 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and reactivity of Ni site models of [NiFeSe] hydrogenases†
Claire
Wombwell
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 9th December 2013
Abstract
A series of structural models of the Ni centre in [NiFeSe] hydrogenases has been developed which exhibits key structural features of the Ni site in the H2 cycling enzyme. Specifically, two complexes with a hydrogenase-analogous four-coordinate ‘NiS3Se’ primary coordination sphere and complexes with a ‘NiS2Se2’ and a ‘NiS4’ core are reported. The reactivity of the complexes towards oxygen and protons shows some relevance to the chemistry of [NiFeSe] hydrogenases. Exposure of a ‘NiS3Se’ complex to atmospheric oxygen results in the oxidation of the selenolate group in the complex to a diselenide, which is released from the nickel site. Oxidation of the selenolate ligand on Ni occurs approximately four times faster than oxidation with the analogous sulfur complex. Reaction of the complexes with one equivalent of HBF4 results in protonation of the monodentate chalcogenolate and the release of this ligand from the metal centre as a thiol or selenol. Unrelated to their biomimetic nature, the complexes serve also as molecular precursors to modify electrodes with Ni–S–Se containing particles by electrochemical deposition. The activated electrodes evolve H2 in pH neutral water with an electrocatalytic onset potential of −0.6 V and a current density of 15 μA cm−2 at −0.75 V vs. NHE.
Introduction
The sustainable generation of the energy vector H2 would provide a possible solution to a fossil-free economy,1 and the improvement of H2 evolution catalysts is therefore attracting much current attention.2 In nature, the ubiquitous metals nickel and iron are employed in the active sites of [NiFe] and [FeFe] hydrogenases to reversibly catalyse the interconversion of protons and electrons to H2 and operate at high rates at modest over-potentials.3 [NiFeSe] hydrogenases are a subclass of the [NiFe] hydrogenases with a selenocysteine (Sec) in place of a cysteine (Cys) residue terminally bound to the Ni centre (Fig. 1).4 These selenium containing enzymes have been shown to display a number of advantageous properties in comparison with the conventional [NiFe] hydrogenases which make them good candidates as H2 evolution biocatalysts: they show higher H2 evolution activities than [NiFe] hydrogenases5 and have fast reactivation times from O2 damage at a low redox potential.5b,6 They were also previously employed in photocatalytic H2 generation schemes5d,7 and even display catalytic H2 production in the presence of O2.5b,7 Therefore [NiFeSe] hydrogenases serve as an attractive blueprint and inspiration for the development of synthetic H2 evolution catalysts.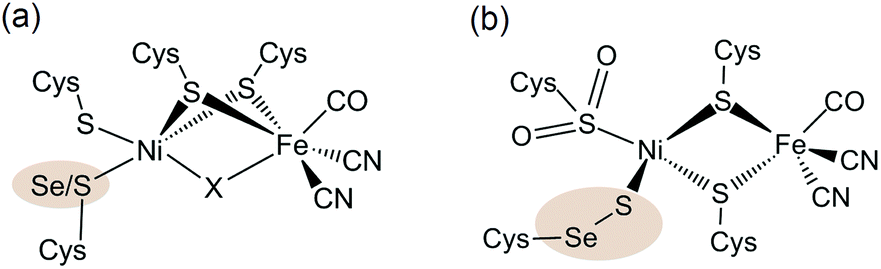 | ||
| Fig. 1 (a) Molecular structure of the [NiFe(Se)] hydrogenase active site. The highlighted atom (Se/S) is S in [NiFe] hydrogenases and Se in [NiFeSe] hydrogenases. A hydride or oxo-group can serve as the bridging moiety X (the latter was only observed in oxidised [NiFe] hydrogenases) and an active reduced state does not contain a bridging ligand X. (b) Composition of a structural conformer in the oxidised [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough.9f,g,10 | ||
The active site structure of the [NiFe(Se)] hydrogenases has been well studied using spectroscopic5a,6a,8 and crystallographic4,9 techniques (Fig. 1). It consists of a dinuclear [NiFe] centre bridged by two cysteine ligands. There is also a Cys and a Sec or Cys residue terminally bound to Ni.
The Ni ion serves as the active redox centre: its oxidation state varies between Ni(I) and Ni(III),8a,d–h whereas Fe remains low spin Fe(II)8h in all active site states. A catalytically active form of the enzyme has been shown to exist as Ni(III) with a bridging hydride between the Ni and Fe centres (position X in Fig. 1a) and a reduced Ni(II) form does not contain a bridging ligand X.8f,g,i The Cys or Sec residue highlighted in Fig. 1a is proposed to act as a proton relay during H2 cycling.4,9a–c The different properties of Sec compared with Cys, such as its higher nucleophilicity and lower pKa,11 may cause the increased H2 production activity of the [NiFeSe] subclass.5 We also note that there are other differences apart from the active site Sec, which could affect the chemistry of the enzyme. For example a medial [3Fe4S] cluster is present in D. baculatum [NiFeSe] hydrogenase rather than a [4Fe4S] cluster in conventional [NiFe] hydrogenases in the electron transfer chain between the active site and the protein surface.4
Despite a large number of structural mimics of the [NiFe] hydrogenase active site,12 no systematic series of Ni subsite models with a stoichiometrically accurate ‘NiS3Se’ core has been previously reported. Noteworthy approaches to mimic selenium in hydrogenases are complexes with a ‘NiS3SeP’ centre such as penta-coordinate [Ni(SePh)(P(o-C6H4S)2(o-C6H4SH))]−,12j,k a number of Ni complexes with nitrogen and selenium-donor ligands,12m,n and a series of diiron complexes containing mixed sulfur/selenium bridging ligands such as [Fe2(μ-SC3H6Se-μ)(CO)6].13
Herein we report on the synthesis, characterisation, reactivity towards O2 and protons, and electrochemical study of four-coordinate Ni complexes with thiolate and selenolate ligands as structural models of the Ni site of reduced [NiFeSe] hydrogenases. The mononuclear model compounds display protonation and O2 reactivity relevant to the hydrogenases. Electrochemical studies reveal that the Ni complexes do not operate as molecular H2 production catalysts. However, they are useful precursors to catalytic Ni-containing particles for H2 production in pH neutral H2O.
Results and discussion
Synthetic pathway to hydrogenase inspired Ni complexes
The synthetic route to structural models of the Ni site in [NiFeSe] hydrogenase is outlined in Scheme 1. The novel tridentate mixed donor ligand ‘LSe’–H2 was synthesised from thianthrene. A catalytic amount of 4,4′-di-tert-butylbiphenyl was used with two equivalents of lithium powder in tetrahydrofuran at −90 °C to give a radical anion which is strong enough to cleave one of the thianthrene carbon–sulfur bonds.14 Introduction of elemental selenium into the lithiated product and acidic workup gave the diselenide (‘LSe–H’)2, which can be subsequently reduced with LiAlH4 to give the selenol ‘LSe’–H2 after acidic workup. Refluxing a solution of ‘LSe’–H2 in tetrahydrofuran with one equivalent of Ni(OCOCH3)2·4H2O in methanol yields the complex [Ni(LSe)]n as an insoluble black precipitate in 87% yield.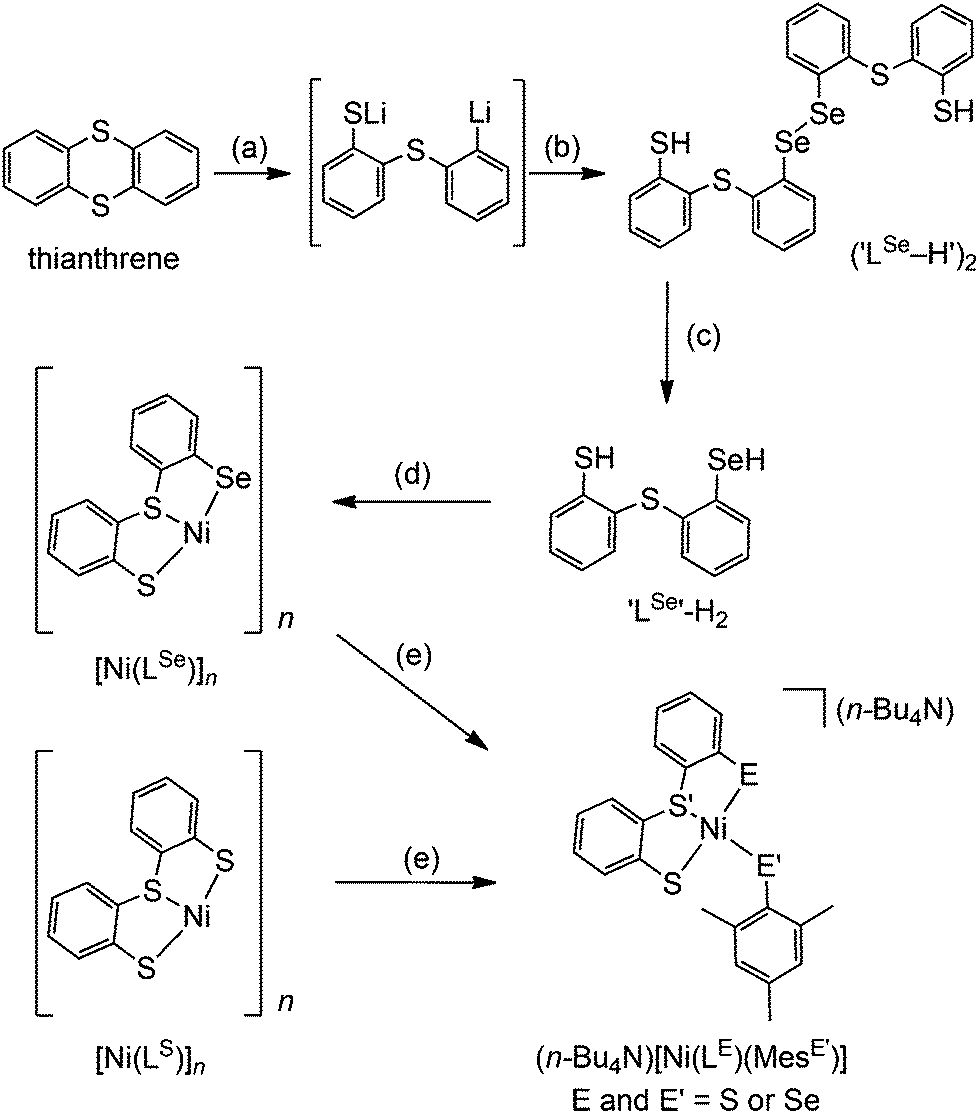 | ||
| Scheme 1 Synthetic pathway to models of the Ni-site in [NiFe(Se)] hydrogenases: (a) Li, 4,4′-di-tert-butylbiphenyl, tetrahydrofuran, −90 °C. (b) (i) Se, −90 °C to −50 °C; (ii) H3O+. (c) (i) NaBH4; (ii) H3O+. (d) Ni(OCOCH3)2·4H2O, tetrahydrofuran–methanol, reflux. (e) NaMesE′ (E′ = S or Se), n-Bu4NOH, tetrahydrofuran–methanol. | ||
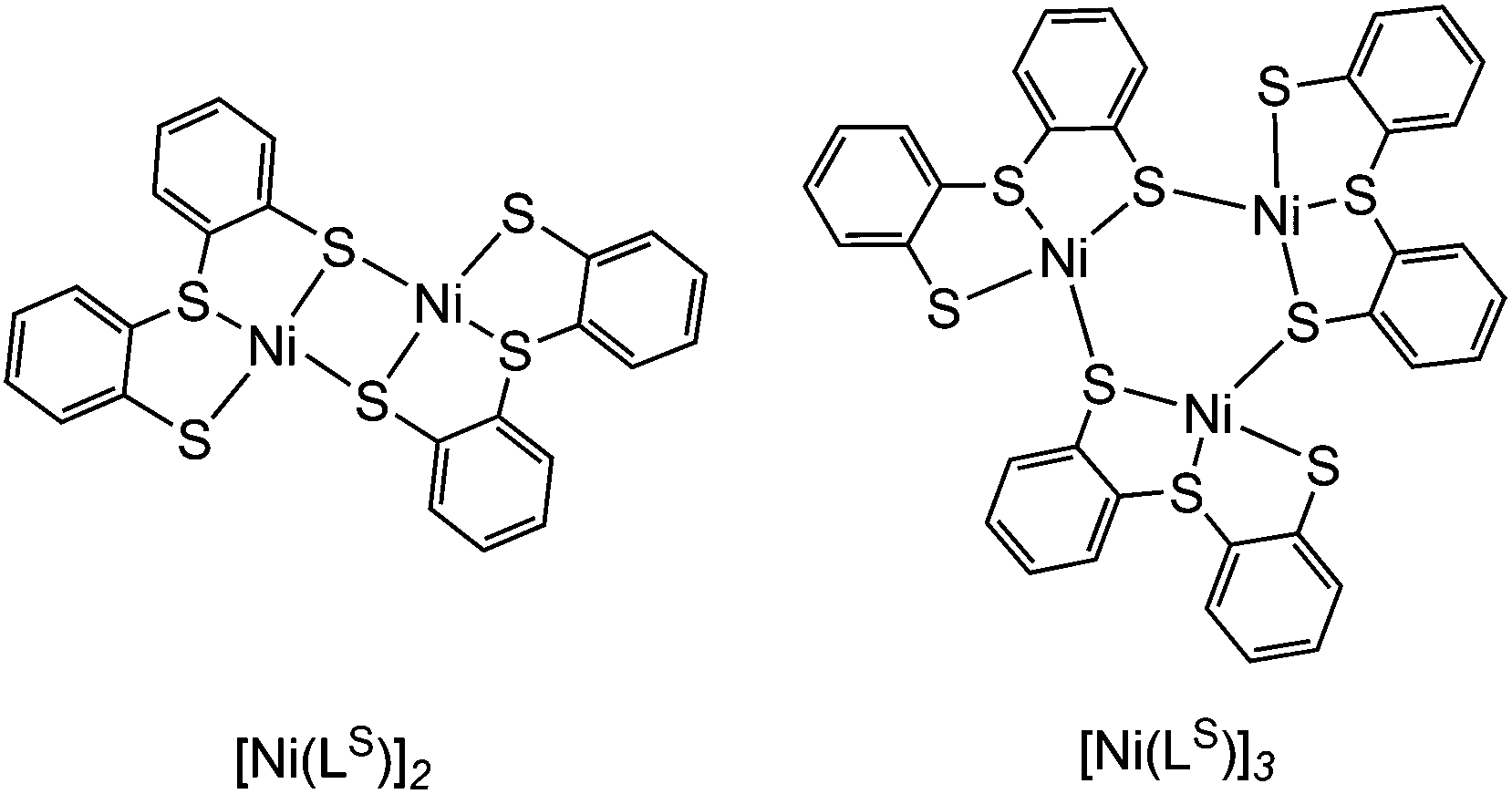
[Ni(LS)]n (Scheme 1) was previously characterised by Sellmann and co-workers using single crystal X-ray analysis and it showed dimeric and trimeric forms, depending on crystal growth conditions (Fig. 2).12i,l This composition is therefore also likely for [Ni(LSe)]n. [Ni(LS)]n has been prepared following a previously reported procedure.12i The oligomeric structure in [Ni(LSe)]n and [Ni(LS)]n can be broken upon reaction with one equivalent of sodium mesityl selenolate or sodium mesityl thiolate in tetrahydrofuran–methanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature. Addition of one equivalent of n-Bu4NOH yields the corresponding four mononuclear Ni complexes (n-Bu4N)[Ni(LE)(MesE′)] (E = S or Se; E′ = S or Se, Scheme 1). Brown crystals of the Ni complexes were grown from a saturated solution in tetrahydrofuran–hexane giving the pure product in good yields (68 to 79%). The compounds have been characterised by 1H NMR (Fig. S1–S8†), mass spectrometry (Fig. S9–S16†), ATR-IR spectroscopy (Fig. S17†) and elemental analysis. The tetraphenyl phosphonium salts of [Ni(LS)(MesS)]− and [Ni(LS)(MesSe)]− have also been prepared and characterised by 1H, 13C, 77Se NMR spectroscopy, ESI-MS and elemental analysis.
:1) at room temperature. Addition of one equivalent of n-Bu4NOH yields the corresponding four mononuclear Ni complexes (n-Bu4N)[Ni(LE)(MesE′)] (E = S or Se; E′ = S or Se, Scheme 1). Brown crystals of the Ni complexes were grown from a saturated solution in tetrahydrofuran–hexane giving the pure product in good yields (68 to 79%). The compounds have been characterised by 1H NMR (Fig. S1–S8†), mass spectrometry (Fig. S9–S16†), ATR-IR spectroscopy (Fig. S17†) and elemental analysis. The tetraphenyl phosphonium salts of [Ni(LS)(MesS)]− and [Ni(LS)(MesSe)]− have also been prepared and characterised by 1H, 13C, 77Se NMR spectroscopy, ESI-MS and elemental analysis.
Structural analyses of Ni complexes
The single crystal X-ray structures of all four complexes were obtained as tetrabutylammonium salts (Fig. 3; selected bond lengths and angles are summarised in Table 1). The complex anions [Ni(LSe)(MesS)]− and [Ni(LS)(MesSe)]− contain a Ni2+ cation bound to three S and one Se donors and reflect the primary ligand sphere of the Ni site in [NiFeSe] hydrogenases.4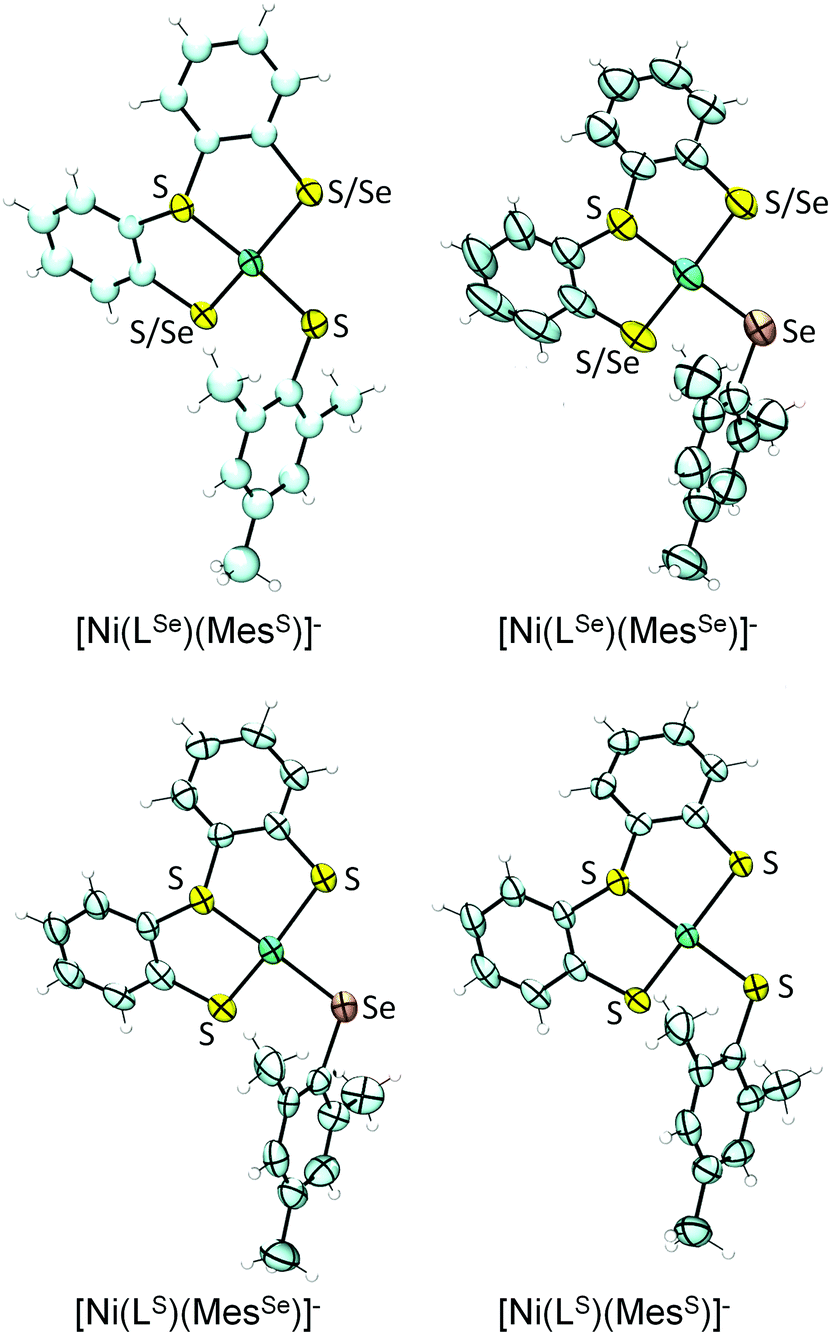 | ||
| Fig. 3 X-ray single crystal structures of complexes (n-Bu4N)[Ni(LE)(MesE′)] showing 50% probability ellipsoids (countercation not shown; the asymmetric units of [Ni(LSe)(MesS)]−, [Ni(LS)(MesSe)]− and [Ni(LS)(MesS)]− contain two independent anions; images created using Ortep 3 for Windows15). S/Se indicates disorder of S and Se over two positions. | ||
| (n-Bu4N)[Ni(LSe)(MesS)] (E = Se, E′ = S) | (n-Bu4N)[Ni(LSe)(MesSe)] (E = Se, E′ = Se) | (n-Bu4N)[Ni(LS)(MesSe)] (E = S, E′ = Se) | (n-Bu4N)[Ni(LS)(MesS)] (E = S, E′ = S) | ||||
|---|---|---|---|---|---|---|---|
| a n.d. = not accurately determined due to crystallographic disorder. | |||||||
| Bond lengths | |||||||
| Ni–S | n.d.a | n.d.a | n.d.a | 2.158(2) | 2.155(2) | 2.1630(12) | 2.19642(14) |
| Ni–S′ | 2.1257(17) | 2.1263(18) | 2.124(1) | 2.124(2) | 2.131(3) | 2.1254(13) | 2.1218(14) |
| Ni–E | n.d.a | n.d.a | n.d.a | 2.197(2) | 2.190(2) | 2.1987(12) | 2.1960(14) |
| Ni–E′ | 2.1930(18) | 2.183(2) | 2.3102(9) | 2.3009(14) | 2.3066(17) | 2.1947(14) | 2.1856(16) |
| Bond angles | |||||||
| S–Ni–E | 153.2a | 157.0a | 155.2a | 153.63(9) | 156.26(10) | 152.78(5) | 156.14(5) |
| S–Ni–S′ | 89.6a | 90.5a | 90.0a | 92.12(9) | 91.39(10) | 91.76(5) | 91.15(5) |
| S–Ni–E′ | 90.9a | 89.7a | 90.6a | 96.51(8) | 95.39(9) | 97.07(5) | 96.05(6) |
| E–Ni–S′ | 92.2a | 91.3a | 91.8a | 89.47(9) | 89.78(10) | 89.24(5) | 89.58(5) |
| E–Ni–E′ | 96.9a | 95.8a | 95.7a | 91.36(7) | 90.67(8) | 91.64(5) | 90.71(6) |
| S′–Ni–E′ | 158.74(7) | 161.21(8) | 160.79(5) | 158.64(7) | 161.97(8) | 158.64(5) | 161.33(6) |
The complexes display a distorted square planar arrangement around the metal centre. The average trans S–Ni–SMes and S–Ni–SeMes bond angle is 160.2 ± 0.6°. The [NiFeSe] hydrogenase displays angles of 168.0° and 106.4° between Se–Ni–(μ-S) and Sterminal–Ni–(μ-S), respectively (PDB 1CC1).4 Crystallographic disorder of the thiolate and selenolate donors in ‘LSe’ occurs in [Ni(LSe)(MesSe)]− (indicated as S/Se in Fig. 3) and [Ni(LSe)(MesS)]− and we therefore only consider the bond lengths from the Ni to the MesS and MesSe ligand in our discussion. The average Ni–Se(Mes) bond length at 2.306 ± 0.003 Å is longer than the Ni–S(Mes) distance at 2.189 ± 0.003 Å. A Ni–Se bond length of 2.46 Å and average Ni–S distance of 2.2 Å (at 2.15 Å resolution) was reported for the reduced [NiFeSe] hydrogenase from Desulfomicrobium baculatum.4
Reactivity of the Ni complexes with atmospheric O2
A distinct difference between the [NiFe] and [NiFeSe] hydrogenases is the reactivity of their active sites with O2. EPR studies showed that upon reactivity with atmospheric O2 the Ni in the conventional [NiFe] hydrogenases is oxidised to Ni(III), in one of two states known as the ‘ready’ (Ni–B) and ‘unready’ (Ni–A) states.8a,d,e,h Crystal structures of the oxidised enzyme show an oxygen containing ligand bridging the Ni and Fe centre (position X, Fig. 1) in both oxidised states.9b,d,e The oxidised active site in [NiFeSe] hydrogenase is quite different; the Ni centre is not oxidised and it remains diamagnetic Ni(II).5a Single crystal X-ray structures of the oxidised [NiFeSe] hydrogenase in D. vulgaris and D. baculatum confirm that there is no bridging oxo ligand between the metal centres. In fact, selenium and/or sulfur are oxidised instead.9f–h This different active site chemistry might explain the different rates for inactivation and re-activation between [NiFe] and [NiFeSe] hydrogenases.5bWe therefore investigated the reactivity of the Ni complexes with O2. As expected, all complexes are air sensitive and convert completely to oxidised products over a maximum of five days. (PPh4)[Ni(LS)(MesSe)] and (PPh4)[Ni(LS)(MesS)] convert cleanly to [Ni(LS)]n and the dichalcogenide (MesSe)2 or (MesS)2 (Scheme 2). The oxidation products were isolated and characterised after exposing the complexes to atmospheric O2 in dichloromethane. [Ni(LS)]n was isolated as a black precipitate and characterised by ATR-IR spectroscopy (Fig. S18†) and elemental analysis. The dichalcogenide was isolated from the supernatant and characterised by 1H NMR and ESI-MS (Fig. S19–S21†).
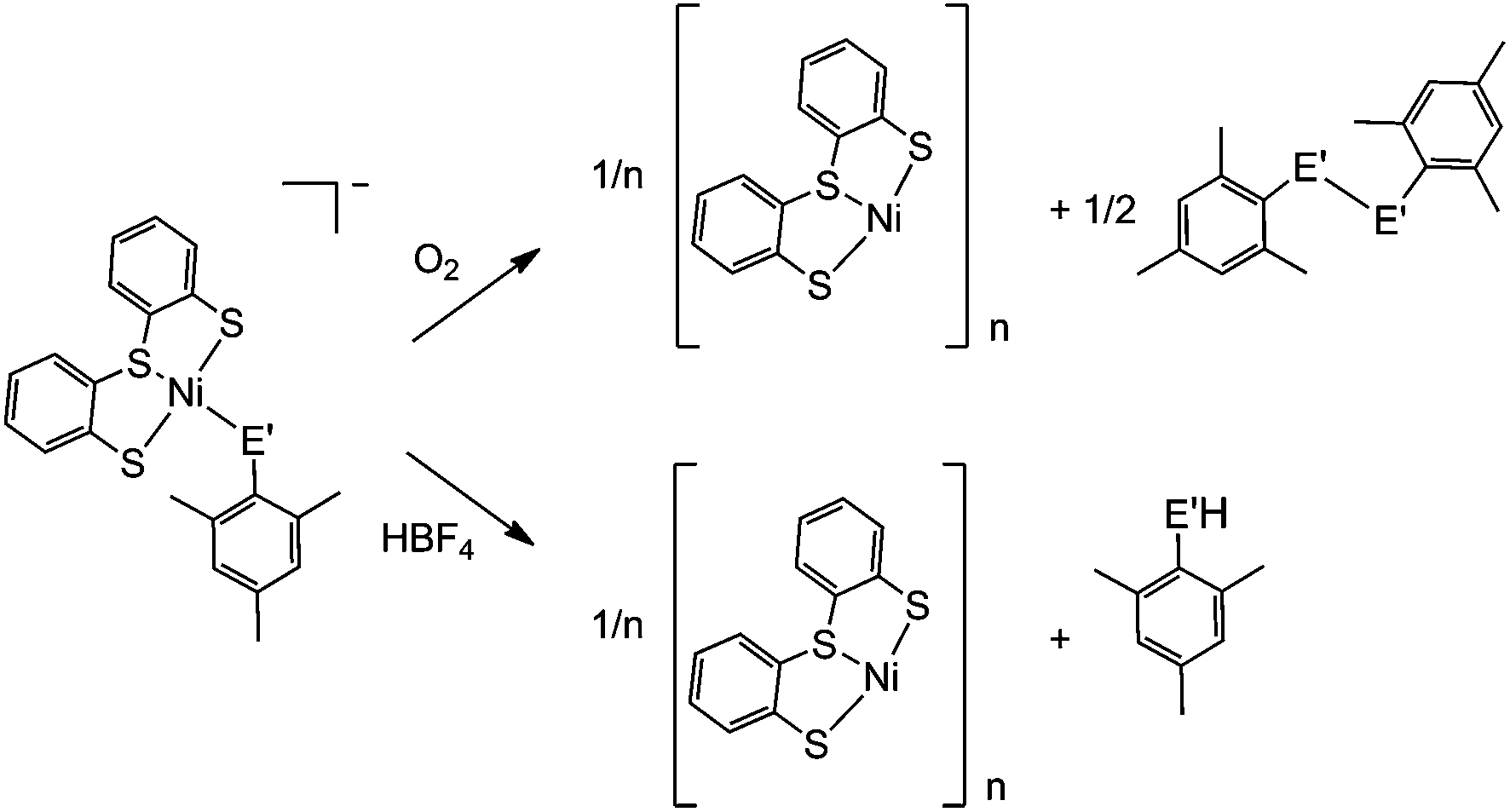 | ||
| Scheme 2 Reactivity of [Ni(LS)(MesSe)]− (E′ = Se) and [Ni(LS)(MesS)]− (E′ = S) with O2 and HBF4. | ||
The oxidation of the Se containing model emulates the reactivity of the enzyme. Oxidation of Se from the −2 to −1 oxidation state, and the formation of a Se–chalcogen bond concomitant with the loss of Se from the metal centre are all observed in the oxidised [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough.9f,g Crystallographic analysis of this oxidised enzyme shows that the selenium is bound to a sulfur atom extrinsic to the active site. The source of the S atom is proposed to be H2S in this sulfate reducing bacterium. In one oxidised conformer the selenium is not bound to the Ni centre and the extrinsic sulfur binds to Ni in its place (Fig. 1b).9f,g
The oxidation of (PPh4)[Ni(LS)(MesSe)] and (PPh4)[Ni(LS)(MesS)] was also followed by 1H NMR spectroscopy in deuterated dichloromethane (Fig. S22†). Over the course of oxidation the signals of the ‘Ls’ ligand at 6.8 to 7.5 ppm decrease as [Ni(LS)]n precipitates from solution and the methyl signals for the mesityl thiolate/selenolate ligand at 2.1 to 2.6 ppm decrease as new methyl signals appear around 2.3 ppm for the dichalcogenide oxidation product. The selenolate containing complex (PPh4)[Ni(LS)(MesSe)] is oxidised more rapidly than the all sulfur complex: it is completely converted within 24 hours, compared with 96 h for (PPh4)[Ni(LS)(MesS)]. This observation supports the hypothesis that the fast reactivity of Se with O2 prevents the nickel centre from being oxidised in [NiFeSe] hydrogenases.9f–h
Reactivity of the Ni complexes with HBF4
Crystallographic evidence suggests that one of the terminal cysteine residues bound to the Ni centre in [NiFe] hydrogenases (highlighted in Fig. 1a) acts as a proton relay during the catalytic cycle. This residue has increased crystallographic disorder compared with the rest of the active site, suggesting various protonation states.9a–c In the D. baculatum [NiFeSe] hydrogenase the selenocysteine residue in the same position exhibits this crystallographic disorder suggesting that it is the proton relay in this case.4Exposure of (n-Bu4N)[Ni(LS)(MesSe)] to one equivalent of HBF4 in dichloromethane results in the protonation of the mesityl selenolate group (Scheme 2), in analogy to protonation in the enzyme active site. Mesityl selenol is released from the metal centre and has been isolated from the solution and characterised using 1H NMR spectroscopy (Fig. S23†). The remaining ‘Ni(LS)’ centre precipitates as the black solid [Ni(LS)]n (separated and characterised by ATR-IR and elemental analysis). Diethyl ether was added to the filtrate to precipitate n-Bu4NBF4, which was separated by filtration and characterised using 1H NMR spectroscopy and mass spectrometry. The solvent was removed from the remaining filtrate to give mesityl selenol. A comparable reactivity pattern is observed with (n-Bu4N)[Ni(LS)(MesS)]: the ‘MesS’ ligand is protonated and is released from the metal centre as mesityl thiol (Scheme 2; Fig. S24†). The products of protonation were separated and characterised in the same way as with (n-Bu4N)[Ni(LS)(MesSe)]. The protonation of these complexes parallels the proposed protonation of the selenocysteine and cysteine residue in the hydrogenase.
Electrochemistry and electrodeposition of Ni complexes
The cyclic voltammograms (CVs) of all four complexes show an irreversible reduction process at approximately −1.4 V vs. NHE at a scan rate of 100 mV s−1 in acetonitrile, dichloromethane and dimethylformamide containing n-Bu4NBF4 as a supporting electrolyte (Fig. 4a black trace, Fig. S25–26†). This wave is assigned to the reduction of Ni(II) to Ni(I) based on previous electrochemical studies with Ni–thiolate complexes.12g,16 A catalytic wave grows at approximately −1.30 V vs. NHE with increasing amounts of triethylammonium chloride (Et3NHCl) in acetonitrile indicating electrocatalytic H2 production (Fig. 4a, Fig. S27†). The overpotential required for the reduction of protons is approximately 0.65 V under these conditions, calculated from the half wave potential for catalytic proton reduction in the presence of 10 equivalents of Et3NHCl (−1.17 V vs. NHE, Fig. S28†) and the standard potential for reduction of protons from Et3NHCl in acetonitrile (−0.51 V vs. NHE).17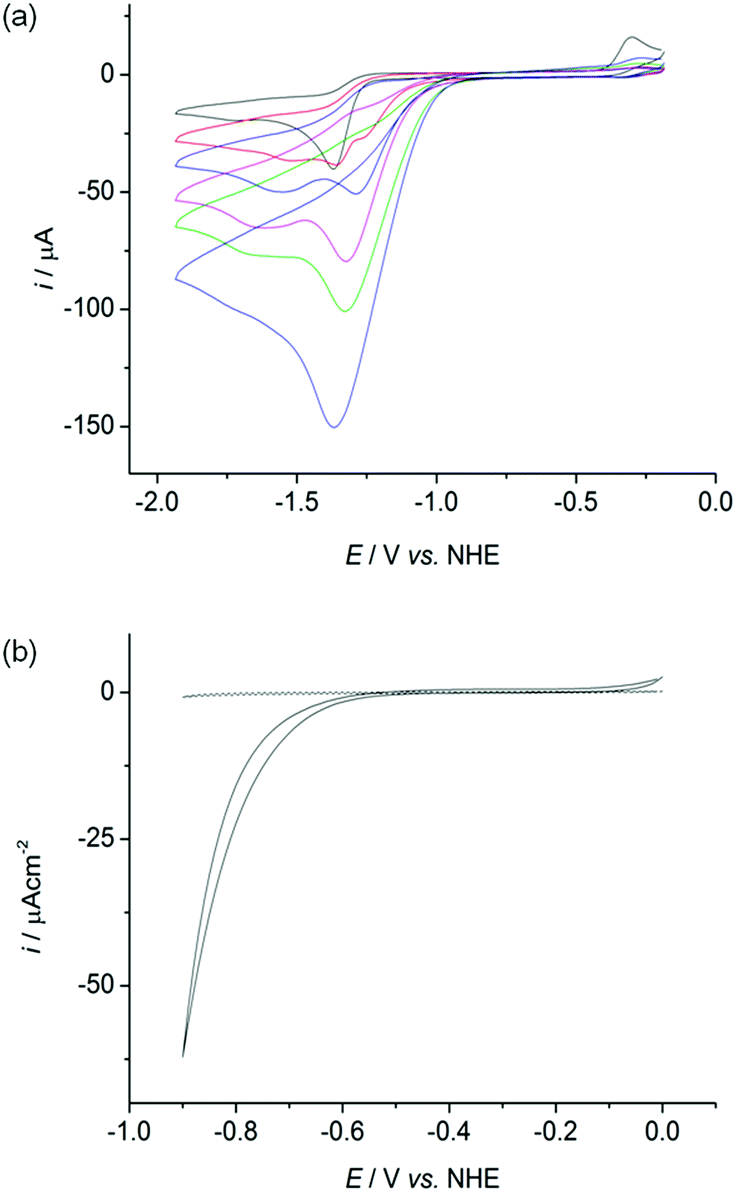 | ||
| Fig. 4 (a) CV of (n-Bu4N)[Ni(LSe)(MesS)] (1 mM) in acetonitrile containing n-Bu4NBF4 (0.1 M) at a glassy carbon electrode with a scan rate of 100 mV s−1 with increasing concentrations of Et3NHCl: no acid (black), 1 (red), 2 (blue), 3 (pink), 5 (green) and 10 (purple) mM. (b) CV of a Ni particle activated (solid trace) and unmodified (dashed trace) glassy carbon electrode in a phosphate buffered aqueous solution (0.1 M) at pH 7 at a scan rate of 100 mV s−1 at room temperature. | ||
However, the proton reduction activity does not stem from a molecular Ni species, but from a solid deposit formed on the electrode surface at the applied negative potential. Here, the Ni complexes act as molecular precursors to catalyst particles. The same electrocatalytic response was observed after removing the working electrode following electrocatalytic proton reduction in n-Bu4NBF4–acetonitrile (0.1 M) with Et3NHCl (10 mM) and a Ni complex, rinsing it with acetonitrile and immersing it in a fresh, Ni complex free electrolyte solution (Fig. S29†). Electronic absorption spectroscopy of the nickel complexes confirmed their stability in the presence of 10 equivalents of Et3NHCl in acetonitrile in the absence of an applied potential, demonstrating that the complexes were deposited electrochemically on the electrode at negative potentials (Fig. S30†).
The oxidation wave at E = −0.25 V in Fig. 4a and S25–S27† is attributed to the oxidation of a soluble product formed during the irreversible reduction of the complexes. The redox process cannot be assigned to the oxidation of deposited material as it is not visible in the CV of a nickel modified glassy carbon electrode in a fresh electrolyte solution.
Although the particle formation has no obvious relevance to the biomimetic features of the complexes, there is currently much interest in assembling heterogeneous electrocatalysts for proton reduction from molecular precursors.18 We therefore decided to investigate the Ni particle formation in more detail. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) analysis of a glassy carbon slide (total surface area 1.6 cm2) modified with (n-Bu4N)[Ni(LSe)(MesS)] (1 mM) at −1.33 V vs. NHE for 0.5 h showed the deposition of nickel containing particles on the glassy carbon surface (Fig. 5a).
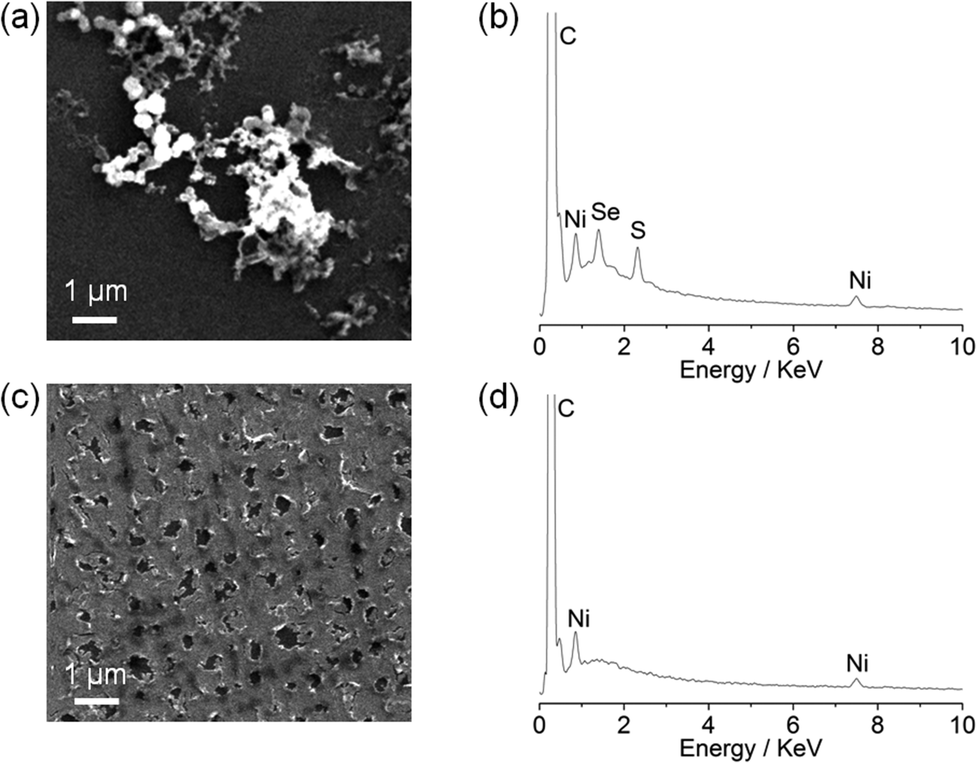 | ||
| Fig. 5 SEM images and EDX spectra of a (a and b) Ni particle modified glassy carbon electrode prepared from the electro-deposition of (n-Bu4N)[Ni(LSe)(MesS)] (1 mM) and (c and d) Ni film on a glassy carbon electrode prepared from the electrodeposition of Ni(NO3)2·6H2O (1 mM). The deposits were formed with the compounds at an applied potential of −1.33 V vs. NHE for 0.5 h in acetonitrile (0.1 M n-Bu4NBF4) with Et3NHCl (10 mM). | ||
EDX of the particles revealed that they contain Ni, S and Se (Fig. 5b), but the exact composition and oxidation state of the Ni–S–Se containing deposit remains unknown. Using Ni(NO3)2·6H2O as a precursor resulted in a Ni film (no sulfur or selenium residues were detected) covering the electrode surface (Fig. 5c and 5d) rather than Ni particle formation as observed with the Ni complexes. Our observations demonstrate that the molecular Ni precursors allow for the incorporation of sulfur and selenium in the solid precipitate and the formation of a distinct morphology compared to using Ni(NO3)2·6H2O.
Electrocatalytic activity of Ni-activated electrodes in water
We subsequently investigated the activity of the Ni particle modified electrode, which was formed by holding the potential at −1.33 V vs. NHE for 0.5 h in a solution of the Ni complexes (1 mM) and Et3NHCl (10 mM) in n-Bu4NBF4–acetonitrile (0.1 M) and washing of the electrode. The Ni particle electrode displayed a respectable activity in a pH neutral Ni free aqueous phosphate solution (0.1 M; Fig. 4b). The onset of a catalytic wave was observed at approximately −0.60 V vs. NHE, which corresponds to a small overpotential of 180 mV (E0, H+/H2 = −0.42 V at pH 7).The production of H2 was confirmed using controlled potential electrolysis at −0.9 V vs. NHE with a glassy carbon rod or fluoride doped tin oxide (FTO) coated glass electrode (both approximately 1.6 cm2) modified with the Ni precursor by the same procedure. SEM/EDX analysis also confirmed the deposition of Ni on the FTO substrate (Fig. S31†). A Faradaic yield of approximately 60 to 71% was observed for all four deposits on glassy carbon and FTO glass modified with the Ni. The total amount of H2 generated varied widely on glassy carbon (10 to 2000 μmol after 20 h bulk electrolysis), but was more reproducible on FTO coated glass (9 to 55 μmol after 10 h electrolysis, Fig. S32 and Table S1†).
Modifying the FTO electrode with Ni(NO3)2·6H2O by the same procedure resulted in the formation of comparable amounts of H2, but with a reduced Faradaic yield of only 32%, suggesting that the molecular precursor does not only have an effect on the composition and morphology of Ni deposit, but also on the activity of the Ni-modified electrodes. We also compared the Ni particle electrodes with the benchmark proton reduction catalyst Pt.19 A metallic Pt foil with the same geometrical surface area (approximately 1.6 cm2) was therefore tested as the working electrode under the same experimental conditions. The activity of the Pt foil is approximately an order of magnitude higher than the Ni particle modified FTO electrodes in an aqueous phosphate solution at pH 7 (Fig. S32†).
Conclusions
We have demonstrated the synthesis and characterisation of the first series of complexes, which resemble the primary coordination sphere of the Ni site in [NiFeSe] hydrogenases. Single crystal X-ray structures are reported for all complexes. Two complexes, [Ni(LS)(MesSe)]− and [Ni(LSe)(MesS)]−, display the key structural features of the Se-containing enzyme such as a distorted four-coordinate ‘NiS3Se’ environment.Aerobic oxidation of the complex [Ni(LS)(MesSe)]− results in the oxidation of the monodentate selenium ligand to form a dichalcogenide, whereas Ni remained in the +2 oxidation state. Comparable chemistry was observed in the active site of D. vulgaris [NiFeSe] hydrogenase after exposure to O2.9f,g Reactivity of [Ni(LS)(MesSe)]− with HBF4 leads to protonation of the selenolate ligand, indicating that the selenium atom is indeed a plausible protonation site during H2 cycling in [NiFeSe] hydrogenases. Although unrelated to the biomimetic composition of the Ni molecules, the complexes also act as precursors to Ni-containing particles on an electrode surface, which show high electroactivity in pH neutral aqueous solution. Work is currently in progress to introduce iron in our Ni site precursors and assemble a full structural [NiFeSe] hydrogenase model complex.
Experimental section
Materials and methods
Unless otherwise stated all compounds were prepared using an anhydrous and anaerobic MBraun glovebox or Schlenk techniques. All starting materials were purchased from commercial suppliers in the highest available purity for all analytical measurements and used without further purification. Organic solvents were dried and deoxygenated prior to use. Mesityl selenol20 and [Ni(LS)]n12i (Scheme 1) were prepared according to literature procedures.
Physical measurements
1H and 13C NMR spectra were recorded on a Bruker DPX-400 MHz spectrometer and the spectra referenced against the solvent peak. The 77Se NMR spectrum was recorded on a Bruker Avance 500 MHz BroadBand NMR spectrometer and referenced against dimethyl selenide in d-benzene as an external reference at 0 ppm. The mass spectrum of (‘LSe–H’)2 was carried out on a Waters ZQ HPLC-MS. The mass spectra of ‘LSe’–H2 and dimesityl diselenide were recorded by the University of Cambridge Mass Spectrometry Service using a Bruker Bio Apex 4.0 FTICR EI MS. The mass spectra of the metal complexes and inorganic salts were recorded on a Waters Quattro LC electrospray ionisation mass spectrometer. Expected and experimental isotope distributions of [Ni(LE)(MesE′)]− were compared. Elemental analysis was carried out by the microanalysis service of the Department of Chemistry, University of Cambridge. IR spectra were recorded on a Perkin Elmer SpectrumOne FTIR spectrometer with an ATR sampling accessory. Electronic absorption spectra were recorded on an Agilent Cary UV-Vis 50 Bio spectrometer. The SEM images were obtained using a Philips XL30 132-10 electron microscope. Energy-dispersive X-ray spectroscopy (edax PV7760/68 ME) was used at a 15 kV acceleration voltage, spot size 4.0 and an acquisition time of at least 100 s. The elements were assigned and atomic ratios were identified using the built in software (EDAX).X-ray crystallographic studies
All data were recorded with Mo Kα radiation (λ = 0.71073 Å) on a Nonius Kappa CCD diffractometer fitted with an Oxford Cryosystems Cryostream cooling apparatus. The single crystals were mounted in Paratone N oil on the tip of a glass fibre and kept under a stream of N2. Structure solution was carried out using direct methods and refined by least squares (SHELXL-97)21 using Chebyshev weights on Fo2. The weighted R-factor, wR and goodness of fit (GOF) are based on F2. The hydrogen atoms were assigned to idealised positions and given thermal parameters of 1.5 (methyl hydrogens) or 1.2 (non-methyl hydrogens) times the thermal parameter of the carbon atom to which they were attached. The tridentate ‘LSe’ in the complexes (n-Bu4N)[Ni(LSe)(MesS)] and (n-Bu4N)[Ni(LSe)(MesSe)] exhibits the same sort of disorder, in each case modelled as two poorly resolved S/Se sites. Crystal data, data collection parameters, and structure refinement details for the complexes are given in Table 2. Selected bond lengths and angles are shown in Table 1. The mean bond lengths and angles for the discussion in the paper were calculated as follows: for a sample of n observations xi, a weighted mean value (xu) with its standard deviation (σ) was calculated using the following equations: xu = Σixi/n, σ = {Σi(xi − xu)2/[n(n − 1)]}1/2.| Complex | (n-Bu4N)[Ni(LSe)(MesS)] | (n-Bu4N)[Ni(LSe)(MesSe)] | (n-Bu4N)[Ni(LS)(MesSe)] | (n-Bu4N)[Ni(LS)(MesS)] |
|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | ||||
| Empirical formula | C37H55NNiS3Se | C37H55NNiS2Se2 | C37H55NNiS3Se | C37H55NNiS4 |
| Formula weight | 747.67 | 794.57 | 747.67 | 700.77 |
| Temperature (K) | 180(2) | 220(2) | 180(2) | 180(2) |
| Space group | P21/c | P21/n | P21/c | P21/c |
| a (Å) | 18.6006(4) | 10.6327(2) | 18.7224(2) | 18.5275(2) |
| b (Å) | 23.1712(5) | 22.5152(5) | 23.0398(2) | 23.1544(2) |
| c (Å) | 18.7401(5) | 16.1237(5) | 18.7818(2) | 18.7038(2) |
| β (°) | 109.268(1) | 94.708(1) | 109.810(1) | 109.270(1) |
| V (Å3) | 7624.5(3) | 3846.95(16) | 7622.27(13) | 7574.26(13) |
| Z | 8 | 4 | 8 | 8 |
| ρ calc (g cm−3) | 1.303 | 1.372 | 1.303 | 1.229 |
| μ (Mo Kα, mm−1) | 1.653 | 2.530 | 1.654 | 0.758 |
| Crystal size (mm) | 0.46 × 0.10 × 0.02 | 0.23 × 0.21 × 0.02 | 0.28 × 0.23 × 0.10 | 0.28 × 0.12 × 0.02 |
| Θ range (°) | 3.51 to 25.34 | 3.55 to 25.66 | 3.51 to 22.49 | 3.52 to 27.50 |
| Total number of data | 43959 |
35314 |
52166 |
73103 |
| Number of unique data | 13739 |
7112 | 9870 | 17283 |
| Number of parameters | 393 | 304 | 602 | 602 |
| Completeness | 0.984 | 0.974 | 0.992 | 0.993 |
|
R
1a |
0.0711 | 0.0452 | 0.0634 | 0.0757 |
| wR2b |
0.1765 | 0.1123 | 0.1672 | 0.1819 |
Electrochemical measurements
CVs were recorded at room temperature under Ar using an IviumStat or CompactStat potentiostat. A standard three electrode cell was used for all measurements with a glassy carbon disc working (3 mm diameter), a platinum foil counter and a Ag/Ag+ (organic solutions) or a Ag/AgCl/KCl(sat) (aqueous solutions) reference electrode. For CVs recorded in acetonitrile containing n-Bu4NBF4 (0.1 M, electrochemistry grade, Sigma Aldrich), the Fc/Fc+ couple was used as an external reference and potentials were converted to the normal hydrogen electrode (NHE) by adding 630 mV in acetonitrile.22 For CVs in a pH 7 aqueous phosphate solution (0.1 M), potentials were converted by adding 0.2 V to the potential against Ag/AgCl/KCl(sat).23Controlled potential electrolysis (CPE)
CPE in phosphate solution (0.1 M, pH 7) was carried out using a fluoride doped tin oxide (FTO) coated glass electrode or glassy carbon rod electrode (geometrical surface area in contact with electrolyte solution approximately 1.6 cm2), a platinum mesh counter electrode and a Ag/AgCl reference electrode. CPE was carried out in an airtight electrochemical cell containing N2 with 2% methane as the internal standard for gas chromatography, GC, analysis. The headspace gas was analysed using an Agilent 7890A GC equipped with a 5 Å molecular sieve column, using N2 carrier gas with a flow rate of approximately 3 mL min−1. The GC columns were kept at 40 °C and a thermal conductivity detector (TCD) was used. Measurements were taken every 2 h and the cell was purged to remove all hydrogen following each GC measurement. All measurements were repeated at least three times. Faradaic efficiency (%) = 100[H2 (mol) × 2F/Q (C)].Synthesis of ‘LSe’–H2
The first step of this reaction must be carried out under argon. Lithium (25% dispersion in oil, Sigma Aldrich) was washed with hexane (3 × 2 mL). To the de-oiled Li metal (0.080 g, 11.53 mmol) was added 4,4′-di-tert-butylbiphenyl (DTBB, 0.115 g, 432 μmol) in tetrahydrofuran (3 mL) and the reaction mixture was cooled to −90 °C (acetone–liquid N2 bath), resulting in the formation of a bright blue solution of a radical anion of DTBB.14 Thianthrene (1.25 g, 5.76 mmol) in tetrahydrofuran (15 mL) was added at −90 °C and the resulting beige solution was stirred for eight hours during which time the temperature of the reaction mixture was allowed to slowly reach −50 °C. Selenium powder (0.455 g, 5.76 mmol) was added in one batch and the orange solution was allowed to reach room temperature slowly overnight. Degassed water (50 mL) was slowly added and the aqueous layer was extracted with diethyl ether (4 × 50 mL) to remove DTBB and any remaining unreacted starting material. The aqueous layer was then acidified with aqueous HCl (2 M, 50 mL) and extracted with dichloromethane (3 × 25 mL). The combined organic layers were washed with aqueous HCl (2 M, 25 mL), and the solvent was removed under vacuum to give a yellow gum of crude (‘LSe–H’)2, which was used for the next step without further purification.24 The crude (‘LSe–H’)2 was dissolved in tetrahydrofuran (15 mL) and added dropwise to a solution of LiAlH4 (68 mg, 1.79 mmol) in tetrahydrofuran (3 mL) with stirring at room temperature. The resulting colourless solution was stirred overnight. Aqueous HCl (2 M, 12 mL) was then added dropwise and the product was extracted with diethyl ether (3 × 20 mL). The combined extracts were washed with aqueous HCl (2 M, 12 mL) and the solvent was removed under high vacuum at room temperature to give the product as a white solid, which was purified by recrystallisation from a saturated solution in tetrahydrofuran at −35 °C. Yield: 494 mg, 29%. 1H NMR (400 MHz, CDCl3) δ/ppm = 7.47 (1H, dd), 7.33 (1H, dd), 7.00–7.19 (6H, m), 4.05 (1H, s, SH), 1.91 (1H, s, SeH); EI-MS (CHCl3) +ive: 297.94 (30%, LSe); elemental analysis calculated (%) for C12H10S2Se C 48.48, H 3.39; found C 48.67, H 3.39.Synthesis of [NiLSe]n
A solution of ‘LSe’–H2 (200 mg, 671 μmol) in tetrahydrofuran (3 mL) was added dropwise to a solution of Ni(OCOCH3)2·4H2O (167 mg, 671 μmol) in methanol (0.5 mL). The dark brown suspension was heated to reflux for four hours, whereupon an insoluble black precipitate was separated by filtration, washed with tetrahydrofuran (3 × 2 mL) and dried under vacuum at room temperature. Yield 208 mg, 87%. Elemental analysis calculated (%) for [C12H8NiS2Se]n C 40.72, H 2.28; found C 40.78, H 2.31. ATR-IR![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 = 3040, 1568, 1441, 1424, 1239, 1154, 1085, 1038, 757, 746. [NiLSe]n is insoluble in any common organic solvent.
/cm−1 = 3040, 1568, 1441, 1424, 1239, 1154, 1085, 1038, 757, 746. [NiLSe]n is insoluble in any common organic solvent.
General procedure for synthesis of (n-Bu4N)[Ni(LE)(MesE′)]
To a solution of NaOMe in methanol was added mesityl thiol or selenol. The solution was stirred for 20 min and then added to a stirred suspension of [NiLE]n in tetrahydrofuran at room temperature. The reaction mixture was stirred for one hour until [NiLE]n fully dissolved giving a brown solution. A solution of n-Bu4NOH·30H2O in methanol was added and the solution was stirred for an additional 20 min. All solvents were removed under high vacuum at room temperature and the brown residue was taken up in tetrahydrofuran and filtered through a Millex FG PTFE microfilter (pore size 20 μm). Hexane was layered on top of the tetrahydrofuran solution and the undisturbed mixture gave brown crystalline needles of the product after several days. The crystals were separated by filtration and washed with hexane. X-ray diffraction quality single crystals were selected directly from the reaction vessel.(n-Bu4N)[Ni(LSe)(MesS)]
NaOMe (28 mg, 525 μmol) in methanol (1 mL), mesityl thiol (79 μL, 525 μmol), [Ni(LSe)]n (186 mg, 525 μmol) in tetrahydrofuran (4 mL), n-Bu4NOH·30H2O (420 mg, 525 μmol) in methanol (0.75 mL). Yield 280 mg, 71%. 1H NMR (400 MHz, THF-d8) δ/ppm = 7.52 (1H, d, LSe) 7.43 (1H, d, LSe), 7.38 (1H, d, LSe), 7.30 (1H, d, LSe), 6.79–6.97 (4H, m, LSe), 6.78 (2H, s, MesS), 3.34 (8H, m, n-Bu4N) 2.79 (6H, s, MesS), 2.46 (3H, s, MesS), 1.59 (8H, m, n-Bu4N), 1.56 (8H, m, n-Bu4N), 1.37 (12H, t, n-Bu4N); ESI-MS (CH2Cl2) −ive: 505 (100%, [Ni(LSe)(MesS)]−), +ive: 242 (100%, n-Bu4N+); elemental analysis calculated (%) for C37H55NiNS3Se C 59.44, H 7.41, N 1.87; found C 59.17, H 7.36, N 1.90.(n-Bu4N)[Ni(LSe)(MesSe)]
NaOMe (7.6 mg, 141 μmol) in methanol (2 mL), mesityl selenol (28.1 mg, 141 μmol), [Ni(LSe)]n (50 mg, 141 μmol) in tetrahydrofuran (3 mL), n-Bu4NOH·30H2O (113 mg, 141 μmol) in methanol (1 mL). Yield 77 mg, 68%. 1H NMR (400 MHz, THF-d8) δ/ppm = 7.59 (1H, d, LSe), 7.52 (1H, d, LSe), 7.26 (1H, d, LSe), 7.17 (1H, d, LSe), 6.78–6.88 (4H, m, LSe), 6.66 (2H, s, MesSe), 3.41 (8H, m, n-Bu4N) 2.71 (6H, s, MesSe), 2.11 (3H, s, MesSe), 1.66 (8H, m, n-Bu4N), 1.35 (8H, m, n-Bu4N), 0.90 (12H, t, n-Bu4N); ESI-MS (CH2Cl2) −ive: 553 (100%, [Ni(LSe)(MesSe)]−), +ive: 242 (100%, n-Bu4N+); elemental analysis calculated (%) for C37H55NiNS2Se2 C 55.93, H 6.98, N 1.76; found C 55.85, H 6.94, N 1.81.(n-Bu4N)[Ni(LS)(MesSe)]
NaOMe (17.6 mg, 326 μmol) in methanol (2 mL), mesityl selenol (64.9 mg, 326 μmol), [Ni(LS)]n (100 mg, 326 μmol) in tetrahydrofuran (3 mL), n-Bu4NOH·30H2O (261 mg, 326 μmol) in methanol (1 mL). Yield 175 mg, 72%. 1H NMR (400 MHz, THF-d8) δ/ppm = 7.60 (2H, d, LS), 7.09 (2H, d, LS), 6.83 (2H, t, LS), 6.72 (2H, t, LS), 6.66 (2H, s, MesSe), 3.41 (8H, m, n-Bu4N) 2.73 (6H, s, MesSe), 2.11 (3H, s, MesSe), 1.67 (8H, m, n-Bu4N), 1.36 (8H, m, n-Bu4N), 0.90 (12H, t, n-Bu4N); ESI-MS (CH2Cl2) −ive: 505 (100%, [Ni(LS)(MesSe)]−), +ive: 242 (100%, n-Bu4N+); elemental analysis calculated (%) for C37H55NiNS3Se C 59.44, H 7.41, N 1.87; found C 59.59, H 7.35, N 1.93.(n-Bu4N)[Ni(LS)(MesS)]
NaOMe (65 mg, 1.21 mmol) in methanol (3 mL), mesityl thiol (182 μL, 1.21 mmol), [Ni(LS)]n (400 mg, 1.30 mmol) in tetrahydrofuran (9 mL), n-Bu4NOH·30H2O (966 mg, 1.21 mmol) in methanol (3 mL). Yield 672 mg, 79%. 1H NMR (400 MHz, THF-d8) δ/ppm = 7.62 (2H, d, LS), 7.12 (2H, d, LS), 6.92 (2H, t, LS), 6.79 (2H, t, LS), 6.71 (2H, s, MesS), 3.49 (8H, m, n-Bu4N), 2.77 (6H, s, MesS), 2.17 (3H, s, MesS), 1.75 (8H, m, n-Bu4N), 1.43 (8H, m, n-Bu4N), 0.98 (12H, m, n-Bu4N); ESI-MS (CH2Cl2) −ive: 457 (100%, [Ni(LS)(MesS)]−), +ive: 242 (100%, n-Bu4N+); elemental analysis calculated (%) for C37H55NiNS4 C 63.41, H 7.91, N 2.00; found C 63.46, H 7.92, N 2.08.Synthesis of (PPh4)[Ni(LS)(MesSe)]
Elemental Se (24 mg, 300 μmol) was added to mesityl magnesium bromide (300 μL of a 1 M solution in diethyl ether). The solution was stirred overnight until the Se powder dissolved and the colour changed from orange to yellow. Tetrahydrofuran (2 mL) was added followed by [Ni(LS)]n (96 mg, 300 μmol) and this was stirred for one hour until [Ni(LS)]n dissolved giving a brown solution. A solution of PPh4Br (126 mg, 300 μmol) in methanol (0.5 mL) was added and the solution was stirred for 20 min. The solution was concentrated to dryness under reduced pressure. The brown residue was dissolved in tetrahydrofuran (3 mL) and filtered through a Millex FG PTFE microfilter (pore size 20 μm). Diethyl ether was added to precipitate the brown product which was separated by filtration, washed with diethyl ether and dried under vacuum. Yield 169 mg, 67%. 1H NMR (400 MHz, CDCl3) δ/ppm = 7.66 (4 H, m, PPh4), 7.52 (8H, m, PPh4), 7.49 (8H, m, PPh4), 7.42 (2H, d, LS), 7.04 (2H, d, LS), 6.79 (2H, t, LS), 6.70 (4H, m, LS and MesSe), 2.52 (6H, s, MesSe), 2.08 (3H, s, MesSe); 13C NMR (500 MHz, CDCl3) δ/ppm = 156.6, 143.8, 134.4 (PPh4), 133.0, 130.8 (PPh4), 129.8, 128.3, 127.0, 126.9, 120.2, 117.8 (PPh4), 117.1 (PPh4), 27.2 (MesSe), 21.1 (MesSe); 77Se NMR (500 MHz, CD2Cl2) δ/ppm = 605.4; ESI-MS (CD2Cl2): −ive: 505 (100%, [Ni(LS)(MesSe)]−), +ive: 339 (100%, PPh4+); elemental analysis calculated (%) for C45H39NiPS3Se C 63.99, H 4.65, P 3.67; found C 63.60, H 4.50, P 3.60.Synthesis of (PPh4)[Ni(LS)(MesS)]
To a solution of NaOMe (17 mg, 309 μmol) in methanol (1 mL) was added mesityl thiol (50 μL, 309 μmol). The solution was stirred for 20 min and then added to a stirred suspension of [NiLS]n (100 mg, 309 μmol) in tetrahydrofuran (3 mL) at room temperature and the solution was stirred for one hour until [NiLS]n fully dissolved giving a brown solution. A solution of PPh4Br in methanol was added and this was stirred for 20 min. The solution was concentrated to dryness under reduced pressure. The brown residue was dissolved in tetrahydrofuran (3 mL) and filtered through a Millex FG PTFE microfilter (pore size 20 μm). Diethyl ether was added to precipitate the brown product which was separated by filtration, washed with diethyl ether and dried under vacuum. Yield 158 mg, 64%. 1H NMR (400 MHz, CD2Cl2) δ/ppm = 7.92 (4H, m, PPh4), 7.78 (8H, m, PPh4), 7.62 (8H, m, PPh4), 7.49 (2H, d, LS), 7.14 (2H, d, LS), 6.89 (2H, t, LS), 6.80 (2H, m, LS and MesS), 2.59 (6H, s, MesS), 2.17 (3H, s, MesS); 13C NMR (500 MHz, CD2Cl2) δ/ppm = 155.4, 142.9, 140.3, 135.7, 134.4 (PPh4), 134.1, 130.7, 130.6 (PPh4), 129.2, 127.3, 127.3, 120.4, 117.8 (PPh4), 117.1 (PPh4), 24.2 (MesS), 20.8 (MesS); ESI-MS (CH2Cl2); −ive: 457 (100%, [Ni(LS)(MesS)]−), +ive: 339 (100%, PPh4+); elemental analysis calculated (%) for C45H39NiPS4 C 67.75, H 4.93, P 3.88; found C 67.40, H 4.95, P 3.91.Reaction of (PPh4)[Ni(LS)(MesSe)] with oxygen
(PPh4)[Ni(LS)(MesSe)] (116 mg, 137 μmol) in dichloromethane (5 mL) was exposed to atmospheric O2 for one day with stirring to give a brown solution containing dimesityl diselenide with a black precipitate characterised as [Ni(LS)]n. Isolation and characterisation of precipitate containing [Ni(LS)]n: The solid was separated by filtration and washed with dichloromethane, methanol and diethyl ether and dried under vacuum. ATR-IR spectroscopy, elemental analysis and single crystal X-ray analysis identified the solid product as [Ni(LS)]n12l (22 mg, 53%). ATR-IR/cm−1 = 3036, 1442, 1425, 1251, 1092, 759, 727. Isolation and characterisation of filtrate containing (MesSe)2: Diethyl ether (15 mL) was added to the filtrate solution to precipitate a tetraphenyl phosphonium salt as a pink solid which was separated by filtration and washed with diethyl ether (42 mg). 1H NMR (400 MHz, CD2Cl2) δ/ppm = 7.66 (4 H, m, PPh4), 7.52 (8H, m, PPh4), 7.49 (8H, m, PPh4), ESI-MS (CD2Cl2): +ive: 339 (100%); λmax/nm (acetonitrile) 226 nm. The solvent was removed from the resulting filtrate to give crude dimesityl diselenide as a yellow solid, which was purified by filtration through silica in diethyl ether (10 mg, 36%). 1H NMR (400 MHz, CDCl3) δ/ppm = 6.77 (2H, s), 2.18 (3H, s), 2.14, (6H, s), EI-MS 398.0; λmax/nm (acetonitrile) 289.
Reaction of (PPh4)[Ni(LS)(MesS)] with oxygen
(PPh4)[Ni(LS)(MesSe)] (30.7 mg, 38.4 μmol) in dichloromethane (5 mL) was exposed to atmospheric O2 for five days under stirring to give a brown solution containing dimesityl disulfide with a black precipitate, characterised as [Ni(LS)]n. Isolation and characterisation of precipitate containing [Ni(LS)]n: The solid was separated by filtration and washed with dichloromethane, methanol and diethyl ether and dried under vacuum. ATR-IR spectroscopy and elemental analysis identified the solid product as [Ni(LS)]n12l (9.6 mg, 86%). Isolation and characterisation of filtrate containing (MesS)2: Diethyl ether (15 mL) was added to the filtrate to precipitate a tetraphenyl phosphonium salt as a pink solid which was separated by filtration, washed with diethyl ether and dried under vacuum (11 mg). The solvent was removed from the resulting filtrate to give crude dimesityl disulfide as a pale yellow solid, which was purified by filtration through silica in diethyl ether (5.8 mg, 80%). 1H NMR (400 MHz, CDCl3) δ/ppm = 6.82 (2H, s), 2.18 (3H, s), 2.14 (6H, s).Reaction of (n-Bu4N)[Ni(LE)(MesE′)] with oxygen
Reaction of (n-Bu4N)[Ni(LE)(MesE′)] with a n-Bu4N cation gives the dichalcogenides (MesE′)2 along with a mixture of insoluble products which could not be unequivocally isolated and characterised.Reaction of (n-Bu4N)[Ni(LS)(MesSe)] with HBF4
To a solution of (n-Bu4N)[Ni(LS)(MesSe)] (20 mg, 26.7 μmol) in dichloromethane (3 mL) was added HBF4·Et2O (3.7 μL, 26.7 μmol) in dichloromethane (2 mL) and the reaction mixture was stirred for 30 min, whereupon [Ni(LS)]n (5.5 mg, 64%) formed as a black precipitate. The solid was separated by filtration and washed with dichloromethane, methanol and diethyl ether, dried under vacuum and characterised by ATR-IR and elemental analysis. Diethyl ether was added to the remaining filtrate to precipitate crude n-Bu4NBF4, which was separated by filtration, washed with diethyl ether and dried under vacuum. 1H NMR (400 MHz, CDCl3) δ/ppm = 3.20 (8H, m), 1.62 (8H, m), 1.44 (8H, m), 1.00 (12 H, t), ESI MS (CHCl3); −ive: 87 (100%, BF4−) +ive: 242 (100%, n-Bu4N+). Diethyl ether was removed from the remaining filtrate under vacuum to yield mesityl selenol (4.3 mg, 79%). 1H NMR (400 MHz, CDCl3) δ/ppm = 6.88 (2H, s), 2.34 (6H, s), 2.23 (3H, s), 1.24 (1H, s).Reaction of (n-Bu4N)[Ni(LS)(MesS)] with HBF4
To a solution of (n-Bu4N)[Ni(LS)(MesS)] (50 mg, 71.3 μmol) in dichloromethane (4 mL) was added HBF4·Et2O (9.7 μL, 71.3 μmol) in dichloromethane (2 mL) and this was stirred for 30 min. [Ni(LS)]n (20.9 mg, 83%) and mesityl thiol (8 mg, 74%) formed in analogy to the reaction of (n-Bu4N)[Ni(LS)(MesSe)] with HBF4 and were characterised by the same methods. 1H NMR of mesityl thiol (400 MHz, CDCl3) δ/ppm = 6.81 (2H, s), 3.04 (1H, s), 2.27 (6H, s), 2.17 (3H, s).Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EP/H00338X/2), the Christian Doppler Research Association and the OMV group. We thank Mr Dirk Mersch for recording SEM images and Dr John Davies for collecting and refining the crystallographic data and providing a letter about refinement of the X-ray structures (see ESI†).Notes and references
- (a) Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed; (b) S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- (a) F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed; (b) E. S. Andreiadis, P.-A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- (a) S. V. Hexter, F. Grey, T. Happe, V. Climent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS PubMed; (b) A. Abou Hamdan, S. Dementin, P.-P. Liebgott, O. Gutierrez-Sanz, P. Richaud, A. L. De Lacey, M. Rousset, P. Bertrand, L. Cournac and C. Léger, J. Am. Chem. Soc., 2012, 134, 8368–8371 CrossRef CAS PubMed.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS.
- (a) F. M. A. Valente, A. S. F. Oliveira, N. Gnadt, I. Pacheco, A. V. Coelho, A. V. Xavier, M. Teixeira, C. M. Soares and I. A. C. Pereira, J. Biol. Inorg. Chem., 2005, 10, 667–682 CrossRef CAS PubMed; (b) A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS PubMed; (c) P. M. Vignais, L. Cournac, E. C. Hatchikian, S. Elsen, L. Serebryakova, N. Zorin and B. Dimon, Int. J. Hydrogen Energy, 2002, 27, 1441–1448 CrossRef CAS; (d) E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed; (e) F. M. A. Valente, C. C. Almeida, I. Pacheco, J. Carita, L. M. Saraiva and I. A. C. Pereira, J. Bacteriol., 2006, 188, 3228–3235 CrossRef CAS PubMed.
- (a) C. Gutiérrez-Sánchez, O. Rüdiger, V. M. Fernández, A. L. DeLacey, M. Marques and I. A. C. Pereira, J. Biol. Inorg. Chem., 2010, 15, 1285–1292 CrossRef PubMed; (b) K. Nonaka, N. T. Nguyen, K.-S. Yoon and S. Ogo, J. Biosci. Bioeng., 2013, 115, 366–371 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- (a) B. Bleijlevens, F. A. van Broekhuizen, A. L. DeLacey, W. Roseboom, V. M. Fernandez and S. P. J. Albracht, J. Biol. Inorg. Chem., 2004, 9, 743–752 CrossRef CAS PubMed; (b) A. J. Pierik, W. Roseboom, R. P. Happe, K. A. Bagley and S. P. J. Albracht, J. Biol. Chem., 1999, 274, 3331–3337 CrossRef CAS PubMed; (c) C. Fichtner, C. Laurich, E. Bothe and W. Lubitz, Biochemistry, 2006, 45, 9706–9716 CrossRef CAS PubMed; (d) M. Carepo, D. L. Tierney, C. D. Brondino, T. C. Yang, A. Pamplona, J. Telser, I. Moura, J. J. G. Moura and B. M. Hoffman, J. Am. Chem. Soc., 2002, 124, 281–286 CrossRef CAS PubMed; (e) M. van Gastel, C. Fichtner, F. Neese and W. Lubitz, Biochem. Soc. Trans., 2005, 33, 7–11 CrossRef CAS PubMed; (f) M. Brecht, M. van Gastel, T. Buhrke, B. Friedrich and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 13075–13083 CrossRef CAS PubMed; (g) S. Foerster, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83–93 CrossRef CAS PubMed; (h) K. K. Surerus, M. Chen, J. W. van der Zwaan, F. M. Rusnak, M. Kolk, E. C. Duin, S. P. J. Albracht and E. Munck, Biochemistry, 1994, 33, 4980–4993 CrossRef CAS; (i) M.-E. Pandelia, P. Infossi, M. Stein, M.-T. Giudici-Orticoni and W. Lubitz, Chem. Commun., 2012, 48, 823–825 RSC.
- (a) P. M. Matias, C. M. Soares, L. M. Saraiva, R. Coelho, J. Morais, J. Le Gall and M. A. Carrondo, J. Biol. Inorg. Chem., 2001, 6, 63–81 CrossRef CAS; (b) A. Volbeda, Y. Montet, X. Vernède, E. C. Hatchikian and J. C. Fontecilla-Camps, Int. J. Hydrogen Energy, 2002, 27, 1449–1461 CrossRef CAS; (c) Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549–556 CrossRef CAS; (d) H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS PubMed; (e) A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2005, 10, 239–249 CrossRef CAS PubMed; (f) M. C. Marques, R. Coelho, A. L. De Lacey, I. A. C. Pereira and P. M. Matias, J. Mol. Biol., 2010, 396, 893–907 CrossRef CAS PubMed; (g) M. C. Marques, R. Coelho, I. A. C. Pereira and P. M. Matias, Int. J. Hydrogen Energy, 2013, 38, 8664–8682 CrossRef CAS PubMed; (h) A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061–7063 RSC.
- The oxidised D. vulgaris [NiFeSe] hydrogenase was reported to exist in three different structural conformers. The source of the extrinsic sulfur atom S is presumably H2S.
- R. E. Huber and R. S. Criddle, Arch. Biochem. Biophys., 1967, 122, 164–173 CrossRef CAS.
- (a) S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed; (b) K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745–20755 CrossRef CAS PubMed; (c) D. Schilter, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 8931–8941 CrossRef CAS PubMed; (d) S. Canaguier, M. Field, Y. Oudart, J. Pecaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876–5878 RSC; (e) S. Tanino, Z. Li, Y. Ohki and K. Tatsumi, Inorg. Chem., 2009, 48, 2358–2360 CrossRef CAS PubMed; (f) A. Perra, E. S. Davies, J. R. Hyde, Q. Wang, J. McMaster and M. Schroder, Chem. Commun., 2006, 1103–1105 RSC; (g) R. Angamuthu and E. Bouwman, Phys. Chem. Chem. Phys., 2009, 11, 5578–5583 RSC; (h) A. Begum, G. Moula and S. Sarkar, Chem.–Eur. J., 2010, 16, 12324–12327 CrossRef CAS PubMed; (i) D. Sellmann, F. Geipel and F. W. Heinemann, Eur. J. Inorg. Chem., 2000, 271–279 CrossRef CAS; (j) C.-H. Chen, G.-H. Lee and W.-F. Liaw, Inorg. Chem., 2006, 45, 2307–2316 CrossRef CAS PubMed; (k) C.-M. Lee, C.-H. Chen, S.-C. Ke, G.-H. Lee and W.-F. Liaw, J. Am. Chem. Soc., 2004, 126, 8406–8412 CrossRef CAS PubMed; (l) D. Sellmann, D. Häußinger and F. W. Heinemann, Eur. J. Inorg. Chem., 1999, 1715–1725 CrossRef CAS; (m) C. A. Marganian, H. Vazir, N. Baidya, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 1995, 117, 1584–1594 CrossRef CAS; (n) N. Baidya, B. C. Noll, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1992, 31, 2999–3000 CrossRef CAS.
- M. K. Harb, H. Görls, T. Sakamoto, G. A. N. Felton, D. H. Evans, R. S. Glass, D. L. Lichtenberger, M. El-khateeb and W. Weigand, Eur. J. Inorg. Chem., 2010, 3976–3985 CrossRef CAS.
- M. Yus, F. Foubelo and J. V. Ferrández, Tetrahedron Lett., 2002, 43, 7205–7207 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- (a) H.-h. Cui, J.-y. Wang, M.-q. Hu, C.-b. Ma, H.-m. Wen, X.-w. Song and C.-n. Chen, Dalton Trans., 2013, 42, 8684–8691 RSC; (b) K. N. Green, S. M. Brothers, R. M. Jenkins, C. E. Carson, C. A. Grapperhaus and M. Y. Darensbourg, Inorg. Chem., 2007, 46, 7536–7544 CrossRef CAS PubMed.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed.
- (a) E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef PubMed; (b) S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed; (c) N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- N. P. Dasgupta, C. Liu, S. Andrews, F. B. Prinz and P. Yang, J. Am. Chem. Soc., 2013, 135, 12932–12935 CrossRef CAS PubMed.
- G. G. Briand, A. Decken and N. S. Hamilton, Dalton Trans., 2010, 39, 3833–3841 RSC.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University Göttingen, Göttingen, Germany, 1997 Search PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2001 Search PubMed.
- We also purified and characterised (‘LSe–H’)2 from the crude product using column chromatography (Sephadex LH-20) under a N2 atmosphere. 1H NMR (400 MHz, CD2Cl2) δ = 7.66 (2H, m), 7.40, (2H, d), 7.15–7.25 (8H, m), 7.07–7.10 (4H, m), 4.14 (2H, s, SH); ESI-MS (CH2Cl2) −ive: 296.9 (‘LSe–H’)−.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting table, figures and X-ray structure refinement details. CCDC 975187 (n-Bu4N)[Ni(LSe)(MesS)], 939413 (n-Bu4N)[Ni(LS)(MesS)], 939414 (n-Bu4N)[Ni(LSe)(MesSe)] and 939415 (n-Bu4N)[Ni(LS)(MesSe)]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52967c |
| This journal is © The Royal Society of Chemistry 2014 |