 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step synthesis of a highly homogeneous SBA–NHC hybrid material: en route to single-site NHC–metal heterogeneous catalysts with high loadings†
Mansuy
Rocquin
a,
Mickaël
Henrion
a,
Marc-Georg
Willinger
b,
Philippe
Bertani
c,
Michael J.
Chetcuti
a,
Benoît
Louis
*d and
Vincent
Ritleng
*ae
aLaboratoire de Chimie Organométallique Appliquée, UMR CNRS 7509, Ecole européenne de Chimie, Polymères et Matériaux, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: vritleng@unistra.fr; Tel: +33 3 6885 2797
bDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, Berlin 14195, Germany
cLaboratoire de RMN et Biophysique des Membranes, Institut de Chimie, UMR CNRS 7177, Université de Strasbourg, 1 rue Blaise Pascal, 67000 Strasbourg, France
dLaboratoire de Synthèse, Réactivité Organiques et Catalyse, Institut de Chimie, UMR CNRS 7177, Université de Strasbourg, 1 rue Blaise Pascal, 67000 Strasbourg, France. E-mail: blouis@unistra.fr
eInstitut d'Etudes Avancées de l'Université de Strasbourg (USIAS), 5 allée du Général Rouvillois, 67083 Strasbourg, France
First published on 17th December 2013
Abstract
The one-step synthesis of a mesoporous silica of SBA type, functionalized with a 1-(2,6-diisopropylphenyl)-3-propyl-imidazolium (iPr2Ar-NHC-propyl) cation located in the pore channels, is described. This material was obtained by the direct hydrolysis and co-condensation of tetraethylorthosilicate (TEOS) and 1-(2,6-diisopropylphenyl)-3-[3-(triethoxysilyl)propyl]-imidazolium iodide in the presence of Pluronic P123 as a non-ionic structure-directing agent and aqueous HCl (37%) as an acid catalyst. Small-angle X-ray diffraction measurements, scanning and transmission electron microscopies, as well as dinitrogen sorption analyses revealed that the synthesized material is highly mesoporous with a 2D hexagonal arrangement of the porous network. 13C and 29Si CP-MAS NMR spectroscopy confirmed that the material contains intact iPr2Ar-NHC-propyl cations, which are covalently anchored via silicon atoms fused into the silica matrix. Moreover, comparison of the latter data with those of an analogous post-synthetic grafted SBA–NHC material allowed us to establish that, as expected, (i) it is most probably more homogeneous and (ii) it shows a more robust anchoring of the organic units. Finally, elemental mapping by energy dispersive X-ray spectroscopy in the scanning electron microscope demonstrated a very homogeneous distribution of the imidazolium units within the one-pot material, moreover with a high content. This study thus demonstrates that a relatively bulky and hydrophilic imidazolium unit can be directly co-condensed with TEOS in the presence of a structure-directing agent to provide in a single step a highly ordered and homogeneous mesoporous hybrid SBA–NHC material, possessing a significant number of cationic NHC sites.
Introduction
Since the first isolation of a stable imidazol-2-ylidene,1 N-heterocyclic carbenes (NHCs) have become a very important class of ligands in organometallic chemistry.2 NHCs behave like typical strong σ-donor ligands with non-negligible π-acceptor abilities.3,4 These electronic characteristics are similar to those of tertiary phosphines, and they show similar abilities to stabilize the various oxidation states and coordinatively unsaturated intermediates that appear in catalytic reactions.2,5 However, NHCs show superior performance in many aspects over traditional phosphine ligands, including versatility, simple preparation, and thermal, air and moisture stability. In particular, NHCs exhibit superior qualities regarding ligand dissociation and degradative cleavage,3,4 both of which are more unlikely compared to tertiary phosphines.6 In addition, carbene complexes have shown unprecedented catalytic activity under homogeneous conditions in a very broad palette of organic reactions.2,7Despite the high activity and selectivity, homogeneous catalysis has been of somewhat limited use in industry,8 mainly because of the difficulty of separating the soluble catalyst from the reaction product, which in turn may lead to economical/environmental problems, especially in the case of expensive or toxic metal catalysts. Among the possible strategies for separating and recycling active catalysts,9 the immobilization of homogeneous catalysts on a solid support appears particularly attractive because it should combine the advantages of conventional heterogeneous catalysis with the versatility of homogeneous catalysis. Compared to expensive organic polymers,10 inorganic silica materials are common supports for the heterogenization of molecular catalysts due to their excellent thermal and chemical stability. They have rigid structures that are not deformed by solvent swelling during catalytic reactions, and can be used at both high and low temperatures and at high pressures.11 In addition, they possess high surface areas and organic groups can be robustly anchored to the surface silanol groups.
Among the variety of silica materials available, ordered mesoporous silicas such as SBA-1512 present advantageous properties due to their narrow pore size distribution, large specific surface area, pore volume and large number of surface silanol groups. Thanks to their thick framework walls, they exhibit very high thermal and hydrothermal stability. This high strength renders them well suited for use as a catalyst support in catalytic processes where thermal treatments and repeated regeneration are frequently encountered.
The above described versatility, unique catalytic properties and stability of NHC–metal catalysts make them perfect candidates for single-site heterogeneous catalysis. The stronger binding of the NHC ligands to the metal center and their ability to remain bound to it throughout many catalytic cycles2 should allow one to avoid the limited lifetimes and levels of recyclability resulting from metal leaching, which are prevalent for phosphine-based complexes. Thus, this research area is rapidly increasing and NHC ligands have now been grafted onto different supports ranging from soluble polymers to monolithic supports.13,14
However if one considers NHCs bound to the inner pore surface of ordered mesoporous materials, it is striking that the chosen immobilization procedure generally involves condensation of Si(OR)3-substituted NHC species with the surface silanols of preformed hexagonal SBA-1515 or MCM-41,16 or cubic SBA-1617 materials. The main advantages of this post-synthetic grafting process lie in its simplicity and in the preservation of the initial texture of the material (despite a slight decrease in the specific surface area). The main drawback is that it typically yields materials with an ill-controlled anchoring mode, loading and spatial distribution of the organic units.18
An alternative approach would consist in the direct introduction of trialkoxy-substituted imidazolium salts or NHC complexes during the synthesis of the material by co-condensation with tetraethylorthosilicate (TEOS) in the presence of a structure-directing agent (SDA).19 In this case, the NHC species would be regularly distributed on the pore surfaces, and robustly anchored via a silicon atom fused into the silica matrix. Inclusion of bis-silylated imidazolium salts in the framework walls of periodic mesoporous organosilica by similar co-condensations in the presence of a SDA is well established.20 However, examples of such direct syntheses for the introduction of mono-silylated imidazolium salts on the inner pore surface of ordered mesoporous silica are extremely scarce,21 probably because imidazolium cations are believed to be too hydrophilic to enter the core of the micelle and too bulky to avoid its perturbation. As a result, Thieuleux et al. recently developed an elegant two-step approach in which they (i) introduce a hydrophobic organic group (that can be readily further functionalized22) during the co-condensation synthesis of the material, and (ii) react the thus introduced organic functionalities with imidazole units to obtain the desired highly organized hybrid mesostructured imidazolium-containing materials with NHC loadings varying between 0.2 and 0.5 mmol g−1.23
Herein we describe: (i) a one-step procedure for the synthesis of a mesoporous silica of the SBA type functionalized with 1-(2,6-diisopropylphenyl)-3-propyl-imidazolium (iPr2Ar-NHC-propyl), as a model imidazolium cation, and (ii) its full characterization and the comparison of its solid state NMR data with those of an analogous post-synthetic grafted material. The obtained results demonstrate that the one-pot material is, as expected, of better quality than the latter, showing a highly ordered mesoporous 2D hexagonal arrangement of the porous network with a high content (0.77 mmol g−1) of robustly anchored and homogeneously distributed iPr2Ar-NHC-propyl cations.
Experimental section
General information
Organic syntheses were carried out using standard Schlenk techniques under an atmosphere of dry argon. Material syntheses were carried out under air. Commercial compounds were used as received. In experiments requiring dry free oxygen solvents, solvents were distilled from appropriate drying agents under argon prior to use. Solution NMR spectra were recorded at 298 K on a FT-Bruker Ultra Shield 300 spectrometer operating at 300.13 MHz for 1H and at 75.47 MHz for 13C{1H}. The assignment of the 13C{1H} NMR spectrum of 1-(2,6-diisopropylphenyl)-3-[3-(triethoxysilyl)propyl]imidazolium iodide 1 was performed with the aid of a DEPT 13C spectrum. The chemical shifts are referenced to residual deuterated solvent peaks. Chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz, respectively. N2 sorption isotherms were obtained at 77 K with a Micromeritics sorptometer Tri Star 3000 apparatus. Prior to measurements, the samples were outgassed under vacuum (0.01 mbar) for 4 h at 250 °C. The Brunauer–Emmet–Teller (BET) surface area was determined from data in the relative pressure range 0.05–0.30. The pore volume was calculated using the nitrogen volume adsorbed when the capillary condensation was ended. Small-angle X-ray powder diffraction patterns were recorded on a Bruker D8 Advance diffractometer, with a Ni detector side filtered Cu Kα radiation (1.5406 Å) over a 2θ range of 0.7–1.8° and a position sensitive detector using a step size of 0.02° and a step time of 2 s. For TEM investigations, the samples were dispersed in CHCl3, ultrasonificated for a few seconds and drop-deposited on a copper TEM grid with a holey carbon film. Bright field TEM and HAADF STEM images were recorded using an image Cs corrected FEI Titan instrument operated at 300 kV. For solid-state NMR spectra, the samples were packed in a 4 mm ZrO2 rotor under air. The CP-MAS experiment spectra were recorded at 298 K on a Bruker Solid State DSX 300 MHz NMR spectrometer operating at 75.52 MHz for 13C and at 69.66 MHz for 29Si and equipped with a Bruker 4 mm 1H/X CP-MAS probe. A shaped cross-polarization pulse sequence with tangential modulation on both channels was used with the following parameters: the spinning speed was set to 10 kHz, the spectral width to 30 kHz, the contact time was in the range of 2 ms for 13C and 1 ms for 29Si, proton RF field was around 55 kHz for decoupling (using the SPINAL 64 sequence) and 40 kHz for contact, with a recycle delay of 3 s for 13C and 5 s for 29Si. The 29Si MAS spectra (see ESI†) were recorded on the same spectrometer and under the same conditions as the CP-MAS using a single pulse experiment with high power proton decoupling, a 29Si 90° pulse of 5 microseconds and a recycling delay of 30 s. The spectra were calibrated with respect to an external adamantane sample (38.2 ppm) for 13C and to an external PDMS sample (−35.1 ppm) for 29Si. The microstructure of the hybrid SBA–NHC 2 was investigated using a Hitachi S4800 SEM Genesis 4000 EDX detector. The X-rays emitted upon electron irradiation were acquired in the range 0–20 keV. Quantification was done by the standard-less ZAF correction method using the Genesis software from Bruker.Reagents
Pluronic P123 (M ∼ 5800 g mol−1), tetraethylorthosilicate (TEOS) and (3-chloropropyl)triethoxysilane were purchased from Sigma-Aldrich. 1-(2,6-Diisopropylphenyl)-1H-imidazole was synthesized according to a published procedure.24Synthesis of 1-(2,6-diisopropylphenyl)-3-[3-(triethoxysilyl)-propyl]imidazolium iodide (1)
A suspension of 1-(2,6-diisopropylphenyl)1-H-imidazole (1.90 g, 8.32 mmol), 3-chloropropyltriethoxysilane (2.00 mL, 8.31 mmol) and KI (2.07 g, 12.47 mmol) in DME (42 mL) was vigorously stirred at reflux for 5 days in the dark. The mixture was cooled to room temperature and the solvent was removed under vacuum. The orange residue was extracted with acetonitrile (20 mL) and filtered through Celite, which was rinsed with acetonitrile (3 × 5 mL). The resulting solution was then evaporated to dryness. The residue was dissolved in a minimum of CH2Cl2 and triturated with a diethylether–pentane mixture to afford 1 as a pale yellow solid (3.96 g, 7.06 mmol, 85%) after filtration and washings with a diethylether–pentane mixture (3 × 5 mL). 1H NMR (CDCl3, 300.13 MHz): δ = 10.05 (t, J = 1.8, 1H, NCHN), 7.84 (t, J = 1.8, 1H, NCH), 7.55 (t, 3J = 7.8, 1H, p-H), 7.32 (d, 3J = 7.8, 2H, m-H), 7.19 (s, J = 1.8, 1H, NCH), 4.83 (t, 3J = 6.9, 2H, NCH2), 3.84 (q, 3J = 7.0, 6H, OCH2), 2.31 (hept, 3J = 6.9, 2H, CHMe2), 2.14 (m, 2H, NCH2CH2), 1.27–1.20 (d + t, 15H, CHMe2 and OCH2CH3), 1.16 (d, 3J = 6.9, 6H, CHMe2), 0.67 (m, 2H, CH2Si). 13C{1H} NMR (CDCl3, 75.47 MHz): δ = 145.5 (o-CAr), 137.6 (NCHN), 132.1 (p- or ipso-CAr), 130.0 (ipso- or p-CAr), 124.8 (m-CAr), 124.3 and 123.5 (NCH), 58.8 (OCH2), 52.4 (NCH2), 28.9 (CHMe2), 24.8 (NCH2CH2), 24.6 (CHMe2), 24.3 (CHMe2), 18.4 (OCH2CH3), 6.8 (CH2Si). MS (ESI+): m/z [M − H+] calcd for C24H42N2O3Si 433.296, found 433.292.One-step synthesis of the hybrid SBA–NHC (2)
Pluronic P123 (1.36 g, 0.23 mmol) was dissolved in a HCl (37%) (8.9 mL, 108 mmol) solution in water (33 mL, 1.83 mol) and stirred vigorously at 40 °C for 30 min to give a clear solution. TEOS (3.40 mL, 15.2 mmol) was then added dropwise. After 30 min, 1 (303 mg, 0.541 mmol) was poured into the reaction mixture (composition: 1 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.036 (1):0.015 P123:7.1 HCl:121 H2O), which was stirred at 40 °C for 20 h. The obtained slurry was then maintained static and heated in a Teflon autoclave at 130 °C for 24 h. The resulting solid was filtered and washed with water (2 × 50 mL), ethanol (2 × 50 mL) and diethylether (2 × 50 mL). The surfactant was removed by extraction with ethanol using a Soxhlet for 24 h. Drying in air at 80 °C for 24 h afforded 2 as a very pale yellow powder (1.06 g). 13C CP-MAS NMR: δ = 146, 137, 131, 124, 70,a 59,a 58,a 52, 28, 23, 16,a 9. 29Si CP-MAS NMR: δ = −60 (T2), −68 (T3), −93 (Q2), −102 (Q3), −111 (Q4).
:0.036 (1):0.015 P123:7.1 HCl:121 H2O), which was stirred at 40 °C for 20 h. The obtained slurry was then maintained static and heated in a Teflon autoclave at 130 °C for 24 h. The resulting solid was filtered and washed with water (2 × 50 mL), ethanol (2 × 50 mL) and diethylether (2 × 50 mL). The surfactant was removed by extraction with ethanol using a Soxhlet for 24 h. Drying in air at 80 °C for 24 h afforded 2 as a very pale yellow powder (1.06 g). 13C CP-MAS NMR: δ = 146, 137, 131, 124, 70,a 59,a 58,a 52, 28, 23, 16,a 9. 29Si CP-MAS NMR: δ = −60 (T2), −68 (T3), −93 (Q2), −102 (Q3), −111 (Q4).
(a) Peaks at δ = 70, 59, 58 and 16 ppm correspond to the residual template agent present in micropores and to Si-OEt signals that arise from surfactant removal by extraction with EtOH; see 13C CP-MAS NMR data of pristine SBA-15 3 below, and the spectrum (Fig. S3a, ESI†).
Synthesis of pristine SBA-15 (3)
Pluronic P123 (1.36 g, 0.23 mmol) was dissolved in a HCl (37%) (8.9 mL, 108 mmol) solution in water (33 mL, 1.83 mol) and stirred vigorously at 40 °C for 30 min to give a clear solution. TEOS (3.40 mL, 15.2 mmol) was then added dropwise and the reaction mixture (composition: 1 TEOS:0.015 P123:7.1 HCl:121 H2O) was stirred at 40 °C for 20 h. The obtained slurry was maintained static and heated in a Teflon autoclave at 130 °C for 24 h. The resulting solid was filtered and washed with water (2 × 50 mL), ethanol (2 × 50 mL) and diethylether (2 × 50 mL). The surfactant was removed by extraction with ethanol using a Soxhlet for 24 h. Drying in air at 80 °C for 24 h afforded 3 as a white powder (0.945 g). 13C CP-MAS NMR: δ = 75, 70, 59, 58, 16. 29Si CP-MAS NMR: δ = −93 (Q2), −102 (Q3), −112 (Q4).
Synthesis of grafted SBA–NHC (4)
3 (100 mg) was suspended in toluene (15 mL) and stirred for 30 min at 50 °C to achieve a homogeneous dispersion. para-Toluenesulfonic acid (5 mg, 0.029 mmol) and 1 (33.6 mg, 0.060 mmol) were then added, and the mixture was stirred at reflux for 2 h. The resulting solid was filtered and washed with ethanol (3 × 15 mL). Drying in air at 100 °C for 16 h afforded 4 as a very pale yellow powder (0.083 g). 13C CP-MAS NMR: δ = 146, 138, 131, 125, 70,a 59,a 53, 28, 23, 17,a 10. 29Si CP-MAS NMR: δ = −49 (T1), −59 (T2), −67 (T3), −91 (Q2), −101 (Q3), −110 (Q4).(a) Peaks at δ = 70, 59 and 17 ppm correspond to the residual template agent present in micropores and/or to Si-OEt signals that arise from surfactant removal by extraction with EtOH; see 13C CP-MAS NMR data of pristine SBA-15 3 above and the spectrum (Fig. S3a, ESI†).
Results and discussion
Choice and preparation of the imidazolium salt (1)
Copéret, Thieuleux et al. recently demonstrated, in an elegant study, that a flexible tether may provide significant advantages during a catalytic reaction compared to a rigid linker, by allowing an eventual low-coordinated catalytic intermediate to have stabilizing metal–surface interactions.25 With that in mind, we chose the 1-(2,6-diisopropylphenyl)-3-[3-(triethoxysilyl)propyl]-imidazolium iodide 1 as a model compound to demonstrate that imidazolium salts can be introduced directly during the co-condensation synthesis of functionalized ordered mesoporous silica. The latter was synthesized according to our published method for the preparation of N-alkyl-N′-arylimidazolium iodides from inexpensive chloro-alkyls (Scheme 1).26 Thus, the reaction of 1-(2,6-diisopropylphenyl)-1H-imidazole24 with (3-chloropropyl)triethoxysilane in the presence of excess potassium iodide in DME at reflux for 5 days afforded 1 as a pale yellow solid in 85% yield after work-up. 1 was characterized by 1H and 13C{1H} NMR spectroscopy and mass spectroscopy. | ||
| Scheme 1 Synthesis of the imidazolium salt 1. | ||
One-step synthesis of the hybrid SBA–NHC (2) and two-step synthesis of the grafted SBA–NHC (4)
The one-step synthesis of the hybrid SBA–NHC material 2 was derived from that reported for the preparation of a periodic mesoporous organosilica containing a covalently bonded organic ion pair, and featuring a SBA-15 structure27 using a more concentrated and more acidic medium than is usually the case for the direct synthesis of SBA materials functionalized with hydrophobic molecules on the inner pore walls.22,23 Thus, the ordered mesoporous hybrid material was obtained by co-condensation at 40 °C under acidic conditions of 1 with TEOS in the presence of Pluronic P123 as the SDA, with a molar composition of the reaction mixture corresponding to: 1 TEOS:0.036 (1):0.015 P123:7.1 HCl:121 H2O (Scheme 2). After proper ageing of the SBA–NHC gel precursor, the surfactant was removed by hot ethanol extraction in a Soxhlet apparatus affording the expected hybrid SBA–NHC material 2 as a very pale yellow powder.
 | ||
| Scheme 2 One-step synthesis of the hybrid SBA–NHC 2. | ||
For comparison purposes, a grafted SBA–NHC analogue of 2 was also prepared. Its synthesis was achieved by condensation at reflux in toluene in the presence of para-toluenesulfonic acid (PTSA) of the imidazolium salt 1 with the surface silanols of a preformed SBA-15 material 3 (Scheme 3). Such an acid-catalyzed condensation on preformed SBA-15 being well documented,28 no particular comment will be made, except that in order for both materials to be really comparable: (i) the pristine SBA-15 3 was synthesized using a molar composition strictly similar to that used for the synthesis of the hybrid SBA–NHC 2, i.e. 1 TEOS:0.015 P123:7.1 HCl:121 H2O, and (ii) the surfactant was removed by ethanolic extraction similarly to what was done for the synthesis of 2.
 | ||
| Scheme 3 Two-step synthesis of the grafted SBA–NHC 4. | ||
Characterization of the hybrid SBA–NHC (2) and comparison with the grafted SBA–NHC (4)
The structure of the as-synthesized hybrid SBA–NHC silica 2 was first investigated by small-angle powder X-ray diffraction. In contrast to small-angle diffraction patterns of pristine SBA-15, which generally display three well-resolved reflections that can be indexed as the (1 0 0), (1 1 0) and (2 0 0) diffractions of the ordered p6mm hexagonal space group,12 the latter only exhibits the intense diffraction peak corresponding to the d100 spacing (Fig. S2a, ESI†). No long-range ordering could be resolved. This result is in line with earlier studies reporting the synthesis of hexagonal-type mesoporous materials containing either organic functionalities22,23a,b or metal oxides within their channels.29 Nevertheless, similar single reflection materials have been shown to still exhibit a high degree of mesoscopic organization and local hexagonal symmetry.30 This is confirmed by the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the hybrid SBA–NHC 2 shown in Fig. 1 and 2, respectively, as well as by the high angle annular dark-field images (HAADF) recorded in scanning transmission electron microscopy (STEM) mode shown in Fig. 3. The nanometer-scaled SEM, TEM and HAADF STEM images (Fig. 1c, d, 2 and 3) indeed clearly evidence the hexagonal arrangement of the porous network within the material and confirm a hexagonal structure over a large area, along with an extremely uniform pore size distribution with an average diameter of 6.7 nm and a wall thickness of 3.3 nm (Fig. 3a). The unit cell dimension in the a and b directions (considering a hexagonal cell) measures 10–10.2 nm. The values were abstracted from the Fourier transform shown as an inset in Fig. 2a. | ||
| Fig. 1 SEM images of the hybrid SBA–NHC 2. | ||
 | ||
| Fig. 2 TEM images of the hybrid SBA–NHC 2. | ||
 | ||
| Fig. 3 HAADF STEM images of the hybrid SBA–NHC 2. | ||
Nevertheless, it is noteworthy that the low-magnification SEM images of SBA–NHC 2 reveal two kinds of morphologies for these particles (Fig. 1a and b). Some of the particles aggregate into micrometer-sized spheres of about 2–3 μm (Fig. 1a). In addition, a classical worm-like morphology with a particle width of less than 1 μm is also obtained (Fig. 1b). The presence of the NHC salt 1, and hence of additional organic material within the synthesis mixture, thus induces a double-type self-assembly mode. This observation is in line with previous studies,31 and can be partly explained by the modulation of surfactant/pore wall interactions by co-adsorption of the NHC cation, thus playing the role of a surface modifying agent.31b However, one cannot completely rule out the existence of ligand–ligand interactions at such a high content of NHC ligand (vide infra) anchored to the surface. The latter interactions could induce vicinal ligand molecules to fall next to the walls and thus reduce their accessibility for further metalation. Besides, micrometer-sized mesoporous silica SBA-15 spheres with a high dispersivity were also observed by Zhao et al. when starch was added in the medium.31a Anyway, as indicated above, these two morphologies, obtained by distinct aggregation models, exhibit the rod-like morphology of their P123-templated mesopores with unchanged hexagonal symmetry (Fig. 1c–d and 2). The synthesis conditions are therefore suitable with an appropriate self-assembling rate of the surfactant and the silica species that provides enough time for the equilibrium-assembly of hundreds of nanometer-sized rod-like silica.
The N2 adsorption–desorption isotherm of the hybrid SBA–NHC 2 is shown in Fig. 4. It is of type IV with H1 hysteresis, following the IUPAC classification.32 This profile is characteristic of mesoporous materials possessing one-dimensional cylindrical channels. Moreover, the presence of a sharp adsorption step in the P/P0 region from 0.6 to 0.8 and of a hysteresis loop supports the existence of a well-defined array of regular mesopores. Regarding the textural properties of the hybrid material 2, it is worth noting that a specific surface area and a pore volume of, respectively, SBET = 555 m2 g−1 and Vp = 0.83 cm3 g−1 were measured. These values are in line with those reported for a similar SBA–NHC material prepared in two steps by Thieuleux et al. (vide supra),25 and nearly 30% and 15% lower compared to the classical values of a non-functionalized SBA-15 (SBET = 785 m2 g−1 and Vp = 0.98 cm3 g−1),12 thus strongly suggesting that the iPr2Ar-NHC-propyl cation is present within the pores, as expected. Therefore, though its presence affects the textural properties of the mesoporous silica, the ordering of regular hexagonal mesopores is kept within the organic–inorganic hybrid 2.
 | ||
| Fig. 4 N2 adsorption–desorption isotherm of the hybrid SBA–NHC 2. | ||
The high degree of mesoscopic organization and local hexagonal symmetry of the hybrid SBA–NHC material 2 being thus established, the presence of intact iPr2Ar-NHC-propyl functionalities was confirmed by 13C CP-MAS NMR. The signals at 146, 137, 131, 124, 52, 28, 23, and 9 ppm indeed closely match with those of the 1-(2,6-diisopropylphenyl)-3-propyl-imidazolium part of 1 in its 13C solution NMR spectrum (Fig. S1b and S2b, ESI†). Moreover, these peaks are relatively sharp when compared to the corresponding signals of the 13C CP-MAS NMR spectrum of the grafted SBA–NHC 4, the peaks of 2 showing a ca. 35% smaller full-width at half-maximum compared to those of 4: for instance 270 Hz (2) vs. 400 Hz (4) for the peak at 145 ppm or 170 Hz (2) vs. 270 Hz (4) for the peak at 28 ppm. This suggests that the one-pot hybrid material 2 is more homogeneous than the latter,33,34 and thus has a more regular distribution of the organic units, as expected (Fig. 5).18
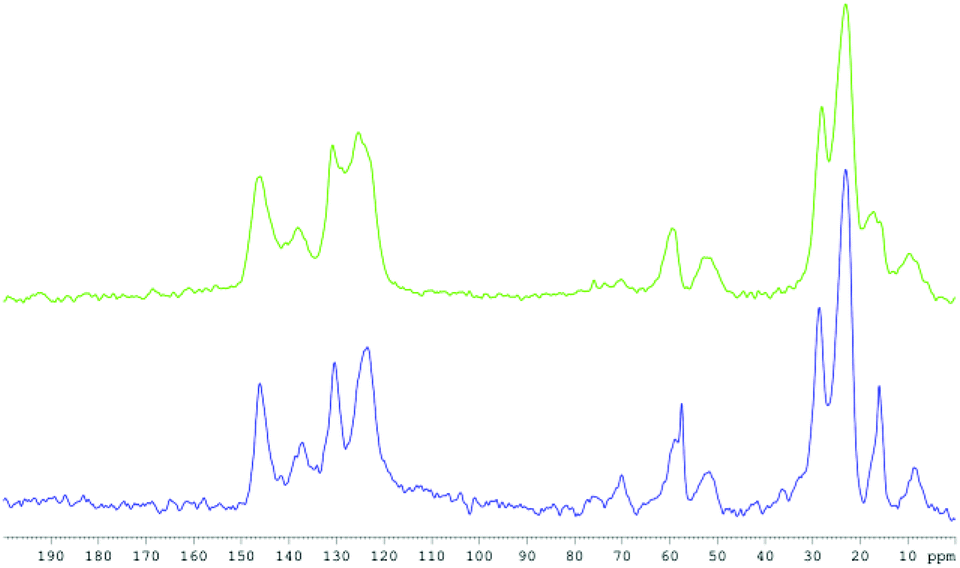 | ||
| Fig. 5 13C CP-MAS NMR spectra of the hybrid SBA–NHC 2 (blue) and of the grafted SBA–NHC 4 (green). | ||
Moreover, the 29Si MAS NMR spectrum of 2 demonstrated a high degree of condensation of the hybrid material with signals at −93, −102, −111 ppm associated with Q2, Q3 and Q4 (major)35 substructures, respectively (Fig. S2d, ESI†). In addition, the 29Si CP-MAS NMR spectrum of 2 reveals signals at −67 and −59 ppm, associated with T3 and T2 substructures, which shows that the iPr2Ar-NHC-propyl units are bonded to the matrix via two or three Si–O bonds (Fig. 6 and S2c, ESI†). This contrasts with the 29Si CP-MAS NMR spectrum of the grafted SBA–NHC 4, which in addition to T3 and T2 signals displays a signal at −49 ppm, associated with a T1 substructure, and thus suggests a lower degree of condensation of the grafted iPr2Ar-NHC-propyl units within the preformed silica matrix,36 as expected (Fig. 6 and S4b, ESI†).18
 | ||
| Fig. 6 29Si CP-MAS NMR spectra of the hybrid SBA–NHC 2 (blue) and of the grafted SBA–NHC 4 (green). | ||
The high homogeneity of the one-pot hybrid material 2 suggested by the relatively sharp signal observed in its 13C CP-MAS NMR spectrum was confirmed by elemental mapping in the scanning electron microscope (SEM) using energy dispersive X-ray spectroscopy (EDX). EDX mapping of Si, O, C and N elements within one SBA–NHC 2 particle indeed supports a very homogeneous distribution of all these elements throughout the mesoporous silica material (Fig. 7). As the nitrogen atoms can only arise from the iPr2Ar-NHC-propyl units, this confirms that the latter are well distributed all over the material. Furthermore, based on EDX analysis, the N-content in 2 was quantified to 2.17 wt%, which led us to sample estimate the degree of NHC incorporation within its inorganic host to 0.77 mmol g−1 (by taking into consideration that 2 N-atoms are incorporated into one NHC ligand). Such values are notably higher than those reported by Thieuleux et al. for similar SBA–NHC hybrids prepared via their two-step approach (vide supra),23 and further strengthen the importance of our strategy to develop one-pot synthesized SBA–NHC inorganic–organic hybrids.
 | ||
| Fig. 7 SEM-EDX elemental mapping of the hybrid SBA–NHC 2. | ||
Finally, based on previous surface characterizations of pristine SBA-15 materials, where the total number of O–H groups present in the mesoporous silica was shown by the H/D isotope exchange technique to be 2.70 mmol g−1,37 one can thus roughly estimate the degree of NHC grafting onto its inorganic host as approximately 30%. All this makes this single-step-synthesized hybrid SBA–NHC material 2 an excellent candidate for further functionalization with any catalyst precursor to give highly desirable single-site heterogeneous NHC–metal catalysts with high catalyst loadings.
Conclusions
In summary, we demonstrate in this study that, a contrario to common belief, a relatively bulky and hydrophilic triethoxysilyl-functionalized imidazolium salt can be directly co-condensed with TEOS in the presence of a structure-directing agent to provide in one step a highly mesoporous hybrid silica material with a 2D hexagonal arrangement of the porous network, functionalized into the pore channels with regularly distributed and robustly anchored NHC pro-ligands. In addition to the fact that it is direct, this approach is particularly interesting in that it provides a remarkably homogeneous material with a high content of NHCs. Provided that they are accessible, their metalation should indeed give access to heterogeneous NHC–metal catalytic materials that should potentially show remarkable efficiencies due to the high number of catalytic sites present within the material.Acknowledgements
We are grateful to the Université de Strasbourg and the CNRS for their financial help. The Agence Nationale de la Recherche is also acknowledged for its support to V.R. and the doctoral fellowship of M.H. (ANR 2010 JCJC 716 1; SBA-15-NHC-NiCat).Notes and references
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (b) L. H. Gade and S. Bellemin-Laponnaz, Coord. Chem. Rev., 2007, 251, 718 CrossRef CAS PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (d) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed; (e) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599 CrossRef CAS PubMed; (f) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (g) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CrossRef CAS PubMed.
- (a) G. Frenkin, M. Solà and S. F. Vyboishchikov, J. Organomet. Chem., 2005, 690, 6178 CrossRef PubMed; (b) U. Radius and F. M. Bickelhaupt, Coord. Chem. Rev., 2009, 253, 678 CrossRef CAS PubMed; (c) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687 CrossRef CAS PubMed.
- (a) T. Strassner, Top. Organomet. Chem., 2004, 13, 1 CrossRef CAS; (b) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS PubMed; (c) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef PubMed.
- N. M. Scott and S. P. Nolan, Eur. J. Inorg. Chem., 2005, 1815 CrossRef CAS.
- Phosphines are often subject to P–C bond cleavage (a) or orthometalation (b); (a) P. E. Garrou, Chem. Rev., 1985, 85, 171 CrossRef CAS; (b) J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science, Mill Valley, CA, 2nd edn, 1987 Search PubMed.
- W. A. Herrmann, M. Elison, J. Fischer, C. Kocher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS.
- (a) J. Hagen, Industrial Catalysis: A practical Approach, Wiley-VCH, Weinheim, 1999 Search PubMed; (b) J. G. de Vries, Can. J. Chem., 2001, 79, 1086 CrossRef CAS; (c) C. E. Tucker and J. G. de Vries, Top. Catal., 2002, 19, 111 CrossRef CAS.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS PubMed.
- (a) N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS PubMed; (b) C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- (a) W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860 CrossRef CAS PubMed; (b) C. S. J. Cazin, C. R. Chim., 2009, 12, 1173 CrossRef CAS PubMed; (c) A. Monge-Marcet, R. Pleixats, X. Cattoën and M. Wong Chi Man, Catal. Sci. Technol., 2011, 1, 1544 RSC; (d) K. V. S. Ranganath, S. Onitsuka, A. K. Kumar and J. Inanaga, Catal. Sci. Technol., 2013, 3, 2161 RSC.
- A. M. Oertel, V. Ritleng and M. J. Chetcuti, Organometallics, 2012, 31, 2829 CrossRef CAS.
- (a) P. Li, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2013, 5, 3324 CrossRef CAS; (b) L. Li and J.-l. Shi, Adv. Synth. Catal., 2005, 347, 1745 CrossRef CAS.
- (a) S. Dastgir, K. S. Coleman and M. L. H. Green, Dalton Trans., 2011, 40, 661 RSC; (b) C. del Pozo, M. Iglesias and F. Sánchez, Organometallics, 2011, 30, 2180 CrossRef CAS.
- H. Yang, X. Han, G. Li and Y. Wang, Green Chem., 2009, 11, 1184 RSC.
- (a) D. J. Macquarrie, D. B. Jackson, J. E. J. Mdoe and J. H. Clark, New J. Chem., 1999, 23, 539 RSC; (b) A. Walcarius and C. Delacôte, Chem. Mater., 2003, 15, 4181 CrossRef CAS.
- (a) F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed; (b) A. Mehdi, C. Reyé and R. Corriu, Chem. Soc. Rev., 2011, 40, 563 RSC.
- (a) T. P. Nguyen, P. Hesemann, P. Gaveau and J. J. E. Moreau, J. Mater. Chem., 2009, 19, 4164 RSC; (b) H. Yang, G. Li, Z. Ma, J. Chao and Z. Guo, J. Catal., 2010, 276, 123 CrossRef CAS PubMed; (c) H. Yang, X. Han, G. Li, Z. Ma and Y. Hao, J. Phys. Chem. C, 2010, 114, 22221 CrossRef CAS; (d) T. P. Nguyen, P. Hesemann and J. J. E. Moreau, Microporous Mesoporous Mater., 2011, 142, 292 CrossRef CAS PubMed; (e) L. Wang, S. Shylesh, D. Dehe, T. Philippi, G. Dörr, A. Seifert, Z. Zhou, M. Hartmann, R. N. Klupp Taylor, M. Jia, S. Ernst and W. R. Thiel, ChemCatChem, 2012, 4, 395 CrossRef CAS.
- (a) G. Liu, M. Hou, T. Wu, T. Jiang, H. Fan, G. Yang and B. Han, Phys. Chem. Chem. Phys., 2011, 13, 2062 RSC; (b) A. Monge-Marcet, R. Pleixats, X. Cattoën and M. Wong Chi Man, claimed that they achieved such a synthesis without giving experimental details, see ref. 13c.
- For a seminal example, see: J. Alauzun, A. Mehdi, C. Reyé and R. Corriu, New J. Chem., 2007, 31, 911 RSC.
- (a) T. K. Maishal, J. Alauzun, J.-M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reyé, L. Veyre and C. Thieuleux, Angew. Chem., Int. Ed., 2008, 47, 8654 CrossRef CAS PubMed; (b) I. Karamé, M. Boualleg, J.-M. Camus, T. K. Maishal, J. Alauzun, J.-M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reyé, L. Veyre and C. Thieuleux, Chem.–Eur. J., 2009, 15, 11820 CrossRef PubMed; (c) T. K. Maishal, M. Boualleg, M. Bouhrara, C. Copéret, E. Jeanneau, L. Veyre and C. Thieuleux, Eur. J. Inorg. Chem., 2010, 5005 CrossRef CAS; (d) M. Baffert, T. K. Maishal, L. Mathey, C. Copéret and C. Thieuleux, ChemSusChem, 2011, 4, 1762 CrossRef CAS PubMed.
- J. Liu, J. Chen, J. Zhao, Y. Zhao, L. Li and H. Zhang, Synthesis, 2003, 2661 CAS.
- M. K. Samantaray, J. Alauzun, D. Gajan, S. Kavitake, A. Mehdi, L. Veyre, M. Lelli, A. Lesage, L. Emsley, C. Copéret and C. Thieuleux, J. Am. Chem. Soc., 2013, 135, 3193 CrossRef CAS PubMed.
- A. M. Oertel, V. Ritleng and M. J. Chetcuti, Synthesis, 2009, 1647 CAS.
- A. El Kadib, P. Hesemann, K. Molvinger, J. Brandner, C. Biolley, P. Gaveau, J. J. E. Moreau and D. Brunel, J. Am. Chem. Soc., 2009, 131, 2282 CrossRef PubMed.
- N. García, E. Benito, J. Guzmán, P. Tiemblo, V. Morales and R. A. García, Microporous Mesoporous Mater., 2007, 106, 129 CrossRef PubMed.
- (a) Ch. Subrahmanyam, B. Louis, F. Rainone, B. Viswanathan, A. Renken and T. K. Varadarajan, Appl. Catal., A, 2003, 241, 205 CrossRef CAS; (b) Y. Sun, S. Walspurger, J.-P. Tessonnier, B. Louis and J. Sommer, Appl. Catal., A, 2006, 300, 1 CrossRef CAS PubMed; (c) G. Laugel, J. Arichi, H. Guerba, M. Moliere, A. Kiennemann, F. Garin and B. Louis, Catal. Lett., 2008, 125, 14 CrossRef CAS PubMed; (d) G. Laugel, J. Arichi, M. Moliere, A. Kiennemann, F. Garin and B. Louis, Catal. Today, 2008, 138, 38 CrossRef CAS PubMed.
- (a) P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS PubMed; (b) P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865 CrossRef CAS PubMed.
- (a) D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275 CrossRef CAS; (b) B. Bharti, M. Xue, J. Meissner, V. Cristiglio and G. H. Findenegg, J. Am. Chem. Soc., 2012, 134, 14756 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. Pauli, B. van Rossum, H. Förster, H. J. M. de Groot and H. Oschkinat, J. Magn. Reson., 2000, 143, 411 CrossRef CAS PubMed.
- The different line broadening between the spectra of 2 and 4 could, in theory, also result from different mobilities (T2). However such a difference in T2 would imply faster movements for the sample with the sharpest peaks, i.e. for the hybrid SBA–NHC 2. This would go against the fact that the imidazolium units are expected to be more embedded into the silica matrix than in the grafted material.
- With the CP-MAS technique, nuclei that are far away from protons are difficult to detect. That is why Q4 peaks appear so small in the 29Si CP-MAS spectra displayed in Fig. 6, whereas it is the major peak in the 29Si MAS spectra (see Fig. S2d and S4c†).
- As the CP-MAS dynamics of 2 and 4 are not the same, no conclusion regarding the respective amounts of the different T groups can be drawn. Nevertheless the fact that, in contrast to the spectrum of 4, that of 2 does not show T1 signals allows us to think with reasonable confidence that the iPr2Ar-NHC-propyl unit is more robustly anchored in 2 than in 4.
- (a) Z. El Berrichi, B. Louis, J. P. Tessonnier, O. Ersen, L. Cherif, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2007, 316, 219 CrossRef CAS PubMed; (b) S. Walspurger and B. Louis, Appl. Catal., A, 2008, 336, 109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 1–4, and powder XRD pattern of 2. See DOI: 10.1039/c3dt52982g |
| This journal is © The Royal Society of Chemistry 2014 |