 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II) and osmium(II) 1,2,3-triazolylidene organometallics: a preliminary investigation into the biological activity of ‘click’ carbene complexes†
Kelly J.
Kilpin
*a,
Stéphanie
Crot
a,
Tina
Riedel
a,
Jonathan A.
Kitchen
b and
Paul J.
Dyson
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
bChemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, United Kingdom
First published on 30th October 2013
Abstract
Taking advantage of the facile and versatile synthetic properties of ‘click’ 1,2,3-triazolylidene N-heterocyclic carbenes (tzNHC's), a range of new organometallic Ru(II) and Os(II) arene complexes containing functionalised tzNHC ligands, [M(η6-p-cymene)(tzNHC)Cl2] [M = Ru(II), Os(II)], have been synthesised and fully characterised, including the X-ray crystal structure of one of the Os(II) complexes. The tzNHC ligands remain coordinated to the metal centres under relevant physiological conditions, and following binding to the model protein, ubiquitin. The in vitro cytotoxicity of the compounds towards human ovarian cancer cells is dependent on the substituent on the tzNHC ligand but is generally <50 μM and in some cases <1 μM, whilst still retaining a high degree of selectivity towards cancer cells over healthy cells (1.85 μM in A2780 ovarian cancer cells versus 435 μM in human embryonic kidney cells in one case).
Introduction
The introduction and concurrent success of cisplatin and other platinum-based drugs in the clinic has undoubtedly transformed cancer chemotherapy.1 However, the problems associated with platinum agents, in particular their lack of selectivity towards cancer cells, has resulted in the search for alternative metal-based agents which overcome this limitation.2 In this regard, ruthenium(III) coordination compounds, notably NAMI-A and KP1019 (Fig. 1), are under clinical development.3,4 As the proposed mechanism of action of such compounds is thought to involve reduction in vivo, organometallic Ru(II) compounds have also started to attract attention.5,6 Within this field, significant efforts have centered around the development of the RAPTA-based (Ru Arene PTA, pta = 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane) compounds7 and ethylene diamine [Ru-arene-en]+ compounds8 (Fig. 1). Although complexes containing ligands other than phosphines and amines have been investigated for their biological applications,9,10 one class of ligands which seems to have been largely neglected are N-heterocyclic carbenes (NHC's), which is somewhat surprising given the large research efforts and success of Ru-NHC complexes in catalytic applications.11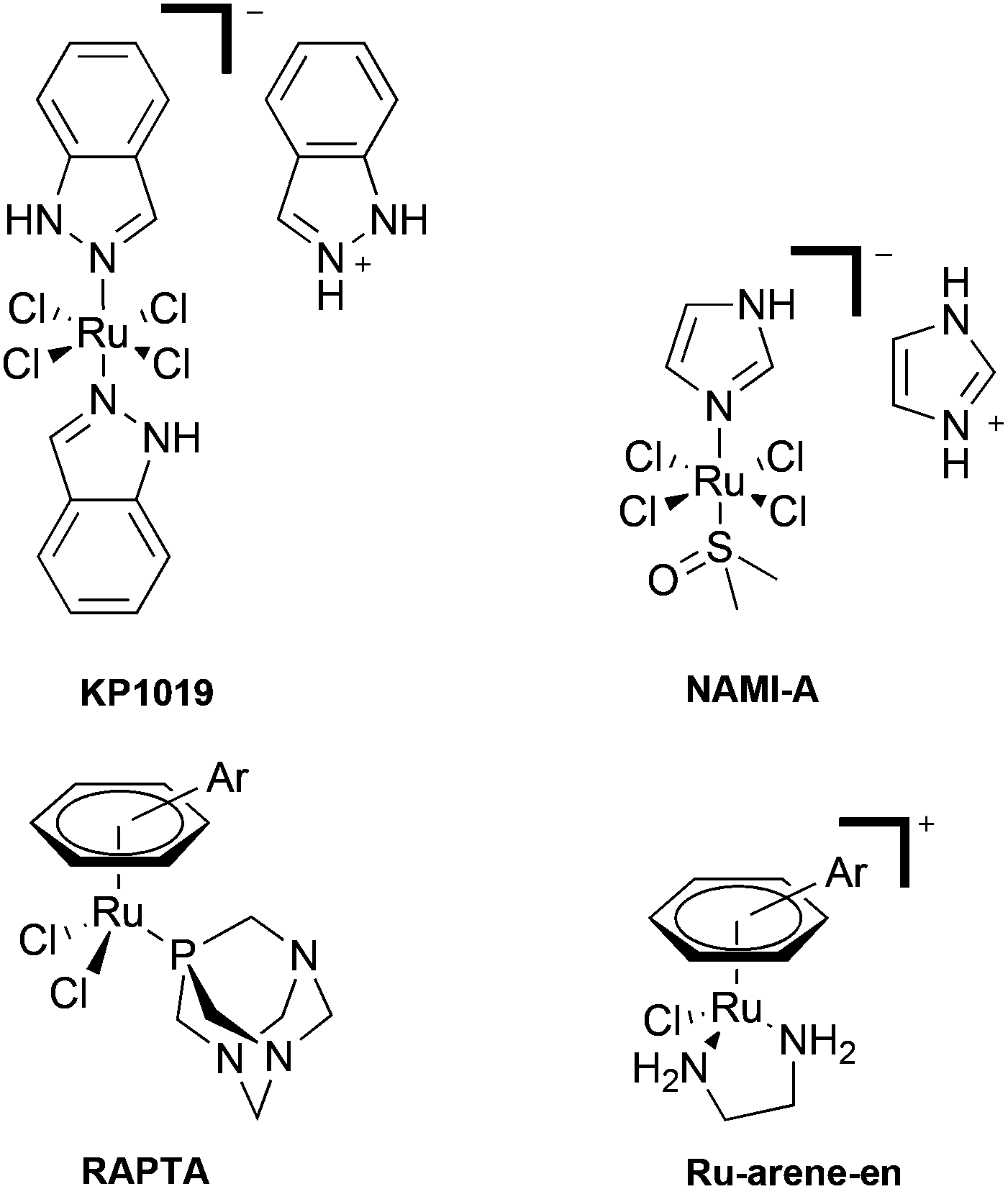 | ||
| Fig. 1 Structures of selected Ru(III) and Ru(II) complexes developed as anti-tumour agents. | ||
The potential of metal-based NHC complexes as anti-cancer agents has been reviewed only recently, with the bulk of the research centered on either Group 10 or 11 metal complexes.12–14 However, Ru(II) NHC complexes have been touched upon, and show promising results in terms of enzyme inhibition and in vitro anti-proliferative effects.15,16 Furthermore, studies on zebrafish embryos have demonstrated that a selection of the complexes are essentially non-toxic,17 a somewhat significant observation considering the high toxicity of the clinically used platinum drugs.
Although relatively new,18 one class of topical NHC ligands, which to the best of our knowledge remain unexplored in biological applications, are the 1,2,3-triazolylidene N-heterocyclic carbenes.19,20 We are particularly interested in this class of NHC as the facile, modular synthetic route via the CuAAC (copper catalysed azide alkyne cycloaddition) ‘click’ reaction offers the ability to readily introduce a number of functional groups onto the parent triazole21,22 and ultimately the metal complex23e.g. for use in cellular targeting or enzyme inhibition.24 Furthermore, the resulting strong metal–ligand bond of the organometallic complex25 may prove advantageous in a highly complex biological environment.
Herein, we report the synthesis, characterisation and, to the best of our knowledge, the first investigation into the biological activity of ruthenium(II) and osmium(II) arene compounds containing 1,2,3-triazolylidene NHC ligands.
Results and discussion
Synthesis and characterisation
Ruthenium(II) and osmium(II) piano stool complexes containing functionalised tzNHC ligands were synthesised (Scheme 1) using an adaption of the procedure reported by Albrecht et al.18 | ||
| Scheme 1 Route used to prepare Ru(II) and Os(II) 1,2,3-triazolylidene NHC complexes. 1a/2a/3a/4a/5a: R = ethyl; 1b/2b/3b/4b/5b: R = n-hexyl; 1c/2c/3c/4c/5c: R = n-dodecyl; 1d/2d/3d/4d/5d: R = benzyl; 1e/2e/3e/4e/5e: R = 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl; 2f/3f/4f/5f: R = 2,6-diisopropylphenyl. | ||
In brief, the facile one-pot CuAAC methodology described by Crowley and co-workers was used to synthesise the 1,4-disubstituted-1,2,3-triazoles 2a–e, eliminating the need to isolate the potentially explosive azide intermediates.26,27 Conversely, 2f was synthesised directly from 1-azido-2,6-diisopropyl benzene. Regioselective methylation of the triazoles 2a–f at the N3 position was carried out using [Me3O][BF4] to afford 3a–f.28 Subsequent reaction of the triazolium salts 3a–f with Ag2O, followed by addition of either [Ru(η6-p-cymene)Cl2]2 or [Os(η6-p-cymene)Cl2]2, afforded the mononuclear piano stool complexes in good yields.18 Using this method we were able to introduce the simple alkyl (ethyl 4a and 5a, hexyl 4b and 5b, dodecyl 4c and 5c), benzyl (4d and 5d), and diisopropylphenyl (4f and 5f) substituents onto the tzNHC metal complex. Furthermore, as a proof of principle that the methodology can be extended to incorporate biologically relevant functional groups such as glucosyl-based moieties, we also synthesised compounds containing a 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl group as a substituent on the tzNHC (4e and 5e). All the metal compounds were isolated as yellow to orange solids which were soluble in a range of solvents (MeOH, CH2Cl2, MeCN, acetone, DMSO), and in addition, the presence of the carbohydrate substituent on 4e and 5e imparted good water solubility to the complexes.
Characterisation data corroborate the expected structures of the metal carbene complexes. Namely, the pre-carbene proton of the triazolium salts 3a–f (H-1, ca. 8.6–8.8 ppm, CDCl3, ESI†) is no longer present in the 1H NMR spectra of the metal complexes 4a–e and 5a–e, and the alpha protons on the N1 triazolylidene substituent show a slight downfield shift (ca. 0.2 ppm) in the 1H NMR spectra relative to the triazolium salt precursors 3a–e. The 13C{1H} chemical shift of the carbene centre (C-1) differs slightly for the ruthenium (ca. 160 ppm) and osmium (ca. 148 ppm) complexes.
The main ion observed in the ESI mass spectra of the complexes arises from fragmentation via the loss of a chloride ligand to give [M − Cl]+ type species.
The molecular structure of 5d was unequivocally verified by single crystal X-ray crystallography on crystals grown at 4 °C by the slow diffusion of diethylether into a dichloromethane solution of 5d. The molecular structure of 5d is shown in Fig. 2 and selected bond parameters are given in the caption.
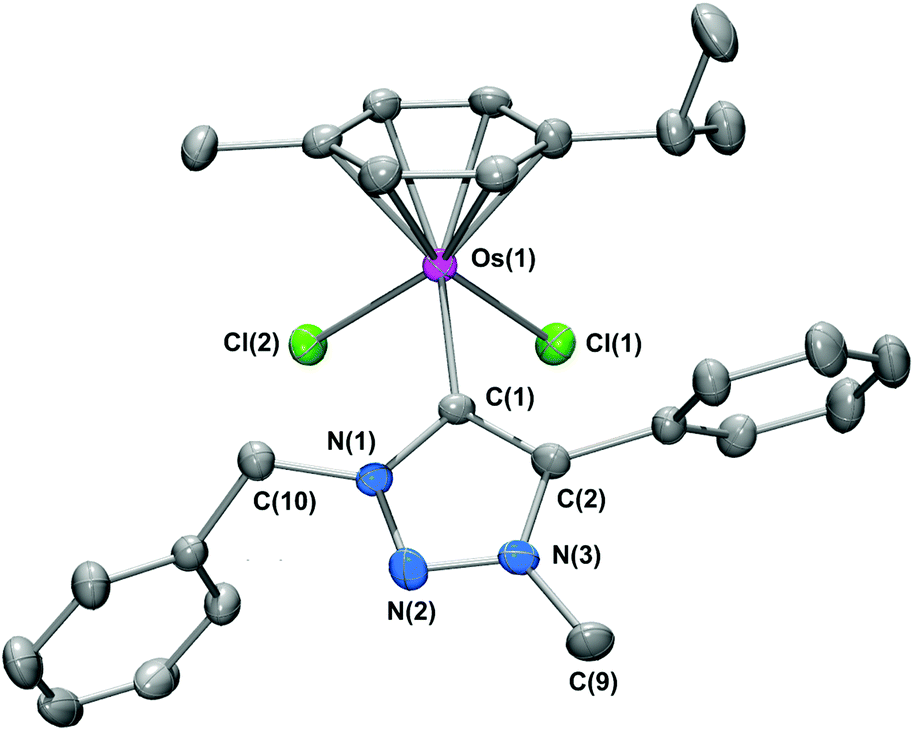 | ||
| Fig. 2 Molecular structure of 5d·CH2Cl2 with selected atom labelling. Hydrogen atoms and the CH2Cl2 solvate have been omitted for clarity. Thermal ellipsoids are drawn at 30% probability. Selected bond lengths (Å) and angles (°): Os(1)–C(1) 2.068(10), Os(1)–Cl(1) 2.420(3), Os(1)–Cl(2) 2.445(3); C(1)–Os(1)–Cl(1) 85.7(3), C(1)–Os(1)–Cl(2) 90.9(3), Cl(1)–Os(1)–Cl(2) 82.53(11). | ||
As expected, the complex adopts a piano-stool motif, comprising the p-cymene ‘seat’, with the two chloride ligands and the tzNHC ligand representing the three ‘legs’. The Os–tzNHC bond length [Os(1)–C(1)] is 2.068(10) Å, with the benzyl N(1) substituent orientated in such a manner to minimize interactions with the p-cymene ring. The Os⋯p-cymene centroid distance is [1.675(5) Å], with no appreciable tilt or offsetting of the p-cymene ring observed.
Behaviour in solution
The chloride ligand(s) of ruthenium arene compounds such as those of the RAPTA and [Ru-arene-en]+ series (Fig. 1) undergo hydrolysis in aqueous solution, giving aquated species, which are proposed to be the active form of the compounds inside cells.29,30 The rate of this hydrolysis process has been suggested to influence the biological activity of ruthenium drugs therefore, it is useful to evaluate the behaviour of new compounds under pseudo-physiological conditions.31,32 As such, the solution characteristics of the new ruthenium(II) and osmium(II) tzNHC complexes were probed by a combination of UV-Vis spectroscopy and 1H NMR spectroscopy, and compared to the RAPTA series of compounds, in particular RAPTA-C, ([Ru(η6-p-cymene)Cl2(PTA)], due to the structural similarity. In aqueous phosphate buffered solution (10 mM, pH 7.4), the UV-Vis spectra of 4e was stable and unchanged from the initial solution over a period of 7 days. However, the UV-Vis trace differed when the spectrum was acquired in saline phosphate buffered solution (10 mM, pH 7.4, [Cl−] 150 mM), indicating that under the former conditions hydrolysis of one, or both, of the chloride ligand(s) had already taken place. Unlike RAPTA-C, which reaches an equilibrium after ca. 20 min,29 hydrolysis of 4e must be very rapid. Moreover, as with RAPTA-C, the process is suppressed under relatively high physiological chloride concentrations. Furthermore, although the 1H NMR spectrum of 4e (D2O) is rather complicated due to the presence of a number of different species (tentatively assigned as hydrolysis products with the loss of either one or both chloride ligands), there was no evidence to suggest the loss of either the p-cymene or tzNHC ligands. The electrospray mass spectra of an aqueous solution of 4e also provides evidence which points towards the loss of one or both of the chloride ligands, with the ions corresponding to both [4e–Cl]+ and [4e–2Cl]2+ species present (see ESI†). As with 4e, the osmium(II) complex 5e also appears to undergo immediate hydrolysis in aqueous phosphate buffered solution.Biological activity
The biological activity of the compounds was initially explored by determining their cytotoxicity against two human tumour (A2780 and A2780R) and non-tumour (HEK) cell lines (Table 1).| A2780a | A2780Rb | HEKc | |
|---|---|---|---|
| a Human ovarian carcinoma cells. b Human ovarian carcinoma cells – acquired resistance to cisplatin. c Human embryonic kidney cells. | |||
| 4a | 1.85 ± 0.29 | 25.6 ± 5.5 | 435 ± 25 |
| 5a | 27.8 ± 12.3 | 112 ± 13 | 168 ± 11 |
| 4b | 0.31 ± 0.08 | 1.77 ± 0.04 | 29.0 ± 1.8 |
| 5b | 4.97 ± 0.03 | 14.2 ± 3.3 | 16.5 ± 1.4 |
| 4c | 0.12 ± 0.02 | 0.23 ± 0.02 | 1.80 ± 0.20 |
| 5c | 0.12 ± 0.02 | 0.33 ± 0.06 | 2.59 ± 0.34 |
| 4d | 4.82 ± 0.12 | 37.8 ± 7.8 | 64.0 ± 6.1 |
| 5d | 1.41 ± 0.06 | 2.36 ± 0.46 | 31.7 ± 5.0 |
| 4e | 41.0 ± 8.2 | >200 | >200 |
| 5e | 19.9 ± 2.3 | 105 ± 5 | >200 |
| 4f | 0.21 ± 0.05 | 1.10 ± 0.12 | 1.85 ± 0.12 |
| 5f | 0.13 ± 0.03 | 0.21 ± 0.01 | 2.89 ± 0.03 |
| Cisplatin | 4.3 ± 0.5 | 18.2 ± 1.0 | 15.3 ± 0.5 |
The new complexes show promising activity profiles, notably, a number of compounds display low micromolar activity against the tumour cell lines (<1 μM), which contrasts with other bifunctional ruthenium arene compounds such as RAPTA-C.33 This difference may be related to the relatively fast hydrolysis process, at least in part, as relationships between the rate of hydrolysis (activation) and cytotoxicity have been demonstrated.34 Importantly, in all cases the tzNHC complexes show selectivity towards the tumour cell lines (A2780 and A2780R) over the non-tumourigenic HEK cell line – this is exemplified by 4a which has a ca. 200-fold selectivity for the A2780 cell line over the HEK cell line. In addition, such a marked in vitro selectivity profile in this assay shows improvements over the selectivity characteristics of cisplatin. It is also evident that increasing the lipophilicity of the complex (brought about by modifying the tzNHC alkyl sidechain) influences the in vitro activity of the complexes,35 with the activity trends being 4a < 4b < 4c and 5a < 5b < 5c, presumably due to increased uptake into cells.36 The relationship between the metal ion employed, i.e. Ru(II) vs. Os(II), and the cytotoxicity for the reported tzNHC complexes follows no obvious trends, and prediction of the level of biological activity upon switching the metal centre from Ru(II) to Os(II) is not straightforward. For example, whilst the in vitro cytotoxicity of RAPTA-C and OsAPTA-C ([Os(η6-p-cymene)Cl2(PTA)] is of a similar order of magnitude,37 when the PTA ligand is replaced by a carbohydrate-based phosphine in a similar series of compounds, the Ru(II) complexes are more active than the Os(II) analogues.38
As RAPTA complexes are believed to interact with proteins in preference to DNA,39 we also probed the interaction of 4b with the model protein ubiquitin (Ub) using HR-ESI mass spectrometry. Complex 4b was incubated with Ub (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 37 °C and the mass spectra were recorded after 1 and 5 days (Fig. 3).
:1) at 37 °C and the mass spectra were recorded after 1 and 5 days (Fig. 3).
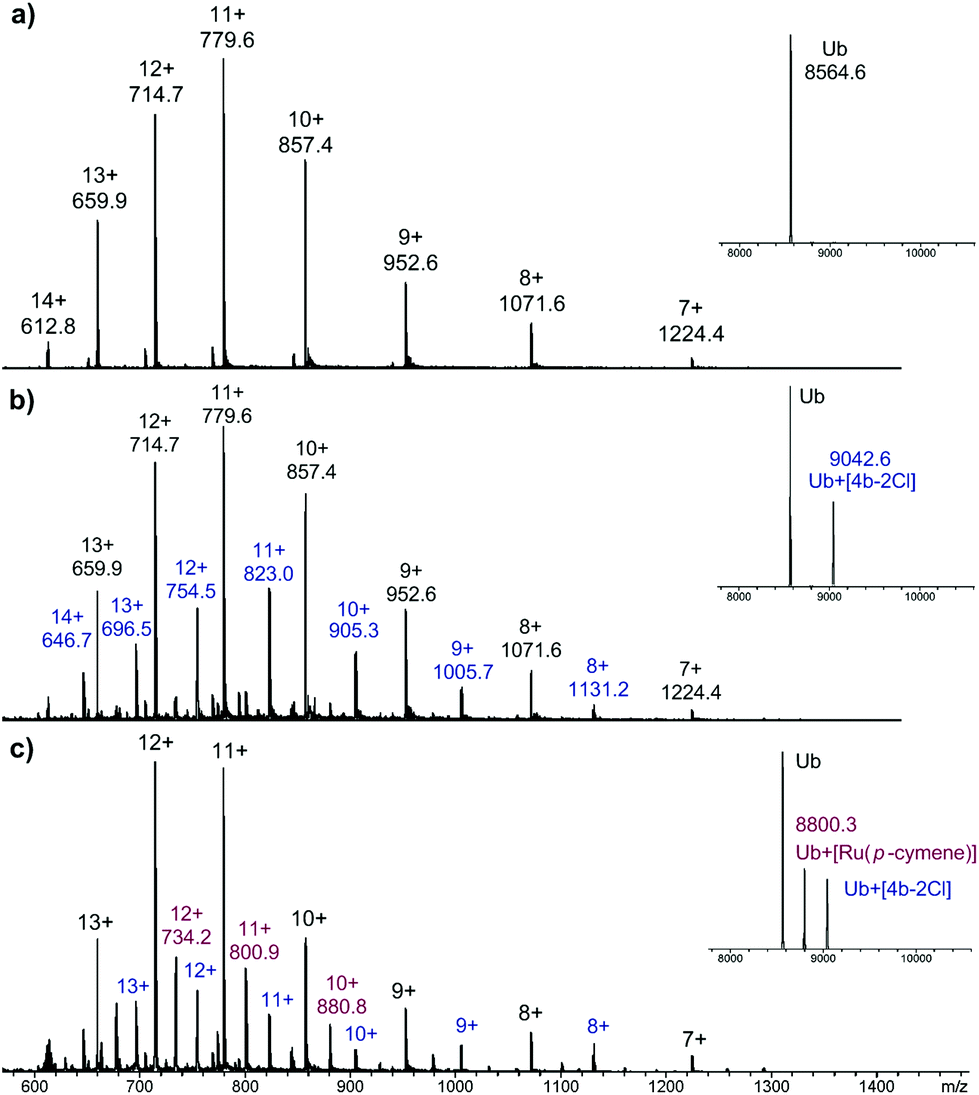 | ||
| Fig. 3 ESI mass spectra of (a) Ub; (b) Ub + 4b, (1:5 ratio) 1 day incubation and (c) Ub + 4b, (1:5 ratio) 5 day incubation, with deconvoluted mass spectra shown as insets. | ||
After 1 day, peaks corresponding to a 1:1 Ub-complex adduct is observed, assigned as Ub + [4b–2Cl], with the tzNHC fragment still bound to the metal centre. After 5 days, partial loss of the tzNHC ligand was observed, and although 4b was present in excess, the mass spectrum still indicated the presence of unmodified Ub. This reactivity is quite distinct to that of RAPTA-C (see ESI†), which reacts far more readily with Ub, with loss of the pta ligand observed after 1 day, and prolonged incubation leading to the complete disappearance of free Ub. The osmium(II) tzNHC complex responded differently again, and was not found to coordinate to Ub under the employed conditions (see ESI†). The low reactivity of the tzNHC compounds towards Ub, and presumably other relevant proteins, may prove advantageous, as with some drugs (in particular cisplatin), interactions with proteins are believed to deactivate the drug and/or lead to adverse side effects.40
Conclusions
For the first time, we have demonstrated that the relatively new tzNHC ligands offer considerable potential in medicinal inorganic/organometallic chemistry. The ruthenium(II) and osmium(II) η6-arene complexes with these ligands show an excellent activity profile against a range of cell lines, and in some instances, such as compound 4a, show excellent selectivity towards tumour cell lines over non tumour lines. In keeping with previously studied Ru(II) arene complexes, the tzNHC examples also undergo hydrolysis in aqueous solution, but in contrast, do not react with model proteins such as Ub as readily. Different groups can be easily introduced onto the tzNHC ligand via the initial CuAAC click reaction. Thus, a number of different functionalities, e.g. targeting groups, enzyme inhibitors, optically active fluorophores/lumophores, may be introduced, allowing diversity to be included in the complexes, an important aspect with respect to drug discovery.Experimental
General
[Ru(η6-p-cymene)Cl2]2,41 and [Os(η6-p-cymene)Cl2]2,42 were prepared using literature procedures, all other reagents and solvents were obtained from commercial sources and used without further purification. The general synthetic protocol for the synthesis of the metal tzNHC complexes 4a–f and 5a–f is outlined below, full synthetic details and characterisation data for all compounds in this study (including 1,4-disubstituted-1-2-3-triazoles 2a–f and the corresponding triazolium salts 3a–f) are included in the ESI.†General synthetic procedure for Ru(II) and Os(II) tzNHC complexes 4a–f and 5a–f
The triazolium salt (1 equiv.), Ag2O (0.5 equiv.) and NMe4Cl (1 equiv.) were stirred in CH2Cl2–MeCN (1:1, 10 mL) in a foil-covered flask for 18 h to give a cloudy white solution. The solvent was removed and the residue redissolved in CH2Cl2. [M(η6-p-cymene)Cl2]2 (M = Ru or Os) (0.5 equiv.) was added and the solution stirred for a further 90 min. The solution was then filtered through a short column of celite to remove the salt by-products, concentrated under reduced pressure and precipitated with either Et2O or hexane at −4 °C. The solid was isolated by filtration, washed with either Et2O or hexane and dried in vacuo.
X-Ray crystallography
Single crystal X-ray diffraction data for 5d·CH2Cl2 were collected using graphite monochromated Cu Kα radiation (λ = 1.54180 Å) on an Agilent Technologies SuperNova dual system in combination with an Atlas CCD detector. Data reduction was carried out using Crysalis PRO.43 The structure was solved by direct methods (SHELXS-97) and refined against all F2 data (SHELXL-97 and Shelxle).44,45 Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were positioned geometrically and refined using a riding model with d(CHaro) = 0.93 Å, Uiso = 1.2Ueq.(C) for aromatic, d(CH) = 0.98 Å, Uiso = 1.2Ueq.(C) for CH, 0.97 Å, Uiso = 1.2Ueq.(C) for CH2 and 0.96 Å, Uiso = 1.2Ueq.(C) for CH3 (Table 2).| CCDC code | 960089 |
| Empirical formula | C27H31Cl4N3Os |
| Formula weight | 729.55 |
| Temperature | 298(2) K |
| Wavelength | 1.54180 Å |
| Crystal system | Monoclinic |
| Space group | P21/n |
| Unit cell dimensions | a = 6.38330(10) Å, b = 28.5636(6) Å, c = 15.9512(3) Å; β = 98.391(2)° |
| Volume | 2877.25(9) Å3 |
| Z | 4 |
| Density (calculated) | 1.684 Mg m−3 |
| Absorption coefficient | 11.946 mm−1 |
| F(000) | 1432 |
| Crystal size | 0.33 × 0.17 × 0.10 mm3 |
| Theta range for data collection | 3.09 to 67.48° |
| Index ranges | −7 ≤ h ≤ 5, −20 ≤ k ≤ 34, −16 ≤ l ≤ 19 |
| Reflections collected | 10726 |
| Independent reflections | 5178 [R(int) = 0.0360] |
| Completeness to theta = 67.48° | 99.8% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.3919 and 0.1083 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 5178/0/317 |
| Goodness-of-fit on F2 | 1.192 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0641, wR2 = 0.1693 |
| R indices (all data) | R 1 = 0.0713, wR2 = 0.1732 |
| Largest diff. peak and hole | 2.700 and −2.486 e Å−3 |
Cell culture conditions and cytotoxicity assay
Human A2780 and A2780cisR ovarian carcinoma and HEK (human embryonic kidney) cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). A2780 and A2780R cells were grown routinely in RPMI-1640 medium, while HEK cells were grown with DMEM medium, with 10% foetal calf serum (FCS) and antibiotics at 37 °C and 5% CO2. Cytotoxicity was determined using the MTT assay (MTT = 3(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide). Cells were seeded in 96-well plates as monolayers with 100 μL of cell solution (approximately 20000 cells) per well and pre-incubated for 24 h in medium supplemented with 10% FCS. Compounds were prepared as DMSO solutions then immediately dissolved in the culture medium and serially diluted to the appropriate concentration, to give a final DMSO concentration of 0.1%. 100 μL of drug solution was added to each well and the plates were incubated for another 72 h. Subsequently, MTT (5 mg mL−1 solution) was added to the cells and the plates were incubated for a further 2 h. The culture medium was aspirated, and the purple formazan crystals formed by the mitochondrial dehydrogenase activity of vital cells were dissolved in DMSO. The optical density, directly proportional to the number of surviving cells, was quantified at 590 nm using a multiwell plate reader and the fraction of surviving cells was calculated from the absorbance of untreated control cells. Evaluation is based on means from at least two independent experiments, each comprising three microcultures per concentration level.
Ubiquitin binding studies
The compound was incubated with Ubiquitin (from bovine red blood cells, min 90%, Sigma Aldrich) in MilliQ water at a molar ratio of 5:1 at 37 °C. Immediately prior to analysis, an aliquot of the incubation solution was diluted with water–acetonitrile–trifluoroacetic acid (50:50:0.1) to yield a final protein concentration of 0.5 μM, and introduced by direct infusion into the ESI source of the mass spectrometer at a flow rate of 5 μL min−1 (source voltage, +4.5 kV; capillary temperature, 180 °C; sheath gas flow, 4 L min−1). Data were processed with DataAnalysis 4.0 SP5 software (Bruker Daltonics).
Acknowledgements
This work was supported by the New Zealand Ministry of Business, Innovation and Employment (formally Foundation of Research, Science and Technology Postdoctoral Fellowship, EPFL1001, to KJK) and the EPFL. We thank Dr R. Scopelliti for the collection of the X-ray data set.Notes and references
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 3993–3993 CrossRef.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CrossRef CAS.
- B. Therrien, Coord. Chem. Rev., 2009, 253, 493–519 CrossRef CAS PubMed.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23–32 RSC.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- L. Oehninger, H. Alborzinia, S. Ludewig, K. Baumann, S. Wölfl and I. Ott, ChemMedChem, 2011, 6, 2142–2145 CrossRef CAS PubMed.
- L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Munoz-Castro, R. W. Koster, K. Baumann, S. Wolfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 1657–1666 RSC.
- J. M. Alfaro, A. Prades, M. del Carmen Ramos, E. Peris, J. Ripoll-Gómez, M. Poyatos and J. S. Burgos, Zebrafish, 2010, 7, 13–21 CrossRef CAS PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- J. D. Crowley, A.-L. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118–1132 CrossRef CAS.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334–6346 CrossRef CAS PubMed.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- D. Yuan and H. V. Huynh, Organometallics, 2012, 31, 405–412 CrossRef CAS.
- J. D. Crowley, P. H. Bandeen and L. R. Hanton, Polyhedron, 2010, 29, 70–83 CrossRef CAS PubMed.
- J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC.
- K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- K. J. Kilpin, S. M. Cammack, C. M. Clavel and P. J. Dyson, Dalton Trans., 2013, 42, 2008–2014 RSC.
- W. H. Ang, E. Daldini, C. Scolaro, R. Scopelliti, L. Juillerat-Jeannerat and P. J. Dyson, Inorg. Chem., 2006, 45, 9006–9013 CrossRef CAS PubMed.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- W. H. Ang, Ecole Polytechnique Federale de Lausanne (EPFL), PhD Thesis, 2007 Search PubMed.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS PubMed.
- A. K. Renfrew, L. Juillerat-Jeanneret and P. J. Dyson, J. Organomet. Chem., 2011, 696, 772–779 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- A. Dorcier, W. H. Ang, S. Bolaño, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- M. Hanif, A. A. Nazarov, C. G. Hartinger, W. Kandioller, M. A. Jakupec, V. B. Arion, P. J. Dyson and B. K. Keppler, Dalton Trans., 2010, 39, 7345–7352 RSC.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS PubMed.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- H. Werner and K. Zenkert, J. Organomet. Chem., 1988, 345, 151–166 CrossRef CAS.
- Crysalis PRO, release 1.171.35.21, Agilent Technologies, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data, selected NMR spectra, mass spectra. CCDC 960089. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52584h |
| This journal is © The Royal Society of Chemistry 2014 |