 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chloride ion impact on materials for light-emitting electrochemical cells†
Gabriel E.
Schneider
a,
Henk J.
Bolink
bc,
Edwin C.
Constable
*a,
Cathrin D.
Ertl
a,
Catherine E.
Housecroft
a,
Antonio
Pertegàs
bc,
Jennifer A.
Zampese
a,
Andreas
Kanitz
d,
Florian
Kessler
d and
Sebastian B.
Meier
d
aDepartment of Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: edwin.constable@unibas.ch
bInstituto de Ciencia Molecular, Universidad de Valencia, Catedrático José Beltrán 2, Paterna, E-46980, Spain. E-mail: henk.bolink@uv.es
cFundació General de la Universitat de Valencia (FGUV), PO Box 22085, Valencia, Spain
dSiemens AG, Corporate Technology CT RTC MAT IEC-DE, Günther-Scharowsky-Str. 1, 91058 Erlangen, Germany
First published on 5th December 2013
Abstract
Small quantities of Cl− ions result in dramatic reductions in the performance of ionic transition metal complexes in light-emitting electrochemical cells. Strong ion-pairing between aromatic protons and chloride has been established in both the solid state and solution. X-ray structural determination of 2{[Ir(ppy)2(bpy)][Cl]}·2CH2Cl2·[H3O]·Cl reveals the unusual nature of an impurity encountered in the preparation of [Ir(ppy)2(bpy)][PF6].
High performance electronic devices have traditionally been prepared by vapour deposition methods involving reactions of ultrapure volatile compounds to generate solid state materials.1 Solution casting offers an alternative fabrication method in which preformed materials are deposited from the solution phase.2 Materials of interest are usually refined to chemical, but not electronic, standards of purity. In this communication we report the dramatic effect of trace components on the device performance of ionic iridium emitters in light-emitting electrochemical cells (LECs) and describe an unusual interaction of chloride with such complexes.
We routinely prepare the complex [Ir(ppy)2(bpy)][PF6] (Hppy = 2-phenylpyridine, bpy = 2,2′-bipyridine), which gives long-lived LECs,3 by the reaction of [Ir2(ppy)4Cl2] with bpy in MeOH under microwave heating, followed by precipitation with excess [NH4][PF6] and multiple chromatographic purification (Scheme 1). The preparation is readily upscaled to gram quantities, but we have found batch-to-batch variability between materials of apparently similar chemical constitution and purity when evaluated in a solid-state LEC (see Fig. 1).4 The device configuration is similar to that previously reported.3 A layer of poly(3,4-ethylenedioxythiophene): polystyrenesulfonate (PEDOT:PSS) (100 nm) was deposited on top of a patterned indium tin oxide (ITO) substrate to increase the device preparation yield. The complex [Ir(ppy)2(bpy)][PF6] together with 1-butyl-3-methylimidazolium hexafluoridophosphate [BMIM][PF6] at a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was then spin-coated from MeCN solution to yield an 80 nm thick layer. The ionic liquid was added to reduce the turn-on time of the device.5 The device was completed by the deposition of 80 nm aluminium as the top electrode contact.
:1 was then spin-coated from MeCN solution to yield an 80 nm thick layer. The ionic liquid was added to reduce the turn-on time of the device.5 The device was completed by the deposition of 80 nm aluminium as the top electrode contact.
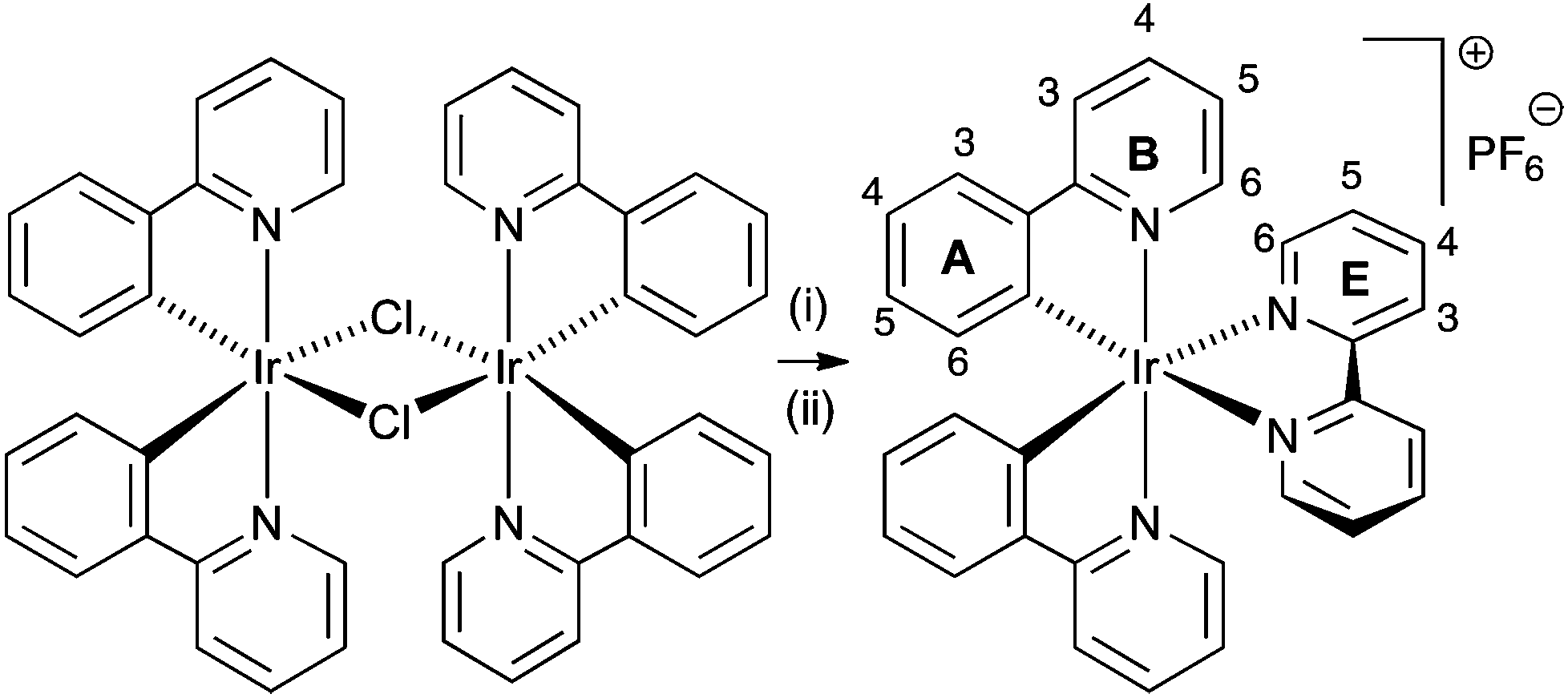 | ||
| Scheme 1 Synthesis of [Ir(ppy)2(bpy)][PF6] with labelling for NMR spectroscopic assignments: (i) bpy, MeOH, microwave, 2 h, 120 °C; (ii) [NH4][PF6]. | ||
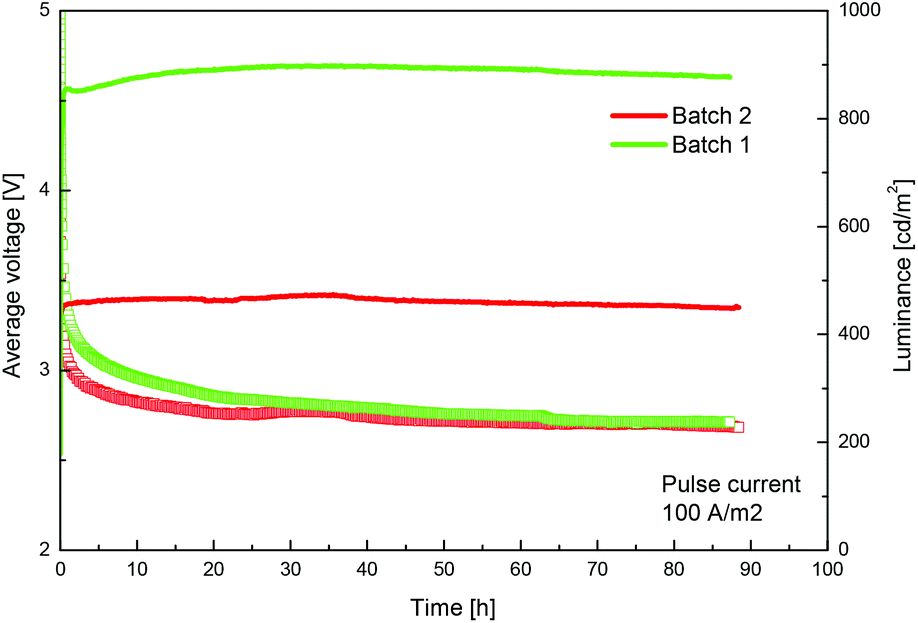 | ||
| Fig. 1 Performance of two batches of [Ir(ppy)2(bpy)][PF6] with a similar chemical history incorporated in a LEC device (ITO/PEDOT:PSS/[Ir(ppy)2(bpy)][PF6] + [BMIM][PF6](4:1)/Al). The performance of batch 2 is significantly inferior due to the presence of small amounts of chloride as counterion. | ||
The 1H NMR spectra of the materials used for the two devices in Fig. 1 were at first sight identical, but a detailed analysis revealed that the signal assigned to proton E3 was shifted by about 0.5 Hz between the two samples (Fig. S1†). Subsequent elemental analysis indicated that the poorly performing materials had typically 1% Cl− and 99% [PF6]− as counter-ion. Further purification of the poorly performing material by precipitation with [NH4][PF6] and Ag[PF6] (to precipitate any Cl− present as AgCl) followed by chromatography gave material with optimal performance, confirming that the presence of small amounts of Cl− has a profound effect on the device properties as smaller counter-ions have been shown to alter the LEC dynamics.6,7 The ESI† provides a protocol for the preparation of chloride-free material with reproducible device performance.
In order to understand the effect of chloride ion, we have performed 1H NMR titrations of [nBu4N]Cl into CD2Cl2 solutions of pure [Ir(ppy)2(bpy)][PF6] (Fig. 2). The signal due to proton E3 (the 3 and 3′ protons of the 2,2′-bipyridine ligand) exhibited dramatic shifts to higher frequency upon the addition of [nBu4N]Cl, from δ 8.505 to 9.326 ppm after the addition of >10 equivalents. Of the remaining protons, only the resonance for proton E4 showed a shift and this was very much less than that observed for proton E3.
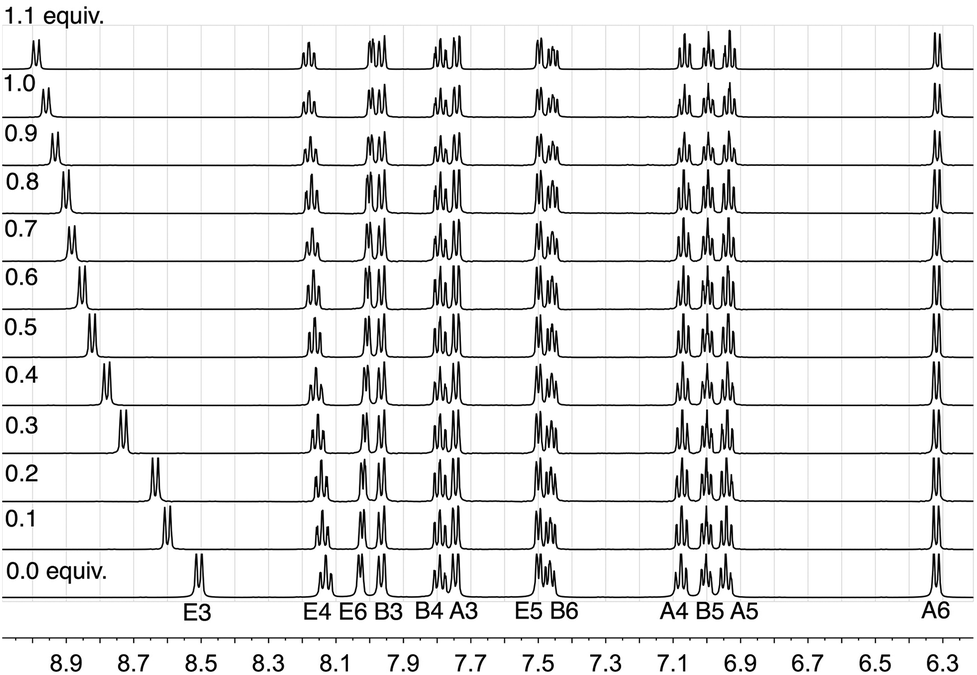 | ||
| Fig. 2 The effect of adding 1.1 equivalents of [nBu4N]Cl in 0.1 aliquots to a CD2Cl2 solution of [Ir(ppy)2(bpy)][PF6] (0.02 mmol of complex in 0.75 mL CD2Cl2). Only the resonance assigned to proton E3 is significantly affected and a limiting shift of δ 9.326 ppm is observed after adding >10 equivalents of [nBu4N]Cl. See Scheme 1 for labelling. | ||
Dichloromethane is a poorly solvating solvent in which relatively tight ion-pairing between cation and anion is expected and we have analysed the data according to the equilibrium:
| {[Ir(ppy)2(bpy)][PF6]} + Cl− ⇌ {[Ir(ppy)2(bpy)]Cl} + [PF6]− |
Non-linear least-squares fitting of the 1H chemical shift of E3 using WinEQNMR28 yielded a log K value of 2.24 (R = 0.001789) for this equilibrium, favouring the formation of the chloride ion-pair. Similar small shifts, of the order of δ 0.7 ppm are also observed in the 19F NMR spectrum upon the addition of [nBu4N]Cl to [Ir(ppy)2(bpy)][PF6] (Fig. S2†); the 31P NMR spectrum is unchanged. As expected, the equilibrium is also affected by the addition of additional [PF6]− (Fig. 3) and on this basis, the chemical shift of the E3 proton can be used as a sensitive qualitative and quantitative probe for the presence of Cl− in supposedly pure samples of [Ir(ppy)2(bpy)][PF6]. For a CD2Cl2 solution, the value of δE3 as a function of the percentage of [Ir(ppy)2(bpy)]Cl versus [Ir(ppy)2(bpy)][PF6] is given by:
| δE3 = 8.505 + {(−0.3289 × 10−5)(%Cl)2 + 0.007843(%Cl)} |
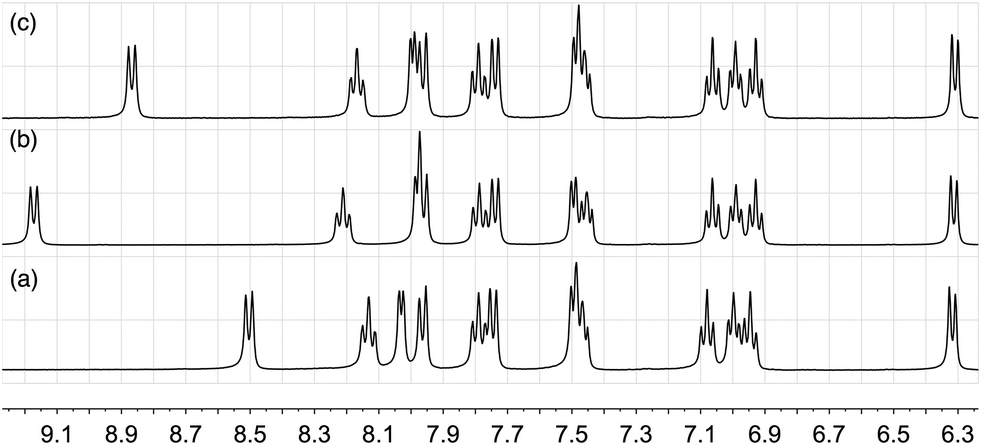 | ||
| Fig. 3 Room temperature 400 MHz 1H NMR spectra of CD2Cl2 solutions of (a) pure [Ir(ppy)2(bpy)][PF6], (b) [Ir(ppy)2(bpy)][PF6] with added [nBu4N]Cl, and (c) after the addition of [nBu4N][PF6] to solution (b). | ||
Not unexpectedly, the photophysical properties of the complex depend on the counterion and pure samples of [Ir(ppy)2(bpy)][PF6] and [Ir(ppy)2(bpy)][Cl] were studied in the solid state and in MeCN solution (to avoid CH2Cl2 as a potential source of Cl−). In solution, the pure complexes and those with mixed counter-ions exhibited the same properties (λem = 600 nm, ϕ = 0.13), whereas in the solid state, [Ir(ppy)2(bpy)][PF6] and [Ir(ppy)2(bpy)]Cl exhibited different emission maxima (λem = 510 nm and 540 nm respectively) although the quantum yields were similar (ϕ = 0.16).
The sensitivity of the E3 protons to the presence of Cl− suggests a highly specific interaction of chloride ion with these positions of the 2,2′-bipyridine rings. This position is relatively acidic and has previously been observed to undergo deuterium exchange in the complex [Ru(bpy)3]2+ in basic conditions9 and is also the site of orthometallation in iridium10 as well as nickel, palladium and platinum11 2,2′-bipyridine complexes. As this manuscript was being prepared, related observations for specific interactions of chloride with the 3- and 3′-protons in [Ru(bpy)3]2+ derivatives were described.12 We have observed analogous shifting of the E3 proton signals in a wide range of [Ir(C^N)2(N^N)]+ complexes, both in CD2Cl2 and CD3CN solutions. For example, in CD3CN, protons E3 and F3† in [Ir(msppy)2(6-Phbpy)][PF6] resonate at δ 8.56 and 8.60 ppm, compared to δ 8.65 and 8.68 ppm in [Ir(msppy)2(6-Phbpy)]Cl (Hmsppy = 2-(4-methylsulfonylphenyl)pyridine, 6-Phbpy = 6-phenyl-2,2′-bipyridine).† While significant, the shift in signals is less dramatic in CD3CN than in the less polar CD2Cl2. The 13C NMR spectra† of [Ir(msppy)2(6-Phbpy)][PF6] and [Ir(msppy)2(6-Phbpy)][Cl] show no significant differences. To understand the origin of the NMR spectroscopic changes, we determined the solid state structures of [Ir(msppy)2(6-Phbpy)]Cl (as a representative bulk-sample chloride salt) and of the chloride-containing material present in gram-scale syntheses of [Ir(ppy)2(bpy)][PF6].
Single crystals† of 2{[Ir(ppy)2(bpy)][Cl]}·2CH2Cl2·[H3O]·Cl were obtained from solutions containing [Ir(ppy)2(bpy)][PF6] and [Ir(ppy)2(bpy)]Cl. The structure of one of the two enantiomeric {[Ir(ppy)2(bpy)]Cl} units in the lattice is shown in Fig. 4. The [Ir(ppy)2(bpy)]+ cation closely resembles that in [Ir(ppy)2(bpy)][PF6]3 with the metal ion in a slightly distorted octahedral environment and with the expected trans-arrangement of ppy− N-donor atoms. The ppy− ligands are near-planar with angles between the least squares planes of the phenyl and pyridine rings of 4.8 and 5.0°. The bpy ligand is more distorted with an interannular angle of 8.7°. The Cl− anion is chelated to the bpy 3- and 3′-protons (crystallographic ally labelled H4a and H7a, Fig. 4a), consistent with solution NMR spectra, and forms hydrogen bonds13 to H4a and H7a with CH⋯Cl distances of 2.79 and 2.87 Å, respectively, (C4⋯Cl1 = 3.627(4) and C7⋯Cl1 = 3.794(4) Å) and the angle H4a–Cl1–H7a = 45.6°. This chloride ion is also hydrogen bonded to the 4-proton of one bpy and the 5-proton of a ppy− (see Scheme 1) of two different {[Ir(ppy)2(bpy)][Cl]} moieties (CH⋯Cl, 2.86 and 2.68 Å, respectively). The [H3O]+Cl− unit is disordered and has been modelled with the O and Cl atoms (O10 and Cl10) in symmetry related sites, each of fractional occupancy 0.5. The O⋯Cl separation of 2.818(6) Å is similar to, but shorter than, that observed in the crystal structure of [H3O]Cl (2.95(1) Å).14 Single crystals of [Ir(msppy)2(6-Phbpy)][Cl] grew after diffusion of Et2O into a MeCN solution of the complex. Fig. 4b shows the structure of the ion pair containing the Δ-[Ir(msppy)2(6-Phbpy)]+ cation; both enantiomers are present in the lattice. The octahedral coordination environment of atom Ir1 is similar to that in [Ir(ppy)2(bpy)]+, and bond parameters are given in the caption to Fig. 4b. The Cl− anion is again bound by the bpy 3- and 3′-protons (H4a and H7a in Fig. 4b) with CH⋯Cl distances of 2.62 and 2.80 Å, respectively, with an H4a–Cl1–H7a angle of 46.0°. Although the CSD (v. 5.34 with three updates, Conquest v. 1.1515) contains 429 examples of metal-bound 2,2′-bipyridine with Cl (either Cl− or covalently bonded Cl) hydrogen-bonded between the 3- and 3′-protons, only nine of these are iridium complexes; of these, only two involve Cl− ion16,17 as opposed to, for example, a Cl–M unit of an adjacent molecule.
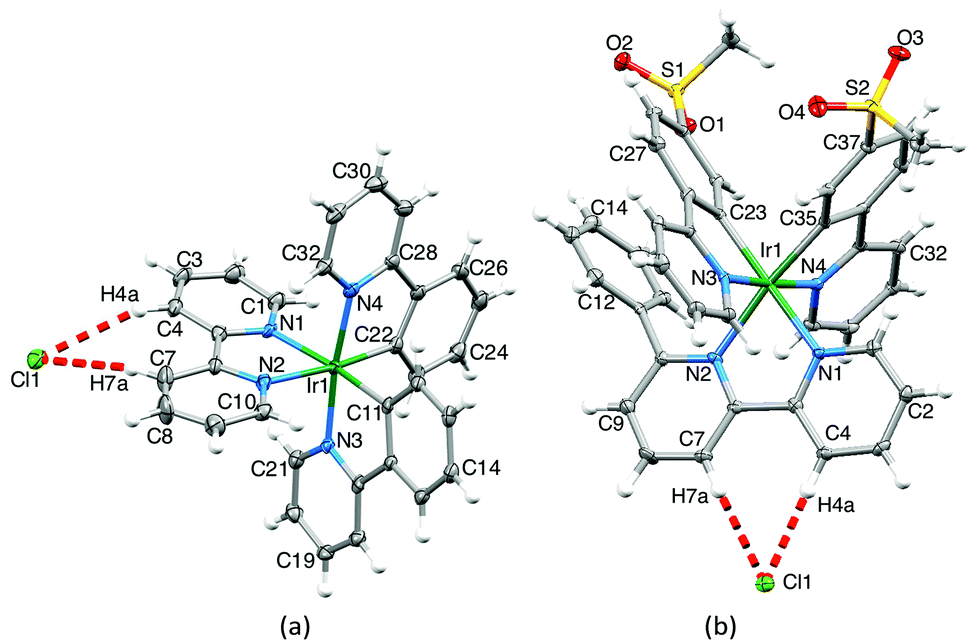 | ||
| Fig. 4 (a) The Δ-cation–Cl− ion pair of 2{[Ir(ppy)2(bpy)][Cl]}·2CH2Cl2·[H3O]·[Cl] Selected bond parameters: Ir1–C11 = 2.011(3), Ir1–C22 = 2.012(3), Ir1–N4 = 2.042(3), Ir1–N3 = 2.050(3), Ir1–N2 = 2.135(3), Ir1–N1 = 2.142(3) Å; N1–Ir1–N2 = 76.5(1), N3–Ir1–C11 = 80.2(1), N4–Ir1–C22 = 80.4(1)°. (b) The Δ-cation–Cl− ion pair in [Ir(msppy)2(6-Phbpy)]Cl. Selected bond parameters: Ir1–C35 = 2.005(4), Ir1–C23 = 2.034(4), Ir1–N1 = 2.119(3), Ir1–N2 = 2.207(3), Ir1–N3 = 2.050(3), Ir1–N4 = 2.050(3) Å; N1–Ir1–N2 = 76.14(12), C23–Ir1–N3 = 80.33(14), C35–Ir1–N4 = 80.43(14)°. Both are shown with 40% probability level ellipsoids. | ||
Conclusions
The presence of chloride can result in a dramatic reduction in the performance of the ionic transition metal complex [Ir(ppy)2(bpy)][PF6] used as the emissive component in LECs. The solution 1H NMR chemical shift of the bpy 3- and 3′-protons is very sensitive to the presence of Cl− (both in CD2Cl2 and more polar CD3CN), and in the solid state, Cl− forms a strong chelating hydrogen bond with this position. The tight ion-pairing in [Ir(C^N)2(N^N][Cl] means that Cl− is easily carried through synthetic steps which start with [Ir2(C^N)4Cl2] dimers. An HCl adduct of the salt [Ir(ppy)2(bpy)]Cl was structurally characterized allowing clarification of the nature of side-products isolated in gram-scale preparations of [Ir(ppy)2(Xbpy)][PF6] materials.Acknowledgements
We thank the Swiss National Science Foundation, University of Basel, European Union (CELLO, STRP 248043) and European Research Council (Advanced Grant 267816 LiLo) for financial support. A.P. acknowledges the Spanish Ministry of Economy and Competitiveness (MINECO) for an FPI grant. Liselotte Siegfried, Dr Colin J. Martin and Dr Collin Morris (University of Basel) are thanked for initial NMR titrations, help with WinEQNMR2, and ESI-MS measurements, respectively.Notes and references
- D. M. Dobkin and M. K. Zuraw, Principles of Chemical Vapor Deposition, Springer, 2010 Search PubMed.
- R. H. Friend, J. H. Burroughes and T. Shimoda, Phys. World, 1999, 12(June), 35 CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2009, 19, 3456 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- S. T. Parker, J. D. Slinker, M. S. Lowry, M. P. Cox, S. Bernhard and G. G. Malliaras, Chem. Mater., 2005, 17, 3187 CrossRef CAS.
- M. Buda, G. Kalyuzhny and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 6090 CrossRef CAS PubMed.
- H. Rudmann, S. Shimada and M. F. Rubner, J. Am. Chem. Soc., 2002, 124, 4918 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- E. C. Constable and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1982, 34 RSC.
- P. S. Braterman, G. A. Heath, A. J. MacKenzie, B. C. Noble, R. D. Peacock and L. J. Yellowlees, Inorg. Chem., 1984, 23, 3425 CrossRef CAS; A. C. Hazell and R. G. Hazell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 806 CrossRef; P. J. Spellane, R. J. Watts and C. J. Curtis, Inorg. Chem., 1983, 22, 4060 CrossRef; G. Nord, A. C. Hazell, R. G. Hazell and O. Farver, Inorg. Chem., 1983, 22, 3429 CrossRef.
- B. Butschke and H. Schwarz, Organometallics, 2010, 29, 6002–6011 CrossRef CAS; B. Butschke, M. Schlangen, D. Schroeder and H. Schwarz, Chem.–Eur. J., 2008, 14, 11050–11060 CrossRef PubMed.
- W. M. Ward, B. H. Farnum, M. Siegler and G. J. Meyer, J. Phys. Chem. A, 2013, 117, 8883 CrossRef CAS PubMed.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91 RSC.
- Y. K. Yoon and G. B. Carpenter, Acta Crystallogr., 1959, 12, 17 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed; I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- T. Moriuchi, C. Katano and T. Hirao, Chem. Lett., 2012, 41, 310 CrossRef CAS.
- M.-T. Youinou and R. Ziessel, J. Organomet. Chem., 1989, 363, 197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: 1H NMR signals for HE3 and HB3 in batches 1 and 2 of materials; Fig. S2: 19F NMR spectra for addition of [nBu4N]Cl to [Ir(ppy)2(bpy)][PF6]. Synthesis of chloride-free [Ir(ppy)2(bpy)][PF6] and of [Ir(msppy)2(6-Phbpy)][PF6] and [Ir(msppy)2(6-Phbpy)][Cl]. Crystallographic data for 2{[Ir(ppy)2(bpy)][Cl]}·2CH2Cl2·[H3O]·Cl and [Ir(msppy)2(6-Phbpy)][Cl]. CCDC 959828 and 971737. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt53229a |
| This journal is © The Royal Society of Chemistry 2014 |