 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homoleptic aminophenolates of Zn, Mg and Ca. Synthesis, structure, DFT studies and polymerization activity in ROP of lactides†
Jakub
Wojtaszak
,
Krzysztof
Mierzwicki
,
Sławomir
Szafert
,
Nurbey
Gulia
and
Jolanta
Ejfler
*
Department of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: jolanta.ejfler@chem.uni.wroc.pl; Tel: +48 (71) 375 73 10
First published on 20th November 2013
Abstract
The reaction of MgBu2, ZnEt2 or Ca(OiPr)2 with 2 eq. of three-coordinating N-[methyl(2-hydroxy-3,5-dimethylphenyl)]-N-methyl-N-methyl-1,3-oxolaneamine (mpoa-H) or N-[methyl(2-hydroxy-3,5-di-tert-butylphenyl)]-N-methyl-N-methyl-1,3-oxolaneamine (tbpoa-H) gave neutral, monomeric [Mg(mpoa)2], [Zn(mpoa)2], [Zn(tbpoa)2], and [Ca(tbpoa)2] as white powders in 58–90% yields. The resulting aminophenolates were characterized in solution by NMR showing, in the case of [Zn(tbpoa)2], interesting dynamics. [Zn(tbpoa)2] and [Ca(tbpoa)2] were characterized by X-ray crystallography to show the Zn atom to be pseudo-octahedrally coordinated and the Ca atom in six-coordination mode. The new homoleptic complexes were tested in the polymerization of lactide with an external alcohol to reveal stable behaviour (during the polymerization process) only in the case of [Zn(tbpoa)2]. The high activity of the catalyst was correlated with a ligand flexibility that was further supported by theoretical studies.
Introduction
Over the past two decades biodegradable polymers have attracted increasing attention as the subject of fundamental research and as products of the chemical industry.1 One of the most prominent examples of such molecules is polylactide (PLA), which is presently developed as a commodity polymer for packaging (bottles and thin films), fibres (tissue and clothes), as well as for biomedical applications as bioresorbable sutures, screws, orthopedic implants, drug delivery agents or scaffolds for tissue engineering.2 Due to its favorable material properties and the fact that it can be produced from inexpensive renewable sources, PLA is qualified to be a viable alternative to petrochemical-based plastics.3Excellent reviews have recently appeared, describing the most effective method for the synthesis of PLAs, i.e. ring-opening polymerization (ROP) of lactides catalyzed by metal alkoxides.4 A wide variety of different kinds of complexes have been used for this purpose, comprising not only compounds of biologically benign metals like lithium, sodium, magnesium, zinc, calcium, and iron, but also more or even highly toxic ones like aluminum, tin, lead, bismuth, and lanthanides. Nevertheless, since it is practically unviable to completely remove catalyst residues from the polymer, which is important for biomedical applications and green packages, the most interesting remain environmentally friendly non-toxic catalysts.
Among the galore of tested catalysts, the so-called well-defined heteroleptic catalysts of LnM-OR type possess a great advantage owing to their ability to facilitate ring-opening polymerization with control of both molecular weight and polymer microstructure. For a specific “single-site” LnM-OR catalyst, the relative rate of ROP correlates well with an M–O bond polarity. For example, for a given ligand environment, the relative rate of ROP changes in the order Ca+2 > Mg+2 > Zn+2.5 An excellent study of a family of “single-site” divalent metal initiators supported by β-diketiminate, trispyrazolylborate or amino/imino-phenolate6 ligands, which included derivatives based on Zn, Mg, Ca, and Sn, has been reported.7 Such initiators illustrate the anticipated trends in polymerization rates, correlating well with the size of an initiating group and the electronic properties of ancillary ligand substituents.8
For the above mentioned LnM-OR initiators, it is important to modify a metal coordination sphere by an ancillary ligand with sufficient steric bulk to prevent bischelation, which process is considered to represent a deactivation pathway by the formation of inactive ML2 compounds.9 Therefore, homoleptic metal complexes have not been qualified as potentially effective initiators for ROP of lactides. However, the efficacy of ligands stabilizing heteroleptic LnM-OR (M = Ca, Mg, Zn) complexes is arguable due to high lability of such species. This seems especially apparent for initiators derived in situ from organometallic LnM-R precursors. It also seems difficult to ensure the stability of such compounds towards an excess of external alcohol in the case of iROP (immortal ROP).
An increasing demand for highly active, non-toxic, colourless, inexpensive, and stable complexes which can be easily handled is forcing the development of an alternative to single-site initiators. Among those, very attractive are catalytic systems for the monomer activated pathway based on homoleptic compounds of ML2 type combined with an external alcohol.10 In this regard, the recent study on homoleptic magnesium and zinc catalyst supported by bulky N,O-donor ligands is very promising.11 As we have recently reported, the aminophenolate ligands are able to form homoleptic zinc and magnesium monomers, which combine great potential as active catalysts in the ROP of lactide with an acceptable stability.12 Additionally, our studies indicate the dynamic behavior of these coordinatively saturated complexes in solution, which can be crucial for both stability and catalytic activity in ML2/ROH systems.
A more in-depth mechanistic understanding of the activation process of aminophenolate complexes in lactide polymerization as well as the way in which the structural “perturbations” of the active centre and reaction conditions influence their catalytic activity constitutes the aim of the current research. Herein we have described the synthesis and characterization of magnesium, calcium, and zinc complexes supported by the aminophenolate ligands with hemilabile arms containing additional ether O-donor and their application as initiators for lactide polymerization. The study extensively correlates the experimental outcomes with DFT calculations to rationalize the results.
Experimental section
General materials, methods and procedures
All the reactions and operations were performed under an inert atmosphere of N2 using a glove-box (MBraun) or standard Schlenk techniques. Reagents were purified by standard methods: THF, distilled from Na/benzophenone; toluene, distilled from Na; CH2Cl2, distilled from P2O5; hexanes, distilled from Na; methanol, distilled from Mg; C6D6, distilled from CaH2. L-LA ((3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione) (98%; Aldrich) was sublimed and recrystallized from toluene prior to use. Benzyl alcohol (Aldrich) was distilled prior to use. ZnEt2 (1.0 M solution in hexanes), MgBu2 (2.5 M solution in hexanes), Ca(OiPr)2 (99.9+%), 2,4-dimethylphenol (98%), 2-methylaminomethyl-1,3-dioxolane (98%), and formaldehyde (37% solution in H2O) were purchased from Aldrich and used as received. The ligand tbpoa-H was prepared according to the literature.12a1H and 13C NMR spectra were detected in the temperature range from 233 K to 333 K using Bruker ESP 300E or 500 MHz spectrometers. Chemical shifts are reported in parts per million and referenced to the residual solvent signal. The weights and number-average molecular weights of PLAs were determined by gel permeation chromatography (GPC) using a HPLC-HP 1090 II with a DAD-UV/Vis and an RI detector HP 1047A and polystyrene calibration. Microanalyses were conducted with an ARL Model 3410+ICP spectrometer (Fisons Instruments) and a VarioEL III CHNS (in-house).
Syntheses
Representative procedure for solution polymerization
In a typical experiment, the monomer L-LA and a solution of a metal complex (M) in CH2Cl2 were placed in a Schlenk flask at a fixed molar ratio. Then, after 10 minutes an external alcohol in stoichiometric amount (M/ROH = 1/1) was added. The reaction was stirred at the desired temperature for a prescribed time. At certain time intervals, about 1 mL aliquots were removed, precipitated with hexanes, and dried in vacuo. A conversion was determined observing 1H NMR resonances of the polymer and the monomer by dissolving the precipitates in C6D6. After the reaction was completed, an excess of hexanes was added to the reaction mixture. Filtration and vacuum drying yielded a white polymer.Details of X-ray data collection and reduction
[Zn(mpoa)2] and [Ca(tbpoa)2] (separately) were dissolved in CH2Cl2 and placed in a freezer at −15 °C. After several days, colourless, good quality crystals had formed. X-ray diffraction data for [Zn(mpoa)2]·CH2Cl2 and [Ca(tbpoa)2]·C6H5CH3 were collected using a KUMA KM4 CCD (ω scan technique) diffractometer equipped with an Oxford Cryosystem-Cryostream cooler.13 The space groups were determined from systematic absences and subsequent least-squares refinement. Lorentz and polarization corrections were applied. The structures were solved by direct methods and refined by full-matrix-least squares on F2 using SHELXTL Package.14 Scattering factors were taken from the literature.15 Non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atom positions were calculated and added to the structure factor calculations, but were not refined. All data (except structure factors) have been deposited with the Cambridge Crystallographic Data Centre or as supplementary publications CCDC-962646 and CCDC-962645. The solvated CH2Cl2 in [Zn(mpoa)2]·CH2Cl2 was distorted and it was refined with FVAR of 0.69.Computational methods
Theoretical calculations of harmonic vibrational frequencies, chemical shifts and energies for calcium, magnesium and zinc complexes were performed using TURBOMOLE 6.316 and Gaussian 09.17 During optimizations and frequency calculations C2 point group symmetry constraints (for Ca-, Mg-, and few Zn-complexes), m4 grid (in TURBOMOLE notation), tight SCF convergence criteria, and density fitting approach (resolution-of-identity, RI) have been used. Geometries from crystal structure investigations were taken as a starting point for the full gas-phase optimization. Two terminal tBu groups were replaced by hydrogen atoms as shown in Fig. 1. | ||
| Fig. 1 Schematic structure (with numbering of selected atoms; H atoms omitted except for the CH protons of dioxolane rings) of [ML2] complexes (M = Ca, Mg, Zn). | ||
Since the X-ray data showed two slightly different isomers for the zinc complex, both structures were considered in subsequent studies. In the first stage, geometry optimization and vibrational analysis were performed at the TPSS-D3/def2-SVPD level18–20 with respective default auxiliary basis sets,21 where TPSS-D3 means the meta-GGA functional with Grimme's D3 dispersion correction.22 Though for like-charged species (for example two ligands in considered [ML2] complexes) the Coulombic repulsion is usually dominant, as was recently shown by Grimm et al., going from point charges model to real ions one should also take into account London dispersion attraction. In some cases the dispersion not only overcomes electrostatic repulsion but also the entropy penalty of complex formation.22 Therefore the dispersion correction seems to be important not only for quantitative but even for correct qualitative description of such molecules. All the vibration frequencies in each studied complex were real. This proves that the obtained structures are true minima. Such calculated molecules were reoptimized at the TPSS-D3/def2-TZVPPD level20,23 (with respective auxiliary basis sets taken from TURBOMOLE library). This functional was chosen because – as we found – for the considered molecules it gives results which are quite close to MP2 ones. The NMR results suggested that for the zinc complex, depending on temperature, dioxolane arms can be bound or non-bound. Therefore we optimized these molecules with different dioxolane ring positions and found additional isomers.
Isotropic 1H chemical shifts were computed (with the Gaussian 09 package) as a difference between chemical shieldings of reference hydrogen atom in tetramethylsilane (TMS, optimized at the TPSS-D3/def2-TZVPPD level of theory) and proton chemical shieldings in considered complexes. All these values were obtained using the gauge independent atomic orbital (GIAO) method24 for the gas-phase geometries. We employed WP04 functional, proposed recently by Wiitala et al.25 and 6-31G(d,p) basis set. ChemCraft package has been used to visualize some of the results.26
Results and discussion
Synthesis and solid-state determination
Monomeric, homoleptic magnesium, calcium or zinc alkoxides, like similar alkoxides of other metals, are rare. Their high tendency for bridging must be suppressed by the steric bulk of an alkoxo ligand and therefore proposed ligands contain obstructed ortho and para positions of the phenol moiety as well as a hemilabile amino-arm with a coordinating dioxolane ring.Aminophenolates mpoa-H and N-[methyl(2-hydroxy-3,5-di-tert-butylphenyl)]-N-methyl-N-methyl-1,3-dioxolaneamine (tbpoa-H) were prepared according to modified Mannich condensation27 using respective disubstituted phenol, paraformaldehyde, and 2-methylaminomethyl-1,3-dioxolane as described in previous literature12 and in the Experimental section. Next, both ligands were used for complex syntheses. In the first thrust, a solution of mpoa-H was slowly treated with 2.5 M solution of MgBu2 (0.5 eq.) in hexanes at room temperature to give a white solid of [Mg(mpoa)2] in 90% yield (after recrystallization from toluene) as shown in Scheme 1.
 | ||
| Scheme 1 Synthesis of homoleptic Zn, Mg, and Ca aminophenolates. | ||
Analogous reactions of mpoa-H or tbpoa-H with ZnEt2 (1 M solution in hexanes) or tbpoa-H with Ca(OiPr)2 gave analytically pure [Zn(mpoa)2], [Zn(tbpoa)2], and [Ca(tbpoa)2] in 84–58% yield as white solids. Only in the case of [Ca(tbpoa)2] a longer reaction time and an elevated temperature were applied. Compounds with a tbpoa ligand are well soluble in toluene, CH2Cl2 or THF, while the solubility of aminophenolates with methyl substituents [Mg(mpoa)2] and [Zn(mpoa)2] appear much poorer in these solvents. All compounds are insoluble in aliphatic hydrocarbons.
The complexes were characterized by 1H and 13C NMR spectroscopy, which showed complicated dynamics in solution. Magnesium compound exhibited (both in 1H and 13C NMR spectra) multiple signals of the OCHO group of the dioxolane ring. In the proton spectrum there were two broad signals positioned at δ: 5.69 and 5.49 ppm. Also the 13C spectrum showed two signals of OCHO carbon, which were located at δ: 101.8 and 102.9 ppm. Moreover, the signal of the neighbouring methylene N–CH2–CH protons appeared as a multiplet in the δ range: 3.40–3.28 ppm. The results suggest the coordination of just one dioxolane arm to the metal center in [Mg(mpoa)2] or the so-called “gorilla” effect (quick coordination and decoordination of both dioxolane rings interchangeably). Interestingly, in [Zn(mpoa)2] the 1H NMR spectrum showed just one well resolved triplet of the OCHO group at δ: 5.25 ppm and the neighbouring methylene N–CH2–CH signal appeared as a doublet at δ: 2.63 ppm. Also the 13C NMR spectrum of [Zn(mpoa)2] exhibited a single peak of the OCHO group at δ: 102.9 ppm. This might be an effect of a slightly higher ionic radius of Zn(II) compared to Mg(II), which enables a better coordination of both dioxolane rings.28 An exchange of the substituents in the phenolic part of the aminophenolate ligand for larger the tBu group resulted in a “tighter” arrangement around the metal center, which affected its coordination mode. In [Zn(tbpoa)2], once again the 1H and 13C NMR spectra showed multiple signals for the OCHO group. The proton spectrum showed two broadened signals at δ: 5.22 and 5.13 ppm and a multiplet of the adjacent methylene N–CH2–CH group in the δ range: 2.53–2.25 ppm. Interestingly the signal of the methylene N–CH2–Ar protons in [Zn(tbpoa)2] appeared as a pair of doublets, which might suggest that they became diastereotopic as shown in Fig. 2. The effect is similar to that observed for [Mg(mpoa)2] and suggests a similar “gorilla” mechanism.
 | ||
| Fig. 2 The signals of methane OCHO (marked with bA, bB) and methylene N–CH2–Ar protons (marked with aA, aB) belonging to possible isomers of [Zn(tbpoa)2] at room temperature (in d6-benzene). The signal of methane OCHO proton of the internal standard (free tbpoa-H) was marked with bC. | ||
The other explanation could be stereochemistry at the N centers (the possible isomers for [Zn(tbpoa)2] are SS, RR and R,S). This could be supported by the appearance of two signals of the methyl N–CH3 groups in 13C NMR at δ: 42.7 and 43.8 ppm. This observation indicates that decoordination of the dioxolane moiety causes stronger coordination of an N arm of the aminophenolate ligand. In order to better understand the nature of the phenomenon, a variable temperature NMR experiment was performed in d8-toluene as shown in Fig. 3. Noticeably, signals of all the methylene and methine groups coalesce at 333 K. This nice triplet of the methine group can be observed, which proves the existence of pendant dioxolane ring. It is worth mentioning that such bond-dangling quick (on the NMR time scale) exchange of the coordination mode may have important implications on the catalytic properties of this complex. Although some other isomers are possible for [Zn(tbpoa)2] arising from different coordinations of dioxolane rings or from the general geometry around the central atom, there was no indication of those in the NMR or X-ray data (similarly as for [Ca(tbpoa)2]).
 | ||
| Fig. 3 Variable temperature 1H NMR spectra for [Zn(tbpoa)2]. The signal of methane OCHO proton of the internal standard (free tbpoa-H) was marked with bC. | ||
In [Ca(tbpoa)2], despite the larger tBu substituent at the phenolic moiety, the larger ionic radius of the calcium ion enabled octahedral coordination. The 1H NMR spectra showed a single broadened signal of the OCHO proton at δ: 5.20 ppm and a multiplet in the δ range: 2.78–2.54 ppm indicating some dynamics. A single signal was observed for the OCHO group in 13C NMR for [Ca(tbpoa)2] at δ: 103.7 ppm.
X-ray crystallography
Needle-shaped, colorless crystals of [Zn(tbpoa)2]·CH2Cl2 and [Ca(tbpoa)2]·C6H5CH3 were grown by slow evaporation of dichloromethane and toluene solution, respectively, and their molecular structures were determined as outlined in Table 1 and summarized in the Experimental section.| [Zn(tbpoa)2]·CH2Cl2 | [Ca(tbpoa)2]·C6H5CH3 | |
|---|---|---|
| Molecular formula | C41Cl2H66N2O6Zn | C47H72CaN2O6 |
| Molecular weight | 819.23 | 801.15 |
| Temp. of collection (K) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Triclinic |
| Space group | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 18.403(4) | 10.293(3) |
| b [Å] | 19.279(4) | 15.167(4) |
| c [Å] | 25.718(5) | 16.176(4) |
| α [°] | 90 | 81.41(3) |
| β [°] | 110.61(2) | 74.00(3) |
| γ [°] | 90 | 71.62(3) |
| V [Å3] | 8540(3) | 2298.2(11) |
| Z | 8 | 2 |
| d c [g cm−3] | 1.274 | 1.158 |
| μ [mm−1] | 0.746 | 0.184 |
| Crystal dimensions [mm] | 0.33 × 0.20 × 0.03 | 0.24 × 0.18 × 0.15 |
| Θ Range [°] | 2.61 ≤ Θ ≤ 25.04 | 2.78 ≤ Θ ≤ 28.62 |
| Range/indices (h,k,l) | −19,21; −22,22; −30,30 | −13,13; −20,20; −21,21 |
| No. of reflections | 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 167 167 |
20255 |
| No. of unique data | 7540 | 10595 |
| No. of observed data | 4984 [I > 2σ(I)] | 4007 [I > 2σ(I)] |
| No. refined parameters | 489 | 508 |
| R int | 0.0695 | 0.0688 |
| R indices [I > 2σ(I)] | R 1 = 0.0765 | R 1 = 0.0688 |
| wR2 = 0.1994 | wR2 = 0.1393 | |
| R indices (all data) | R 1 = 0.1143 | R 1 = 0.1862 |
| wR2 = 0.2274 | wR2 = 0.1799 | |
| Goodness of fit | 1.048 | 0.900 |
| Largest diff. peak hole [e Å−3] | 1.607, −0.716 | 0.812, −0.598 |
Interatomic distances and angles have been provided in Table 2 and a view of each structure is given in Fig. 4 and 5.
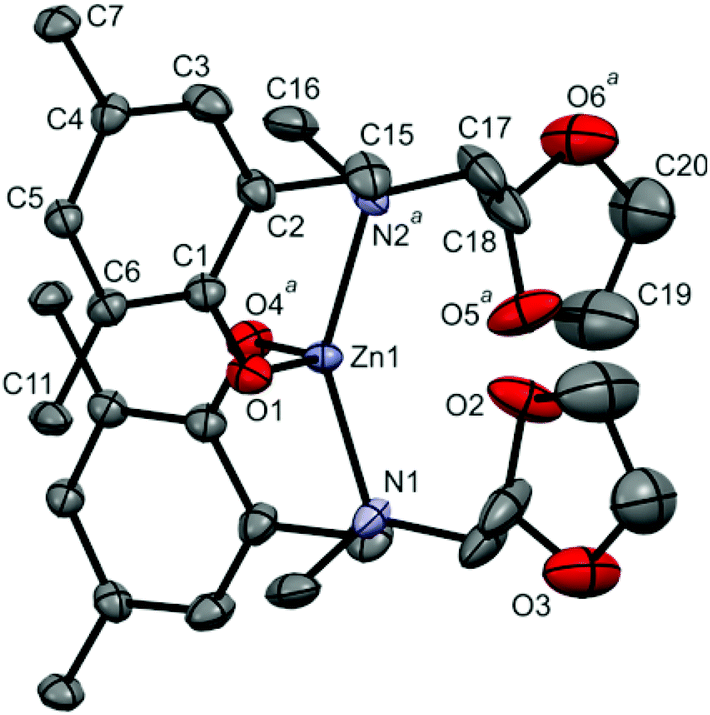 | ||
| Fig. 4 The view of [Zn(tbpoa)2]·CH2Cl2 molecule (solvated CH2Cl2, H-atoms and methyl group carbons from tBu substituents were omitted for clarity; atoms with superscript a are symmetry related, see Table 2 caption). | ||
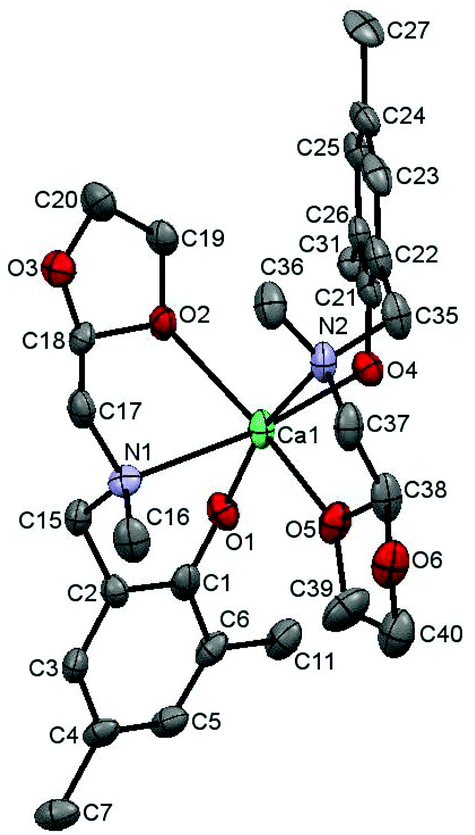 | ||
| Fig. 5 The view of [Ca(tbpoa)2]·C6H5CH3 molecule (solvated toluene, H-atoms and methyl group carbons from tBu substituents were omitted for clarity). | ||
| Atoms | Angles | ||
|---|---|---|---|
| a Atoms N(2), O(4), and O(5) are related to N(1), O(1), and (O2), respectively, by symmetry operation: 1 − x, y, 1.5 − z. b Values in parentheses are for the non-bonding interactions. | |||
| N(1)–M–N(2)a | 147.4(2) | 148.5(3) | 100.04(9) |
| N(1)–M–O(1) | 96.20(16) | 96.25(17) | 78.70(9) |
| N(1)–M–O(2) | (71.03)b | (71.68)b | 68.74(9) |
| N(1)–M–O(4)a | 98.99(16) | 100.0(2) | 171.73(9) |
| N(1)–M–O(5)a | (82.01)b | (84.32)b | 91.64(9) |
| N(2)a–M–O(1) | 98.99(16) | 100.0(2) | 165.81(10) |
| N(2)a–M–O(2) | (82.01)b | (84.32)b | 86.71(9) |
| N(2)a–M–O(4)a | 96.20(16) | 96.25(17) | 80.16(9) |
| N(2)a–M–O(5)a | (71.03)b | (71.68)b | 67.07(9) |
| O(1)–M–O(2) | (151.07)b | (159.58)b | 105.68(9) |
| O(1)–M–O(4)a | 123.8(2) | 117.1(4) | 103.14(9) |
| O(1)–M–O(5)a | (84.52)b | (81.87)b | 98.77(10) |
| O(2)–M–O(4)a | (84.52)b | (81.87)b | 103.07(9) |
| O(2)–M–O(5)a | (68.34)b | (80.60)b | 144.36(9) |
| O(4)a–M–O(5)a | (151.07)b | (159.58)b | 96.01(9) |
The zinc and calcium solvates [Zn(tbpoa)2]·CH2Cl2 and [Ca(tbpoa)2]·toluene crystallize in C2/c (monoclinic) and P (triclinic) space groups, respectively. No internal or external hydrogen bonds were observed for both structures. There are two independent molecules of [Zn(tbpoa)2]·CH2Cl2 in the unit cell, each lying at the two fold axis. In both, zinc(II) centers possess analogous four coordinated arrangement, where each metal is surrounded by two pairs of N,O atoms from two aminophenolates.12b,29
Nevertheless, a closer look at the positions of the cis-sited dioxolane substituents shows that although the arms are dangling they remain in close proximity to the metal center to form a pseudooctahedral arrangement. Interestingly, the dioxolane O atoms being closer to the metal atoms differ significantly in the M–O distance of 3.007(4) and 2.725(5) Å. The pseudooctahedral arrangement is further supported by the bond angles around the metal centers, as shown in Table 2. Also here the O–M–O angles formed by O atoms from dioxolane rings are markedly different for the two independent molecules (68.3 vs. 80.6°). The observations – although made for a solid state – support the NMR data, which showed important dynamics of the ligands in [Zn(tbpoa)2]. The Zn(1)–N(1) and Zn(2)–N(2) distances of 2.092(4) and 2.106(4) Å and Zn(1)–O(1) and Zn(2)–O(2) distances of 1.923(4) and 1.933(5) Å are similar to those found in Zn(tbpca)2 (L = N-[methyl(2-hydroxy-3,5-di-tert-butylphenyl)]-N-methyl-N-cyclohexylamine; Zn–O(1) = 1.909(2) and Zn–N(1) = 2.130(2) Å), [Zn(L)]·H2O (L = 1-ethyl-4,7-bis(3-tert-butyl-5-methoxy-2-hydroxybenzyl)-1,4,7-triazacyclononane; Zn–O = 1.963(1) and 1.934(1) Å; Zn–N = 2.113(1) and 2.277(1) Å)27 and [ZnL2] (L = N-(2-hydroxy-5-nitrobenzyl)-(R)-a-methylbenzylamine; Zn–O = 1.935(2) and 1.933(2) Å; Zn–N = 2.0426(19) and 2.0458(19) Å).29
Although the solid structure of [Ca(tbpoa)2]·C6H5CH3 is also a molecular monomer, it substantially differs from that of [Zn(tbpoa)2]·CH2Cl2. As has already been revealed by the NMR data, both dioxolane rings in [Ca(tbpoa)2]·C6H5CH3 are coordinated to the metal center and remain in trans arrangement. The Ca–O(2) and Ca–O(5) distances are 2.401(2) and 2.377(3) Å, respectively. The nitrogen atoms are cis one to the other and so are the phenoxo oxygens. The Ca–N(1) and Ca–N(2) distances are 2.608(3) and 2.666(3) Å and Ca–O(1) and Ca–O(4) are 2.189(3) and 2.210(2) Å. The octahedron around the central atom is substantially distorted, which can be concluded from the bond angles in Table 2.
Lactide polymerization
As has already been mentioned, a catalytic system based on a homoleptic complex ML2/ROH combination for “the activated monomer pathway” has been proposed for ROP of cyclic esters, as an alternative to “single-site” LnM-OR catalysts. As shown above, the aminophenolate ligands are able to form, with M(II) ions, labile monomeric complexes whose dynamic behaviour in solution can be crucial for catalytic activity in ML2/ROH systems. The focus of our attention has now been shifted towards verification of the reactivity of [M(tbpoa)2] (M = Zn, Ca) and [M(mpoa)2] (M = Mg, Zn) as initiators in lactide polymerization. After running several trials of the polymerization in THF, CH2Cl2, and toluene at 298–323 K, the latter became the best choice for these systems. Nevertheless, poor solubility of [M(mpoa)2] and low activity of [Ca(tbpoa)2] excluded them from further experiments. Moreover, the calcium complex in the presence of alcohols loses aminophenolate ligands very easily and an insoluble mixture of calcium compounds is formed. Ligand displacement is a prominent feature of calcium complexes and the aminophenolate [Ca(tbpoa)2] complex appeared to be no exception. Instead, the most labile (according to the NMR data) compound [Zn(tbpoa)2] appeared as efficient initiators for the polymerization of L-LA.Regardless of its low solubility, the magnesium complex [Mg(mpoa)2] was reacted with 50 equiv. of L-LA and BzOH (BzOH = benzyl alcohol) was added (1 eq.). It achieved high conversion in 15 min and gave out polymers with moderate Mw = 8700 and PDI = 1.1. The polymerization process is living and polymer chains are terminated by aminophenolate and hydroxyl groups. The polymerization results and NMR study are consistent with the DFT study (see below), indicating a possibility of equilibrium of five and six coordinated magnesium species. Usually an octahedral environment of magnesium atoms is preferred and therefore a coordination gap (after decoordination of a hemilabile arm) is immediately substituted by lactide, which is the first step in the polymerization process. These structural perturbations caused rending of one aminophenolate ligand as a polymer end-group.
Experimental results showed [Zn(tbpoa)2] to be an efficient and the most interesting initiators for the polymerization of L-LA. Representative results are collected in Table 3. The choice of the propargyl alcohol was determined by the possibility of conducting synthesis of end-functionalized oligomers, which could later be applied as building blocks in molecular engineering.
| Entry | ROH | [I]/[L-LA]/ROH | Time [min] | C [%] |
M
n,calcb [g mol−1] |
M
n,obsc [g mol−1] |
M
w/Mnd |
|---|---|---|---|---|---|---|---|
| Reaction conditions: Vsolvent = 25 mL, toluene; T = 58 °C.a Obtained from 1H NMR.b Calculated from Mn,theo = [L-LA]0/[ROH]0 × C% × 144.13 + MROH unless otherwise specified.c Determined by GPC calibrated versus polystyrene standards and corrected by a factor of 0.58 according to literature recommendations.30d Obtained from GPC. | |||||||
| 1 | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OH CCH2OH |
1/5/1 | 2 | 98 | 762 | 908 | 1.081 |
| 2 | HCCCH2OH |
1/10/1 | 5 | 92 | 1382 | 1280 | 1.012 |
| 3 | HCCCH2OH |
1/20/1 | 15 | 99 | 2881 | 3333 | 1.083 |
| 4 | HCCCH2OH |
1/30/1 | 20 | 99 | 4337 | 4912 | 1.040 |
| 5 | HCCCH2OH |
1/50/1 | 30 | 98 | 7118 | 7612 | 1.081 |
| 6 | C18H37OH | 1/30/1 | 45 | 94 | 4335 | 4530 | 1.031 |
Based on the narrow PDI values (1.012–1.083) complex [Zn(tbpoa)2] behaves in a controlled manner. The linear relationship between the Mn and the conversion exhibited by [Zn(tbpoa)2] implies the living character of the polymerization process as shown in Fig. 6.
 | ||
| Fig. 6 Polymerization of L-LA catalyzed by [Zn(tbpoa)2] in toluene at 58 °C. The relationship between Mn (blue line) or PDI (red line) vs. the initial molar ratio [L-LA]0/[ROH]0. | ||
The end-group analysis is demonstrated by the 1H NMR spectrum of the PLA produced by initiators [Zn(tbpoa)2]/ROH (ROH – propargyl alcohol, octadecanol) indicated that polymer chains are terminated by hydroxyl and appropriate ester groups as demonstrated in Fig. 7.
 | ||
| Fig. 7 1H NMR spectra of poly(L-lactide) with acetylene end-group. | ||
The polymerization conditions essentially determine the activity of [Zn(tbpoa)2], which is in agreement with theoretical calculations. At room temperature either the closed structure or the structure with one pendant dioxolane arm is dominant. The latter form after the coordination of lactide molecules shows low polymerization activity. Instead, the change of conditions to a fully open structure (both dioxolane arms pendant) and the addition of lactide creates an active complex. As proven by theoretical calculations, there is no big difference which – of the many possible open conformations – is formed after the dioxolane arms decoordinate, since they all easily transform into each other. The most important thing is to keep those reaction conditions that retain open forms. The shutter of hemilabile arms at room temperature gave stable and easy to store slipping catalysts.
DFT study
In order to gain a more detailed picture of the processes taking place during the polymerization, theoretical calculations have been performed as described in the Experimental section. First the interaction energies have been calculated:| ΔE(ML-L) = −(E(ML2) − E(ML+) − E(L−)) |
| ΔE(M-L) = −(E(ML+) − E(M2+) − E(L−)) |
To calculate these values, in the first place we performed a gas-phase geometrical optimization. The optimized structures are shown in Fig. 8 and the major conformers are marked 1 and 8′.
 | ||
| Fig. 8 Optimized structures of [Zn(tbpoa)2] complexes (H atoms omitted except for the CH protons of dioxolane rings) and their relative energies (in kcal mol−1) calculated at the TPSS-D3/TZVPPD level of theory. | ||
Although 8′ has a structure which is close to the crystallographic one (or rather its mirror image), we found two similar lower energy conformers: 8 and 8′′, which differ mainly in dioxolane rings arrangement (pseudo-rotation of this ring; see ESI† for more detailed explanation).
Unlike Ca and Mg, which prefer octahedral arrangement, zinc is usually tetrahedrally coordinated. The analysis of the optimized geometrical parameters (Table 1S in ESI†) of calcium and magnesium complexes show that Ca and Mg cations are 6-coordinated and they are surrounded by six (four O and two N) nearest neighbors in a distorted octahedral arrangement. On the other hand, the optimized Zn initiator appeared to have octahedral or rather pseudo-octahedral coordination, but the analysis of M–N and M–O distances (see Table 4 and 1S†), as well as ionic radii (0.74 Å, 4-coordinated ion; 0.88 Å, 6-coordinated ion) led to the conclusion that its structure more closely resembles a distorted tetrahedron. Calculated and experimentally measured M–N and M–O (phenyl oxygen) distances are the smallest in the Zn complex and the largest in the Ca one. These results are consistent with ionic radii of Ca (1.14 Å, 6-coordinated ion), Mg (0.86 Å, 6-coordinated ion), and Zn (0.74 Å, 4-coordinated ion) cations.
| ML2 | ML+ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecule (for M) [e] | ΔE(ML-L) [kcal mol−1] | q(M) [e] | q(OPh) [e] | q(OOx) [e] | q(N) [e] | Natural electron configurations (for M) [e] | r(M–OOx) r(M–OPh) r(M–N) [Å] | ΔE(M-L) [kcal mol−1] | q(M) [e] | q(OPh) [e] | q(OOx) [e] | q(N) [e] | Natural electron configurations |
| a Contributions of dioxolane-, amine-, and phenyl-fragments to the total interaction energies. b Free anion L (in complex geometry) partial NPA charges. | |||||||||||||
| [Ca(tbpoa)2] | 192 (24, 22, 147)a | 1.80 | −0.92 (−0.78)b | −0.63 (−0.51)b | −0.49 (−0.38)b | 4s(0.12), 3d(0.08), 4d(0.01) | 2.408 | 352 (31, 36, 284)a | 1.83 | −0.95 | −0.67 | −0.51 | 4s(0.05), 3d(0.13) |
| 2.204 | |||||||||||||
| 2.683 | |||||||||||||
| [Mg(tbpoa)2] | 217 (20, 31, 167) | 1.83 | −0.97 (−0.77) | −0.60 (−0.50) | −0.52 (−0.38) | 3s(0.14), 3p(0.01), 3d(0.02) | 2.172 | 418 (34, 49, 335) | 1.82 | −1.01 | −0.65 | −0.55 | 3s(0.17), 3p(0.01) |
| 1.970 | |||||||||||||
| 2.300 | |||||||||||||
| [Zn(tbpoa)2] (1) | 213 (14, 36, 163) | 1.76 | −0.98 (−0.80) | −0.53 (−0.52) | −0.56 (−0.38) | 4s(0.23), 4p(0.01), 3d(9.98), 4d(0.01) | 2.675 | 459 (21, 47, 391) | 1.38 | −0.87 | −0.51 | −0.53 | 4s(0.61), 4p(0.03), 3d(9.98) |
| 1.952 | |||||||||||||
| 2.153 | |||||||||||||
| [Zn(tbpoa)2] (8) | 216 (3, 38, 175) | 1.77 | −0.97 (−0.79) | −0.57 (−0.51) | −0.55 (−0.37) | 4s(0.21), 4p(0.01), 3d(9.98), 4d(0.02) | 4.094 | 443 (2, 51, 391) | 1.43 | −0.86 | −0.57 | −0.53 | 4s(0.56), 4p(0.02), 3d(9.98) |
| 1.912 | |||||||||||||
| 2.117 | |||||||||||||
Table 4 presents also ML-L and M-L interaction energies for the ML2 complexes ad ML+ ions (M = Ca, Mg, Zn). The lowest ΔE(ML-L) and ΔE(M-L) values have been found for calcium. In the case of magnesium, ΔE(MgL-L) is over 20 kcal mol−1 larger than for the Ca complex. The ΔE(Mg-L) value is much greater than that for ΔE(Ca-L) and reaches 62 kcal mol−1. But the difference between ΔE(MgL-L) and ΔE(ZnL-L) seems to be too small to explain why in contrast to the zinc complex the magnesium initiator loses one ligand during the polymerization process. There may be different reasons for this: (i) in the Mg-complex both ligands have conformations that are significantly distinct from those for Zn initiator (Fig. S1†), which may facilitate the incorporation of one ligand into the growing polymer and (ii) one should remember that structures of these molecules in solution are not necessarily the same as in crystal. As mentioned before the changes observed in NMR signals for the Zn complex can be interpreted as a rotation of dioxolane rings and hence the lack of coordination to the metal cation. It can be seen from Fig. 9 that interconversion between closed 1 and half-opened complex 2 though it is not energetically favorable is possible.
 | ||
| Fig. 9 TPSS-D3/TZVPPD energy profile for the transition between 1 and 3. In () and [ ] parentheses are values calculated in dichloromethane and toluene, respectively. All values in kcal mol−1. | ||
Opening of the second dioxolane arm is also possible with an even slightly smaller barrier. The influence of solvents on calculated relative energies is not very significant (in all cases obtained values were smaller). For the fully opened complex we found additional lower energy conformer 3′ (Fig. 8). 3 and 3′ differ mainly in the distance of aromatic rings (see Fig. 2S and Table 2S†). Interestingly, the energy difference between 3 and 3′ decreases going from gas phase to toluene (ε = 2.4) and next to dichloromethane (ε = 8.9). In the case of the magnesium complex such an opening of dioxolane arms is probably less favorable. To confirm this supposition we estimated the contributions of individual fragments of the ligand (dioxolane ring, amine part, and phenyl ring) to the total interaction energy (see Fig. 3S in ESI† for more detailed explanation). From Table 4, the contribution of dioxolane ring–metal interaction to the ligand–metal interaction energy is less than 7% for Zn and 9–13% for Mg and Ca. As expected, the phenyl ring oxygen–metal interaction is the largest part of the ΔE(M-L) and it grows going from Ca to Mg and to the Zn complex. These values are so large mainly due to the strong attractive Coulombic interaction, and the observed trend is consistent with decreasing M–OPh distances. A similar trend is observed for the amine part–metal interaction, which is understandable, taking into account the fact that zinc prefers coordination to “softer” atoms like nitrogen.
In all considered complexes the ΔE(ML-L) is much smaller than ΔE(M-L). It is worth noting that ΔE(M-L) − ΔE(ML-L) difference is the smallest for CaL2 (∼160 kcal mol−1), medium for MgL2 (∼201 kcal mol−1), and the largest for ZnL2 molecules (∼227–246 kcal mol−1). Because magnesium complex loses only one ligand during polymerization, a calculated larger ΔE(M-L) − ΔE(ML-L) value for Mg in comparison to Ca is consistent with experimental observations.
Table 4 lists also partial NPA charges and metal cation natural electron configurations for the ML2 complexes and ML+ ions. It is evident from this table that the most (negative) charge is transferred to Zn and most of the additional electron density goes into the 3s and 4s orbitals for magnesium and zinc, respectively. For the calcium complex a significant portion of the electron density transferred to the metal ion resides in the 4s and 3d orbitals (Zn loses some electron density in this orbital). In all cases, the presence of the metal cation affects the electron density distribution of neighboring N and O atoms. As one can guess, the smallest change of this distribution is observed for dioxolane oxygen atoms coordinated to the zinc.
For the Zn-complex, analysis of the experimental 1H NMR spectrum indicates two signals of the methine protons of dioxolane rings, which appear at 5.22 and 5.13 ppm. But from theoretical calculations we obtained two much more distant values: 5.26 ppm (conformer 1) and 5.76 ppm (conformer 8). This may mean that instead of form 8 (or rather, as it was mentioned before, its mirror image form) some other conformer(s) is(are) present in solution. As mentioned before the solution NMR studies indicate that bound and non-bound dioxolane rings can readily exchange. Rotation of these groups may lead to interconversion of 1 and 8. Fig. 10 shows such a possible rotational path which passes through several minima.
 | ||
| Fig. 10 TPSS-D3/TZVPPD energy profile for the transition between 1 and 8. In () and [ ] parentheses are values calculated in dichloromethane and toluene, respectively. All values in kcal mol−1. | ||
It should be stressed that this is only one of the possible rotational paths since this complex is quite flexible. During the optimization we have obtained nineteen structures for the Zn-complex (Fig. 8). The overall shape of these molecules is dependent on the combined effects of such factors as the arrangement of dioxolane groups and distances between the phenyl rings. The conformational flexibility of dioxolane and other five-membered ring molecules is a very well-known fact.31–33 Therefore, in addition to the rotations of dioxolane ring around the N28–C33 (N72–C77) and C33–C36 (C77–C80) bonds it was important to consider pseudorotational motions.34,35 In this way, we obtained, for example, two additional conformers of 1 and 8 (Fig. 8; see also Table 2S† for selected geometrical parameters of all Zn-complex conformers). The energy differences between 1, 1′, and 1′′ molecules are rather small and do not exceed 3 kcal mol−1. In the case of 8, 8′, and 8′′ they are even smaller. Since the reported in literature values of the pseudorotation barrier for different oxolanes are rather small, probably a multitude of conformers coexists at room temperature.31,32,36,37 A similar difference in relative energies was found for two other conformers: 3 and 3′. However, as mentioned before, in contrast to 1, 1′, and 1′′ or 8, 8′, and 8′′, they differ in the mutual arrangement of aromatic rings. In higher energy conformer 3 these rings are closer to each other than in 3′.
In Table 5 methine protons H45 and H81 (Fig. 1) chemical shifts for all conformers which we found are reported.
| δ(H45) | δ(H81) | ||
|---|---|---|---|
| [Ca(tbpoa)2] | 5.70 | 5.70 | |
| [Mg(tbpoa)2] | 6.45 | 6.45 | |
| [Zn(tbpoa)2] | |||
| 1 | 5.26 | 5.26 | |
| 1′ | 5.26 | 5.39 | |
| 1′′ | 5.40 | 5.40 | |
| 2 | 5.21 | 5.26 | |
| 2′ | 5.18 | 5.38 | |
| 2′′ | 5.18 | 5.23 | |
| 3 | 5.21 | 5.21 | |
| 3′ | 5.23 | 5.23 | |
| 4 | 5.54 | 5.27 | |
| 5 | 5.86 | 5.31 | |
| 6 | 6.53 | 5.25 | |
| 7 | 6.57 | 5.81 | |
| 8 | 5.94 | 5.70 | |
| 8′ | 5.76 | 5.76 | |
| 8′′ | 6.44 | 6.44 | |
| 9 | 5.46 | 5.31 | |
| 10 | 5.27 | 5.23 | |
| 11 | 5.38 | 5.24 | |
| 12 | 5.42 | 5.42 |
Our calculations show that the lowest energy forms of the zinc initiator (1, 1′, 1′′, 5, 8, 8′, and 8′′) have both dioxolane arms closed. However, the comparison of experimental and calculated 1H chemical shifts may suggest rather the presence of some half-opened or fully opened structures in solution. In the opened 3, 3′, 11, and 12 forms the chemical shifts for H45 and H81 have the values of 5.21–5.42 ppm. For half-opened molecules 2, 2′, 2′′ signals in the similar range (5.18–5.38 ppm) have been observed. Although, for other conformers H45 and/or H81 chemical shifts have larger values, nevertheless, their presence in solution cannot be ruled out.
Conclusions
In summary, four Mg, Zn, and Ca complexes of aminophenolate ligands with a hemilabile (flexible) segment were described. The experimental data verified by theoretical studies suggest that lability of dioxolane fragment is essential to ensure a suitable structure of an active centre for zinc, which is not the case for the calcium complex. Under optimized conditions zinc bis(chelate)complex in the presence of alcohol demonstrates efficient activities for living ROP of lactides. The subtle changes observed in the calculated structure of the complex allowed for optimization of the reaction condition in order to improve the catalyst. Additionally these calculations can elucidate the low stereocontrol in the ROP of racemic lactide by complexes with prochiral ligands. Although heteroleptic complexes are the most explored as initiators for ROP of cyclic esters, yet, the combination of homoleptic ones and external donors can provide an alternative way for catalysts design based on kinetically labile complexes.Acknowledgements
The authors would like to thank the National Science Centre in Poland (Grants N204 200640 and N204 136339) and the Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl; grant no. 162 and no. 011) for support of this research.Notes and references
- For key references on application of biodegradable polymers see references: (a) C.-S. Ha and A. J. Gardella, Chem. Rev., 2005, 105, 4205 CrossRef CAS PubMed; (b) R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198 CrossRef CAS PubMed; (c) J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Faerokhzad, Biomaterials, 2007, 28, 869 CrossRef CAS PubMed; (d) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (e) C. M. Thomas and J. F. Lutz, Angew. Chem., Int. Ed., 2011, 40, 9244 CrossRef PubMed; (f) H. Sun, F. Meng, A. A. Dias, M. Hendriks, J. Feijen and Z. Zhong, Biomacromolecules, 2011, 12, 1937 CrossRef CAS PubMed; (g) R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, Macromol. Biosci., 2012, 12, 286 CrossRef CAS PubMed; (h) H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237 CrossRef CAS PubMed.
- For key references on application of PLA see references: (a) P. U. Rakkanen, O. Bostman, E. Hirvensalo, E. A. Makela, W. K. Partio, H. Patiala, S. Vainionpaa, K. Vihtonen and P. Tormala, Biomaterials, 2000, 21, 2607 CrossRef; (b) V. Piddubnyak, P. Kurcok, A. Matuszowicz, M. Głowala, A. Fiszer-Kierzkowska, Z. Jedliński, M. Juzwa and Z. Krawczyk, Biomaterials, 2004, 25, 5271 CrossRef CAS PubMed; (c) R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS PubMed; (d) C. Chen, C. H. Yu, Y. C. Cheng, P. H. F. Yu and M. K. Cheung, Biomaterials, 2006, 27, 4804 CrossRef CAS PubMed; (e) Z. Wang, H. Hu, Y. Wang, Y. Wang, Q. Wu, L. Liu and G. Chen, Biomaterials, 2006, 27, 2550 CrossRef CAS PubMed; (f) E. L. Rickert, J. P. Trebley, A. C. Peterson, M. M. Morrell and R. V. Weatherman, Biomacromolecules, 2007, 8, 3608 CrossRef CAS PubMed; (g) M. Murariu, A. Doumbia, L. Bonnaud, A. L. Dechief, Y. Paint, M. Ferreira, C. Campagne, E. Devaux and P. Dubois, Biomacromolecules, 2011, 12, 1762 CrossRef CAS PubMed; (h) H. Kim, H. Park, J. Lee, T. H. Kim, E. S. Lee, K. T. Oh, K. C. Lee and Y. S. Youn, Biomaterials, 2011, 32, 1685 CrossRef CAS PubMed; (i) P. Zhang, H. Wu, H. Wu, Z. Lu, C. Deng, Z. Hong, X. Jing and X. Chen, Biomacromolecules, 2011, 12, 2667 CrossRef CAS PubMed.
- J. Wang, W. Zhu, H. Zhang and C. Park, Chem. Eng. Sci., 2012, 75, 390 CrossRef CAS PubMed.
- For leading and recent reviews, see: (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC; (b) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS PubMed; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed; (d) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS PubMed; (e) K. Williams, Chem. Soc. Rev., 2007, 36, 1573 RSC; (f) B. N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef PubMed; (g) A. P. Dove, Chem. Commun., 2008, 6446 RSC; (h) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS; (i) C. A. Wheaton, P. G. Hayes and B. J. Irealand, Dalton Trans., 2009, 4832 RSC; (j) N. Ajellah, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helon, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363 RSC.
- (a) J. E. Kasperczyk, Macromolecules, 1995, 28, 3937 CrossRef CAS; (b) K. A. M. Thakur, R. T. Kean, E. S. Hall, J. J. Kolstad, T. A. Lindgren, M. A. Doscotch, J. I. Siepmann and E. J. Munson, Macromolecules, 1997, 30, 2422 CrossRef CAS; (c) M. H. Chisholm, S. S. Iyer, M. E. Matison, D. G. McCollum and M. Pagel, Chem. Commun., 1997, 1999 RSC; (d) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS; (e) Ref. 6b ; (f) Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510 CrossRef CAS; (g) M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 32, 7700 CrossRef; (h) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291 CrossRef CAS PubMed; (i) J. Belleney, M. Wisniewski and A. Le Borgne, Eur. Polym. J., 2004, 40, 523 CrossRef CAS PubMed; (j) A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS PubMed.
- For key references on β-diketiminate, see references: (a) M. Cheng, A. B. Attygalle, E. B. Lobovskya and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583 CrossRef CAS; (b) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS PubMed; (c) M. H. Chisholm, J. C. Huffman and K. Phomphrai, Dalton Trans., 2001, 3, 222 RSC; (d) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS PubMed; (e) M. H. Chisholm and K. Phomphrai, Inorg. Chim. Acta, 2003, 350, 121 CrossRef CAS; (f) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48 RSC; (g) A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Wiliams, Dalton Trans., 2004, 570 RSC; (h) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2005, 44, 8004 CrossRef CAS PubMed; (i) H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400 CrossRef CAS; (j) E. L. Marshall, V. C. Gibson and H. S. Rzepa, J. Am. Chem. Soc., 2005, 127, 6048 CrossRef CAS PubMed; (k) L.-F. Hsueh, N.-T. Chuang, C.-Y. Lee, A. Datta, J.-H. Huang and T.-Y. Lee, Eur. J. Inorg. Chem., 2011, 36, 5530 CrossRef ; for trispyrazolylborate, see references: ; (l) R. Han and G. Parkin, J. Am. Chem. Soc., 1992, 114, 748 CrossRef CAS; (m) M. H. Chisholm and N. W. Eilerts, Chem. Commun., 1996, 853 RSC; (n) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS; (o) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48 RSC; (p) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717 CrossRef CAS PubMed ; for amino/imino-phenolate ligands, see references: ; (q) Ref. 4e ; (r) Ref. 7d, magnesium; (s) Ref. 12a ; (t) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350 CrossRef CAS PubMed; (u) S. Groysman, E. Sergeeva, I. Goldberg and M. Kol, Eur. J. Inorg. Chem., 2006, 14, 2739 CrossRef; (v) H. Y. Chen, H. Y. Tang and C. C. Lin, Macromolecules, 2006, 39, 3745 CrossRef CAS; (w) Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande Jr. and J.-F. Carpentier, New J. Chem., 2008, 32, 2279 RSC; (x) J. Wu, Y. Z. Chen, W. C. Hung and C. C. Lin, Organometallics, 2008, 27, 4970 CrossRef CAS; (y) G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova and P. Mehrkhodavandi, Organometallics, 2009, 28, 1309 CrossRef CAS; (z) W. C. Hung and C. C. Lin, Inorg. Chem., 2009, 48, 728 CrossRef CAS PubMed; (a a) N. Ikpo, L. N. Saunders, J. L. Walsh, J. M. B. Smith, L. N. Dawe and F. M. Kerton, Eur. J. Inorg. Chem., 2011, 35, 5347 CrossRef; (a b) J. Weil, R. T. Mathers and Y. D. Y. L. Getzler, Macromolecules, 2012, 45, 1118 CrossRef CAS.
- For a leading and recent references on initiators for lactide references, see: lithium and sodium: (a) A. K. Sutar, T. Maharana, S. Dutta, C. T. Chen and C. C. Lin, Chem. Soc. Rev., 2010, 39, 1724 RSC; (b) B. Calvo, M. G. Davidson and D. Garcia-Vivo, Inorg. Chem., 2011, 50, 3589 CrossRef CAS PubMed; (c) L. Wang, X. Pan, L. Yao, N. Tang and J. Wu, Eur. J. Inorg. Chem., 2011, 5, 632 CrossRef; (d) W. Y. Lu, M. W. Hsiao, S. C. N. Hsu, W. T. Peng, Y. J. Chang, Y. C. Tsou, T. Y. Wu, Y. C. Lai, Y. Chen and H. Y. Chen, Dalton Trans., 2012, 41, 3659 RSC . Magnesium: ; (e) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 12, 11845 CrossRef; (f) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS PubMed; (g) M. L. Shueh, Y. S. Wang, B. H. Huang, C. Y. Kuo and C. C. Lin, Macromolecules, 2004, 37, 5155 CrossRef CAS; (h) J. Ejfler, M. Kobyłka, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2005, 2047 RSC; (i) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC; (j) W. C. Hung and C. C. Lin, Inorg. Chem., 2009, 48, 728 CrossRef CAS PubMed; (k) L. Wang and H. Ma, Macromolecules, 2010, 43, 6535 CrossRef CAS; (l) H. J. Chuang, S. F. Weng, C. C. Chang, C. C. Lin and H. Y. Chen, Dalton Trans., 2011, 40, 9601 RSC; (m) L. F. Hsueh, N. T. Chuang, C. Y. Lee, A. Datta, J. H. Huang and T. Y. Lee, Eur. J. Inorg. Chem., 2011, 36, 5530 CrossRef . Zinc: ; (n) M. Bero, J. Kasperczyk and G. Adamus, Macromol. Chem., 1993, 194, 907 CrossRef CAS; (o) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr., M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350 CrossRef CAS PubMed; (p) E. Grunova, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2009, 9010 RSC; (q) L. Wang and H. Ma, Dalton Trans., 2010, 39, 7897 RSC; (r) D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360 CrossRef CAS PubMed; (s) C. A. Wheaton and P. G. Hayes, Chem. Commun., 2010, 46, 8404 RSC; (t) H. Y. Chen, Y. L. Peng, T. H. Huang, A. K. Sutar, S. A. Miller and C. C. Lin, J. Mol. Catal. A: Chem., 2011, 339, 61 CrossRef CAS PubMed; (u) M. T. Chen and C. T. Chen, Dalton Trans., 2011, 40, 12886 RSC . Aluminium: ; (v) E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469 RSC; (w) K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026 CrossRef CAS PubMed; (x) J. Lewiński, P. Horeglad, M. Dranka and I. Justyniak, Inorg. Chem., 2004, 43, 5789 CrossRef PubMed; (y) D. J. Darensbiurg, O. Karroonnirun and S. J. Wilson, Inorg. Chem., 2011, 50, 6775 CrossRef PubMed . Tin: ; (z) K. B. Aubrecht, M. A. Hillmyer and W. B. Tolman, Macromolecules, 2002, 35, 644 CrossRef CAS; (a a) A. P. Dove, V. C. Gibson, E. L. Marchall, H. S. Rzepa, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2006, 128, 9834 CrossRef CAS PubMed; (a b) N. Nimitsiriwat, V. C. Gibson, E. L. Marshall and M. R. J. Elsegood, Inorg. Chem., 2008, 47, 5417 CrossRef CAS PubMed; (a c) N. Nimitsiriwat, V. C. Gibson, E. L. Marshall and M. R. J. Elsegood, Dalton Trans., 2009, 3710 RSC.
- (a) M.-L. Shueh, Y.-S. Wang, B.-H. Huang, C.-Y. Kuo and C.-C. Lin, Macromolecules, 2004, 37, 5155 CrossRef CAS; (b) C. M. Silvernail, L. J. Yao, L. M. R. Hill, M. A. Hillmyer and W. B. Tolman, Inorg. Chem., 2007, 46, 6565 CrossRef CAS PubMed.
- (a) E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004 RSC; (b) C.-Y. Sung, C.-Y. Li, J.-K. Su, T.-Y. Chen, C.-H. Lin and B.-T. Ko, Dalton Trans., 2012, 41, 953 RSC; (c) H. Sun, J. S. Ritch and P. G. Hayes, Dalton Trans., 2012, 41, 3701 RSC.
- (a) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS; (b) P. Dubois, O. Coulembier and J.-M. Raquez, Handbook of Ring-Opening Polymerization, Wiley-VCH, 2009, 53–63 Search PubMed.
- (a) Ref. 6g, amino/imino-phenolate ligands; ; (b) W. Liying and M. Haiyan, Dalton Trans., 2010, 39, 7897 RSC.
- (a) Ref. 7d, magnesium; ; (b) J. Ejfler, S. Szafert, K. Mierzwicki, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2008, 6556 RSC.
- CrysAlisRED Software, Oxford Diffraction, Wrocław, Poland, 1995–2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- D. T. Cromer and J. T. Waber, International Tables for X-ray Crystallography, ed. J. A. Ibers and W. C. Hamilton, Kynoch, Birmingham, England, 1974 Search PubMed.
- TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652 CrossRef CAS; (b) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 CrossRef CAS; (c) F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175 CrossRef CAS.
- S. Grimme and J.-P. Djukic, Inorg. Chem., 2011, 50, 2619 CrossRef CAS PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143 CrossRef CAS.
- (a) F. London, J. Phys. Radium, 1937, 8, 397 CrossRef CAS; (b) R. McWeeny, Phys. Rev., 1962, 126, 1028 CrossRef; (c) R. Ditchfield, Mol. Phys., 1974, 27, 789 CrossRef CAS; (d) K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS; (e) J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 114, 5497 CrossRef.
- K. W. Wiitala, T. R. Hoye and C. J. Cramer, J. Chem. Theor. Comput., 2006, 2, 1085 CrossRef CAS.
- Zhurko G. A. ChemCraft 1.6. http://www.chemcraftprog.com.
- (a) E. Y. Tshuva, N. Gendeziuk and M. Kol, Tetrahedron Lett., 2001, 42, 6405 CrossRef CAS; (b) T. Toupance, S. R. Duberley, N. H. Rees, B. R. Tyrrell and P. Mountford, Organometallics, 2002, 21, 1367 CrossRef CAS.
- (a) R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef; (b) J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, HarperCollins, New York, USA, 4th edn, 1993 Search PubMed.
- V. K. Muppidi, S. Z. Panthapally and S. Pal, Inorg. Chem. Commun., 2005, 8, 543 CrossRef CAS PubMed.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114 CrossRef CAS.
- J. A. Greenhouse and H. L. Strauss, J. Chem. Phys., 1969, 50, 124 CrossRef CAS.
- Q. Shen, T. L. Mathers, T. Raeker and R. L. Hilderbrandt, J. Am. Chem. Soc., 1986, 108, 6888 CrossRef CAS.
- J. Makarewicz and T.-K. Ha, J. Mol. Struct., 2001, 599, 271 CrossRef CAS.
- J. E. Kilpatrick, K. S. Pitzer and R. Spitzer, J. Am. Chem. Soc., 1947, 64, 2483 CrossRef.
- H. J. Geise, C. Altona and C. Romers, Tetrahedron Lett., 1967, 15, 1383 CrossRef.
- D. Cremer, Isr. J. Chem., 1983, 23, 72 CrossRef CAS.
- P. A. Baron and D. O. Harris, J. Mol. Spectrosc., 1974, 49, 70 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tables and figures of DFT calculations. CCDC 962645 and 969646. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52868e |
| This journal is © The Royal Society of Chemistry 2014 |