 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structural characterisation of Pd(II) and Pt(II) complexes with a flexible, ferrocene-based P,S-donor amidophosphine ligand†
Jiří
Tauchman
,
Ivana
Císařová
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz; Fax: +420 221 951 253
First published on 30th October 2013
Abstract
1′-Diphenylphosphino-1-{[(2-(methylthio)ethyl)amino]carbonyl}ferrocene (1), accessible via amidation of 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf) with 2-(methylthio)ethylamine, reacts with [PdCl2(cod)] (cod = cycloocta-1,5-diene) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal-to-ligand ratio to give trans-[PdCl2(1-κ2P,S)] (trans-2) as the sole product. A similar reaction with [PtCl2(cod)] affords a mixture of cis- and trans-[PtCl2(1-κ2P,S)] (cis- and trans-3), which can be separated by fractional crystallisation. Complexation reactions performed with 2 equiv. of the ligand are less selective, yielding mixtures of the expected bis-phosphine complexes (i.e., trans-[PdCl2(1-κP)2], or a mixture of cis- and trans-[PtCl2(1-κP)2]) with the respective monophosphine complexes. The structures of 1, trans-2, cis-3 and trans-3 determined by X-ray diffraction demonstrate the ability of the title ligand to act as a flexible cis- or trans-P,S-chelate donor (the ligand bite angles are 174.03(2)/173.05(2)° for trans-2/3 and 92.86(2)° for cis-3).
:1 metal-to-ligand ratio to give trans-[PdCl2(1-κ2P,S)] (trans-2) as the sole product. A similar reaction with [PtCl2(cod)] affords a mixture of cis- and trans-[PtCl2(1-κ2P,S)] (cis- and trans-3), which can be separated by fractional crystallisation. Complexation reactions performed with 2 equiv. of the ligand are less selective, yielding mixtures of the expected bis-phosphine complexes (i.e., trans-[PdCl2(1-κP)2], or a mixture of cis- and trans-[PtCl2(1-κP)2]) with the respective monophosphine complexes. The structures of 1, trans-2, cis-3 and trans-3 determined by X-ray diffraction demonstrate the ability of the title ligand to act as a flexible cis- or trans-P,S-chelate donor (the ligand bite angles are 174.03(2)/173.05(2)° for trans-2/3 and 92.86(2)° for cis-3).
Introduction
Ligands capable of acting as trans-chelate donors are attractive research targets. They not only represent structurally interesting molecules but are also useful tools for the elucidation of structural effects, reaction mechanisms, etc.1 The most often studied trans-chelating ligands still remain diphosphines, whose donor moieties are brought into appropriate positions with the aid of a rigid organic backbone.1,2 Symmetrical ligands featuring other donor atoms (e.g., N,N-donors3 and bis-carbenes4) and, particularly, their donor-asymmetric counterparts are much less common.For instance, the rare trans-chelating P,N-donors are represented by pyridyl-phosphines Ph2PC6H4CH2O(CH2)n(C5H4N-2)5 (n = 1–3) and by (diphenylphosphino)ferrocene derivatives bearing N-heterocyclic substituents in position 1′ of the ferrocene unit.6 As far as we are aware, however, no similar P,S-bidentate donors have been described in the literature.7
Recently, we found that carboxamides I–IV8–10 (Scheme 1), prepared from 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf)11 and simple amines equipped with a donor substituent at the terminal position readily form trans-chelate complexes with palladium(II) ions. This prompted us to design an analogous, P,S-donor ligand.12 In this contribution, we describe the preparation and structural characterisation of 1′-diphenylphosphino-1-{[(2-(methylthio)ethyl)amino]carbonyl}ferrocene (1), a congener of the recently described phosphinocarboxamide13 donors III and IV,10 and Pd(II) and Pt(II) complexes obtained thereof.
 | ||
| Scheme 1 | ||
Results and discussion
Synthesis and characterisation of ligand 1
Phosphino-amide 1 was obtained upon reacting 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf) with 2-(methylthio)ethylamine in the presence of a slight excess of 1-hydroxybenzotriazole and 1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide as the coupling agents14 (Scheme 2). The compound was isolated by column chromatography, resulting as an air-stable, rusty orange solid in 84% yield. The chromatography also afforded a small amount of a more polar side-product, which was identified as the corresponding phosphine oxide 1O (isolated yield: 7%).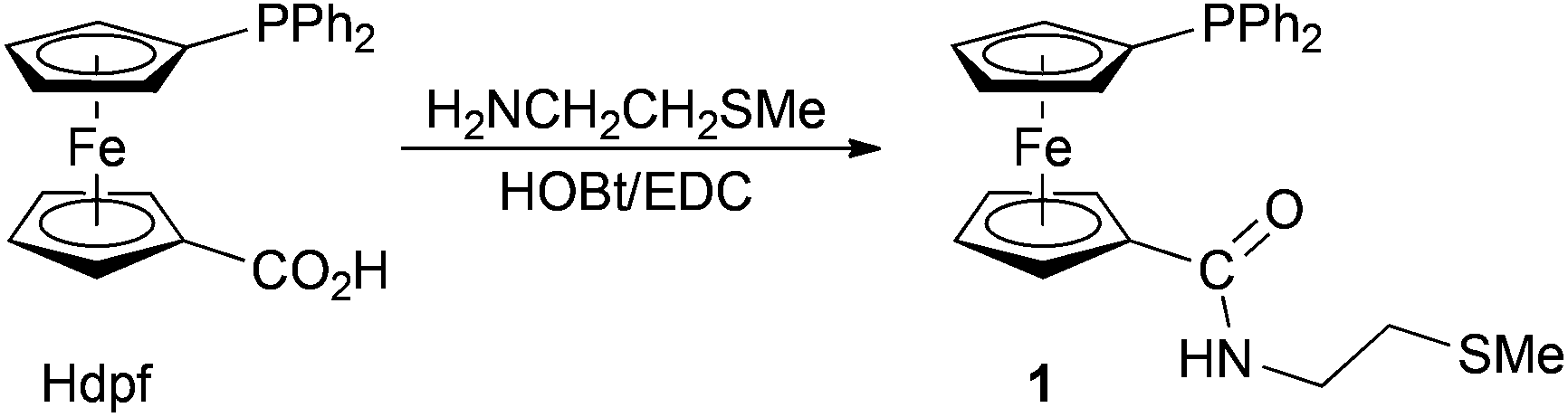 | ||
| Scheme 2 Preparation of amidophosphine 1 (HOBt = 1-hydroxybenzotriazole, EDC = 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide. | ||
NMR spectra of 1 and 1O show signals of the terminal SMe groups (δH/δC: 2.16/14.97 for 1, and 2.06/14.96 for 1O) and the ethane-1,2-diyl linkers. Signals due to the PPh2-substituted ferrocene units are observed at the expected positions and with the characteristic JPC values (cf. ref. 8–10). The 31P NMR shifts are −16.9 and 31.6 for 1 and 1O, respectively. The compounds display typical amide bands (namely νNH and amide I/II bands) in their IR spectra and give rise to pseudomolecular ions in the ESI mass spectra.
The molecular structures of 1 and 1O determined by single-crystal X-ray diffraction analysis are depicted in Fig. 1 and 2. Selected geometric data are given in Table 1. The ferrocene moieties in the molecules of 1 and 1O are regular showing tilt angles below ca. 5° and, accordingly, similar individual Fe–C distances (2.030(2)–2.059(2) Å for 1, and 2.024(2)–2.062(2) Å for 1O). However, their substituents assume different mutual orientations (see τ angles in Table 1). While the conformation of 1 is anticlinal eclipsed, the substituents in 1O are moved closer to a synclinal eclipsed conformation by the intramolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P hydrogen bond. In both cases, the amide planes and the planes of their parent cyclopentadienyl ring are twisted (see φ in Table 1) so that the amide nitrogen is directed towards the side of the ferrocene unit. The amide substituents in 1 and 1O adopt similar orientations pointing toward the other Cp ring and have the substituents at the C24–C25 bond at ca. 60° or in gauche conformation.
P hydrogen bond. In both cases, the amide planes and the planes of their parent cyclopentadienyl ring are twisted (see φ in Table 1) so that the amide nitrogen is directed towards the side of the ferrocene unit. The amide substituents in 1 and 1O adopt similar orientations pointing toward the other Cp ring and have the substituents at the C24–C25 bond at ca. 60° or in gauche conformation.
 | ||
| Fig. 1 View of the molecular structure of 1 showing the atom labelling scheme. Displacement ellipsoids are scaled to the 30% probability level. | ||
 | ||
| Fig. 2 View of the molecular structure of phosphine oxide 1O showing the atom labelling scheme. Displacement ellipsoids enclose the 30% probability level. The N–H1N⋯O1P hydrogen bond is indicated with a dotted line (N⋯O1P = 2.920(2) Å, angle at H1N = 167°). | ||
| Parametera | 1 | 1O |
|---|---|---|
| a The ring planes are defined as follows: Cp1 = C(1–5), Cp2 = C(6–10). Cg1/2 are the respective ring centroids. b Torsion angle C1–Cg1–Cg2–C6. c The range of C6–P–C(12/18) and C12–P–C18 angles. d Dihedral angle subtended by the amide unit (C11, O, N) and the ring Cp1. e Additional data: P–O1P = 1.496(2) Å; O1P–P–C(6/12/18) = 112.5(1)–114.5(1)°. | ||
| Fe–Cg1 | 1.6492(9) | 1.650(1) |
| Fe–Cg2 | 1.6427(9) | 1.646(1) |
| ∠Cp1, Cp2 | 4.5(1) | 2.4(1) |
| τ | 144.9(1) | 71.7(2) |
| P–C6 | 1.808(2) | 1.786(2) |
| P–C12/18 | 1.837(2)/1.834(2) | 1.803(2)/1.809(2) |
| C–P–Cc | 101.16(7)–104.53(7) | 104.8(1)–106.1(1) |
| C11–O | 1.228(2) | 1.233(3) |
| C11–N | 1.348(3) | 1.348(3) |
| O–C11–N | 122.9(2) | 123.8(2) |
| φ | 30.3(2) | 23.7(3) |
| N–C24–C25–S | 61.9(2) | 63.2(2) |
| C25–S–C26 | 100.9(1) | 100.4(1) |
Preparation of Group 10 metal complexes
Divalent Group 10 metal ions were chosen for an evaluation of the coordination properties of compound 1 mainly because they include the borderline Ni(II) as well as the typical soft Pd(II)/Pt(II) metal ions showing different kinetic properties.15,16 Unfortunately, repeated attempts at obtaining some Ni(II)-1 complexes failed. The reactions of [NiCl2(dme)] (dme = 1,2-dimethoxyethane) or NiCl2·6H2O with 1 equiv. of 1 afforded only rapidly decomposing mixtures, from which no defined product could be isolated. This may reflect a mismatch situation resulting from a relatively smaller size of the Ni(II) ion and the soft character of the donor atoms present in 1.In contrast, the reaction of 1 with [PdCl2(cod)] (cod = cycloocta-1,5-diene; Pd:1 = 1:1) proceeded rapidly and cleanly to provide a single product showing its 31P NMR signal at a lower field (δP 23.6, ΔP = 40.5 ppm) compared with the free ligand. This shift, together with a low-field shift of the SMe resonance (ΔH = 0.13 ppm) and its splitting with the 31P nucleus (4JPH = 4.7 Hz), already suggested chelate coordination of ligand 1. Indeed, this was corroborated by X-ray crystallography showing the product to be the trans-chelate complex trans-[PdCl2(1-κ2S,P)] (trans-2 in Scheme 3).
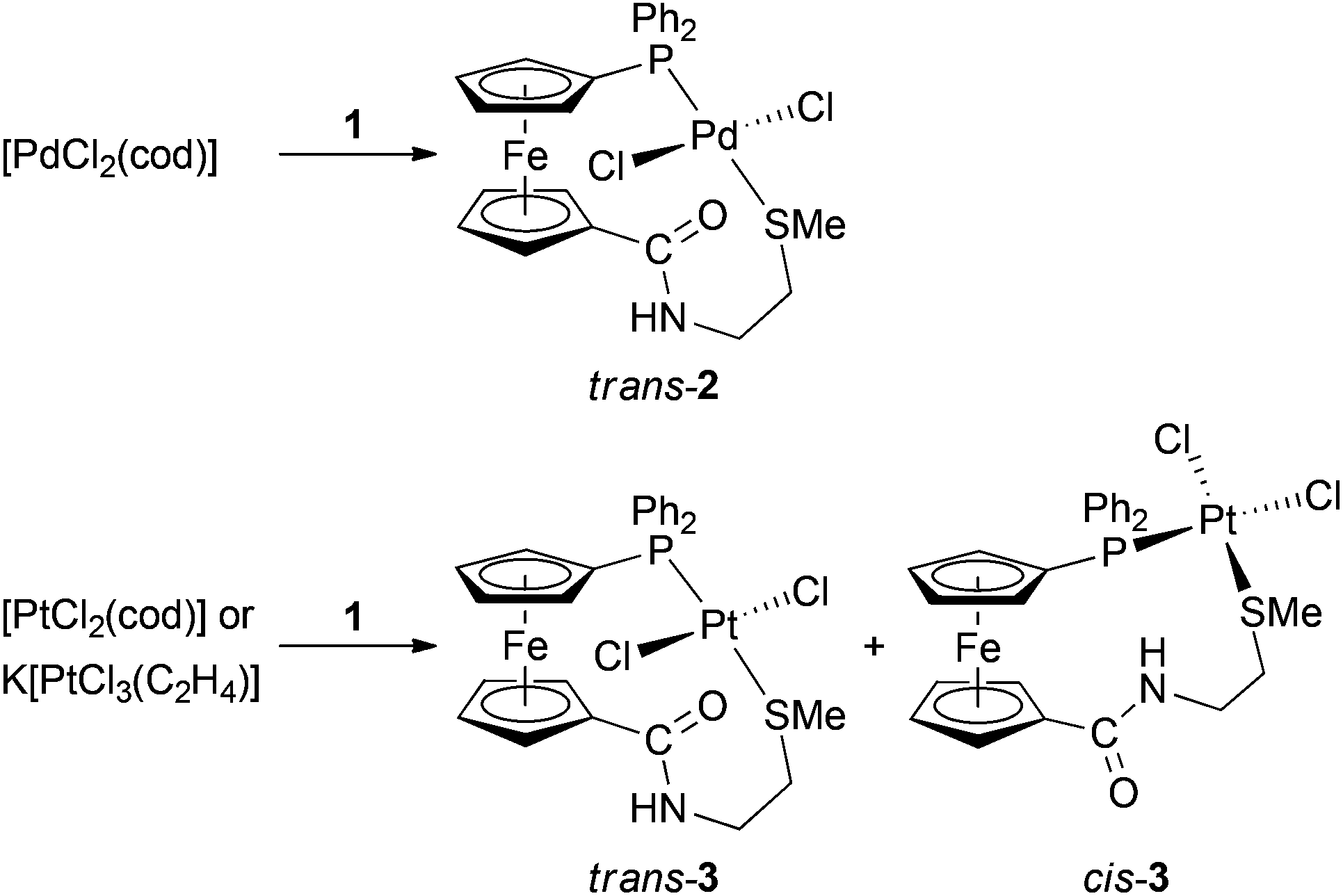 | ||
| Scheme 3 Preparation of complexes 2 and 3 (cod = cycloocta-1,5-diene). | ||
Compound trans-2 shows broad 1H NMR signals for protons at the ferrocene unit and in the ethane-1,2-diyl bridge at room temperature.17 Nonetheless, this broadening is in line with the formulation as it reflects a limited molecular mobility resulting from spatial constraints imposed by a tight chelate coordination. The amide vibrations of trans-2 are observed shifted vs. free 1 (amide I and II by ca. +30 and −10 cm−1, respectively) whilst the amide 13C NMR resonance (CO) remains nearly unaffected. The complex displays a pseudomolecular ion ([M + Na]+) and fragment ions attributable to [M − Cl − HCl]+ and [Pd(Ph2Pfc)]+ (fc = ferrocene-1,1′-diyl) in its ESI MS spectrum.
An analogous reaction of 1 with [PtCl2(cod)] (Scheme 3; CHCl3/refluxing for 3 h) afforded a mixture of two compounds showing different 31P NMR shifts and 1JPtP coupling constants. Considering the fact that even the signals of the SMe protons were flanked with 195Pt satellites, the products were formulated as isomeric P,S-chelate complexes cis-3 (δP 5.3, 1JPtP = 3695 Hz; ca. 35%) and trans-3 (δP 6.6, 1JPtP = 3495 Hz; ca. 65%),18 being distinguished through the 1JPtP coupling constants.19 The isomers differ also in their 1H NMR spectra displaying four degenerate signals and eight anisochronic (diastereotopic) signals for the ferrocene protons in trans-3 and cis-3, respectively.
Fortunately, the isomeric complexes trans-3 and cis-3 could be separated by fractional crystallisation (isolated yields: cis-3 51%, trans-3 26%) and were subjected to X-ray diffraction analysis, which confirmed the assigned structures (see below).
Complexation reactions performed at 1:2 metal-to-ligand ratios proved more complicated. The crude reaction mixture obtained after mixing [PdCl2(cod)] with two molar equivalents of 1 (90 min/room temperature) contained three different compounds, which were identified by 31P NMR spectroscopy as trans-2, 1O (both minor components) and the expected bis-phosphine complex, trans-[PdCl2(1-κP)2] (trans-4). Repeated attempts to isolate this major product (ca. 85% in the reaction mixture) by crystallisation failed and the compound could therefore only be characterised by NMR spectroscopy in a solution.
A similar reaction between [PtCl2(cod)] and two equivalents of 1 (at room temperature for 90 min) afforded a mixture of five different compounds. 31P NMR monitoring revealed the presence of 1O, cis- and trans-3 (all minor components) and two isomeric bis-phosphine complexes, cis- and trans-[PtCl2(1-κP)2] (5). Likewise cis- and trans-3, the isomers of complex 5, can be clearly differentiated by means of 31P NMR spectroscopy19a,b (cis-5: δP 10.2, 1JPtP = 3790 Hz; trans-5: δP 10.9, 1JPtP = 2615 Hz). In accordance with the geometry of the leaving ligand and kinetic inertness of Pt(II) species, the bis-phosphine complexes were formed in cis:trans ratio of ca. 2:1 which, however, changed in favour of the thermodynamically preferred trans-isomer after refluxing for 18 h (cis:trans = 1:2). When the Pt-precursor was replaced with K[PtCl3(η2-C2H4)], whose alkene ligand destabilises the chloride in mutual trans position owing to its large trans-influence,20 the product ratio determined initially (room temperature/90 min) was inverted (cis:trans = 1:2) and did not change upon refluxing for 18 h. Even in this case, however, minor amounts of 1O, cis- and trans-3 were detected in the reaction mixture.
The molecular structures of the Pd(II) and Pt(II) complexes
As indicated above, the structures of trans-2, trans-3 and cis-3 were determined by X-ray diffraction analysis. Only the latter compound was isolated in an unsolvated form while the trans-complexes accommodated molecules of adventitious water (trans-2) or the crystallisation solvent (trans-3) in their structures (for a discussion of the crystal packing, see ESI†). The molecular structures of cis- and trans-3 are presented in Fig. 3. A structural diagram for trans-2 is available as ESI (Fig. S2†). Relevant geometric data are summarised in Table 2.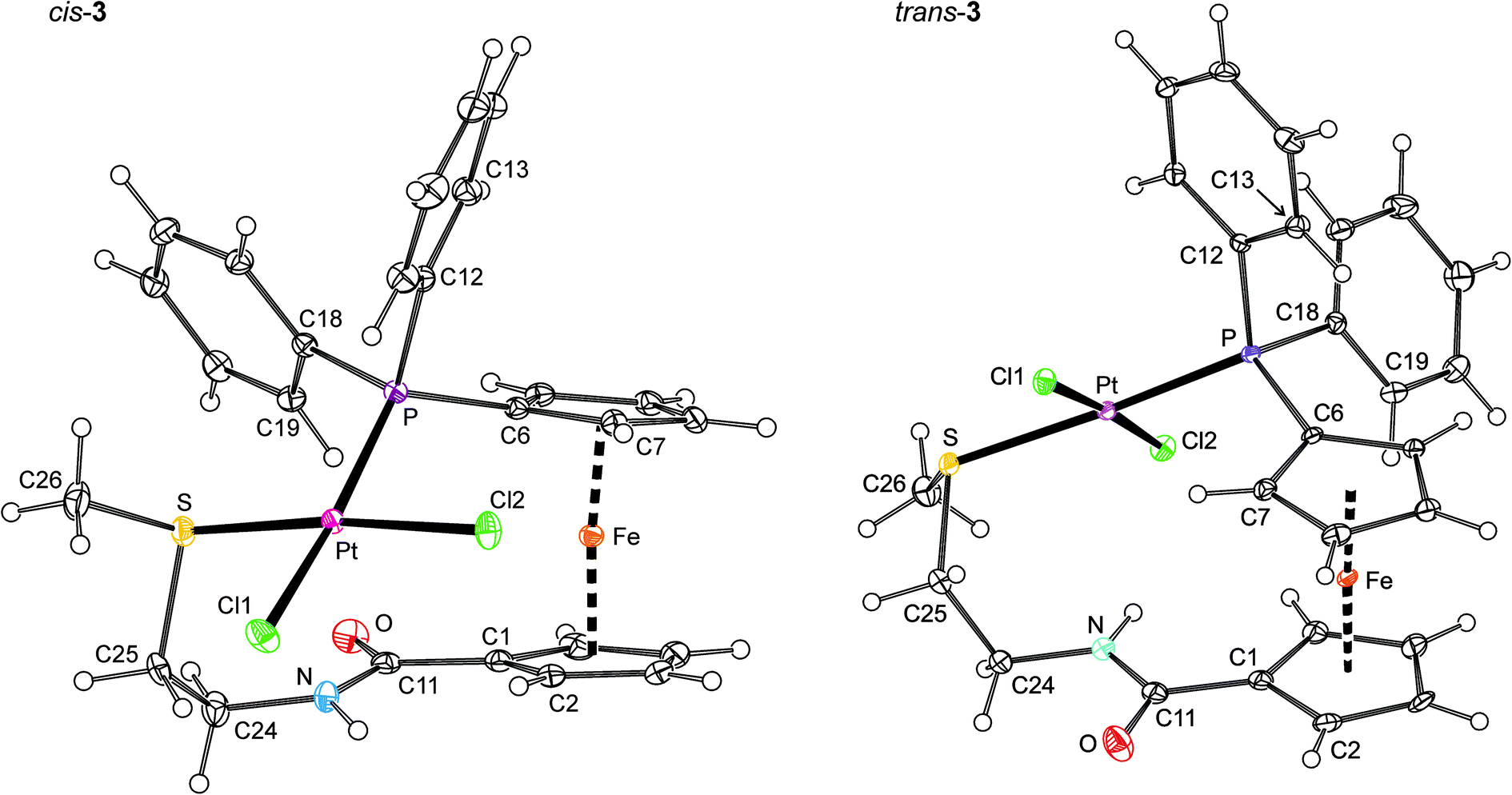 | ||
| Fig. 3 View of the complex molecules in the structures of cis-3 (left) and trans-3·1/2CH3CO2C2H5 (right). Displacement ellipsoids enclose the 30% probability level. | ||
| Parametera | trans-2·1/2H2O | trans-3·1/2CH3CO2Et | cis-3 |
|---|---|---|---|
| a The ring planes are defined as follows: Cp1 = C(1–5), Cp2 = C(6–10). Cg1 and Cg2 stand for the respective ring centroids. b Torsion angle C1–Cg1–Cg2–C6. c Dihedral angle subtended by the amide plane (C11, O, N) and its bonding ring Cp1. | |||
| M | Pd | Pt | Pt |
| M–P | 2.2628(4) | 2.2601(6) | 2.2436(5) |
| M–S | 2.3992(4) | 2.3677(6) | 2.2809(5) |
| M–Cl1 | 2.2931(5) | 2.3051(5) | 2.3564(6) |
| M–Cl2 | 2.2894(5) | 2.3043(5) | 2.3153(5) |
| P–M–S | 174.03(2) | 173.05(2) | 92.86(2) |
| P–M–Cl1 | 90.11(2) | 90.08(2) | — |
| S–M–Cl1 | 83.95(2) | 83.00(2) | 89.84(2) |
| P–M–Cl2 | 91.66(2) | 93.05(2) | 90.37(2) |
| S–M–Cl2 | 94.28(2) | 93.88(2) | — |
| Cl1–M–Cl2 | — | — | 87.87(2) |
| Fe–Cg1 | 1.6547(8) | 1.657(1) | 1.651(1) |
| Fe–Cg2 | 1.6517(8) | 1.651(1) | 1.647(1) |
| ∠Cp1, Cp2 | 3.5(1) | 4.4(1) | 5.5(1) |
| τ | −66.9(1) | −67.7(2) | 33.4(2) |
| C11–O | 1.233(2) | 1.219(3) | 1.234(3) |
| C11–N | 1.347(2) | 1.360(4) | 1.349(3) |
| O–C11–N | 122.4(2) | 122.8(2) | 123.1(2) |
| φ | 7.6(2) | 7.7(3) | 11.3(2) |
| N–C24–C25–S | 68.6(2) | 64.7(3) | 60.7(2) |
| C25–S–C26 | 102.10(8) | 102.2(1) | 99.5(1) |
The complexes adopt the expected square-planar geometry so that the central atoms and their four ligating atoms are coplanar within less than 0.001 Å in trans-2, ca. 0.03 Å in trans-3 and ca. 0.16 Å in cis-3. Rather surprisingly, the largest deviation from a coplanar arrangement detected for the latter complex is associated with the smallest departure of the individual inter-ligand angles from the ideal 90° (ca. 88–93°). Inter-ligand angles in the trans-complexes range ca. 83–94°, with the extreme values pertaining to the S–M–Cl1/2 angles.
The M-donor distances are found within the usual ranges,21 yet they nicely demonstrate the different trans-influence of the ligands (PR3 > SR2 ≫ Cl−)20 and the fact that two soft donor moieties (with a high trans-influence) bonding to a soft metal ion in mutually trans positions destabilise each other.22,23 Thus, the Pt–P and, particularly, the Pt–S bonds are longer in trans-3 than in cis-3,24 while for cis-3, the Pt–Cl distance trans to P is significantly longer than that trans to S. It is also noteworthy that because of practically identical radii of Pd(II) and Pt(II),25 and identical coordination environments, the overall structures of trans-2 and trans-3 are very similar. The M-donor (M = Pd, Pt) bond distances and inter-ligand angles in these complexes differ by no more than ca. 0.03 Å and 1°, respectively.
In order to comply with the demands of the coordinated metal ions, the ligand molecule undergoes a pronounced conformational reorganisation. The major changes are seen at the ferrocene unit, whose substituents are rotated to positions more proximal than in the structure of uncoordinated 1. In trans-2 and 3, the ferrocene units are synclinal eclipsed, and in cis-3 they assume an even closer position near to staggered synclinal. The amide units are also rotated from positions observed in free 1 (by ca. 40°) so that the N atoms are forced away from the ferrocene unit, slightly more in cis-3 than in the trans-complexes. On the other hand, the tilt angles remain rather low, the largest tilting being 5.5(1)° for cis-3. The orientation of the substituents at the C24–C25 are virtually the same as that in the free ligand, but the bond is differently orientated towards the plane of the amide-substituted cyclopentadienyl ring.26 The C–P–C angles and C25–S–C26 angles change only marginally.
Electrochemistry
The electrochemical behaviour of ligand 1, phosphine oxide 1O and Pd(II) complex trans-2 was studied by cyclic and differential pulse voltammetry at a glassy carbon disc electrode in 1,2-dichloroethane containing 0.1 M Bu4N[PF6] as the supporting electrolyte.Compound 1 showed three consecutive oxidations within the accessible potential range (Fig. 4). The first oxidation observed at Epa = 0.28 V vs. ferrocene/ferrocenium27 was quasi-reversible. At relatively lower scan rates or when the switching potential was set to more positive values, it appeared electrochemically irreversible. However, when scanned separately and with higher scan rates, a reductive counter-wave could be detected (see inset in Fig. 4).28
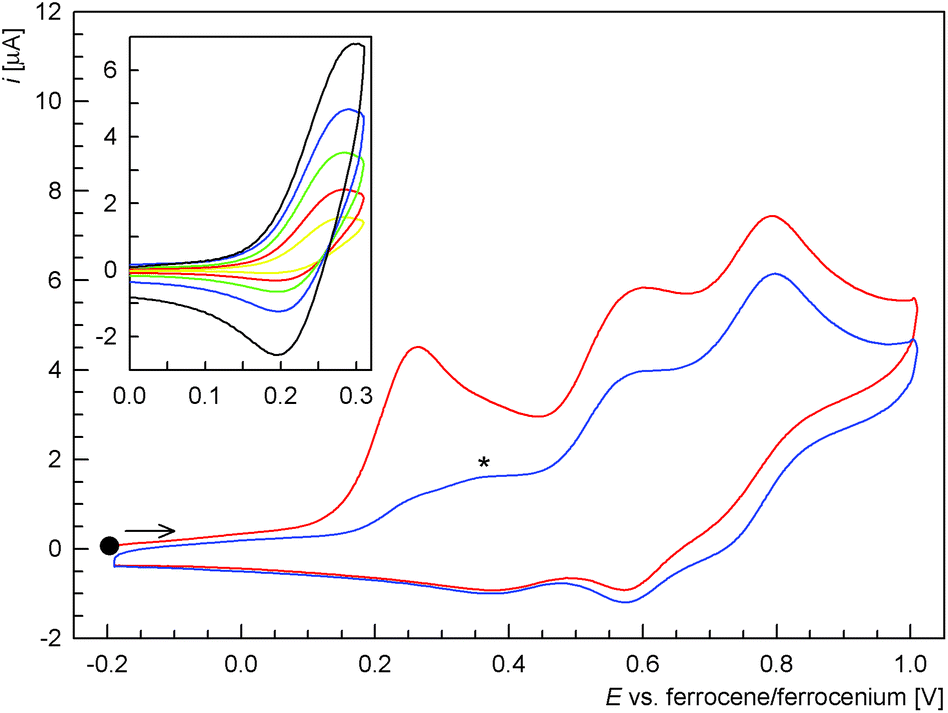 | ||
| Fig. 4 Cyclic voltammogram of 1 recorded in 1,2-dichloroethane on a glassy carbon electrode (first scan in red, second scan in blue). The signal marked with an asterisk is presumably due to 1O. The inset shows partial cyclic voltammograms recorded at different scan rates (yellow 0.02 V s−1, red 0.05 V s−1, green 0.10 V s−1, blue 0.20 V s−1, and black 0.50 V s−1). | ||
The subsequent oxidations seen at Epa = 0.60 and 0.80 V were also irreversible and probably composite in nature. Their complex nature was manifested in the difference pulse voltammograms (Fig. 5). It is also noteworthy that another wave, partly replacing the first oxidation wave, emerged upon repeated scanning (see Fig. 4, signal marked with an asterisk). Based on a comparison of the redox potentials and in view of our previous work,11a this wave could be tentatively assigned to the oxidation of 1O resulting from primary, ferrocene-based oxidation and the associated follow-up chemical reactions of the unstable ferrocenium intermediate.29
 | ||
| Fig. 5 Differential pulse voltammograms of 1 recorded in 1,2-dichloroethane on a glassy carbon electrode and with different modulation amplitudes (given in the figure). | ||
Unlike 1, the redox behaviour of the corresponding phosphine oxide 1O was simple (Fig. 6). The compound, lacking the lone pair of phosphorus as a reactive site, underwent a single, one-electron reversible oxidation at E°′ = 0.40 V. The separation of the peaks in cyclic voltammograms was ca. 80 mV at a scan rate of 0.1 V s−1.30 The anodic and cathodic peak currents increased linearly with the square root of the scan rate (ip ∝ ν1/2) and their ratio remained close to unity over the range 0.02–1.0 V s−1. This oxidation, attributed to the ferrocene/ferrocenium couple, appeared shifted to more positive potentials than the ferrocene reference, in accordance with the electron-withdrawing nature of the substituents attached to the ferrocene unit (cf. the Hammett's σp constants:31 CONHMe 0.35, PPh2 0.19, P(O)Ph2 0.53) that render the oxidation more difficult.
 | ||
| Fig. 6 Representative cyclic (left; scan rate 0.1 V s−1) and differential pulse (right) voltammograms of 1O recorded in 1,2-dichloroethane on a glassy carbon electrode. The voltammograms are presented with the same scale; modulation amplitude for the differential pulse voltammograms is given in the figure. | ||
Similarly to 1O, complex trans-2 (Fig. 7) displayed a one-electron, reversible oxidation at E°′ = 0.46 V.32 This wave was shifted with respect to the free ligand, most likely owing to an electron density transfer from the ligand (ferrocene unit) upon coordination. An additional irreversible, multi-electron reduction was observed at Epc = −1.34 V27vs. ferrocene/ferrocenium. Together with an associated, small oxidation wave at Epcca. −0.61 V, this probably reflects an irreversible reductive removal of one Pd-bound chloride as described previously for [PdCl2(PPh3)2].33
 | ||
| Fig. 7 Cyclic voltammogram of trans-2 recorded in 1,2-dichloroethane on a glassy carbon electrode (first scan in red, second scan in blue). | ||
Conclusions
Functional ferrocene-based phosphino-amide 1 is readily accommodated as a P,S-donor in both the cis- and trans-MCl2 square-planar fragments of the heavier Group 10 metal ions. The latter coordination mode appears particularly remarkable because the ligand is structurally flexible and donor-asymmetric, unlike the majority of trans-spanning donors known to date. Furthermore, the results presented earlier for the structurally related ligands I–IV (see Introduction) and herein for compound 1, demonstrate that the formation of trans-chelate complex donors does not necessarily require the prospective trans-spanning donor to comprise a rigid organic backbone if this is replaced with appropriately selected structural fragments. Further work focusing on other metal ions and a comparison of the structurally related donors 1, III and IV is currently underway.Experimental
Materials and methods
All manipulations were performed under an argon atmosphere and with the exclusion of direct daylight. Chloroform and dichloromethane were dried by standing over K2CO3 and distilled under argon. Methanol was distilled from MeONa. Solvents (from Lachner) for work-up and crystallisations were used without any additional purification. Hdpf,11a [MCl2(cod)] (M = Pd, Pt),34 and K[PtCl3(C2H4)]35 were prepared according to the literature procedures. All other chemicals were obtained from Sigma-Aldrich.NMR spectra were recorded at 25 °C using a Varian UNITY Inova 400 MHz spectrometer operating at 399.95 MHz for 1H, at 100.58 MHz for 13C, and at 161.90 for 31P, if not specified otherwise. Chemical shifts (δ/ppm) are given relative to internal tetramethylsilane (1H and 13C) and to external 85% H3PO4 (31P). Infrared spectra were recorded with an FT IR Nicolet Magna 760 spectrometer in the range 400–4000 cm−1. Electrospray ionisation (ESI) mass spectra were obtained with an Esquire 3000 (Bruker; low resolution) or an LTQ Orbitrap XL instrument (Thermo Fisher Scientific; high resolution).
Syntheses
:1 (v/v) as the eluent. Two orange-yellow bands were collected and evaporated to afford analytically pure phosphine 1 as an orange solid (409 mg, 84%; the first band) and the respective phosphine-oxide 1O (yellow-brown solid, 33 mg, 7%; second band).
1H NMR (CDCl3, 50 °C): δ 2.29 (d, 4JPH = 4.7 Hz, 3 H, SMe), 3.13 (q, J = 5.3 Hz, 2 H, SCH2), 3.79 (br s, 2 H, HNCH2), 4.52 (virtual t, J = 2.0 Hz, 2 H, fc), 4.72 (s, 2 H, fc), 4.80 (br s, 2 H, fc), 5.36 (virtual t, J = 1.7 Hz, 2 H, fc), 7.32–7.47 (m, 6 H, PPh2), 7.51 (t, 3JHH = 5.2 Hz, 1 H, NH), 7.61–7.71 (m, 4 H, PPh2). 31P{1H} NMR (CDCl3): δ 23.6 (s). IR (Nujol): νNH 3366 m, amide I 1659 vs, amide II 1529 vs, 1436 vs, 1306 m, 1284 m, 1170 m, 1098 m, 1056 w, 1033 m, 1000 w, 966 w, 847 w, 821 w, 769 w, 746 m, 708 w, 692 s, 623 w, 548 w, 521 s, 505 m, 475 s, 440 w, 417 w cm−1. MS (ESI+): m/z 688 ([M + Na]+), 526 ([M − HCl − Cl]+), 475 (presumably [Ph2PfcPd]+). Anal. Calcd for C26H26NOPSCl2FePd·0.25CHCl3 (694.5): C 45.39, H 3.81, N 2.02. Found: C 45.44, H 3.82, N 1.84.
A chloroform solution (ca. 1 mL) of the crude product was layered with hexane. Subsequent crystallisation at room temperature for several days afforded orange crystals of cis-3, which were filtered off, washed with hexane (3 mL) and pentane (3 × 3 mL) and dried under vacuum. Yield after two crystallisations: 25 mg (51%), orange crystals. The mother liquor was evaporated and the residue was dissolved in ethyl acetate. The solution was layered with pentane and allowed to crystallise by liquid-phase diffusion for several days. The yellow-orange crystals of trans-3 that formed were filtered off, washed with pentane and dried under vacuum. Yield of trans-3: 12 mg, 26%.
:1 ratio. This ratio changed to 1:2 (inverted) during refluxing for 18 h.
:2 as the major products. The ratio did not change during refluxing for 18 h.
X-Ray crystallography
Single-crystals suitable for X-ray diffraction analysis were grown from hexane–toluene (1), hexane–chloroform (trans-2·1/2H2O, cis-3) and pentane–ethyl acetate (trans-3·1/2CH3CO2Et) by liquid-phase diffusion at room temperature. Crystals of 1O were obtained similarly from hexane–ethyl acetate at −18 °C.The diffraction data (±h ±k ±l; θmax = 27.0 or 27.5°, data completeness ≥99.5%) were collected with an Apex 2 (Bruker) diffractometer equipped with Cryostream Cooler (Oxford Cryosystems) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and were corrected for absorption by the methods incorporated in the diffractometer software.
The structures were solved by direct methods (SHELXS9736) and refined by full-matrix least-squares based on F2 (SHELXL9736). The non-hydrogen atoms were refined with anisotropic displacement parameters and the CH hydrogens were included at their calculated positions and refined as riding atoms. The OH and NH hydrogens were identified on the difference electron density maps and refined as riding atoms with Uiso(H) set to 1.2Ueq(O/N). Relevant crystallographic data and structure refinement parameters are summarised in Table S1 (see ESI†).
Geometric data and all structural drawings were obtained with a recent version of the PLATON program.37 The numerical values were rounded with respect to their estimated deviations (ESDs) given to one decimal place. Parameters pertaining to atoms in constrained positions are given without ESDs.
Acknowledgements
This work was financially supported by the Czech Science Foundation (project no. 13-08890S) and is a part of the long-term research plan of the Faculty of Science, Charles University in Prague supported by the Ministry of Education, Youth and Sports of the Czech Republic (project no. MSM0021620857).Notes and references
- (a) C. A. Bessel, P. Aggarwal, A. C. Marschilok and K. J. Takeuchi, Chem. Rev., 2001, 101, 1031 CrossRef CAS PubMed; (b) Z. Freixa and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2008, 252, 1755 CrossRef CAS.
- For selected recent examples, see: (a) B. P. Morgan and R. C. Smith, J. Organomet. Chem., 2008, 693, 11 CrossRef CAS; (b) R. Schuecker, K. Mereiter, F. Spindler and W. Weissensteiner, Adv. Synth. Catal., 2010, 352, 1063 CrossRef CAS; (c) N. Nasser, D. J. Eisler and R. J. Puddephatt, Chem. Commun., 2010, 46, 1953 RSC; (d) L. Kaganovsky, D. Gelman and K. Rueck-Braun, J. Organomet. Chem., 2010, 695, 260 CrossRef CAS; (e) Y. Canac, N. Debono, C. Lepetit, C. Duhayon and R. Chauvin, Inorg. Chem., 2011, 50, 10810 CrossRef CAS PubMed; (f) K. Ding, D. L. Miller, V. G. Young and C. C. Lu, Inorg. Chem., 2011, 50, 2545 CrossRef CAS PubMed; (g) R. Gramage-Doria, D. Armspach, D. Matt and L. Toupet, Chem.–Eur. J., 2012, 18, 10813 CrossRef CAS PubMed; (h) P. D. Newman, K. J. Cavell and B. M. Kariuki, Dalton Trans., 2012, 41, 12395 RSC; (i) C. Barthes, C. Lepetit, Y. Canac, C. Duhayon, D. Zargarian and R. Chauvin, Inorg. Chem., 2013, 52, 48 CrossRef CAS PubMed; (j) A. G. Jarvis, P. E. Sehnal, S. E. Bajwa, A. C. Whitwood, X. Zhang, M. S. Cheung, Z. Lin and I. J. S. Fairlamb, Chem.–Eur. J., 2013, 19, 6034 CrossRef CAS PubMed.
- (a) N. Wang, T. M. McCormick, S.-B. Ko and S. Wang, Eur. J. Inorg. Chem., 2012, 4463 CrossRef CAS; (b) M. Jung, Y. Suzaki, T. Saito, K. Shimada and K. Osakada, Polyhedron, 2012, 40, 168 CrossRef CAS; (c) P. D. Zeits, G. P. Rachiero, F. Hampel, J. H. Reibenspies and J. A. Gladysz, Organometallics, 2012, 31, 2854 CrossRef CAS.
- (a) J. Gil-Rubio, V. Cámara, D. Bautista and J. Vicente, Inorg. Chem., 2013, 52, 4071 CrossRef CAS PubMed; (b) J. D. Blakemore, M. J. Chalkley, J. H. Farnaby, L. M. Guard, N. Hazari, C. D. Incarvito, E. D. Luzik and H. W. Suh, Organometallics, 2011, 30, 1818 CrossRef CAS; (c) B. P. Morgan, G. A. Galdamez, R. J. Gilliard and R. C. Smith, Dalton Trans., 2009, 2020 RSC.
- K. Tani, M. Yabuta, S. Nakamura and T. Yamagata, J. Chem. Soc., Dalton Trans., 1993, 2781 RSC.
- (a) I. R. Butler, M. Kalaji, L. Nehrlich, M. Hursthouse, A. I. Karaulov and K. M. A. Malik, J. Chem. Soc., Chem. Commun., 1995, 459 RSC; (b) A. Hildebrandt, N. Wetzold, P. Ecorchard, B. Walfort, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2010, 3615 CrossRef CAS.
- The only related examples appear to be complexes containing P,S,X-terdentate ligands, in which the trans-position of the phosphorus and sulphur atoms results from the particular ligand geometry. For representative examples, see: (a) H. A. Ankersmit, N. Veldman, A. L. Spek, K. Vrieze and G. van Koten, Inorg. Chim. Acta, 1996, 252, 339 CrossRef CAS; (b) J. D. G. Correia, Â. Domingos, I. Santos and H. Spies, J. Chem. Soc., Dalton Trans., 2001, 2245 RSC; (c) S. Bonnet, J. Li, M. A. Siegler, L. S. von Chrzanowski, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Chem.–Eur. J., 2009, 15, 3340 CrossRef CAS PubMed.
- J. Kühnert, M. Dušek, J. Demel, H. Lang and P. Štěpnička, Dalton Trans., 2007, 2802 RSC.
- P. Štěpnička, M. Krupa, M. Lamač and I. Císařová, J. Organomet. Chem., 2009, 694, 2987 CrossRef.
- P. Štěpnička, B. Schneiderová, J. Schulz and I. Císařová, Organometallics, 2013, 32, 5754 CrossRef.
- (a) J. Podlaha, P. Štěpnička, J. Ludvík and I. Císařová, Organometallics, 1996, 15, 543 CrossRef CAS; (b) P. Štěpnička, Eur. J. Inorg. Chem., 2005, 3787 CrossRef.
- Ferrocene-based phosphine-thioether donors reported to date include phosphinoferrocenes modified with various SR and CH2SR substituents (R = an organyl group) in positions 1′ or 2 of the ferrocene moiety. For representative examples, see: (a) N. J. Long, J. Martin, G. Opromolla, A. J. P. White, D. J. Williams and P. Zanello, J. Chem. Soc., Dalton Trans., 1999, 1981 RSC; (b) J. E. Aguado, S. Canales, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, Dalton Trans., 2005, 3005 RSC; (c) V. C. Gibson, N. J. Long, A. J. P. White, C. K. Williams, D. J. Williams, M. Fontani and P. Zanello, J. Chem. Soc., Dalton Trans., 2002, 3280 RSC; (d) L. Routaboul, S. Vincendeau, J.-C. Daran and E. Manoury, Tetrahedron: Asymmetry, 2005, 16, 2685 CrossRef CAS; (e) R. Malacea, L. Routaboul, E. Manoury, J.-C. Daran and R. Poli, J. Organomet. Chem., 2008, 693, 1469 CrossRef CAS; (f) E. M. Kozinets, O. Koniev, O. A. Filippov, J.-C. Daran, R. Poli, E. S. Shubina, N. V. Belkova and E. Manoury, Dalton Trans., 2012, 41, 11849 RSC; (g) J. Priego, O. G. Mancheño, S. Cabrera, R. G. Arrayás, T. Llamas and J. C. Carretero, Chem. Commun., 2002, 2512 RSC; (h) O. G. Mancheño, J. Priego, S. Cabrera, R. G. Arrayás, T. Llamas and J. C. Carretero, J. Org. Chem., 2003, 68, 3679 CrossRef PubMed; (i) O. G. Mancheño, R. G. Arrayás and J. C. Carretero, Organometallics, 2005, 24, 557 CrossRef; (j) S. Cabrera, R. G. Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2004, 43, 3944 CrossRef CAS PubMed.
- P. Štěpnička, Chem. Soc. Rev., 2012, 41, 4273 RSC.
- (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- (a) F. R. Hartley, The Chemistry of Platinum and Palladium, Applied Science, London, 1973 Search PubMed; (b) L. Sacconi, F. Mani and A. Bencini, in Comprehesive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 5, ch. 5, p. 1 Search PubMed; (c) C. F. J. Barnard and M. J. H. Russell, in Comprehesive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 5, ch. 51, p. 1099 Search PubMed; (d) A. T. H. Hutton and C. P. Morley, in Comprehesive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 5, ch. 51.9, p. 1157 Search PubMed; (e) D. M. Roundhill, in Comprehesive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 5, ch. 52, p. 351 Search PubMed.
- Some of the signals become well resolved at 50 °C.
- Reaction performed under milder conditions (room temperature/1 h) afforded a mixture containing cis-3 and trans-3 in an approximate 75:25 ratio, which apparently reflects kinetic inertness of Pt(II) and the geometry of the leaving ligand. Also seen was a signal attributable to cis-5 at δP 10.15 with 1JPtP ≈ 3800 Hz, which was not detected in the reaction mixture obtained after refluxing for 3 h.
- In analogy to bis-phosphine complexes of the type [PtCl2L2], the 1J(195Pt,31P) coupling constants in cis isomers can be expected to be larger than in trans isomers: (a) Ref. 16a, ch. 7, pp. 138–140 ; (b) P. S. Pregosin and R. W. Kunz, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer, Berlin, 1979, vol. 16, ch. E, p. 65 Search PubMed.
- (a) T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS; (b) Ref. 16a, ch. 11, p. 299 ff.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., Suppl., 1989, S1 RSC.
- R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS.
- Complexes of the type [PtCl2(PR3)(SR2)] (without chelating P,S-donors), whose crystal structure is known, adopt cis-configuration: (a) H.-P. Abicht, K. Jurkschat, K. Peters, E.-M. Peters and H. G. von Schnering, J. Organomet. Chem., 1989, 364, 415 CrossRef CAS; (b) M. N. I. Khan, C. King, J. P. Fackler Jr. and R. E. P. Winpenny, Inorg. Chem., 1993, 32, 2502 CrossRef CAS; (c) I. Michaud-Soret, J. Kozelka and C. Bois, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 589 CrossRef; (d) P. Kapoor, K. Lövqvist and Å. Oskarsson, J. Mol. Struct., 1998, 470, 39 CrossRef CAS.
- Steric strain resulting from trans-chelate coordination can be expected to operate in accord with trans-influence in this case.
- Compare, for instance, the covalent radii being 1.39 Å for Pd, and 1.36 Å for Pt. This similarity is attributed to lanthanide contraction affecting the heaviest Group 10 metal. The radii are quoted from: B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- The orientation of the C24–C25 bond is nearly the same in all three complexes. Compare the angles subtended by the vector of the C24–C25 bond and the Cp1 plane being 33.0(1)° in 1, 54.7(1)° in trans-2, 55.4(2)° in trans-3, and 56.3(2)° in cis-3.
- E pa and Epc are anodic and cathodic peak potentials, respectively. Potentials determined at the scan rate 0.1 V s−1 are quoted for all irreversible processes.
- The anodic peak current (ipa) was directly proportional to the square root of the scan rate (ipa ∝ ν1/2), which suggests the oxidation to be a standard diffusion-controlled redox transition.
- Chemical reactions following an electron transfer process are well documented for phosphinoferrocenes. For examples, see: (a) J. H. L. Ong, C. Nataro, J. A. Golen and A. L. Rheingold, Organometallics, 2003, 22, 5027 CrossRef CAS; (b) P. Zanello, G. Opromolla, G. Giorgi, G. Sasso and A. Togni, J. Organomet. Chem., 1996, 506, 61 CrossRef CAS; (c) G. Pilloni, B. Longato and B. Corain, J. Organomet. Chem., 1991, 420, 57 CrossRef CAS . See also ref. 11a.
- Ferrocene itself showed practically identical response under the experiment conditions. The observed peak separations are higher than the theoretically predicted values (59 mV at 25 °C), presumably due to a high resistance of the analysed solution. No iR drop correction was applied to the data.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- The process is controlled by diffusion (ip ∝ ν1/2); the separation of the peaks in cyclic voltammogram is 80 mV at a scan rate of 0.1 V s−1.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed and references cited therein.
- D. Drew and J. R. Doyle, Inorg. Synth., 1972, 13, 47 Search PubMed.
- P. B. Chock, J. Halpern and F. E. Paulik, Inorg. Synth., 1990, 28, 349 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal packing diagram for 1, a view of the molecular structure of trans-2·½H2O, description of the crystal packing of the Pd(II) and Pt(II) complexes, and a summary of the crystallographic data. CCDC 962382–962386. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52760c |
| This journal is © The Royal Society of Chemistry 2014 |