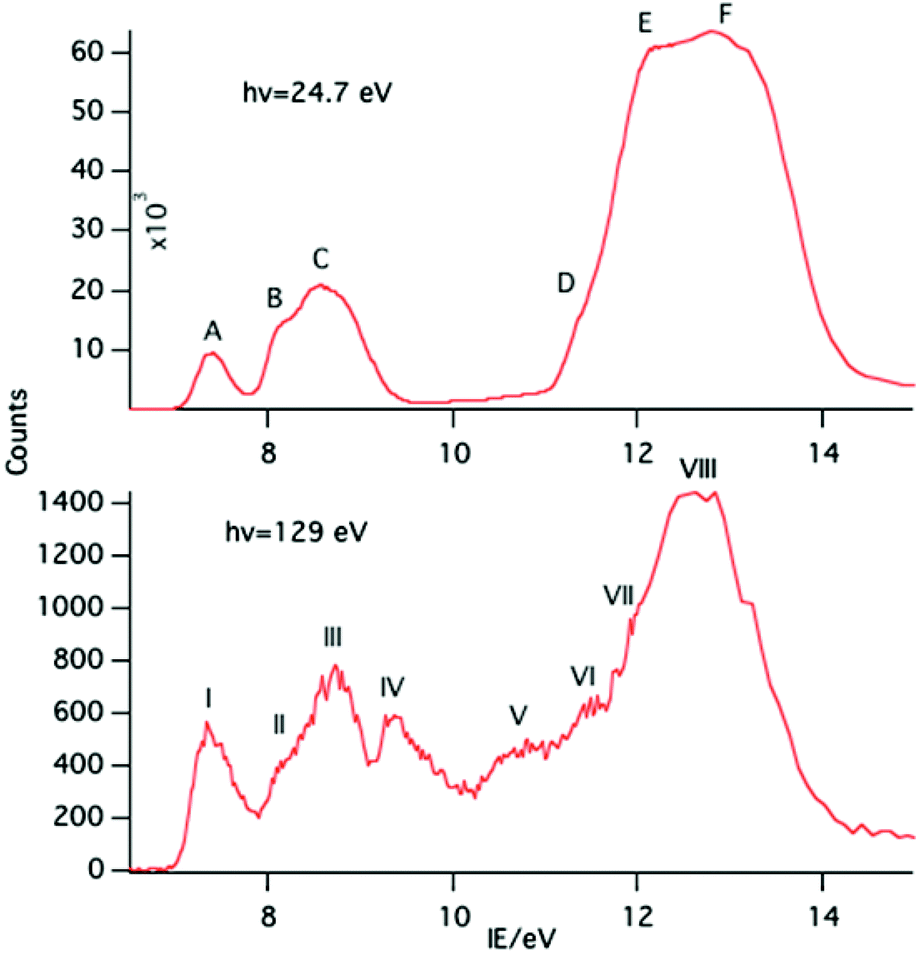 | ||
| Fig. 2 PE spectra of NdCp3 with photon energies of 24.7 and 129 eV. | ||
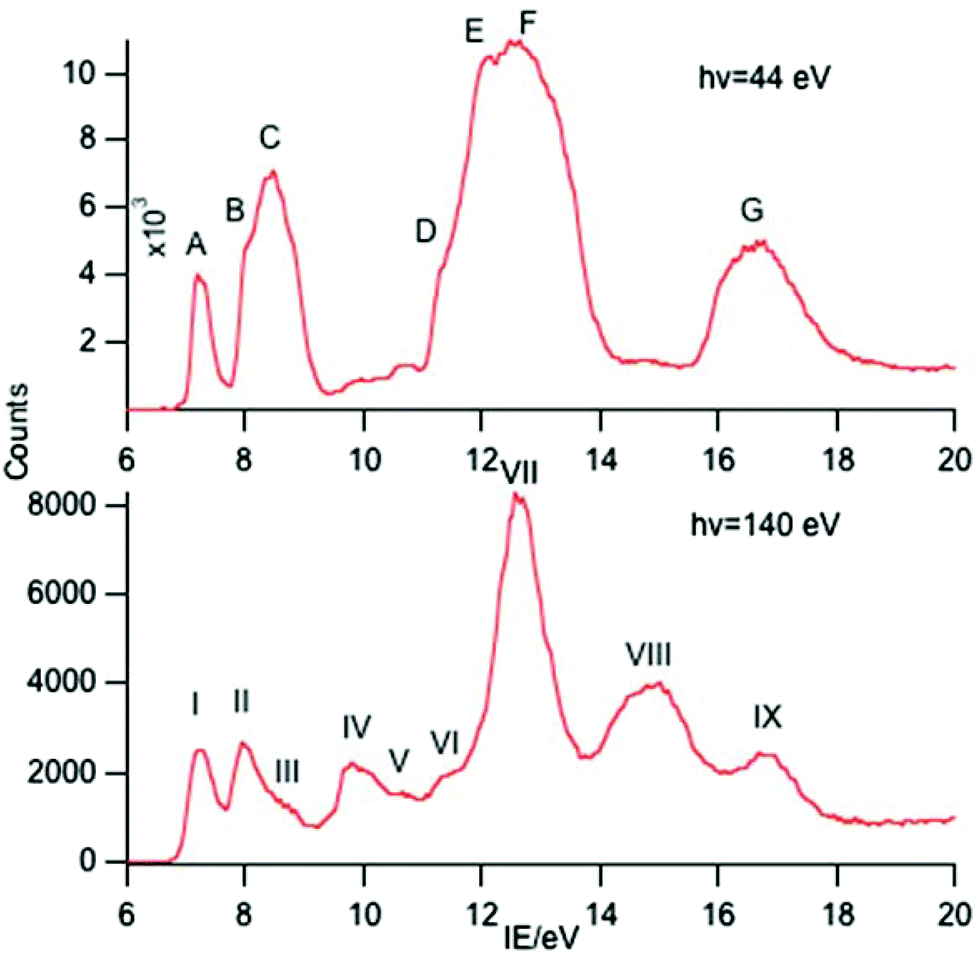 | ||
| Fig. 3 PE spectra of SmCp3 with photon energies of 44 and 140 eV. | ||
Key features are tabulated (Table 1). The ionization bands fall into two distinct types. Those labelled A–G, identifiable for all three compounds, show a decay in intensity relative to the other bands with increasing photon energy. (Although only two photon energy spectra are presented in the figures, many more were recorded which show a gradual evolution.) Those bands labelled with roman numerals increase in relative intensity with photon energy and show resonances in the photon energy region of the respective lanthanide 4d to 4f excitation energies. The emergence of the f-bands over the resonance region is shown for PrCp3 in Fig. 4. Although the f bands dominate in the resonance region they are also visible in the pre-resonance region for example with a photon energy of 100 eV.
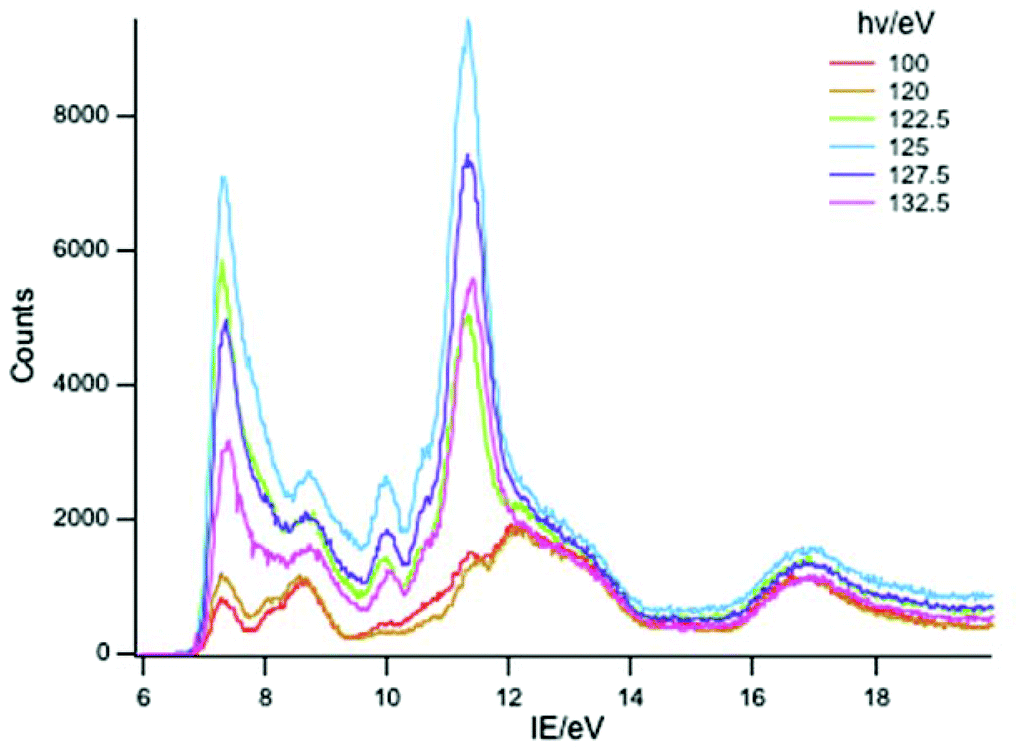 | ||
| Fig. 4 PE spectra of PrCp3 with photon energies 100–132.5 eV. | ||
| Band\Ln | Ce | Pr | Nd | Sm | Yb | Lu | Associated MO |
|---|---|---|---|---|---|---|---|
| A | 7.45 | 7.34 | 7.39 | 7.21 | 7.16 | 7.28 | a′ |
| B | 8.13 | 8.04 | 8.17 | 8.08 | a′′ | ||
| C | 8.58 | 8.46 | 8.57 | 8.50 | 8.46 | 8.81 | e′ + e′′ |
| D | 11.53 | 11.46 | 11.57 | 11.50 | |||
| E | 12.25 | 12.19 | 12.22 | 12.07 | 12.4 | 12.4 | |
| F | 12.80 | 12.94 | 12.79 | 13.00 | |||
| G | 16.53 | 16.49 | 16.53 | 16.60 | 16.7 | 17.2 | |
| I | 6.77 | 7.33 | 7.34 | 7.21 | 7.26 | 14.30 | |
| II | 9.97 | 7.76 | 8.14 | 7.96 | 8.58 | 15.74 | |
| III | 8.77 | 8.74 | 8.61 | 12.93 | |||
| IV | 9.97 | 9.39 | 9.86 | 13.73 | |||
| V | 10.61 | 10.79 | 10.66 | 14.91 | |||
| VI | 11.34 | 11.49 | 11.46 | 15.98 | |||
| VII | 11.99 | 12.56 | 16.71 | ||||
| VIII | 12.64 | 14.96 | 17.66 | ||||
| IX | 16.81 |
In the spectra of all three compounds bands A and I are coincident indicating that the ground ion states are accessible by two different ionization processes.
Discussion
Band assignment
The origin of the bands A–G and their assignment has been discussed previously.6,14–16 The associated ion states have holes in the ligand shells and are accessed from the parent molecules by ionization of ligand based orbitals. PE ionization cross sections of carbon and hydrogen based electrons have a maximum near threshold and decay with increasing photon energy,2,7 thus the ready identification of their ionization bands. Individual assignments are based on comparison with the PE spectra of other cyclopentadienyl compounds and theoretical calculations.Most crucially bands A, B and C arise from ionization of the highest occupied π orbitals of the cyclopentadienyl rings. In the C3h symmetry of the gas-phase LnCp3 molecules these transform as a′ + a′′ + e′ + e′′. The Ln 5d transform as a′, e′, and e′′, and the 6p as a′′ and e′. However, the net overlap of the cylindrically symmetrical 5dz2 (a′) with the Cp-π (a′) MO, which has six nodes in the horizontal plane (Fig. 5), is negligible; in the effective D3h symmetry they would belong to different irreducible representations. The Cp-π a′ combination does find a nodal match in the cos 3ϕ component of the Ln 4f±3 orbitals but lack of substantive radial overlap with the core–like 4f orbitals leads to it being less stabilized by interaction with the lanthanide than the other combinations, which can overlap with Ln 5d and 6p orbitals.
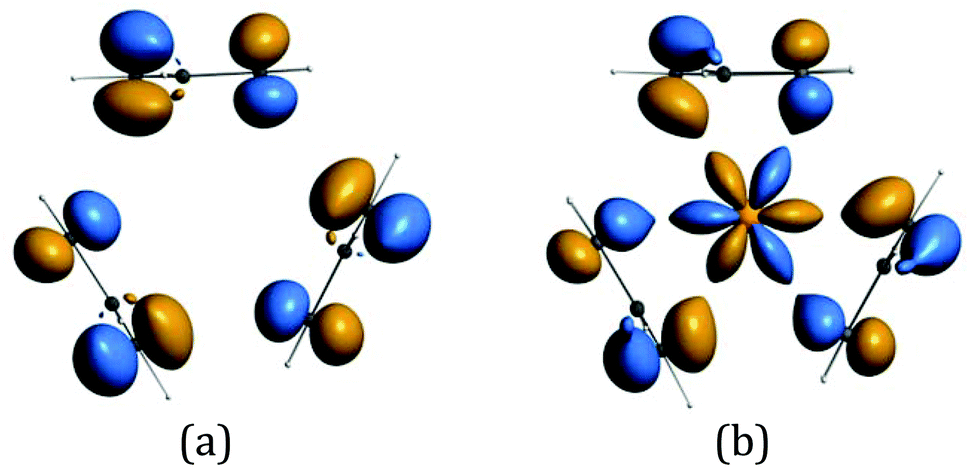 | ||
| Fig. 5 (a) The Cp-π a′ symmetry adapted linear combination (b) possible overlap with cos 3ϕ component of the Ln 4f±3 orbitals. (These isosurfaces at value 4.8 were generated for CeCp3+ using the ADF program suite BP/TZP.)19 | ||
Band A is assigned to ionization from this Cp-π a′ orbital and lies at a lower IE, well separated from bands B and C. Band B is assigned to ionization from the Cp-π a′′ orbital which can mix with Ln 5p and band C to the e′ and e′′ orbitals which form more effective bonding combinations with Ln 5d orbitals. Bands D–F are associated with orbitals based on C 2p and H 1s orbitals whereas band G is principally C 2s in character.
As discussed in the introduction, the cross sections of 4f ionizations are very low near threshold with the consequence that the associated ionization bands are not readily detectable with low photon energies.2,7 They rise in intensity with photon energy passing through a maximum. In addition 4f ionizations commonly show resonances when the photon energy promotes 4d to 4f excitation. Intensity borrowing from this transition may lead to considerable enhancement of a 4f band intensity.2 The ionizations labelled with roman numerals show such cross section features and are associated with ionizations from the 4f orbitals. The number and intensity of the 4f bands is characteristic of the number of f electrons in the molecule.
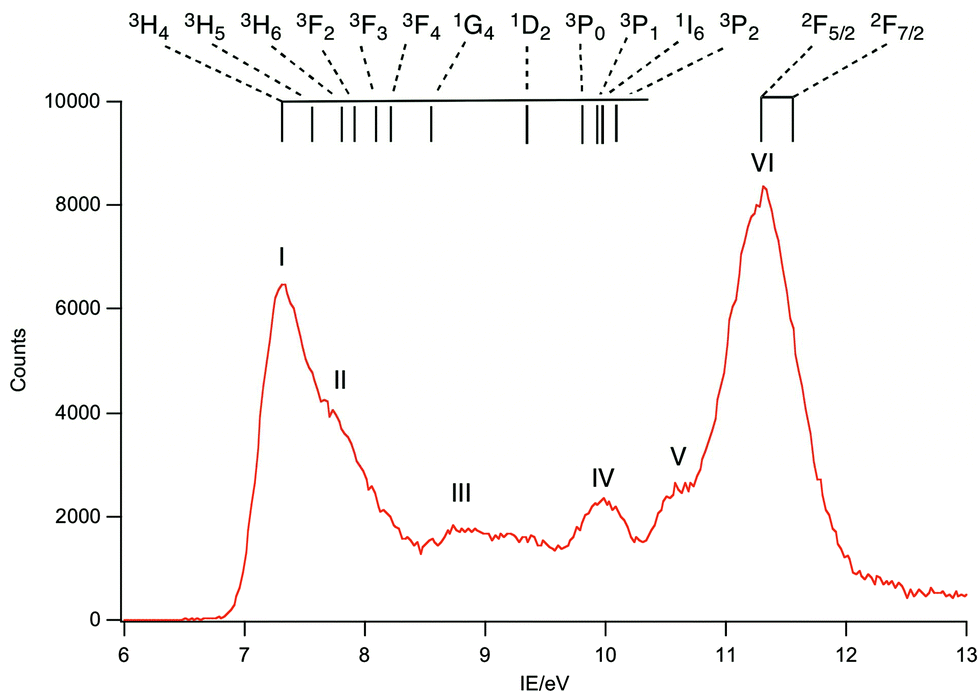 | ||
| Fig. 6 Difference of PE spectrum at 125 eV minus PE spectrum at 110 eV for PrCp3. The highest energy 4f band, VI, is assigned to the 2F states. | ||
Band I, being coincident with band A, corresponds to an ion state with a hole in the Cp-π a′ orbital and Pr(III) in its 3H4 ground state. We denote this ion state |4f2(3H4)a′−1>. Bands II–V are assigned to excited states of the 4f2 configuration and are assigned by comparison with electronic spectra of Pr(III) compounds and associated calculations.21,22 Such calculations include consideration of the crystal field, and that splitting of the spin–orbit states can cover an energy range of up to 0.2 eV. In Fig. 6 we indicate the relative energies of the lowest crystal field term of a particular spin orbit state of Pr3+. It is evident that the spin–orbit states span the energy range of bands I–V.
:0.424:0.563. In PrCp*3 (Cp* = η-C5Me5) these states span an energy range of 0.7 eV.21 For Nd4+ an expansion of the range is anticipated but all three states are likely to occur within the 1.5 eV range covered by band VIII, which is consequently assigned to these 3H4, 3H5 and 3F2 states.
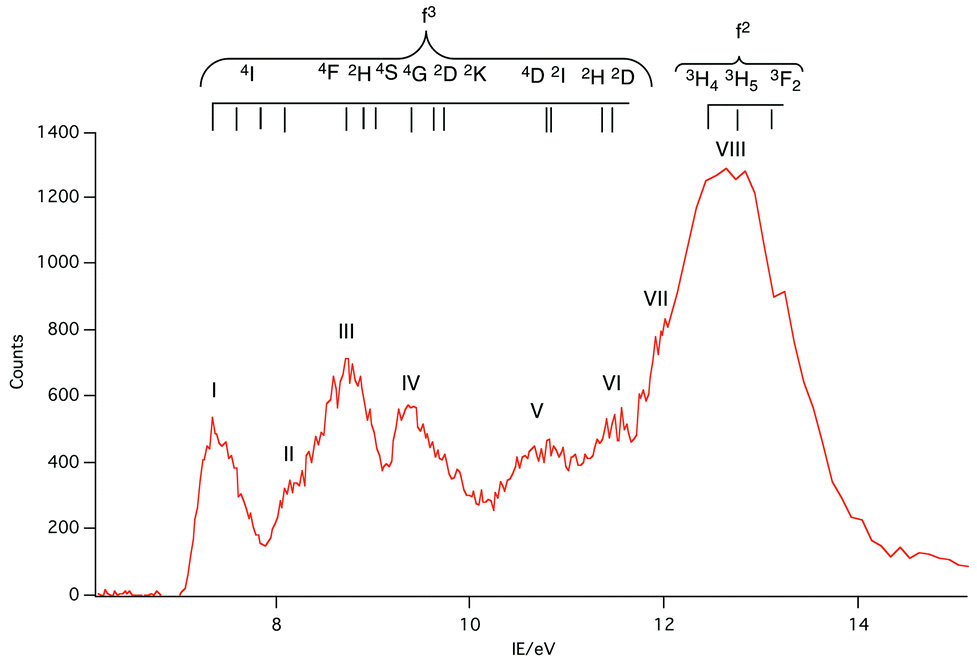 | ||
| Fig. 7 Difference of PE spectrum at 129 eV minus PE spectrum at 110 eV for NdCp3 showing the energy range of identified 4f3 spin–orbit states for Nd3+ and shows the likely energy range of 4f2 states for NdCp3+ accessible by direct photoionization. | ||
Like PrCp3, NdCp3 has many more 4f PE bands than can be accounted for by direct photoionization. The lowest energy 4f band, I, is again coincident with the lowest energy ligand band, A, and can be assigned to the molecular ion ground state |4f3(4I9/2)a′−1>. Nd(III) has a plethora of spin–orbit states. In Fig. 7 we indicate the energy range of identified states for Nd3+.23,24 Bands II–VII all lie within this range.
 | ||
| Fig. 8 Difference of PE spectrum at 140 eV minus PE spectrum at 130 eV for SmCp3. The lower energy 4f bands are assigned to states from a 4f5 configuration and those attributed to accessible sextet states from a 4f4 configuration are labelled with their (2S+1) L symbols. Bands III, V and VI are likely to arise from the many quartet states of Sm3+. | ||
Sm(III) has a 4f5 configuration and a 6H5/2 ground state. Direct ionization is predicted to give four LS states, 5I, 5G, 5F and 5D with relative intensities 2.758:1.266:0.500:0.476.3 Bands VI, VII and VIII have relative intensities 2.76:2.1:0.13 suggesting that band VII incorporates both the 5F and 5G states as has been assigned previously.25 Bands I, II and IV are clearly visible in the spectra of Sm(II) compounds and are assigned to the 6H, 6F and 6P states of the 4f6 configuration.10,25–27 Other states from this configuration are also visible as bands III, V and VI which to the best of our knowledge haven't been reported previously in photoemission studies. Assignment of these states is difficult. There is a plethora of excited states for the Sm3+ ion. Various quartet states lie between the 6F and 6P levels and above the 6P levels28 and we hesitate to make a definite assignment.
Electronic structure and state access
It is helpful at this juncture to propose two orbital schemes for the ground states of tris-cyclopentadienyl lanthanide systems (Fig. 9). Both include as the basis set the highest occupied cyclopentadienyl π orbitals, the lanthanide 4f, 5d and 6p orbitals. Mixing with the 5d and 6p orbitals stabilizes the a′′, e′ and e′′ linear combination of the ring orbitals. Although, formally, in the C3h symmetry of the molecule the ring a′ orbital may interact with the dz2 orbital, their nodal properties are incompatible (Fig. 5). In the first case (Fig. 9a) the 4f orbitals are sufficiently separated in energy from the ring a′ orbitals, such that the interaction is not applicable. This is the case in CeCp3.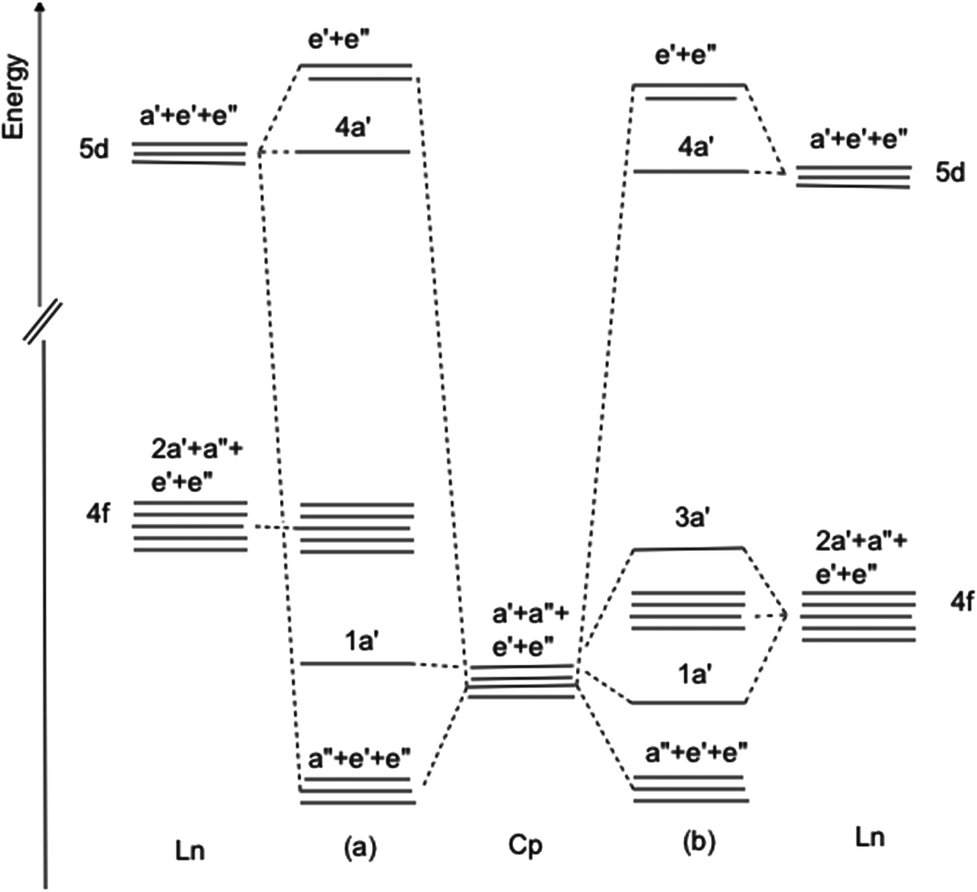 | ||
| Fig. 9 Orbital interaction diagram for LnCp3 (a) 4f orbitals non-interacting (b) 4f orbitals close in energy to ring π a′ orbital. | ||
In the second case (Fig. 9b) the 4f orbitals are sufficiently close in energy to the ring a′ orbital for interaction and charge transfer to take place resulting in a stabilized ring orbital and a destabilized f orbital, specifically one of the real components of the f±3 pair. It is noteworthy in the latter case that even with minimal overlap as a consequence of the radially contracted 4f orbitals significant interaction may occur when the interacting orbitals are close in energy.29 Case 9b pertains to CeCp3+ cation and YbCp3. For CeCp3+ 1a′ is filled and is the HOMO while for YbCp3 3a′ is the SOMO occupied by one unpaired electron.
The resulting ground state wave function is most conveniently described by configuration interaction between two terms (L represents the remaining electrons) where one configuration has n f electrons as expected for a Lnx+ ion and the other has a hole in the ring a′ orbital and one more f electron.
| ΨGS = c0ψ0 + c1ψ1 = c0|fna′2L〉 + c1|fn+1a′1L〉 | (1) |
The two terms must have the same symmetry so the crucial 4f orbital is of a′ symmetry as shown in Fig. 5b.
For the LnCp3+ cations the PE spectra inform as to the relative energies of the ground and excited states. In the cases of Ln = Ce, Pr, Nd, Sm, Yb and Lu the states associated with an fn−1a′2L configuration, where one f electron has been ionized, are unambiguously identified. Removal of a 4f electron results in significant stabilization of the other f electrons and states from this configuration lie at higher energies than the molecular ion ground state. For Ln = Pr, Nd, Sm, Yb and Lu the cationic ground states correspond simply to the removal of one electron from the Cp π a′ orbital and belong to the configuration fna′1L. For Ln = Ce there is a lower energy cationic ground state with a mixed configuration as described by eqn (1). The ionization energies of the lowest states associated with a configuration are plotted in Fig. 10.
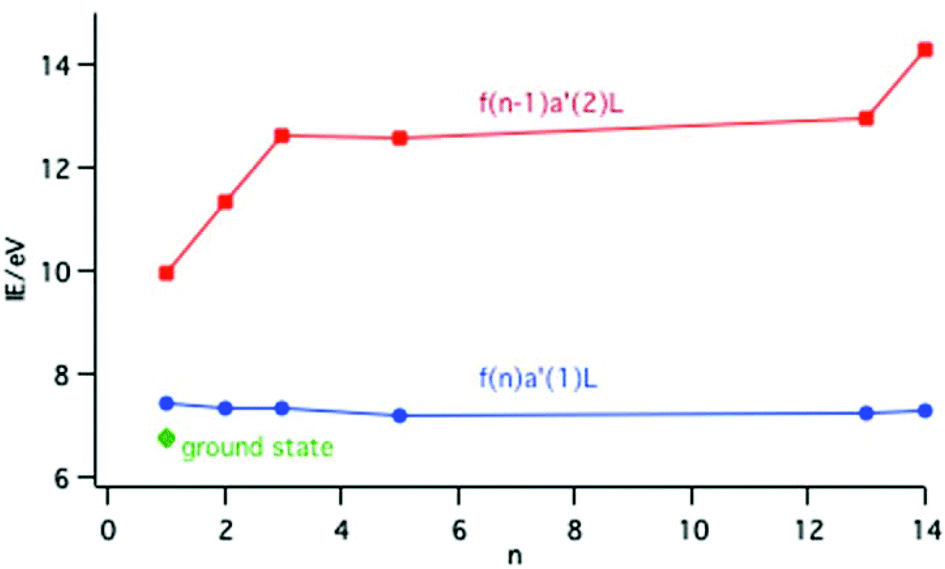 | ||
| Fig. 10 Plot of the IE to the lowest energy state of a configuration across the lanthanide series; red points are associated with an fn−1a′2L configuration and blue points with an fna′1L configuration. The green point represents the ground state of CeCp3+. | ||
The ionization energy to the fna′1L configuration remains fairly constant across the series; removal of an electron from the Cp π a′ orbital is insensitive to the Ln nuclear charge. In contrast the ionization energy to the fn−1a′2L configuration rises across the series because the 4f electrons are not effectively shielded from the increase in Ln nuclear charge. Calculations on the CeCp3+ cation have shown that in the f0a′2L state there is a considerable increase in the population of the metal 5d orbitals compared with the ground state as the ligand metal covalency increases with f ionization.15 This is described as screening in solid state studies.
The ground state of CeCp3+ is 0.68 eV (65 kJ mol−1) more stable than the excited state with an f1a′1L configuration. This separation provides a convenient measure of the binding energy afforded by the configuration interaction (CI) described by eqn (1). Delocalizing the f electron into the hole in the ligand a′ orbital rather than maintaining pure f character provides a significant covalent interaction. Modeling of the electronic spectrum of YbCp3 gave a CI matrix element of a similar order of magnitude, namely −3900 cm−1 (47 kJ mol−1).16 Thus in these situations of close energy proximity significant covalent binding can occur with minimal overlap.
The matrix element governing the band intensity for direct f ionization is given by
| 〈Lfn|O|Lfn−1ε〉 |
Conclusions
The gas phase PE spectra of LnCp3 (Cp = η-C5H5; Ln = Pr, Nd, Sm) have been measured using synchrotron radiation. Variation of band intensity with photon energy, including in the region of 4d to 4f excitation, has enabled unambiguous assignment of the ion states. In all three cases, ion states associated with both 4fn and 4fn−1 configurations are identified, where n is the number of f electrons in the molecular ground state. The data are discussed in the context of the previous results for Ln = Ce, Yb and Lu. The cationic ground state of LnCp3 for all members of the series except Ce has a fn configuration with a hole in the ligand shell, specifically in an orbital of a′ symmetry. Ce is exceptional in having an intermediate valence ground state comprised of a mixture of f0a′2 and f1a′1 configurations, where a′ is the highest occupied orbital of the Cp3 ligand set. The f electron in the latter occupies the cos 3ϕ component of the Ln 4f±3 orbitals giving a singlet state overall. The 4f band intensities are similar to those observed previously in solid state studies but in general are better resolved. The 4fn−1 states are accessed by direct ionization or by a 4d–4f resonance process coupled with super Koster–Kronig decay. The more intense of the 4fn states are observable outside the resonance region but become most clearly visible within the resonance region. The mechanisms of their access are discussed. SmCp3 is the most likely candidate to have an intermediate valence ground state as has been established for YbCp3. However, the intensity of the 4fn bands suggest that an alternative mechanism for their access on 4f ionization must be present.References
- J. C. Green, Comp. Organomet. Chem., 3, 2006, 1, 381–406 Search PubMed.
- J. C. Green and P. Decleva, Coord. Chem. Rev., 2005, 249, 209 CAS.
- P. A. Cox, Struct. Bonding, 1975, 24, 59 CAS.
- J. N. Chazaviel, M. Campagna, G. K. Wertheim and P. Y. Schmidt, Solid State Commun., 1976, 19, 725 CrossRef.
- N. Beatham, P. A. Cox, A. F. Orchard and I. P. Grant, Chem. Phys. Lett., 1979, 63, 69 CrossRef CAS.
- M. Coreno, S. M. de, R. Coates, M. S. Denning, R. G. Denning, J. C. Green, C. Hunston, N. Kaltsoyannis and A. Sella, Organometallics, 2010, 29, 4752–4755 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1 CrossRef CAS.
- U. Fano, Phys. Rev., 1961, 124, 1866 CrossRef CAS.
- U. Fano and J. W. Cooper, Phys. Rev. A, 1965, 137, 1364 CrossRef CAS.
- J. W. Allen, L. I. Johansson, I. Lindau and S. B. Hagstrom, Phys. Rev. B: Condens. Matter, 1980, 21, 1335–1342 CrossRef CAS.
- S. Hüfner, J. Phys. F: Met. Phys., 1986, 16, L31 CrossRef.
- S. Hüfner, F. Schumann, E. Rotenberg, J. Tobin, S.-H. Yang, B. S. Mun, S. Morton, J. Schäfer and D. Ehm, Phys. Rev. B: Condens. Matter, 2001, 63, 85106 CrossRef.
- K. C. Prince, R. R. Blyth, R. Delaunay, M. Zitnik, J. Krempasky, J. Slezak, R. Camilloni, L. Avaldi, M. Coreno, G. Stefani, C. Furlani, M. de Simone and S. Stranges, J. Synchrotron Radiat., 1998, 5, 565 CrossRef CAS PubMed.
- M. Coreno, M. de Simone, J. C. Green, N. Kaltsoyannis, N. Narband and A. Sella, Chem. Phys. Lett., 2006, 432, 17–21 CrossRef CAS PubMed.
- R. Coates, M. Coreno, S. M. De, J. C. Green, N. Kaltsoyannis, A. Kerridge, N. Narband and A. Sella, Dalton Trans., 2009, 5943–5953 RSC.
- R. G. Denning, J. Harmer, J. C. Green and M. Irwin, J. Am. Chem. Soc., 2011, 133, 20644–20660 CrossRef CAS PubMed.
- J. M. Birmingham and G. Wilkinson, J. Am. Chem. Soc., 1956, 78, 42 CrossRef CAS.
- R. R. Blyth, R. Delaunay, M. Zitnik, R. Krempasky, J. Krempaska, J. Slezak, K. C. Prince, R. Camilloni, L. Avaldi, M. Coreno, G. Stefani, C. Furlani, M. de Simone, S. Stranges and M.-Y. Adam, J. Electron Spectrosc. Relat. Phenom., 1999, 101–103, 959 CrossRef CAS.
- S. ADF2012.01, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com, 2012 Search PubMed.
- W. C. Martin, R. Zalubas and L. Hagan, Atomic Energy Levels {The Rare Earth Elements, NSRDS-NBS 60, U.S. Government Printing Office, Washington, DC, 1978 Search PubMed.
- H.-D. Amberger, H. Reddmann, T. J. Mueller and W. J. Evans, Organometallics, 2010, 29, 1368–1373 CrossRef CAS.
- D. A. Wensky and W. G. Moulton, J. Chem. Phys., 1970, 53, 423–435 CrossRef CAS PubMed.
- H.-D. Amberger, H. Reddmann, T. J. Mueller and W. J. Evans, J. Organomet. Chem., 2011, 696, 2829–2836 CrossRef CAS PubMed.
- M. Karbowiak and C. Rudowicz, Chem. Phys., 2011, 383, 68–82 CrossRef CAS PubMed.
- J. N. Chazalviel, M. Campagna, G. K. Wertheim and P. H. Schmidt, Phys. Rev. B: Condens. Matter, 1976, 14, 4586–4592 CrossRef CAS.
- M. Campagna, E. Bucher, G. K. Wertheim and L. D. Longinotti, Phys. Rev. Lett., 1974, 33, 165–168 CrossRef CAS.
- P. Wang, J.-F. Ni, H.-N. Li, W.-H. Zhang and J.-F. Zhu, Surf. Sci., 2008, 602, 3728–3732 CrossRef CAS PubMed.
- J. B. Gruber, B. Zandi and M. F. Reid, Phys. Rev. B: Condens., 1999, 60, 15643–15653 CrossRef CAS.
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407 CrossRef CAS PubMed.
- C.-H. Wong, T.-Y. Lee and Y.-T. Lee, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 2580 CrossRef CAS.
- S. H. Eggers, J. Kopf and R. D. Fischer, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2288–2290 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |