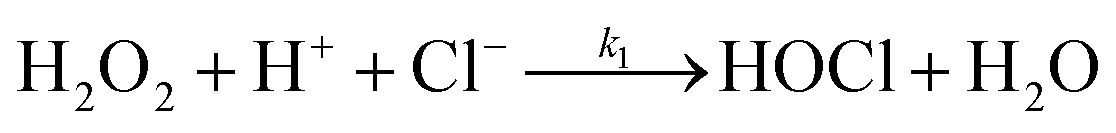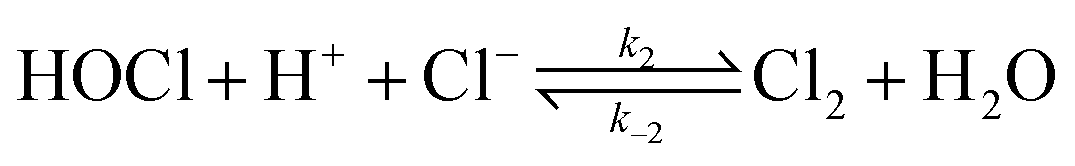DOI:
10.1039/C3DT53057D
(Paper)
Dalton Trans., 2014,
43, 6308-6314
Mechanism of tetrachloroplatinate(II) oxidation by hydrogen peroxide in hydrochloric acid solution†
Received
29th October 2013
, Accepted 6th December 2013
First published on 20th December 2013
Abstract
Oxidation of tetrachloroplatinate(II) by hydrogen peroxide in hydrochloric acid was studied by UV-Vis spectrophotometry. Oxidation takes place via two parallel reactions with hypochlorous acid and hydrogen peroxide, respectively, according to the overall rate law d[Pt(IV)]/dt = (k0 + kH2O2[Pt(II)])[H2O2]. For oxidation of [PtCl4]2− at relatively low concentrations, [PtCl4]2− ≪ 0.5 mM, hypochlorous acid formation is fast relative to the oxidation of [PtCl4]2− by hydrogen peroxide, as a result of the rate determining reaction H2O2 + H+ + Cl− → HOCl + H2O, resulting in a rate law d[Pt(IV)]/dt = k0[H2O2] with a value k0 = (8 ± 2) × 10−7 s−1 at 35 °C. For concentrations of [PtCl4]2− > 0.5 mM, oxidation by hydrogen peroxide becomes dominant, resulting in the pseudo-first order rate law d[Pt(IV)]/dt = kH2O2[Pt(II)][H2O2] with the value kH2O2 = (1.5 ± 0.1) × 10−2 M−1 s−1 at 35 °C. The final oxidation product is a mixture of [PtCl5(H2O)]− and [PtCl6]2−, with [PtCl6]2− formed as a result of [PtCl4]2− assisted chloride anation reactions.
Introduction
Oxidation of Pt(II) square-planar complexes by hydrogen peroxide has been exploited in many areas of research, particularly as a strategy towards the design of new complexes.1 Many of these studies rely on the formation of hydroxido complexes by the oxidation with hydrogen peroxide, which provides more stability and control. The square-planar configuration of the original Pt(II) complex is retained furnishing a Pt(IV) product with new ligands coordinated trans to each other. For instance, oxidation of [PtCl4]2− by hydrogen peroxide in water yields trans-[PtCl4(OH)2]2− quantitatively according to eqn (1).2| | | [PtCl4]2− + H2O2 → trans-[PtCl4(OH)2]2− | (1) |
195Pt NMR indicates that the trans coordinated hydroxido ligands originate from hydrogen peroxide and solvent water respectively.3 Inert hydroxido ligands can be protonated after oxidation to render the aqua ligands that are labile, promoting substitution reactions.4
By comparison, oxidation of [PtCl4]2− by hydrogen peroxide in acidic medium yields trans-[PtCl4(H2O)2], while relatively fast Pt(II) assisted ligand scrambling reactions cause a redistribution of the oxidation product(s) to form a mixture of [PtCl6−n(H2O)n]2−n (n = 0–4) complexes.5 The large-scale separation of platinum from other platinum group metals (PGMs) depends, amongst other factors, on the efficient oxidation of Pt(II) to Pt(IV) in solution. The oxidation states of the various PGMs dissolved in the hydrochloric acid process solutions are manipulated to allow for their separation by inter alia solvent extraction (SX), oxidative distillation and/or classical ion-exchange methods.5 Although hydrogen peroxide is not used in the refining industry, as part of ongoing work in this context, we examined in detail the oxidation of [PtCl4]2− by hydrogen peroxide in hydrochloric acid as a benchmark system. In the oxidation of Pt(II) to Pt(IV) in solution, Pearson and Basolo proposed a reaction mechanism involving Pt(II) assisted ligand exchange more than 50 years ago.6 This mechanism was later revised to account for direct formation of [PtCl6]2− from trans-[PtCl4(H2O)2] in a chloride rich solution.4 The origin of the difference in behaviour between trans-Pt(IV) aqua and hydroxido analogues is attributed to the labile trans aqua ligands which are susceptible to bond breakage.7 Kinetic studies dealing with oxidation of Pt(II) complexes by hydrogen peroxide have been neglected, particularly in the presence of free chloride. A kinetic study dealing with the oxidation of [PtCl4]2− by hydrogen peroxide in perchloric acid has been reported.8
In the present study, oxidation of [PtCl4]2− by hydrogen peroxide is revisited, with the added complexity of free chloride ions in solution, presenting a more complex mechanism. Formation of [PtCl5(H2O)]− and possibly trans-[PtCl4(H2O)2] coincides with [PtCl4]2− assisted ligand exchange. Apart from the complications associated with such reactions, hydrochloric acid catalyses the decomposition of hydrogen peroxide to form hypochlorous acid according to eqn (2).
| | 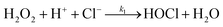 | (2) |
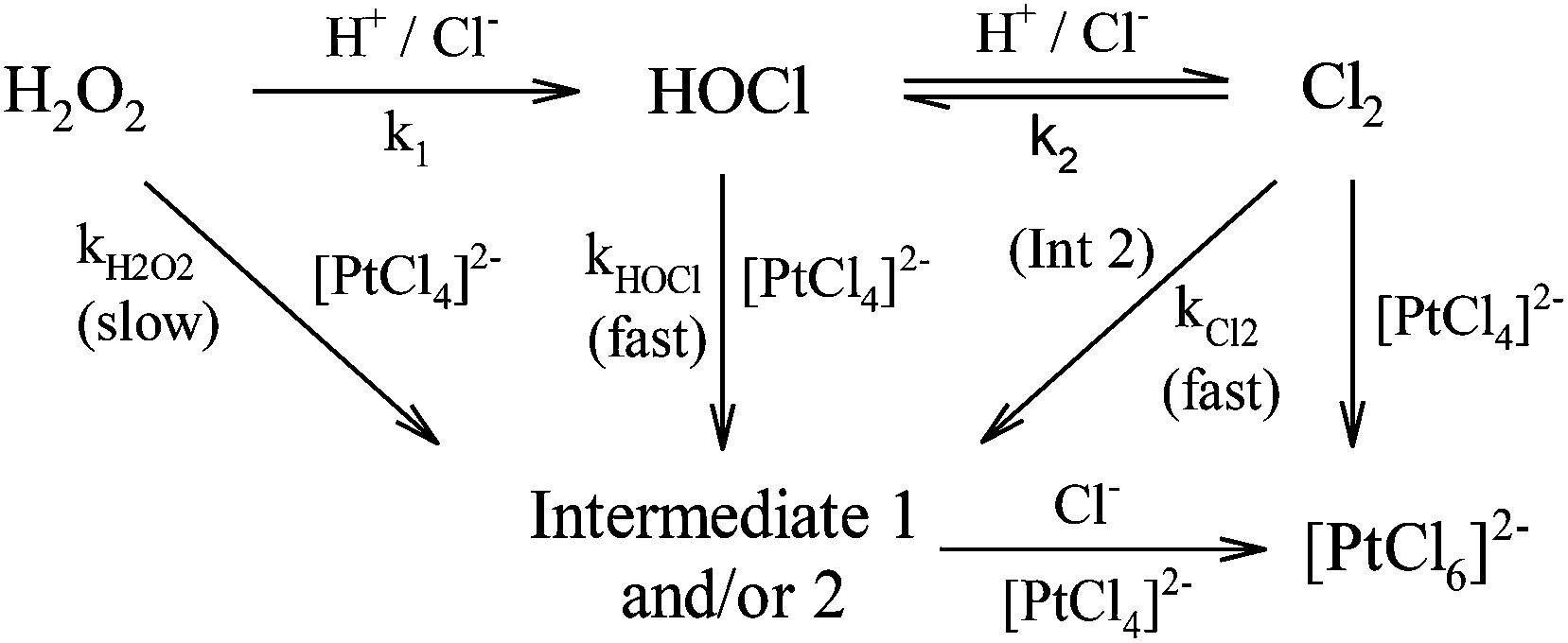
Oxidation by hydrogen peroxide and hypochlorous acid may therefore coincide to yield two parallel reactions kH2O2 and kHOCl as depicted in the scheme in (3), where the intermediates are 1 = trans-[PtCl4(H2O)2] and 2 = [PtCl5(H2O)]−.
| | 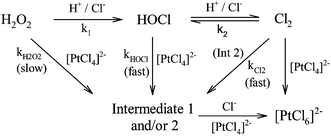 | (3) |
Oxidation of [PtCl4]2− by hydrogen peroxide is slow, whereas oxidation by hypochlorous acid is rapid in comparison, but yields the same product.8,9 Furthermore, depending on the reaction conditions, hypochlorous acid may form chlorine which can also act as an oxidant, further complicating this process. These aspects are explored and discussed in the present study to elucidate the overall oxidation mechanism of [PtCl4]2− by hydrogen peroxide in the presence of an excess of hydrogen cations and chloride anions.
Experimental section
Chemicals and solutions
All chemicals were of reagent grade quality and used without further purification. Potassium tetrachloroplatinate(II) (99.9+%, K2PtCl4), sodium chloride (99+%, NaCl) and sodium perchlorate (99+%, NaClO4) were obtained from Sigma-Aldrich. Hydrogen peroxide (30% w/w, H2O2, Sigma-Aldrich) was of reagent grade quality and used as received. Solutions of hydrogen peroxide were prepared immediately before use. Analytically pure concentrated perchloric acid (70% w/w, HClO4, 1 L = 1.68 kg, Merck) and hydrochloric acid (HCl, Sigma-Aldrich) were used to prepare solutions. Stock solutions of 1 mM or 10 mM [PtCl4]2− were prepared in 1 M or 2 M HCl from which further dilutions were made. Solutions containing Pt(II) were kept in the dark to eliminate photo-induced aquation reactions.10,11 Concentrations of [PtCl4]2− were evaluated by UV-Vis spectrophotometry at 331 nm (ε331 = 59 M−1 cm−1) or 390 nm (ε390 = 56 M−1 cm−1).12 Chloride concentrations >0.1 M are sufficient to suppress aquation of [PtCl4]2− → [PtCl3(H2O)]−, and in 1 M HCl, all Pt(II) essentially exists as [PtCl4]2−.13 All aqueous solutions were made with ultra-pure de-ionised water.
Spectrophotometry
Photo-induced reactions necessitate kinetic measurements in UV-Vis absorption spectra at a specific wavelength. UV-Vis spectra in the range 200–600 nm were recorded after the oxidation was complete. Measurements were performed on a Shimadzu UV-2010PC spectrophotometer. The instrument was equipped with a thermoelectrically controlled cell holder using 1 cm tandem quartz cuvettes. Activation volume measurements for slow reactions were performed on a Shimadzu UV-2010PC spectrophotometer equipped with a high pressure cell fitted with a 1.5 cm pill-box quartz-cuvette.14 Activation volume measurements for relatively fast reactions were performed on a laboratory-made high-pressure stopped-flow instrument.15 The temperature was controlled and maintained in these instruments at 35.0 ± 0.1 °C using a circulating water bath (Julabo MP-5).
Kinetics
Observed rate constants for pseudo-zero order reactions were obtained directly from the slope of concentration versus time plots. Observed rate constants for reactions showing pseudo-first order character were calculated directly from absorbance versus time plots using a least-squares program. The ionic strength was kept constant at 1 M in all experiments by using the correct ratios of hydrogen chloride, sodium chloride, sodium perchlorate and perchloric acid. Hydrogen peroxide was always present in large excess (>15 times) with regard to the substrate ensuring pseudo-order reaction conditions.
Spectra
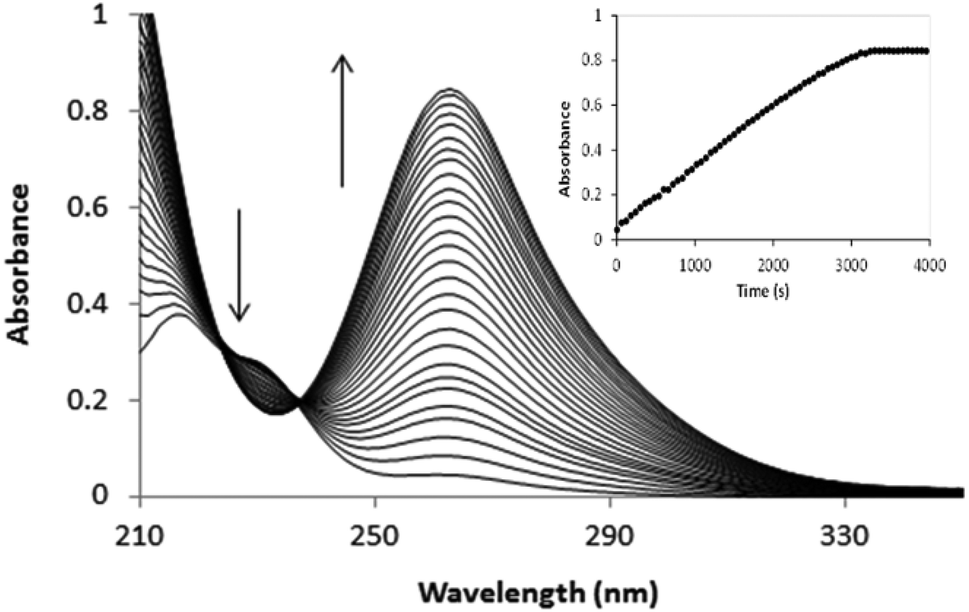
Both [PtCl5(H2O)]− and [PtCl6]2− were identified in the final spectra after oxidation of [PtCl4]2− by hydrogen peroxide. The [PtCl5(H2O)−]/[PtCl62−] ratio is proportional to the oxidation rate which in turn is influenced by the concentration of [PtCl4]2−, H2O2, acid and chloride. Slow reactions led to almost complete conversion of [PtCl4]2− → [PtCl6]2− (>90% of Pt(IV) identified as [PtCl6]2−), while for fast oxidation reactions [PtCl5(H2O)]− is the major product. Since chloride anation of [PtCl5(H2O)]− is slow in the absence of [PtCl4]2−, conversion of [PtCl5(H2O)]− → [PtCl6]2− is effectively quenched after oxidation when [PtCl4]2− is depleted.4 For the oxidation of [PtCl4]2− an absorbance increase is observed between 200 and 500 nm except for the region between 222 and 236 nm in which isosbestic points occur and an absorbance decrease is observed, as shown in Fig. 1.
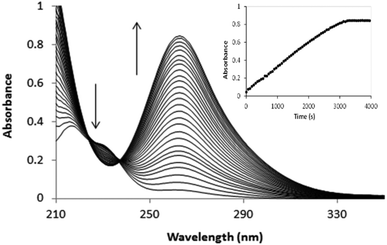 |
| | Fig. 1 Spectral changes recorded for the oxidation of 0.04 mM [PtCl4]2− by 4 mM H2O2 in 1 M HCl. | |
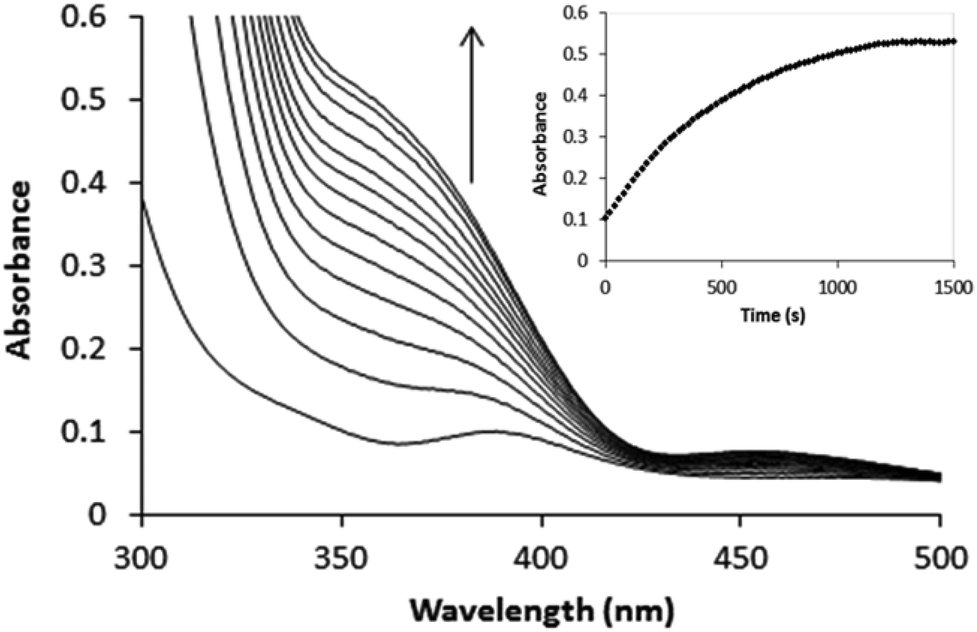
The [PtCl5(H2O)−]/[PtCl62−] ratio for Pt(II) ≤ 0.07 mM was determined at 230 and 262 nm where these complexes show absorbance maxima and minima, respectively. The UV spectra of these complexes are known and have been reported elsewhere.2,16 For [PtCl5(H2O)]−, ε230 = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1 and ε262 = 11600 M−1 cm−1, whereas for [PtCl6]2−, ε230 = 3300 M−1 cm−1 and ε262 = 24500 M−1 cm−1. Since the visible spectra of [PtCl5(H2O)]− and [PtCl6]2− are very similar (ε353 = 490 M−1 cm−1 for both complexes), it was not possible to determine accurate values for the [PtCl5(H2O)−]/[PtCl6]2− concentration ratio from the visible spectra for concentrations of Pt(II) ≥ 0.2 mM. Dilution of such solutions, however, indicated that [PtCl6]2− is the dominant species. Typical spectral changes observed for the oxidation of [PtCl4]2− at relatively high concentration are illustrated in Fig. 2.
500 M−1 cm−1 and ε262 = 11600 M−1 cm−1, whereas for [PtCl6]2−, ε230 = 3300 M−1 cm−1 and ε262 = 24500 M−1 cm−1. Since the visible spectra of [PtCl5(H2O)]− and [PtCl6]2− are very similar (ε353 = 490 M−1 cm−1 for both complexes), it was not possible to determine accurate values for the [PtCl5(H2O)−]/[PtCl6]2− concentration ratio from the visible spectra for concentrations of Pt(II) ≥ 0.2 mM. Dilution of such solutions, however, indicated that [PtCl6]2− is the dominant species. Typical spectral changes observed for the oxidation of [PtCl4]2− at relatively high concentration are illustrated in Fig. 2.
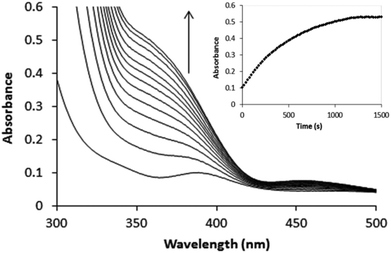 |
| | Fig. 2 Spectral changes recorded for the oxidation of 1 mM [PtCl4]2− by 100 mM H2O2 in 1 M HCl. | |
Small differences in absorbance versus time plots collected at a specific wavelength were noticed when compared to spectra collected in the wavelength range 200–500 nm on a rapid-scan UV-Vis spectrophotometer. It has long been known that Pt(II/IV) complexes are sensitive to light resulting in photo-induced reactions.10,11 The rates of [PtCl4]2− and/or [PtCl6]2− aquation as well as [PtCl5(H2O)]− chloride anion reactions are enhanced when spectra are recorded in the 200–500 nm wavelength range, which accounts for the small differences observed in the kinetic traces. To suppress such effects, changes in absorbance were monitored at 262 or 353 nm, close to the absorbance maxima of [PtCl6]2−.
Oxidation of [Pt(CN)4]2− by chlorine yields trans-[Pt(CN)4Cl(H2O)]− as the primary product even when the free chloride ion concentration in solution is as high as 1 M.9 A preference for solvent water to coordinate the negatively charged Pt(II) precursor complex, trans to the oxidant, was considered here. The spectrum of trans-[PtCl4(H2O)2] is known and has been reported before.2 No evidence could be found for the presence of significant concentrations of trans-[PtCl4(H2O)2] under the reaction conditions and in the time scale studied here. Associations between [PtCl4]2− and trans-[PtCl4(H2O)2], rapidly resulting in the formation of [PtCl5(H2O)]− and/or [PtCl6]2− within the time scale investigated here, is possible as suggested by the rate constants estimated in this study (vide infra).
Results
Oxidation of [PtCl4]2− ≤ 0.07 mM
Oxidation of [PtCl4]2− by excess hydrogen peroxide quantitatively converts all Pt(II) → Pt(IV) so that −d[PtII]/dt = d[Pt(IV)]/dt. Oxidation reactions of [PtCl4]2− ≤ 0.07 mM with [H2O2] (5–100 mM) generate predominantly linear absorbance vs. time plots at 262 nm (Fig. 1; data summarized in Table 1), indicating that the reaction is pseudo-zero order with respect to Pt(II) and the observed rate is defined by rate law eqn (4).| | | d[Pt(IV)]/dt = k0[H2O2] = k0obs (M s−1) | (4) |
Table 1 Experimental conditions and kinetic data for the oxidation of 0.02–0.07 mM [PtCl4]2− yielding pseudo-zero order kinetics at 35 °C
| [PtCl4]2− (mM) |
H2O2 (mM) |
k
0obs (M s−1) |
k
0 = k0obs/[H2O2] (s−1) |
| 0.02 |
100 |
6.49 × 10−8 |
6.49 × 10−7 |
| 0.02 |
70 |
4.64 × 10−8 |
6.63 × 10−7 |
| 0.02 |
50 |
3.19 × 10−8 |
6.38 × 10−7 |
| 0.02 |
30 |
1.97 × 10−8 |
6.56 × 10−7 |
| 0.02 |
10 |
6.83 × 10−8 |
6.83 × 10−7 |
| 0.02 |
5 |
3.67 × 10−8 |
7.34 × 10−7 |
| 0.03 |
100 |
5.49 × 10−8 |
5.49 × 10−7 |
| 0.03 |
70 |
3.84 × 10−8 |
5.49 × 10−7 |
| 0.03 |
50 |
2.89 × 10−8 |
5.78 × 10−7 |
| 0.03 |
30 |
1.68 × 10−8 |
5.60 × 10−7 |
| 0.03 |
10 |
0.67 × 10−8 |
6.74 × 10−7 |
| 0.03 |
5 |
0.39 × 10−8 |
7.74 × 10−7 |
| 0.04 |
100 |
6.68 × 10−8 |
6.68 × 10−7 |
| 0.04 |
70 |
4.86 × 10−8 |
6.94 × 10−7 |
| 0.04 |
50 |
3.34 × 10−8 |
6.68 × 10−7 |
| 0.04 |
30 |
2.06 × 10−8 |
6.87 × 10−7 |
| 0.04 |
10 |
0.85 × 10−8 |
8.52 × 10−7 |
| 0.04 |
5 |
0.46 × 10−8 |
9.12 × 10−7 |
| 0.05 |
100 |
7.96 × 10−8 |
7.96 × 10−7 |
| 0.05 |
70 |
6.54 × 10−8 |
9.34 × 10−7 |
| 0.05 |
60 |
5.51 × 10−8 |
9.18 × 10−7 |
| 0.05 |
50 |
4.52 × 10−8 |
9.04 × 10−7 |
| 0.05 |
40 |
3.82 × 10−8 |
9.55 × 10−7 |
| 0.06 |
100 |
8.69 × 10−8 |
8.69 × 10−7 |
| 0.06 |
70 |
6.58 × 10−8 |
9.4 × 10−7 |
| 0.06 |
60 |
5.83 × 10−8 |
9.72 × 10−7 |
| 0.06 |
50 |
4.96 × 10−8 |
9.92 × 10−7 |
| 0.06 |
40 |
4.27 × 10−8 |
10.68 × 10−7 |
| 0.07 |
100 |
8.75 × 10−8 |
8.75 × 10−7 |
| 0.07 |
70 |
6.47 × 10−8 |
9.24 × 10−7 |
| 0.07 |
60 |
6.14 × 10−8 |
10.23 × 10−7 |
| 0.07 |
50 |
4.85 × 10−8 |
9.70 × 10−7 |
| 0.07 |
40 |
4.45 × 10−8 |
11.13 × 10−7 |
|
|
|
Mean value = (8 ± 2) × 10−7 s−1 |
The observed rate constants (k0obs) were obtained directly from the slope of concentration versus time plots. Plots of k0obsversus [H2O2] are linear with zero intercept for the [PtCl4]2− concentration range 0.02–0.04 mM (ESI Fig. S1A†). A small intercept ∼1.0 × 10−8 M s−1 (ESI Fig. S1B†) was observed for [PtCl4]2− concentrations between 0.05 and 0.07 mM. Pseudo-zero order rate constants k0 (s−1) calculated from eqn (5) are listed in Table 1, from which it follows that k0 = (8 ± 2) × 10−7 s−1 over the entire concentration range investigated.
The activation parameters ΔH‡ and ΔS‡ were estimated from the variation of k0obs in the temperature range 15–35 °C (Table 2), and plots of ln (k0/T) versus 1/T according to the Eyring equation gave linear correlations (ESI Fig. S2†). Values for ΔH‡ and ΔS‡ were subsequently calculated from the slope and intercept of such plots according to eqn (6) and (7), where kB is Boltzmann's constant and h is Planck's constant. The calculated parameters are listed in Table 2.
| | | Intercept = ln(kB/h) + ΔS‡/R | (7) |
Table 2 Kinetic data for the oxidation of [PtCl4]2− by H2O2 as a function of temperature with calculated activation parameters
| [PtCl4]2− (mM) |
[H2O2] (mM) |
Temp. (°C) |
k
0 (s−1) |
ΔH‡ (kJ mol−1) |
ΔS‡ (J K−1 mol−1) |
| 0.04 |
100 |
15 |
0.76 × 10−7 |
78 ± 5 |
−109 ± 7 |
| 0.04 |
100 |
20 |
1.53 × 10−7 |
|
|
| 0.04 |
100 |
25 |
2.75 × 10−7 |
|
|
| 0.04 |
100 |
30 |
4.57 × 10−7 |
|
|
| 0.04 |
100 |
35 |
6.68 × 10−7 |
|
|
| |
|
|
k
H2O2 (M−1 s−1) |
|
|
| 1 |
300 |
15 |
3.18 × 10−3 |
52 ± 1 |
−111 ± 4 |
| 1 |
300 |
20 |
4.43 × 10−3 |
|
|
| 1 |
300 |
25 |
6.81 × 10−3 |
|
|
| 1 |
300 |
30 |
9.57 × 10−3 |
|
|
| 1 |
300 |
35 |
13.83 × 10−3 |
|
|
The activation volume of the oxidation reaction (ΔV‡) for the zero-order process was estimated by varying the pressure in the range 10–132 MPa. A plot of ln k0versus pressure is linear (Fig. S3, ESI†) and ΔV‡ was obtained from the slope of the plot according to eqn (8). The calculated activation parameters are included in Table 3.
| | | [d(ln k0)/dP]T = −ΔV‡/RT | (8) |
Table 3 Kinetic data for the oxidation of [PtCl4]2− by H2O2 as a function of pressure with calculated activation volumes at 35 °C
| [PtCl4]2− (mM) |
[H2O2] (mM) |
Pressure (MPa) |
k
0 (s−1) |
ΔV‡ (cm3 mol−1) |
| 0.04 |
100 |
10 |
1.38 × 10−6 |
−8 ± 2 |
| 0.04 |
100 |
51 |
1.59 × 10−6 |
|
| 0.04 |
100 |
91 |
1.79 × 10−6 |
|
| 0.04 |
100 |
132 |
2.02 × 10−6 |
|
| |
|
|
k
H2O2 (M−1 s−1) |
|
| 1 |
15 |
5 |
1.67 × 10−2 |
−7.8 ± 0.4 |
| 1 |
15 |
51 |
1.86 × 10−2 |
|
| 1 |
15 |
101 |
2.23 × 10−2 |
|
| 1 |
15 |
152 |
2.58 × 10−2 |
|
Oxidation of [PtCl4]2− ≥ 0.2 mM
Oxidation of [PtCl4]2− at concentration levels ≥0.2 mM by [H2O2] in the concentration range 15–300 mM resulted in absorbance vs. time traces at 353 nm which exhibited first-order character. These kinetic traces show inflection points once equilibrium is reached (ESI Fig. S4†), reminiscent of zero-order kinetics prevailing at relative low concentrations of [PtCl4]2−. The observed rate constants (kobs) were estimated from the second-order rate law eqn (9) and are listed in Table 4. Only absorbance data points prior to the inflection point were used for the estimation of kobs values, as obtained from least-squares fits.| | | d[Pt(IV)]/dt = kH2O2[Pt(II)][H2O2] | (9) |
Table 4 Experimental conditions and kinetic data for the oxidation of 0.2–1.0 mM [PtCl4]2− yielding pseudo-first order kinetics at 35 °C
| [PtCl4]2− (mM) |
[H2O2] (mM) |
k
obs (s−1) |
k
H2O2 = kobs/[H2O2] (M−1 s−1) |
| 1.0 |
300 |
3.94 × 10−3 |
1.31 × 10−2 |
| 1.0 |
100 |
1.52 × 10−3 |
1.52 × 10−2 |
| 1.0 |
50 |
0.81 × 10−3 |
1.62 × 10−2 |
| 1.0 |
15 |
0.30 × 10−3 |
2.00 × 10−2 |
| 0.6 |
300 |
4.49 × 10−3 |
1.50 × 10−2 |
| 0.6 |
100 |
1.57 × 10−3 |
1.57 × 10−2 |
| 0.6 |
50 |
0.78 × 10−3 |
1.56 × 10−2 |
| 0.6 |
15 |
0.23 × 10−3 |
1.53 × 10−2 |
| 0.2 |
300 |
3.84 × 10−3 |
1.28 × 10−2 |
| 0.2 |
100 |
1.27 × 10−3 |
1.27 × 10−2 |
| 0.2 |
50 |
0.74 × 10−3 |
1.48 × 10−2 |
| 0.2 |
15 |
0.23 × 10−3 |
1.53 × 10−2 |
|
|
|
Mean value = (1.5 ± 0.1) × 10−2 M−1 s−1 |
Plots of kobsversus [H2O2] demonstrate a linear dependence (ESI Fig. S5†), suggesting that the reaction is first order with respect to hydrogen peroxide. The second order rate constants kH2O2 were calculated from eqn (10) to give the average value (1.5 ± 0.1) × 10−2 M−1 s−1.
Oxidation of 1 mM [PtCl4]2− with 300 mM H2O2 generated traces illustrating pseudo-first order character. Activation parameters were estimated under these reaction conditions from the variation of kH2O2 in the temperature range 15–35 °C and eqn (6) and (7) (Table 2). Values of ΔV‡ were estimated by varying the pressure between 5 and 152 MPa for the oxidation of 1 mM [PtCl4]2− with 15 mM H2O2. A plot of ln kH2O2versus pressure is linear (Fig. S6, ESI†) and allowed the estimation of ΔV‡ from eqn (8) listed in Table 3.
Effect of acid and chloride concentrations on the oxidation rate and reaction order
The effect of variation of acid and/or chloride concentration on the reaction rate and order was evaluated in the ranges 0.6–1 M and 0.02–1 M, respectively. Concentrations of Pt(II) and hydrogen peroxide were kept constant at 0.04 mM and 80 mM, respectively. For the high end concentration range, zero-order kinetics predominated (ESI Fig. S7, ESI,† for the variation of [Cl−]), while a gradual change to first-order kinetics was observed for sufficiently low concentrations of [H+] and/or [Cl−]. Rate constants (k0obs) were estimated from the slope of concentration versus time plots. Traces that display a degree of first-order character were evaluated for the first 500 s (Fig. S7,† plots 3 and 4). The values for k0obs, obtained in this manner, are listed in Table 5. Plots of k0obsversus [H+] and k0obsversus [Cl−] are linear (R2 > 0.99) with a small intercept in both cases (Fig. S8, ESI,† −3.4 × 10−7 M s−1 and 20.3 × 10−7 M s−1, respectively), suggesting that k0obs varies with [H+] and [Cl−] according to eqn (11):where Q represents [H+] or [Cl−], a = 3.4 × 10−7 M s−1/20.3 × 10−7 M s−1 and b = 8.6 × 10−7 s−1/82.3 × 10−7 s−1, respectively. These values were obtained directly from the intercept and gradient of the plots in Fig. S8.†
Table 5 Oxidation of [PtCl4]2− performed to determine the effect of chloride and acid concentrations on the observed reaction order at 35 °C
| HCl (M) |
HClO4 (M) |
NaClO4 (M) |
k
0obs (M s−1) |
k
0 = k0obs/[H2O2] (s−1) |
| 0.5 |
0.5 |
— |
7.71 × 10−7 |
9.64 × 10−6 |
| 0.5 |
0.3 |
0.2 |
5.92 × 10−7 |
7.40 × 10−6 |
| 0.5 |
0.1 |
0.4 |
4.27 × 10−7 |
5.34 × 10−6 |
| 1 |
0 |
— |
1.02 × 10−7 |
1.28 × 10−6 |
| 0.6 |
0.4 |
— |
7.07 × 10−8 |
8.84 × 10−7 |
| 0.2 |
0.8 |
— |
3.65 × 10−8 |
4.56 × 10−7 |
| 0.02 |
0.98 |
— |
2.17 × 10−8 |
2.71 × 10−7 |
Discussion
Rate law for hypochlorous acid formation
An oxidation mechanism for [PtCl4]2− displaying zero-order kinetics can be envisaged as a result of the rapid formation of hypochlorous acid according to eqn (2). Catalytic decomposition of hydrogen peroxide in hydrochloric acid has been studied in detail. It was concluded that hydrogen peroxide decomposes via the acid-dependent reaction eqn (2), in addition to an acid-independent pathway eqn (12).17–19| | | H2O2 + Cl−→ H2O + ClO− | (12) |
The general rate law eqn (13) was established to define the overall rate of decomposition via the parallel reactions eqn (2) and (12).
| | | −d[H2O2]/dt = k1[H2O2][Cl−][H+] + k01[H2O2][Cl−] | (13) |
Reaction (12) is slow relative to the acid-dependent decomposition of hydrogen peroxide, reaction (2), especially in an acidic matrix which will enhance the acid-dependent reaction. The second term in eqn (13) becomes negligible in 1 M H+, as used in this study, so that the rate law for decomposition of hydrogen peroxide reduces to eqn (14).
| | | −d[H2O2]/dt = k1[H2O2][Cl−][H+] | (14) |
Zero-order reaction mechanism
If a [PtCl4]2− solution is mixed with excess hydrogen peroxide and hydrochloric acid, hydrogen peroxide is consumed via reactions denoted by kH2O2 and kHOCl in the scheme outlined in eqn (3). Since oxidation of [PtCl4]2− with hypochlorous acid is rapid, the k1 reaction is expected to be the major oxidation reaction under the condition of eqn (15).9,19| | | kH2O2[PtCl42−] ≪ k1[Cl−][H+] | (15) |
The condition is fulfilled where [PtCl4]2− ≪ 0.5 mM for [Cl−] = [H+] = 1 M.‡ Under such conditions hydrogen peroxide will disappear via the consecutive reactions k1 → kHOCl depicted in the scheme in (3), where k1 is the rate-determining reaction for the conditions of eqn (16).
| | | kHOCl[PtCl42−] ≫ k1[Cl−][H+] | (16) |
Eqn (16) is fulfilled if [PtCl4]2− ≫ 4.6 × 10−9 mM.§ Hence, oxidation proceeds mainly via hypochlorous acid formation in the concentration range 4.6 × 10−9 mM ≪ [PtCl4]2− ≪ 0.5 mM, with [Cl−] = [H+] = 1 M. Oxidation via hydrogen peroxide under these conditions becomes negligibly slow, and the conversion of H2O2 → HOCl is the rate-determining step. Under such conditions the reaction appears to be pseudo-zero-order with respect to [PtCl4]2−. The rate of hypochlorous acid formation (k1) in the hydrogen peroxide concentration range 5–100 mM varies from 1.04 × 10−8 to 2.07 × 10−7 M s−1 according to eqn (14). These values are in the same range as values of k0 determined for the oxidation of [PtCl4]2− reported in Table 1.
First-order reaction mechanism
If the concentration range of [PtCl4]2− > 0.5 mM, with [Cl−] = [H+] = 1 M, oxidation of [PtCl4]2− by hydrogen peroxide becomes dominant and the rate of hypochlorous acid formation is small by comparison. Under such reaction conditions, oxidation via the consecutive reactions k1 → kHOCl competes for the oxidation of [PtCl4]2− with reaction kH2O2 depicted in the scheme given in eqn (3). In the absence of free chloride ions, oxidation of [PtCl4]2− by hydrogen peroxide is first-order with respect to both [PtCl4]2− and H2O2 according to the rate law in eqn (9).8 The “curvature” observed in kinetic traces for oxidation of [PtCl4]2− ≥ 0.2 mM (Fig. 2) thus illustrates this parallel pseudo-first-order oxidation mechanism by hydrogen peroxide. The calculated kobs values according to eqn (9) for the hydrogen peroxide concentration range 15–300 mM vary between 6.38 × 10−5 and 1.28 × 10−3 M s−1. The experimentally determined kobs values for oxidation of [PtCl42−] ≥ 0.2 mM in this work as listed in Table 4 are at the upper limit of this range. A deviation between expected and calculated values may be reasonable, since estimation of kobs values from rate law eqn (9) does not account for any oxidation by hypochlorous acid, the parallel reaction under these reaction conditions.
Rate dependence on chloride and acid concentrations
The oxidation rate and order of [PtCl4]2− is directly proportional to both chloride and acid concentrations as illustrated by the kobs values shown in Table 5. Under conditions where [Cl−][H+] ≫ 0.082 M2 for concentrations of [PtCl4]2− = 0.04 mM and [H2O2] = 80 mM, the condition of eqn (15) will be valid.‡ On the other hand, if [Cl−][H+] ≪ 0.082 M2, the first-order mechanism becomes dominant resulting in more prominent “curvature” in the kinetic trace 4 as indicated in Fig. S7, ESI.†
Under these conditions, oxidation is first-order with respect to both [Cl−] and [H+] as suggested by the linear dependence of k0obs on [Cl−] and [H+] (Fig. S8, ESI†). In order to keep the rate law consistent for the range of reaction conditions investigated, values of k0obs were obtained directly from concentration versus time plots. This approach does not account for the parallel first-order oxidation reaction, a possible consequence of the small intercept in these graphs. The overall rate law accounting for both reaction pathways is expressed by eqn (17), and is the sum of eqn (4) and (9).
| | | d[Pt(IV)]/dt = (k0 + kH2O2[Pt(II)])[H2O2] | (17) |
Chlorine as an oxidant
Several attempts to determine a rate constant for the oxidation of [PtCl4]2− by chlorine have failed.9,20,21 Nevertheless, the rapid oxidation of Pt(II) complexes by chlorine is well established. Oxidation of [Pt(CN)4]2− by chlorine for instance exceeds the oxidation rate of [Pt(CN)4]2− by hypochlorous acid by five orders of magnitude.9 Hypochlorous acid and chlorine are in rapid equilibrium in aqueous solution according to eqn (18).| | 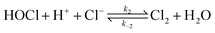 | (18) |
Equilibrium (18) has been responsible for some of the confusion in the literature, where apparent oxidation by chlorine was in fact oxidation by hypochlorous acid.9,20,21 If the formation of chlorine as a result of eqn (18) and subsequent oxidation of [PtCl4]2− exceeds the rate of formation and oxidation by hypochlorous acid as defined by eqn (19), the overall oxidation reaction will proceed via consecutive reactions depicted by k1 → k2 → kCl2 in the scheme given in eqn (3) and chlorine must be considered as an oxidant in the mechanism discussed here.
| | | kHOCl[PtCl42−] ≪ k2[H+][Cl−] | (19) |
Chlorine is the dominant species present in hypochlorous acid/chlorine solutions at equilibrium (> 99.9%, Cl2 = 1 mM). However, since [PtCl4]2− is present in large excess relative to the catalytic concentrations of hypochlorous acid formed by eqn (2), chlorine hydrolysis via k−2 according to eqn (19) will be negligibly slow by comparison. The value of k2 is given as 2.66 × 104 M−2 s−1 in the literature.9 Substituting this value into eqn (19) gives the condition that [Pt(II)] ≪ 60 mM when [H+] and [Cl−] are 1 M. This is the case in our solutions where [Pt(II)] = 1–20 mM, and implies that Cl2 must be considered as an oxidant here if we assume that kCl2 ≥ kH2O2, which is not unreasonable. Oxidation of [PtCl4]2− by chlorine will favour formation of [PtCl6]2− as opposed to the formation of [PtCl5(H2O)]− when hypochlorous acid is the major oxidant. Since the [PtCl5(H2O)]−/[PtCl6]2− ratio increases under conditions that favour the pseudo-zero order mechanism, it supports the idea that hypochlorous acid is the major oxidant and not chlorine.
Conclusions and final comments
The mechanism of oxidation of aqueous [PtCl4]2− by H2O2 in the presence of excess hydrochloric acid is remarkably complex. Our results obtained show that oxidation takes place at least via two parallel reactions with hypochlorous acid and hydrogen peroxide. The overall rate law d[Pt(IV)]/dt = (k0 + kH2O2[Pt(II)])[H2O2] accounts for this process. For oxidation of [PtCl4]2− at relatively low concentrations, [PtCl4]2− ≪ 0.5 mM, hypochlorous acid formation is faster relative to oxidation of [PtCl4]2− by hydrogen peroxide, as a result of the rate determining reaction H2O2 + H+ + Cl− → HOCl + H2O, such that the rate law d[Pt(IV)]/dt = k0[H2O2] gives the value k1 = (8 ± 2) × 10−7 s−1 at 35 °C. For oxidation of [PtCl4]2− ≫ 0.5 mM, oxidation by hydrogen peroxide becomes dominant resulting in a pseudo-first order rate law d[Pt(IV)]/dt = kH2O2[Pt(II)][H2O2], which results in values kH2O2 = (1.5 ± 0.1) × 10−2 M−1 s−1 at 35 °C.
The ΔH‡ and ΔS‡ values support bond formation prior to electron transfer for oxidation by both H2O2 and HOCl (Table 2). These values are comparable to values reported for the oxidation of [PtCl4]2− by H2O2 in 1 M HClO4 (viz. ΔH‡ = 76 ± 3 kJ mol−1 and ΔS‡ = −35 ± 9 J K−1 mol−1) pointing toward a similar mechanism.8 Activation volumes estimated here for the first-order and zero-order mechanisms are almost identical (Table 3). The negative ΔV‡ values are characteristic of oxidative addition reactions, i.e. H2O2–Pt and/or HOCl–Pt bond formation prior to electron transfer, indicating that oxidation takes place via a similar mechanism for both oxidants.22 These observations are in line with an inner-sphere one-step two-electron transfer mechanism typical of Pt(II) square planar complexes. Since square-planar Pt(II) complexes have a vacant coordination site in the axial plane, formation of an inner-sphere complex prior to electron transfer seems reasonable.
Acknowledgements
We thank the Deutscher Akademischer Austauschdienst (DAAD) for financial support to PM to perform this work at the University of Erlangen-Nürnberg. Financial assistance from Stellenbosch University and Anglo American Platinum Limited (bursary support to PM) is gratefully acknowledged.
References
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS.
- L. E. Cox, D. G. Peters and E. L. Wehry, J. Inorg. Nucl. Chem., 1972, 34, 297–305 CrossRef CAS.
- S. O. Dunham, R. D. Larsen and E. H. Abbott, Inorg. Chem., 1993, 32, 2049–2055 CrossRef CAS.
- L. I. Elding and L. Gustafson, Inorg. Chim. Acta, 1976, 19, 31–38 CrossRef CAS.
- P. Murray and K. R. Koch, J. Coord. Chem., 2010, 63, 2561–2577 CrossRef CAS.
- F. Basolo, P. H. Wilks, R. G. Pearson and R. G. Wilkins, J. Inorg. Nucl. Chem., 1958, 6, 161 CrossRef CAS.
- W. R. Mason, Coord. Chem. Rev., 1972, 7, 241–255 CrossRef CAS.
- K. Hindmarsh, D. A. House and R. van Eldik, Inorg. Chim. Acta, 1998, 278, 32–42 CrossRef CAS.
- L. Drougge and L. I. Elding, Inorg. Chem., 1985, 24, 2292–2297 CrossRef CAS.
- R. L. Rich and H. Taube, J. Am. Chem. Soc., 1954, 76, 2608–2611 CrossRef CAS.
- I. V. Znakovskaya, Y. A. Sosedova, E. M. Glebov, V. P. Grivin and V. F. Plyusnin, Photochem. Photobiol. Sci., 2005, 4, 897–902 CAS.
- L. I. Elding and L. F. Olsson, J. Phys. Chem., 1978, 82, 69–74 CrossRef CAS.
- L. I. Elding, Inorg. Chim. Acta, 1978, 28, 255–262 CrossRef CAS.
- M. Spitzer, F. Gartig and R. van Eldik, Rev. Sci. Instrum., 1988, 59, 2092–2093 CrossRef CAS PubMed.
- R. van Eldik, W. Gaede, S. Wieland, J. Kraft, M. Spitzer and D. A. Palmer, Rev. Sci. Instrum., 1993, 64, 1355–1357 CrossRef CAS PubMed.
- L. E. Cox and D. G. Peters, Inorg. Chem., 1970, 9, 1927–1930 CrossRef CAS.
- R. S. Livingston and W. C. Bray, J. Am. Chem. Soc., 1925, 47, 2069–2082 CrossRef CAS.
- A. Mohammad and H. A. Liebhafsky, J. Am. Chem. Soc., 1934, 56, 1680–1685 CrossRef CAS.
- C. Bender and H.-D. Brauer, J. Chem. Soc., Perkin Trans. 2: Phys. Org. Chem., 1999, 2579–2587 RSC.
- L. I. Elding and L. Gustafson, Inorg. Chim. Acta, 1976, 19, 165–171 CrossRef CAS.
- M. M. Jones and K. A. Morgan, J. Inorg. Nucl. Chem., 1972, 34, 259–274 CrossRef CAS.
- R. van Eldik, T. Asano and W. J. le Noble, Chem. Rev., 1989, 89, 549–688 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI:10.1039/c3dt53057d |
| ‡ Calculated from eqn (15) with kH2O2 = 4.25 × 10−3 M−1 s−1 and k1 = 2.07 × 10−6 M−2 s−1.8,18 |
| § Calculated from eqn (16) with kHOCl = (4.5 ± 0.5) × 105 M−1 s−1.9 |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a