 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of a lithium magnesiate for use in the non-cryogenic asymmetric deprotonation of prochiral ketones†
Javier
Francos
,
Silvia
Zaragoza-Calero
and
Charles T.
O'Hara
*
WestCHEM, University of Strathclyde, Department of Pure and Applied Chemistry, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: charlie.ohara@strath.ac.uk; Tel: +44 (0)141 548 2667
First published on 1st November 2013
Abstract
A study has been conducted to determine whether lithium magnesiates are feasible candidates for the enantioselective deprotonation of 4-alkylcyclohexanones. The commercially available chiral amine (+)-bis[(R)-1-phenylethyl]amine (2-H) was utilised to induce enantioselection. When transformed to its lithium salt and combined with nBu2Mg, improved enantioselective deprotonation of 4-tert-butylcyclohexanone (with respect to the monometallic lithium amide) at 20 °C was observed. In an attempt to optimise the reaction further, different additives were added to the lithium amide. The best performing deprotonations at 0 °C were those in which (Me3SiCH2)2Mg (er pro-S 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26) and (Me3SiCH2)2Mn (er pro-S 72:28) were added, hence the lithium magnesiate “LiMg(2)(CH2SiMe3)2” was used in the remainder of the study. The optimum solvent for the reaction was found to be THF. NMR spectroscopic studies of a D8-THF solution of “LiMg(2)(CH2SiMe3)2” appear to show that this mono-amide bis-alkyl species is in equilibrium with a bis-amide mono-alkyl compound (and a tris-alkyl lithium magnesiate). When a genuine bis-amide lithium magnesiate solution is used, the deprotonation results were essentially identical to those obtained for “LiMg(2)(CH2SiMe3)2”. By adding LiCl to “LiMg(2)(CH2SiMe3)2” the er at 0 °C improved to 81:19. At −78 °C good yields and an er of 93:7 were obtained. This LiCl-containing base was used to successfully deprotonate other 4-alkylcyclohexanones.
:26) and (Me3SiCH2)2Mn (er pro-S 72:28) were added, hence the lithium magnesiate “LiMg(2)(CH2SiMe3)2” was used in the remainder of the study. The optimum solvent for the reaction was found to be THF. NMR spectroscopic studies of a D8-THF solution of “LiMg(2)(CH2SiMe3)2” appear to show that this mono-amide bis-alkyl species is in equilibrium with a bis-amide mono-alkyl compound (and a tris-alkyl lithium magnesiate). When a genuine bis-amide lithium magnesiate solution is used, the deprotonation results were essentially identical to those obtained for “LiMg(2)(CH2SiMe3)2”. By adding LiCl to “LiMg(2)(CH2SiMe3)2” the er at 0 °C improved to 81:19. At −78 °C good yields and an er of 93:7 were obtained. This LiCl-containing base was used to successfully deprotonate other 4-alkylcyclohexanones.
Introduction
One of the most fundamentally important reactions in modern day synthesis is metallation, that is the replacement of a relatively unreactive C–H bond with a more reactive (more useful) C–metal one.1 Over the past 60 years, the reagents of choice to carry out such reactions have generally been from the organolithium family, primarily due to their high Brønsted basicity. However, these reagents have their drawbacks including that their carbanions are often so basic that they attack common solvents (particularly ethers such as THF) and they are frequently nucleophilic; hence they generally exhibit poor functional group tolerance. To counteract these pitfalls, lithiations are mostly performed at sub-ambient temperatures (often approaching −100 °C), proving a massive financial burden for the chemical industry. Kerr has recently stated that by carrying out metallations at temperatures below −40 °C, the additional energetic cost to industry is in the region of £250000 per year per batch tonne process.2 Therefore one priority in modern day synthesis is to provide solutions to this problem by designing new metallating reagents that are capable of producing excellent results akin to their lithium counterparts but at temperatures closer to ambient. Over the past decade the concept of ate chemistry in synthesis has come to the fore.3 When an alkali metal organometallic reagent is combined with a magnesium one, the new ‘synergic’ bimetallic entity (an alkali metal magnesiate) has often been shown to function as a highly efficient base at temperatures approaching ambient temperature. By combining LiCl with conventional Grignard (RMgX) or Hauser (R2NMgX) reagents, Knochel has demonstrated the tremendous scope achievable when these new turbo reagents are used in a multitude of deprotonations.3c,4 Mongin has utilised lithium tri(n-butyl)magnesiate to regioselectively metallate an array of substrates, including thiophenes, fluoroaromatics, oxazoles, chloropyridines at temperatures close to ambient temperature.5 The subsequent metallo-intermediate can undergo electrophilic quenching to generate substituted derivatives. In special cases, unique reactivity, including multideprotonations and unprecedented regiochemistries with respect to ‘normal’ lithium reagents can also be achieved.6 Until recently, the domain of ate reagents has been essentially confined to achiral anions. Gros has shown that the use of chiral magnesiates specifically heteroleptic lithium magnesium alkyl TADDOLates, can induce good levels of enantioselection, most recently in the enantioselective addition of chiral pyrazyl magnesiate intermediates across aldehydes.7
Results and discussion
Previous foundation work from our group has focused on determining the structures (both in solution and as solids) of magnesiates (and zincates) that incorporate chiral donor ligands [such as (−)-sparteine and (R,R)-TMCDA] or the chiral amide [(R)-N-benzyl-α-methylbenzylamide].8 Building on this foundation, here we systematically study the use of lithium magnesiates in the enantioselective deprotonation of a class of yardstick reagents, namely some prochiral 4-alkylcyclohexanones.9 We decided to thoroughly investigate whether lithium magnesiates are feasible candidates for performing these metallations. The approach we chose was to co-complex a chiral lithium amide with another additive, ultimately an organomagnesium reagent. Firstly, we compared the performance of two commercially available chiral amines (Scheme 1) [(R)-N-benzyl-α-methylbenzylamine (1-H) and (+)-bis[(R)-1-phenylethyl]amine (2-H)] to generate the desired chiral lithium amide (1-Li or 2-Li). When the respective lithium amides were utilised in the deprotonation of 4-tert-butylcyclohexanone at ambient temperature in THF solution, the quenched silylenol ether was obtained in moderate yields (32 and 76% respectively) and in poor enantiomeric ratios (er) [(S:R); 50:50 and 60:40 respectively)] (entries 1 and 3, Table 1). On adding nBu2Mg to the lithium amides, encouragingly gave vastly improved yields (95 and 83%) and most significantly enhanced er [(S:R); 65:35 and 70:30] (entries 2 and 4, Table 1). As the latter experiment showed most promise in terms of enantioselection, we decided to use 2-Li in our subsequent reactions but altering the second metal.
 | ||
| Scheme 1 Reaction of lithium chiral amides and amido (bis)alkyl lithium magnesiates with 4-tert-butylcyclohexanone. | ||
At 0 °C, the additive-free reaction produced an enantiomeric ratio (S:R) of 60:40 in a yield of 76%. Inorganic salts are known to have an effect – advantageous or detrimental – on the course of reactions.10 When simple inorganic salts such as MgBr2 and CoBr2 were utilised (entries 2–4, Table 2), essentially none of the desired product was formed, although addition of ZnCl2 produced a yield of 25% with a S:R ratio of 65:35. If an organometallic reagent was added instead of the salt, the main trend observed was that the yields improved (in excess of 78%) although enantiomeric ratios were variable.
| Entry | Metal reagent added to 2-Li | Yieldb (%) | er (S:R)b |
|---|---|---|---|
| a CoBr2 and ZnCl2; reaction time was 16 h. b Determined by GC analysis. | |||
| 1 | — | 76 | 60:40 |
| 2 | MgBr2 | 0 | — |
| 3a | CoBr2 | 4 | — |
| 4a | ZnCl2 | 25 | 65:35 |
| 5 | n Bu2Mg | 83 | 70:30 |
| 6 | (Me3SiCH2)2Mg | 96 | 74:26 |
| 7 | Me2Zn | 99 | 58:42 |
| 8 | t Bu2Zn | 98 | 60:40 |
| 9 | Me3Al | 89 | 64:36 |
| 10 | iBu3Al | 78 | 60:40 |
| 11 | (Me3SiCH2)2Mn | 99 | 72:28 |
As the n-butyl ligand contains β-hydrogen atoms, and is therefore prone to decomposition via a β-hydride elimination pathway we switched our attention to the trimethylsilylmethyl anion (Me3SiCH2−). The best S:R enantiomeric ratio (74:26) was obtained when the dialkylmagnesium (Me3SiCH2)2Mg was used (entry 6, Table 2). When Zn or Al organometallics were used (entries 7–10, Table 2) the er observed were essentially identical to those obtained when 2-Li was utilised without an additional metal (entry 1, Table 2). Interestingly, on adding the transition metal alkyl (Me3SiCH2)2Mn to the lithium reagent, essentially quantitative conversion to the silyl enol ether was observed with a high degree of enantioselection (S:R, 72:28; entry 11, Table 3). Thus far our best enantioselection was achieved using 2-Li and (Me3SiCH2)2Mg (entry 6, Table 3). Therefore we focused our attention on trying to optimise this reaction (and that involving commercially available nBu2Mg) further. By adding a deficit (0.5 equivalents) or an excess (two equivalents) of nBu2Mg to 2-Li we discovered that the yield of product increased for the former, but decreased for the latter, and the enantiomeric ratio in both cases dropped to 62:38 (entries 2–4, Table 3). When 1:1, 1:2 and 2:1 ratios of 2-Li and (Me3SiCH2)2Mg are utilised, the product yields are excellent (>89%) and all the er are improved from the optimum nBu2Mg case, the best obtained was for the magnesium-rich reaction (pro the S enantiomer, 78:22) although it was modestly better than the 1:1 case (74:26) (entries 5–7, Table 3).
| Entry | Metal reagent added to 2-Li | Yielda (%) | er (S:R)a |
|---|---|---|---|
| a Determined by GC analysis. | |||
| 1 | — | 76 | 60:40 |
| 2 | 1 equivalent nBu2Mg | 83 | 70:30 |
| 3 | 2 equivalents nBu2Mg | 52 | 62:38 |
| 4 | 0.5 equivalent nBu2Mg | 99 | 62:38 |
| 5 | 1 equivalent (Me3SiCH2)2Mg | 96 | 74:26 |
| 6 | 2 equivalents (Me3SiCH2)2Mg | 89 | 78:22 |
| 7 | 0.5 equivalent (Me3SiCH2)2Mg | 99 | 72:28 |
The next variable to be considered was ascertaining what effect changing the solvent medium from THF would have on the results. In carrying out the reaction of 2·Li and (Me3SiCH2)2Mg in neat toluene, only a trace of the desired product formed. In diethyl ether, the GC-yield was 40% and the er was only 67:33 in favour of the S enantiomer. To aid solubility in hexane, one molar equivalent of THF was added; however, the product yield was low (23%) and the er was 65:35; hence it appears that for the systems tested THF remains the optimum solvent choice.
At this juncture it is appropriate to detail some of the NMR spectroscopic work that was performed in an attempt to shed light on the active species that is likely to carry out the deprotonation. Full details and spectroscopic data can be found in the ESI.† The 1H (and where appropriate 7Li) NMR spectra of: (a) 2-H; (b) 2-Li; (c) (2-)2Mg; (d) 1:1 2-Li and (Me3SiCH2)2Mg; and, (e) Me3SiCH2Li in D8-THF were compared. The NCH methylene H-atom proved an important handle for comparing the spectra of (a)–(d). For (a)–(c) the respective 1H NMR shifts were 3.50, 3.64 and 3.84 ppm. For (d) – the active base system – two distinct resonances in approximately equal proportions were observed (3.53 and 3.69 ppm). It was initially envisaged that these two resonances belonged to the diastereotopic H atoms present in the organometallic complex. However, a DOSY NMR experiment on a D8-THF solution of (d) revealed that the solution contained two distinct amido-containing species.† Thus we believe that the expected mono(amido)-bis(alkyl) species “LiMg(2)(CH2SiMe3)2” undergoes ligand reorganisation and is in equilibrium with a bis(amido)-mono(alkyl) relative “LiMg(2)2(CH2SiMe3)” and the tris(alkyl) “LiMg(CH2SiMe3)3” (Scheme 2 and Fig. 1). To provide evidence of this equilibrium, we prepared genuine D8-THF solutions of “LiMg(2)2(CH2SiMe3)” and “LiMg(CH2SiMe3)3” and obtained their respective 1H NMR spectra corroborating that these species were present in the initial 1:1 2-Li and (Me3SiCH2)2Mg reaction (Fig. 2).
 | ||
| Scheme 2 Proposed equilibrium which occurs when a 1:1 mixture of 2-Li and (Me3SiCH2)2Mg is combined in D8-THF. | ||
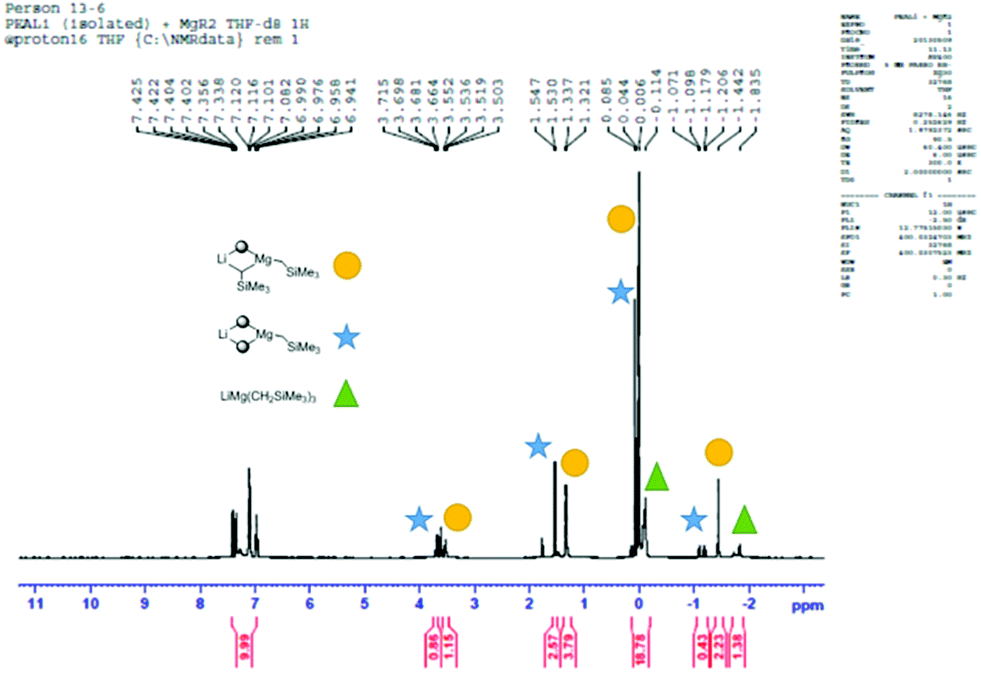 | ||
| Fig. 1 1H NMR spectrum of a mixture of 2-Li and (CH2SiMe3)2Mg in D8-THF showing that co-complexation occurs (star); however, reorganisation of ligands also takes place to give the bis(amido) mono alkyl product (circle) and the tris(alkyl) product (triangle). | ||
 | ||
| Fig. 2 1H NMR spectrum of a mixture 2-Li and (CH2SiMe3)2Mg with an excess of 4-H in D8-THF, showing the presence of the bis(amido) mono alkyl product (star), 2-H (circle) and the generation of SiMe4 (triangle). | ||
Interestingly, when the bis(amido) magnesiate “LiMg(2)2(CH2SiMe3)” was utilised in the enantioselective deprotonation reaction at 0 °C, an identical enantiomeric ratio (74:26 pro-S) was obtained when compared to the 1:1 2-Li and (Me3SiCH2)2Mg reaction (Table 3, entry 5).† The key finding from this experiment is that the addition of a second equivalent of the chiral amine does not improve enantioselection. This begged the question could a simple homometallic alkylmagnesium chiral amide function well in this enantioselective deprotonation? By reacting a 1:1 nBu2Mg–2-H mixture in THF with the ketone for one hour, followed by work-up, only a 3% yield of the desired silylenol ether was obtained. By increasing the reaction time to 16 hours, the yield increased to 72% but the er was 53:47. Hence, for a homometallic magnesium reagent to function it appears that two amides (i.e., the absence of alkyl groups) are required (vide infra).2
Returning to the 1:1 mixture of 2-Li and (Me3SiCH2)2Mg, in an attempt to improve the enantioselectivity of the reaction further, we decided to introduce some common additives (Table 4). Focusing on the reactions involving 2-Li and (Me3SiCH2)2Mg, it was discovered that high yields were maintained using additives; however, TMEDA (N,N,N′,N′-tetramethylethylenediamine) addition caused a slight drop-off in er (from 74:26 to 71:29), but DMPU [1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone] and LiCl both enhanced selectivity (to 78:22 and 81:19 respectively). The same 81:19 ratio was obtained when the LiCl was generated in situ by reaction of 2·HCl with two equivalents of nBuLi.
:1 mixture of 2-Li and (Me3SiCH2)2Mg in the asymmetric deprotonation of 4-tert-butylcyclohexanone at 0 °C
Although the primary purpose of this work was to find an optimum system to operate at close to ambient temperature, for completeness we also investigated the reaction at various temperatures down to −78 °C (Table 5). As expected the optimum er was obtained at the lowest temperature (93:7) with an isolated yield of 87%. When compared with literature work2 which utilised the chiral magnesium bis(amide) (2)2·Mg, it was noted that an identical er was obtained; however, using the magnesiate system gave an increased isolated yield (87% versus 66%). As alluded to earlier, in the monometallic magnesium system two equivalents of chiral amide are required, as one equivalent would produce an alkylmagnesium amide that has an inherently different reactivity and is also subject to Schlenk-type equilibria and perhaps oligomerisation in solution. In our magnesiate system, only one equivalent of chiral amine is required and our sole additive is LiCl i.e., there is no need for DMPU. A final advantage is that the magnesiate reaction time is 3 hours; whereas for the magnesium bis(amide) it is 16 hours.
:1:1 mixture of 2-Li, (Me3SiCH2)2Mg and LiCl (reaction time 1 hour)
To summarise, the optimal results that we have obtained thus far are when we utilise a 1:1:1 mixture of 2-Li, (Me3SiCH2)2Mg and LiCl. As such we tested this reaction mixture with other 4-alkylcyclohexanones (Table 6). In all cases attempted, moderate to good yields were obtained (52–89%). The best er obtained was when the alkyl group's steric bulk was reduced to isopropyl, producing an er of 82:18. Good enantioselection was also obtained for nPr, Ph and Me which were 78:22, 77:23 and 75:25 respectively (Table S1†). Finally, to ascertain whether the opposite enantioselection is possible we employed (−)-bis[(S)-1-phenylethyl]amine in our optimum reaction instead of its enantiomer 2. We obtained 75% isolated yield of the compound with an S:R er of 18:82 at 0 °C (Table 6).
:1:1 mixture of 4-Li, (Me3SiCH2)2Mg and LiCl at 0 °C
Experimental
General methods
1H, 13C NMR and 7Li NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer. All 13C NMR spectra were proton decoupled. Reagents were obtained from commercial suppliers and were used without further purification unless stated below. Purification was carried out according to standard laboratory methods. Diethyl ether, tetrahydrofuran, hexane and toluene were distilled from sodium-benzophenone. (CH2SiMe3)2Mg was prepared from the Grignard reagent (Me3SiCH2)MgCl by manipulation of the Schlenk equilibrium via the dioxane precipitation method. The resultant off-white solid was purified via sublimation at 175 °C (10−2 Torr) to furnish pure (CH2SiMe3)2Mg. tBu2Zn11 and (CH2SiMe3)2Mn12 were prepared according to literature methods and all synthetic work was carried out under an inert argon atmosphere. Gas chromatography was carried out using a Perkin Elmer Clarus 500 Gas Chromatograph.Achiral G.C. analysis: (i) CP Chirasil-DEX CB column; (ii) carrier gas, H2 (45 cm s−1): (i) injector/detector temperature, 250 °C; (ii) initial oven temperature, 90 °C; (iii) temperature gradient, 45 °C min−1; (iv) final oven temperature, 220 °C; and (v) detection method, FID.
Chiral G.C. analysis: (i) CP Chirasil-DEX CB column; (ii) carrier gas, H2 (45 cm s−1): (i) injector/detector temperature, 250 °C; (ii) initial oven temperature, 70 °C; (iii) temperature gradient, 1.7 °C min−1; (iv) final oven temperature, 120 °C; and (v) detection method, FID.
Representative experimental procedure
To a flame-dried and Ar-purged Schlenk flask, (CH2SiMe3)2Mg (0.19 g, 1.0 mmol) was added and dissolved in anhydrous THF (5 mL) and the solution stirred for 5 min. BuLi (1.6 M in hexanes, 1.0 mmol) was added and then the solution was cooled to 0 °C. Bis[(R)-1-phenylethyl]amine (0.22 mL, 1.0 mmol) was added and the cold solution is stirred for 1 hour. After that, 4-tert-butylcyclohexanone (1a) (152 mg, 1.0 mmol) was added to the mixture, and the resulting suspension was allowed to stir for 1 hour. TMSCl (0.256 mL, 2.0 mmol) was added and regular sampling and analysis by gas chromatography monitored the progress of the reaction. After that the reaction is quenched with aq. NH4Cl solution (10 mL) and extracted with AcOEt (3 × 15 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give a residue that was purified by combi-flash chromatography [hexanes–Et2O = 99:1] to afford (4-tert-butylcyclohexen-1-enyloxy)trimethylsilane as a colourless oil (195 mg, 87%).
Conclusions
Our ‘optimised’ lithium magnesiate appears to have the high activity of a lithium reagent, good selectivity of a magnesium one. This promising outcome is in keeping with the previously expressed view that mixed-metal formulations can often operate synergically.13 Future studies will focus on transforming this stoichiometric reaction to a catalytic one.We gratefully acknowledge the support of the EPSRC (J001872/1 and L001497/1) for the award of a Career Acceleration Fellowship to CTOH.
Notes and references
- (a) M. Schlosser, in Organometallics in Synthesis, ed. M. Schlosser, Wiley, Chichester, UK, 2002 Search PubMed; (b) M. Schlosser, Angew. Chem., Int. Ed., 2005, 44, 376 CrossRef CAS PubMed; (c) N. S. Simpkins, Top. Stereochem., 2010, 26, 1 CrossRef CAS.
- L. S. Bennie, W. J. Kerr, M. Middleditch and A. J. B. Watson, Chem. Commun., 2011, 47, 2264 RSC.
- (a) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802 CrossRef CAS PubMed; (b) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS PubMed; (c) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed.
- C. T. O'Hara, in Specialist Periodical Reports: Organometallic Chemistry, ed. I. J. S. Fairlamb and J. M. Lynam, The Royal Society of Chemistry, Cambridge, UK, 2011, vol. 37, p. 1 Search PubMed.
- (a) S. Dumouchel, F. Mongin, F. Trecourt and G. Queguiner, Tetrahedron, 2003, 59, 8629 CrossRef CAS PubMed; (b) F. Mongin, A. Bucher, J. P. Bazureau, O. Bayh, H. Awad and F. Trecourt, Tetrahedron Lett., 2005, 46, 7989 CrossRef CAS PubMed; (c) D. Catel, F. Chevallier, F. Mongin and P. C. Gros, Eur. J. Inorg. Chem., 2012, 53 CrossRef CAS.
- W. Clegg, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and D. M. Tooke, Angew. Chem., Int. Ed., 2001, 40, 3902 CrossRef CAS.
- (a) G. Dayaker, D. Tilly, F. Chevallier, G. Hilmersson, P. C. Gros and F. Mongin, Eur. J. Inorg. Chem., 2012, 6051 CrossRef CAS; (b) O. Payen, F. Chevallier, F. Mongin and P. C. Gros, Tetrahedron: Asymmetry, 2012, 23, 1678 CrossRef CAS PubMed; (c) D. Tilly, K. Snegaroff, G. Dayaker, F. Chevallier, P. C. Gros and F. Mongin, Tetrahedron, 2012, 68, 8761 CrossRef CAS PubMed.
- (a) A. R. Kennedy and C. T. O'Hara, Dalton Trans., 2008, 4975 RSC; (b) D. R. Armstrong, W. Clegg, S. H. Dale, J. Garcia-Alvarez, R. W. Harrington, E. Hevia, G. W. Honeyman, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Chem. Commun., 2008, 187 RSC.
- (a) K. Aoki, H. Noguchi, K. Tomioka and K. Koga, Tetrahedron Lett., 1993, 34, 5105 CrossRef CAS; (b) K. Aoki and K. Koga, Chem. Pharm. Bull., 2000, 48, 571 CrossRef CAS; (c) M. J. Bassindale, J. J. Crawford, K. W. Henderson and W. J. Kerr, Tetrahedron Lett., 2004, 45, 4175 CrossRef CAS PubMed; (d) M. Majewski, A. Ulaczyk-Lesanko and F. Wang, Can. J. Chem., 2006, 84, 257 CrossRef CAS; (e) M. Majewski and F. Wang, Tetrahedron, 2002, 58, 4567 CrossRef CAS; (f) M. Majewski, A. Ulaczyk and F. Wang, Tetrahedron Lett., 1999, 40, 8755 CrossRef CAS; (g) M. Majewski and P. Nowak, Tetrahedron Lett., 1998, 39, 1661 CrossRef CAS; (h) M. Majewski, R. Lazny and P. Nowak, Tetrahedron Lett., 1995, 36, 5465 CAS; (i) C. D. Graf, C. Mulan and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 3014 CrossRef CAS.
- (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1988, 27, 1624 CrossRef; (b) B. Tchoubar and A. Loupy, Salt Effects in Organic and Organometallic Chemistry, VCH, New York, 1992 Search PubMed; (c) P. Caubère, Chem. Rev., 1993, 93, 2317 CrossRef; (d) G. T. Achonduh, N. Hadei, C. Valente, S. Avola, C. J. O'Brien and M. G. Organ, Chem. Commun., 2010, 46, 4109 RSC; (e) M. Hatano, O. Ito, S. Suzuki and K. Isihara, Chem. Commun., 2010, 46, 2674 RSC; (f) M. Hatano, O. Ito, S. Suzuki and K. Ishihara, J. Org. Chem., 2010, 75, 5008 CrossRef CAS PubMed; (g) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681 CrossRef CAS PubMed; (h) H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376 CrossRef CAS PubMed; (i) A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040 CrossRef CAS PubMed; (j) E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 6448 CrossRef CAS PubMed.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184 CrossRef CAS PubMed.
- A. Alberola, V. L. Blair, L. M. Carrella, W. Clegg, A. R. Kennedy, J. Klett, R. E. Mulvey, S. Newton, E. Rentschler and L. Russo, Organometallics, 2009, 28, 2112 CrossRef CAS.
- (a) A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706 CrossRef CAS PubMed; (b) R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, and NMR data. See DOI: 10.1039/c3dt52577e |
| This journal is © The Royal Society of Chemistry 2014 |