 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperparamagnetic iron oxide nanoparticles stabilized by a poly(amidoamine)-rhenium complex as potential theranostic probe†
Daniela
Maggioni
*ae,
Paolo
Arosio
be,
Francesco
Orsini
be,
Anna M.
Ferretti
c,
Tomas
Orlando
de,
Amedea
Manfredi
a,
Elisabetta
Ranucci
ae,
Paolo
Ferruti
ae,
Giuseppe
D'Alfonso
ae and
Alessandro
Lascialfari
*be
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133, Milano, Italy. E-mail: daniela.maggioni@unimi.it; Fax: +39-02-50314305
bDipartimento di Fisica, Università degli Studi di Milano, Via Celoria 16, 20133, Milano, Italy. E-mail: alessandro.lascialfari@unimi.it
cLaboratorio di Nanotecnologie, CNR-Istituto di Scienze e Tecnologie Molecolari, via G. Fantoli 16/15 I, 20138, Milano, Italy
dDipartimento di Fisica, Università degli Studi di Pavia and CNISM, Via Bassi 6, 27100, Pavia, Italy
eUdR Milano Consorzio INSTM, via G. Giusti 9, 50121 Firenze, Italy
First published on 16th October 2013
Abstract
Three-component nanocomposites, constituted by a superparamagnetic iron oxide core coated with a polymeric surfactant bearing tightly bound Re(CO)3 moieties, were prepared and fully characterized. The water soluble and biocompatible surfactant was a linear poly(amidoamine) copolymer (PAA), containing cysteamine pendants in the minority part (ISA23SH), able to coordinate Re(CO)3 fragments. For the synthesis of the nanocomposites two methods were compared, involving either (i) peptization of bare magnetite nanoparticles by interaction with the preformed ISA23SH-Re(CO)3 complex, or (ii) “one-pot” synthesis of iron oxide nanoparticles in the presence of the ISA23SH copolymer, followed by complexation of Re to the SPIO@ISA23SH nanocomposite. Full characterization by TEM, DLS, TGA, SQUID, and relaxometry showed that the second method gave better results. The magnetic cores had a roundish shape, with low dispersion (mean diameter ca. 6 nm) and a tendency to form larger aggregates (detected both by TEM and DLS), arising from multiple interactions of the polymeric coils. Aggregation did not affect the stability of the nano-suspension, found to be stable for many months without precipitate formation. The SPIO@PAA-Re nanoparticles (NPs) showed superparamagnetic behaviour and nuclear relaxivities similar or superior to commercial MRI contrast agents (CAs), which make them promising as MRI “negative” CAs. The possibility to encapsulate 186/188Re isotopes (γ and β emitters) gives these novel NPs the potential to behave as bimodal nanostructures devoted to theranostic applications.
1. Introduction
In the last decade, superparamagnetic iron oxide (SPIO) NPs were intensively investigated as negative T2 contrast agents for magnetic resonance imaging (MRI) and multimodal imaging,1–17 and as agents for magnetic fluid hyperthermia (MFH) treatment and drug delivery.18–25 This is evident from the large number of SPIO NPs commercialized or under clinical investigation (Feridex, Endorem, Resovist, Ferumoxtran-10, Clariscan, AMI-121 and OMP) as contrast agents (CAs).26 The key factor determining the SPIO NPs’ efficiency as CAs is to possess large values of transverse nuclear relaxivity r2, which, in turn, is governed by the size of the NPs, their aggregation state, the diffusion time of the host solution molecules near the NPs and, to a minor extent, by other parameters such as the NP shape, the type of magnetic ion and the coating27,28 (for instance, it has been predicted that the maximum relaxivity for magnetite is reached for particles having a diameter of 25 nm). It should be also remarked that clustering small magnetic nanoparticles was shown to provide an efficient strategy for improving relaxivity without affecting water solubility.27,29Despite the wide effort of the scientific community, up to now no effective synthetic method for biocompatible NPs has been shown to optimize all the above microscopic requirements for obtaining the highest relaxivity, keeping at the same time additional functionalities suitable for e.g. γ- or β-radioemission (to be used in diagnostic SPECT and in radiotherapy, respectively), luminescence, drug delivery or MFH.1–24
The most widely used method for the synthesis of iron oxide based SPIO NPs consists of the co-precipitation of Fe3+ and Fe2+ salts in basic aqueous solution, a major drawback of this synthesis being the poorly achievable monodispersity. For this reason many authors in the last decade investigated alternative synthetic strategies,30–32 which, however, in many cases require extensive post-synthesis treatments to impart hydrophilicity to the NPs.
Actually, it should also be taken into account that for any biomedical application, the nanoparticles should possess high water solubility, biocompatibility and stability at physiological pH. Water soluble polymers, such as dextran and its derivatives, starch or polyethylene glycol, have been largely employed for SPIO stabilization.3,25,33 The use of polymer surfactants directly in the coprecipitation step has been shown to prevent aggregation and achieve a better control of the NP size, as was recently reported.34 On the other hand, poly(amidoamine)s (PAAs) are a family of water soluble polymers which possess many favourable properties for biomedical uses:35 they are biodegradable and biocompatible. Moreover, some PAAs, such as the amphoteric but predominantly anionic one named ISA23, when injected in the bloodstream, exhibit a “stealth-like” behaviour; that is, they circulate for a long time without preferentially localizing in the liver and can be used as carriers for drugs or, if properly labeled, as diagnostic probes.36 The repeating unit of ISA23 contains a carboxyl group and, therefore, holds great potential for coating and clustering SPIO cores, since the carboxyl group is one of the most widely used binding units for surface-capping iron oxide NPs.37–42
In this work we report on the synthesis, by two different routes, and the full characterization of Fe3O4–ISA23SH-Re nanocomposites constituted by a magnetic iron oxide (Fe3O4) core, covered by a copolymer strictly related to ISA23 (namely ISA23SH, Chart 1a),43 which contains a minor fraction (ca. 10%) of repeating units bearing a cysteamine pendant, able to firmly bind to a “Re(CO)3” fragment44 (the resulting stable polymer complex is ISA23SH-Re, Chart 1b).
 | ||
| Chart 1 Schematic drawing of (a) the ISA23SH10% copolymer, in which the majority fraction (0.9) is constituted by the same repeating unit of the ISA23 homopolymer; (b) the ISA23SH-Re complex, in which the sixth coordination position around Re (marked with X) is mainly occupied by a carboxylate group of the polymer backbone.44 | ||
The above SPIO@PAA-Re nanocomposite models a potential diagnostic radiopharmaceutical, since: (a) rhenium has two radioisotopes, 186Re and 188Re, which are both γ and β emitters, with lifetimes and energies compatible with applications in diagnosis (γ) and therapy (β)45 (the use of Re(CO)3 units as a rhenium source is advantageous, because the preparation of the starting material [Re(CO)3(H2O)3]+ marked with 188Re is well established);46–48 (b) the SPIO core allows the NPs to act as contrast agents for MRI. As a consequence, the SPIO@PAA-Re nanocomposites herein described are potential dual probes49 for theranostic applications. It is worth mentioning that in vitro and in vivo biological studies using ISA23SH-Re containing cold rhenium isotopes revealed neither hemolytic activity, nor cytotoxic effects on HeLa cells nor apparent toxic effects after injection in mice.44
2 Experimental
2.1 Reagents and materials
FeCl2·4H2O (Fluka), anhydrous FeCl3 (Merck), NaOH (32%, Fluka), HCl (30%, Suprapur, Merck), and fetal bovine serum (FBS, Sigma) were used as received. Ultrapure water (Milli-Q, Millipore, resistivity 18 MΩ cm−2) was used for the preparation of the aqueous solutions; it was previously deoxygenated by several vacuum/N2 cycles just before use. The copolymer ISA23SH10% (containing ca. 10% of the cysteamine bearing repeating units, Mn = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw = 24800, PD = 1.78) was synthesized as previously reported.44 The ISA23SH-Re copolymer was prepared by reacting ISA23SH with the precursor complex [fac-Re(CO)3(H2O)3](CF3SO3) for 30 min under microwave (MW) activation at 80 °C, as previously reported.44 [fac-Re(CO)3(H2O)3](CF3SO3) was synthesized according to literature procedures,50 starting from Re2(CO)10 (Aldrich).
000, Mw = 24800, PD = 1.78) was synthesized as previously reported.44 The ISA23SH-Re copolymer was prepared by reacting ISA23SH with the precursor complex [fac-Re(CO)3(H2O)3](CF3SO3) for 30 min under microwave (MW) activation at 80 °C, as previously reported.44 [fac-Re(CO)3(H2O)3](CF3SO3) was synthesized according to literature procedures,50 starting from Re2(CO)10 (Aldrich).
2.2 Synthesis of the Fe3O4–ISA23SH-Re nanocomposite by peptization of Fe3O4 nanoparticles: SPIO@PAA-Re(1)
Magnetite nanoparticles were prepared by a slightly modified Massart's coprecipitation method,51 in aqueous medium from a solution of Fe(III) and Fe(II) chloride salts. FeCl3 (38.9 mg, 0.24 mmol) and FeCl2·4H2O (24.3 mg, 0.121 mmol) were dissolved in 400 μL of 0.32 M HCl, and added to 3.0 mL of a 1.5 M NaOH solution, under N2 bubbling to favour the formation of small nanoparticles,3 resulting in the instantaneous formation of a dark brown magnetic precipitate. The precipitate was decanted with an external magnet and washed several times with water until neutral pH, and then it was treated with a water solution of the ISA23SH-Re copolymer containing 0.5 equivalent of Re(CO)3 moieties with respect to the SH groups (112.9 mg in 1.2 mL, 0.3 M with respect to the COOH groups), and sonicated for 2 h at 60 °C. A brown homogeneous suspension was formed without a visible precipitate. The sample of SPIO@PAA(1) thus obtained was dialyzed for three days against deoxygenated water under N2, using a membrane cut-off of 50 kDa, and finally lyophilized.2.3 One-pot synthesis of Fe3O4 nanoparticles coated with ISA23SH: SPIO@PAA(2)
A sample of the copolymer ISA23SH10% (56.2 mg, 0.18 mmol with respect to the repeating monomer) was dissolved in 90 mL water, in a three necked round bottom flask equipped with a mechanical stirrer, and the pH was adjusted to 2 by addition of HCl. Then, 12.2 mg of FeCl2·4H2O (0.06 mmol) and 19.5 mg of anhydrous FeCl3 (0.12 mmol) were added to the acidic solution and stirred for 10 min at 300 rpm. The stirring rate was increased to 500 rpm, and fast injection of 400 μL of 32% NaOH caused a sudden change of the pH (from 2 to 11.5) and of the colour (from colourless to brownish). The clear suspension was stirred at room temperature for 1 h, and then the pH was lowered to neutrality by addition of HCl. The suspension was dialyzed against water under N2 with a membrane cut-off of 50 kDa for three days and finally lyophilized. The lyophilized samples were stored at 4 °C and found to be stable over a period of more than 8 months.2.4 Complexation of fac-Re(CO)3 groups to the Fe3O4-ISA23SH nanocomposites: SPIO@PAA-Re(2)
A clear suspension of the SPIO@PAA(2) (23 mL) at pH 5.1 was treated with 47.2 μL of a 0.1 M solution of [Re(CO)3(H2O)3](CF3SO3), corresponding to 1 equivalent with respect to the SH groups. The mixture was heated by MW irradiation for 30 min at 50 °C. The sample was ultracentrifuged to eliminate unreacted Re starting material (twice, at 7000 rpm, 35 min, 20 °C using an Amicon Ultra Centrifugal Filter (Millipore) with a membrane cutoff of 3000 Da). The occurrence of the reaction was monitored by IR spectra of the retained and filtrate fractions (Bruker Vector22 FTIR instrument, 0.1 mm CaF2 cells): νCO in the retained fractions: 2019s, 1902vs cm−1; in the filtrate 2037s, 1916vs cm−1.2.5 Stability test
The stability of SPIO@PAA-Re(2) nanocomposites was checked in a saline solution (0.150 M NaCl) containing 20 equivalents of the ubiquitous amino acid cysteine. After 24 h at 37 °C neither precipitate formation was observed, nor changes of the νCO IR bands of the retained portion after ultracentrifugation (twice at 7000 rpm, 35 min, 20 °C using an Amicon Ultra Centrifugal Filter (Millipore) with 3000 Da membrane cutoff) were observed. Moreover the filtrate portions did not show any νCO absorption. A further stability test was performed by monitoring by DLS the size of SPIO@PAA-Re(2) nanoparticles, in a suspension added with 10% (v/v) FBS, over 24 h after serum addition. The sample of SPIO@PAA-Re(2) used for the test was suspended for 9 months without significant signs of aggregation.2.6 Thermogravimetric and elemental analyses
TGA measurements were carried out with a heating rate of 3 °C min−1 in the temperature range from 50 °C to 700 °C, under air flux, on 10–20 mg of lyophilized samples, using a Perkin Elmer TGA7HT instrument. The Fe content of the nanocomposites was determined by atomic absorption analyses on an AAnalyst 100 PerkinElmer instrument. The Re and S content of SPIO@PAA-Re(2) was determined by ICP-OES (ICAP 6300, Thermo Electron) on a sample (4.3 mg) dissolved in 1 mL milli-Q water, 0.5 mL 30% HCl (Suprapur) and 0.5 mL 65% HNO3 (Suprapur), digested overnight at room temperature. The S content: calculated 2.72 ppm (on the basis of 1H NMR); measured 2.84 ± 0.04 ppm. Measured Re content 2.49 ± 0.05 ppm (corresponding to a Re–S ratio = 0.16).2.7 Dynamic light scattering (DLS) and ζ-potential measurements
DLS and ζ-potential measurements were carried out on a Zetasizer Nano ZS instrument (Malvern Instruments Corp., Malvern, Worcestershire, UK) at a wavelength of 633 nm with a solid state He–Ne laser at a scattering angle of 173°, at 298 K on diluted samples at pH 7. Each hydrodynamic diameter was averaged from at least three measurements. Standard deviations of the diffusion coefficients were obtained by the non-linear fitting of the correlation function G(t) = 0.15 , where q is the wave vector and is equal to 4πn/λ sin(θ/2), λ is the wavelength of the incident light (633 nm), θ is the detecting angle (173°), and n is the refracting index of the solution approximated to that of the pure solvent (1.33).52 The standard deviations of the hydrodynamic diameters were computed accordingly using the Origin© data analysis software package. From the diffusion coefficients, the hydrodynamic diameters, dH, were obtained through the Stokes–Einstein equation, Dt = (kT)/cπηrH, where k is Boltzmann's constant, T is the absolute temperature, η is the solution viscosity (approximated to that of pure water), and c is a factor depending on the size of the diffusing species with values ranging between 4 (slip boundary conditions) and 6 (stick boundary conditions).53
, where q is the wave vector and is equal to 4πn/λ sin(θ/2), λ is the wavelength of the incident light (633 nm), θ is the detecting angle (173°), and n is the refracting index of the solution approximated to that of the pure solvent (1.33).52 The standard deviations of the hydrodynamic diameters were computed accordingly using the Origin© data analysis software package. From the diffusion coefficients, the hydrodynamic diameters, dH, were obtained through the Stokes–Einstein equation, Dt = (kT)/cπηrH, where k is Boltzmann's constant, T is the absolute temperature, η is the solution viscosity (approximated to that of pure water), and c is a factor depending on the size of the diffusing species with values ranging between 4 (slip boundary conditions) and 6 (stick boundary conditions).53
2.8 Transmission electron microscopy (TEM)
TEM images were recorded using a Zeiss LIBRA EFTEM FEG TEM, operating at 200 kV and equipped with an in-column omega filter for energy selective imaging and diffraction. Samples were prepared by drop drying a water diluted suspension of the studied samples. The average diameter and size distribution of the studied samples were determined by statistical analysis over 500–600 NPs. Images were processed by means of the iTEM TEM Imaging Platform software (Olympus).2.9 Magnetic characterization, relaxivity and MRI measurements
Magnetic measurements were carried out on powders and suspensions using a Quantum Design SQUID MPMS XL-7 magnetometer. The zero field-cooled and field-cooled (ZFC/FC) curves were obtained with an applied magnetic field of 50 Oe in the temperature range 2–300 K, while the field dependent magnetization measurements were recorded in the range of ±5 T at both 2 K and room temperature.The 1H NMR relaxometry characterization (NMR-dispersion profile) was performed at physiological temperature (37 °C) and room temperature by measuring the longitudinal and transverse nuclear relaxation times T1 and T2, in the frequency range 10 kHz–240 MHz. It should be noted that the measurements at room and physiological temperatures gave the same results within 10% (corresponding to viscosity change of water). The range of frequency ν has been chosen in order to cover the most widely used clinical fields, i.e. 0.2 T (ca. 8.5 MHz), 0.5 T (ca. 21 MHz) and 1.5 T (ca. 64 MHz), and to study the mechanisms that led to the nuclear relaxation, through the analysis of the curves of r1 and r2vs. ν (NMRD profiles). The NMR signal detection and generation was obtained with a Smartracer® Fast-Field-Cycling relaxometer (Stelar, Mede, Italy) in the range 10 kHz–10 MHz, with a Stelar Spinmaster spectrometer for 10 MHz–60 MHz and with a Tecmag Apollo spectrometer equipped with an Oxford TH9/88/15 superconductor, working in the range 100–240 MHz. In the second and the third case, standard radio frequency excitation sequences were used for T1 and T2 measurements, respectively saturation-recovery for T1, Carr Purcell Meiboom Gill (CPMG) for T2. To determine the efficiency of MRI contrast agents, we calculated the longitudinal (r1) and transverse (r2) nuclear relaxivities, defined as the increase of relaxation rates of the solvent induced by 1 mmol L−1 of iron:
| ri = [(1/Ti)meas − (1/Ti)dia]/CFe i = 1,2 | (1) |
In vitro MRI experiments were performed on vials containing SPIO solutions at 8.5 MHz using an Artoscan Imager by Esaote SpA. The employed pulse sequence was a high resolution spin echo sequence with TR/TE/NEX = 500 ms/18 ms/2, matrix = 256 × 192, FOV = 180 × 180 for the T1-weighted image, and with TR/TE/NEX = 1000 ms/80 ms/2, matrix = 256 × 192, FOV = 180 × 180 for the T2-weighted image. Here TE is the echo time, TR the repetition time, NEX the number of averages and FOV the field of view.
3. Results and discussion
3.1 Synthesis of the SPIO@PAA-Re nanocomposites
For the synthesis of the nanocomposites two methods were tested. The first one (1 in Scheme 1) involved two steps: bare magnetite nanoparticles were prepared by coprecipitation51 and then peptized by interaction with the water-soluble PAA copolymer ISA23SH-Re (shown in Chart 1b). The rhenium loading of the copolymer corresponded to about one-half of the copolymer SH groups. The IR spectra in the region of the ν(CO) absorptions (Fig. S1†) showed that the interaction with iron oxide did not change the rhenium coordination environment. In particular the position of the bands was in agreement with the presence of a carboxylate group in the sixth coordination position of Chart 1b, as in the starting ISA23SH-Re copolymer.44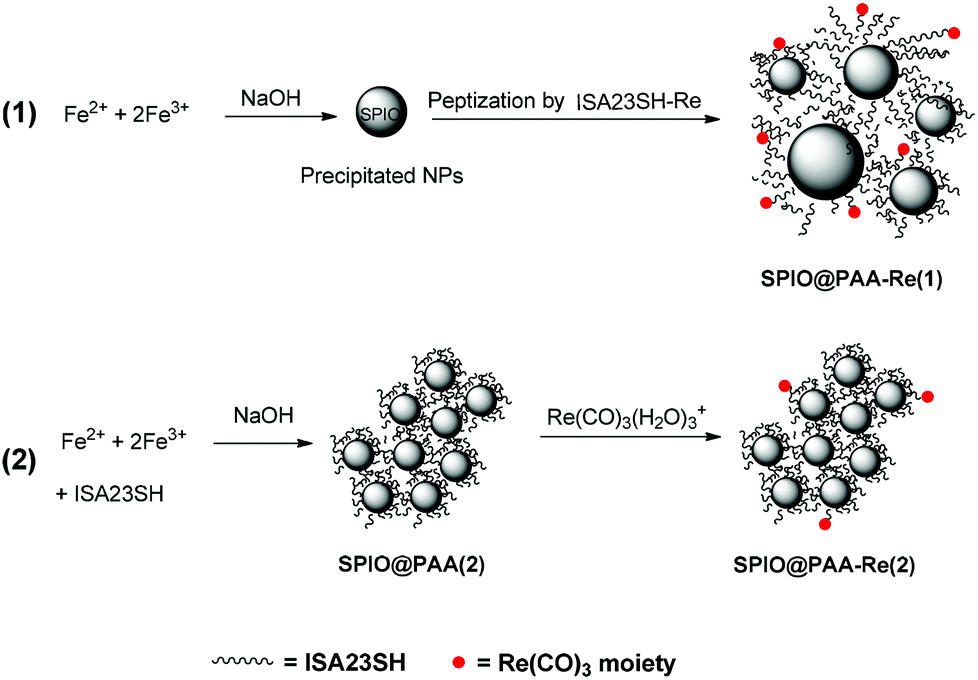 | ||
| Scheme 1 The synthetic routes used in this work for preparing the SPIO@PAA nanomaterials, containing iron oxide nanocrystals coated with the poly(amido)amine (PAA) ISA23SH, decorated with Re(CO)3 units. | ||
The peptization of the precipitated magnetite nanoparticles was effective when performed at pH ca. 3, to favour electrostatic interactions between the COOH groups of the polymer and iron oxide NPs. Indeed, at this pH the surface charge of the NPs is highly positive,54 while the carboxyl groups of the polymer, which are strongly acidic, are deprotonated.55 The peptization process required heating and sonicating (60 °C, 2 h), and produced clear brown suspensions of the nanocomposite, labelled as SPIO@PAA-Re(1) in Scheme 1, which were purified by dialysis (3 days against water) and lyophilized. Thermogravimetric analysis (TGA, Fig. S2†) showed that ca. 20% of the weight was attributable to the iron oxide core (corresponding to ca. 14 wt% of Fe), in good agreement with the analysis by atomic absorption spectroscopy (13 wt% of Fe). This implies the presence of about 150 polymer coils (Mn = 1.4 × 104 Da) per nanoparticle (on the basis of the mean NP size indicated by TEM analysis, see below).
The light-brown lyophilized solid easily dissolved in water, affording suspensions stable over a period of months, without separation of the precipitate. The covering of the nanoparticles by the PAA was further confirmed by ζ potential measurements, which showed the isoelectric point at pH ca. 5, roughly corresponding to that of ISA23,55 but well below the value reported for bare magnetite NPs (pH 8).54 In general the trend of ζ potential values at variable pH (Fig. 1) was very similar to that measured for ISA23SH alone, which is known to spontaneously self-assemble in spherical nanosized aggregates (dHca. 10 nm).44 The ζ potential value at neutral pH was far enough from zero to prefigure stability of the suspensions under physiological conditions.
 | ||
Fig. 1
ζ Potential versus pH curves recorded for SPIO@PAA-Re(1) (■) and for the copolymer ISA23SH alone ( ). ). | ||
However, the physical characterization (see the next paragraph) showed that the magnetic properties were negatively affected by the relatively high polydispersity of the sample. Moreover, the time-consuming purification by dialysis of the SPIO@PAA-Re(1) nanocomposite was poorly compatible with the relatively short lifetimes of the radioactive Re isotopes (17 h 188Re, 88 h 186Re).
Therefore, an alternative synthetic approach was investigated which involved a different kind of two-step procedure with respect to method 1 (route 2 in Scheme 1). At first a SPIO@PAA nanocomposite (devoid of rhenium) was prepared, and then, after proper extensive purification, it was conjugated to rhenium by a fast reaction. The nanocomposite produced in the first step, labelled as SPIO@PAA(2) in Scheme 1, was obtained by a one-pot reaction involving NaOH addition to an acidic solution containing Fe(II), Fe(III) and ISA23SH at room temperature under vigorous mechanical stirring with no need for heating or sonicating. The resulting clear suspension showed high stability, even in the presence of the magnetic field, and favourable morphological and magnetic properties of the nanocomposites (see below). The presence of the polymer during the nanoparticle formation has therefore a beneficial effect on the morphology of the product, as previously reported using different stabilizing polymers.34
For the subsequent conjugation of rhenium, microwave irradiation was used, since it is known that this heating source can promote and accelerate many kinds of reactions.56 In spite of the high dilution, the low temperature and the very short reaction time employed (2 × 10−4 M with respect to Re, 55 °C, 0.5 h, respectively), a satisfactory amount of rhenium was captured by the nanoparticles, corresponding to about one-sixth of the cysteamine pendants of ISA23SH (on average 0.75 rhenium atom per polymer coil and then more than 100 rhenium atoms per NP, according to the ICP analysis of the rhenium content). Such a rhenium loading is adequate, since very low dosages are usually necessary for radioemitter drugs,45,57 whereas high quantities of SPIO NPs have to be administered to provide effective contrast.58 IR spectroscopy of these SPIO@PAA-Re(2) NPs confirmed the uptake of Re(CO)3 groups, showing ν(CO) absorption in the fraction retained after ultracentrifugation. The position of these bands (2019s, 1902vs, br cm−1, Fig. S3†) was different with respect to the nanocomposite SPIO@PAA-Re(1), suggesting the presence of a water molecule in the sixth coordination position of the Re(CO)3–cysteamine complex.44 Indeed, in this case the carboxylate groups of the polymer are supposed to be already interacting with the NP surface in the SPIO@PAA(2) starting material, and are then unable to coordinate to the metal.
The thermal profile by TGA analysis (Fig. S2†) was slightly different with respect to that of NPs obtained by peptization, but the amount of iron oxide was strictly comparable (23%). Moreover, no significant change after rhenium addition was observed.
It is worth pointing out that magnetite cores are produced by the coprecipitation method, but partial subsequent oxidation (in spite of the use of a nitrogen atmosphere for NP handling) might produce different iron oxide nanophases. Among these, maghemite NPs (γ-Fe2O3) have magnetic properties comparable with magnetite and can hardly be differentiated from magnetite NPs by diffraction experiments. The electron diffraction pattern reported below showed a pattern compatible with these two phases only, which throughout this paper will be referred to as iron oxide.
3.2. Size and morphological characterization
The SPIO@PAA-Re(1) nanocomposite was morphologically characterized using a transmission electron microscope (TEM, Fig. 2a). The nanocrystal shape was irregular, varying from quasi-spherical to strongly elongated. An estimation of the mean dimension was done, thus obtaining an equivalent mean diameter = 7.3 ± 3.1 nm and a distribution median = 6.9 nm (Fig. 2b). The difference between the mean and median values highlights the asymmetry and the amplitude of size distribution. The polymer surrounding the NPs was not detectable in these TEM images, which however showed the presence of large aggregates (highlighted by arrows in Fig. 2a). This accounts for the results of the DLS analysis of the same sample, performed at pH 7, which showed the presence of highly polydispersed nanoaggregates, much larger than the single SPIO NPs detected by TEM. The distribution by numbers stated that the most relevant component was represented by NPs with hydrodynamic diameter dH centred at 104 nm. | ||
| Fig. 2 (a) TEM images of SPIO@PAA-Re(1) prepared by peptization (the white arrow shows the aggregates), (b) histogram of size distribution and (c) electron diffraction pattern. | ||
At variance with this, the TEM images of the SPIO@PAA(2) sample (Fig. 3a) revealed that the magnetic cores had a roundish shape with a low dispersion, mean diameter = 6.6 ± 1.8 nm and distribution median = 6.6 nm (Fig. 3b), which stress the sharper and more symmetric distribution of NP dimensions, with respect to SPIO@PAA-Re(1). It is worth noting the presence of small and relatively regular aggregates, having a length of 40–60 nm and a width of 10–30 nm, surrounded by a thin layer of PAA, well visible like a halo in TEM image (Fig. 4). This TEM information is qualitative, but well agrees with DLS data.59 Indeed the analysis of the dialyzed suspension of SPIO@PAA(2) showed a size distribution (numbers) centred at ca. 70 nm (Fig. S4†), which remained constant over several months. The analysis of the correlograms by a non-linear bi-exponential fitting estimated two populations centred at ca. 70 and 170 nm, with the first one being largely dominant (ratio 1000:1), in line with the numbers distribution size analysis.
 | ||
| Fig. 3 (a) TEM images of SPIO@PAA(2) prepared by the “one-pot” synthesis, (b) histogram of size distribution and (c) electron diffraction pattern. | ||
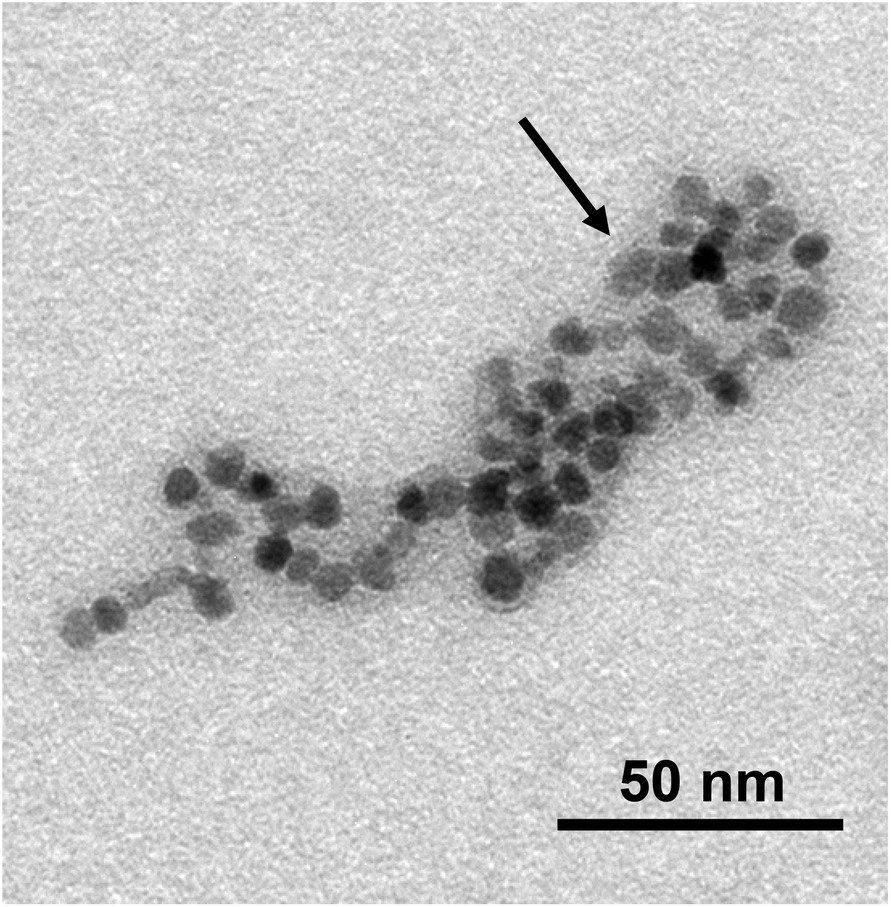 | ||
| Fig. 4 Magnification of a TEM image of SPIO@PAA(2) NPs. The black arrow indicates the polymer presence. | ||
The electron diffraction (ED) analysis (Fig. 2c and 3c) revealed the typical diffraction pattern of magnetite/maghemite crystals. The rings were easily assigned to the low index crystal planes. They were sharper for SPIO@PAA(2) than for SPIO@PAA-Re(1), because of the smaller size distribution, and were slightly blurry, because of the presence of PAA surrounding nanocrystals, as the TEM images pointed out.
The comparison of TEM images acquired before and after the complexation of fac-Re(CO)3 moieties on SPIO@PAA(2) (Fig. S5†) showed that shape, size and size distributions were conserved after microwave irradiation at 50 °C for 30 min for rhenium conjugation. Moreover, the powder diffraction pattern confirmed that the spinel crystal structure of iron oxide nanoparticles was preserved. The only significant effect was a modification in the aggregation of the nanocomposites, since after the complexion reaction, aggregates were no longer clearly detectable in TEM images. In agreement with this, DLS showed a decrease in the size, which after MW irradiation was centred at ca. 25 nm (according to distribution by numbers, Fig. S6†). However, this did not affect the magnetic properties (see the next paragraph).
The formation of aggregates, revealed both by DLS and TEM, likely results not only from non-bonding interactions between polymer coils coating different cores, but also from interactions of single polymer coils with the surfaces of different iron oxide NPs (each coil bearing dozens of carboxylate groups regularly distributed along its chain). Such aggregation does not compromise the stability of the nano-suspensions, since no precipitate formation was observed for very long times.
3.3. Magnetic and relaxivity properties
Fig. 5(a) shows the zero-field-cooled (ZFC) and field-cooled (FC) magnetization of a powder sample of SPIO@PAA-Re(1). The ZFC magnetization curve increases with temperature, reaching a maximum at temperatures T = 75–85 K, which can be assumed, as a first approximation, to coincide with the blocking temperature TB. The FC curve increases as the temperature decreases and never reaches saturation at low temperature, suggesting that interparticle interactions do not significantly affect the relaxation dynamics. The ZFC and FC curves start to diverge at a temperature far above the TB estimated with the ZFC curve. This is due to relatively broad size and shape distributions of the sample as observed in the TEM micrographs. Indeed for this sample, according to the Néel model, the transition from the blocked to the superparamagnetic regime occurs at different temperatures for different particle sizes. The magnetization curve as a function of the field in Fig. 5b shows an open hysteresis loop at 2 K with a coercive field Hc = 480 Oe. The hysteresis curve is substantially saturated at the highest applied field (>3 T).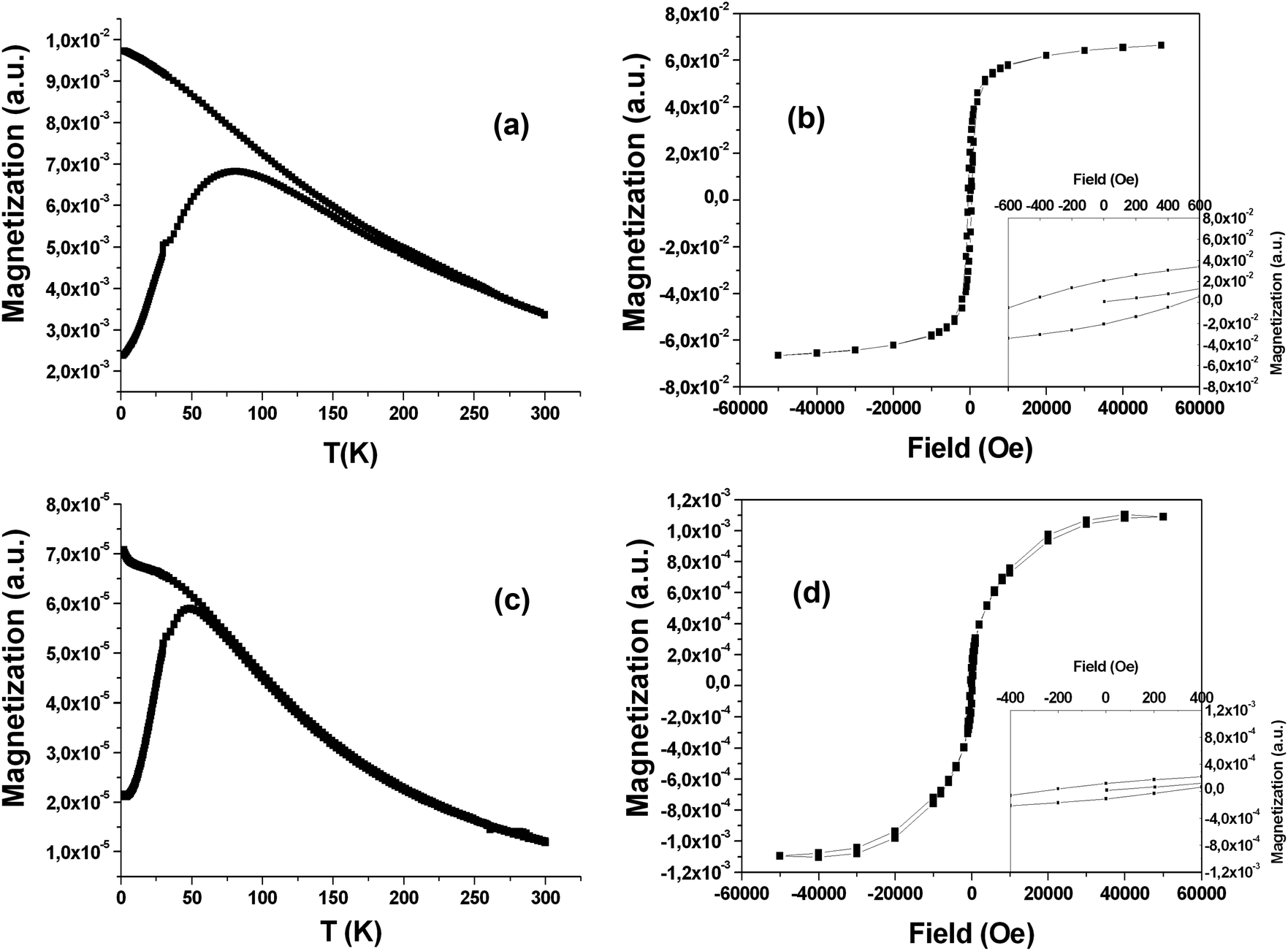 | ||
| Fig. 5 (a) ZFC and FC magnetization curves of SPIO@PAA-Re(1) at 50 Oe; (b) hysteresis cycle recorded at low temperature (2 K) for SPIO@PAA-Re(1); inset: zoom of the hysteresis cycle at low fields; (c) ZFC and FC magnetization curves of SPIO@PAA(2) at 50 Oe; and (d) hysteresis cycle recorded at low temperature (2 K) for SPIO@PAA(2); inset: zoom of the hysteresis cycle at low fields. | ||
On the other hand, magnetic measurements on a sample of SPIO@PAA(2) confirmed the higher monodispersity of the magnetic core, with respect to SPIO@PAA-Re(1), as indicated by TEM micrographs. Indeed the ZFC–FC magnetization curves reported in Fig. 5c show that their divergence occurs at a temperature close to the blocking temperature TB (T = 50 K). In addition a lower TB is indicative of a smaller nanoparticle size as observed in TEM, while the increase of magnetization in the FC curve at low temperature is probably due to paramagnetic impurities. The field dependent magnetization curve for SPIO@PAA(2) confirms that the sample is superparamagnetic at room temperature with zero coercivity, whereas at 2 K an open hysteresis loop is observed, Fig. 5d, with a coercive field Hc = 270 Oe. The hysteresis curve is saturated at 4 T.
SPIO@PAA-Re(1) NPs were characterized by NMR relaxometry and their r2 relaxivity, compared to the negative commercial contrast agent Endorem®, is reported in Fig. 6a. r2(ν) Values were higher than commercial compounds in the whole frequency range and interestingly showed a continuous increase at high frequencies (ν ≥ 10 MHz). The longitudinal nuclear magnetic relaxation dispersion (NMRD) curve (r1vs. ν) obtained for the SPIO@PAA-Re(1) NPs (see Fig. S7†) displayed a typical behaviour for a superparamagnetic CA: it stays flat for low frequencies, and then reaches a maximum and finally decreases rapidly at higher frequencies, showing a profile qualitatively similar to Endorem®, except for a shift towards lower frequencies and an increased height, of the peak. In the case of SPIO@PAA(2) the NMRD longitudinal relaxivity curve (r1vs. ν), reported in Fig. 6b, follows the profile of Endorem®, except for the height of the peak. In general the shape of the r1(ν) NMRD curve can be explained through the known mechanisms of nuclear longitudinal relaxation in superparamagnetic particles.2,3,60,61 The mechanism dominating at low frequency is the Néel relaxation, induced by the reversal of the magnetic moment through the anisotropy energy barrier. The other mechanism, predominating at higher frequencies, is the Curie relaxation, ascribed to the progressive orientation of the magnetic moment with increasing the field. According to Langevin's law, this mechanism depends on the core size and magnetization of the nanoparticles and on the temperature. Curie's mechanism is responsible for the maximum of r1(ν) at higher frequencies and the frequency shift of the peak is directly explained by the change in the nanoparticles size. Therefore the different position of r1 maxima for SPIO@PAA-Re(1) and SPIO@PAA(2), see Fig. S7† and Fig. 6b, is correlated to the different NP sizes, as also evidenced by TEM and magnetic measurements. Measurements on SPIO@PAA-Re(2) NPs (Fig. 6b) at selected frequencies (in the range 1–42 MHz, of interest for the chemico-physical properties of the sample and clinical applications) demonstrated that the subsequent conjugation of rhenium on SPIO@PAA(2) does not affect the mechanisms of longitudinal nuclear relaxation in the system. In Fig. 6c the NMRD transverse relaxivity curve (r2vs. ν) of SPIO@PAA(2) is reported, together with the data for SPIO@PAA-Re(2). Values similar to Endorem were found in the low frequency region, while for ν > 10 MHz a clear enhancement of r2 values was observed. The transverse nuclear relaxation rate (1/T2) is partially described by an approximate heuristic model in the framework of the so-called motional averaging regime (MAR), in which the protons of water molecules surrounding the material are sensitive to the magnetic dipolar field created by the electronic magnetic moments.2,3 The model in the MAR regime (valid when the Redfield condition is verified, i.e. ΔωτD < 1) predicts that the transverse relaxation rate at high field is described by the equation: R2 = 1/T2 = (16/45)fτD(Δω)2, where f is the volume fraction occupied by the nanoparticles in the suspension, Δω = γμ0MV/3 is the angular frequency shift experienced by a proton at the equator of the particle, γ is the gyromagnetic factor of the proton, μ0 is the magnetic permeability of vacuum, MV is the saturation magnetization divided by the particle volume and τD = d2/4D is the translational diffusion time of the protons in the magnetic field inhomogeneities created by the nanoparticles (D being the water translational diffusion constant and d the particle diameter). Recently Gossuin, Sandre et al.62 proposed a universal scaling law to predict the efficiency of magnetic nanoparticles as T2-contrast agents revising the model in the MAR regime. The authors proposed to modify the r2 relaxivity, obtained within the MAR regime, by means of the intra-aggregate volume fraction of magnetic materials ϕintra to derive a corrected relaxivity r2′. In this way the authors state that it is possible to properly compare the obtained relaxation data of different kinds of nanoparticles “as if they were filling the same volume fraction of suspensions of single USPIO nanoparticles”. Our data at a fixed frequency (20 and 60 MHz, near clinical ones) follow this model which, on the other hand, cannot describe the whole transverse NMRD curve (r2vs. ν) or explain the relaxation mechanisms as a function of the applied magnetic field. Nevertheless being the transverse relaxivity r2 the fundamental parameter to test the MRI efficiency for a superparamagnetic material, the reported experimental data demonstrate that our nanoparticles act as promising negative contrast agents. The lowering of r2 values for SPIO@PAA(2) (30–40%) with respect to SPIO@PAA-Re(1) is probably due to the different core size and the different aggregation of the two samples.
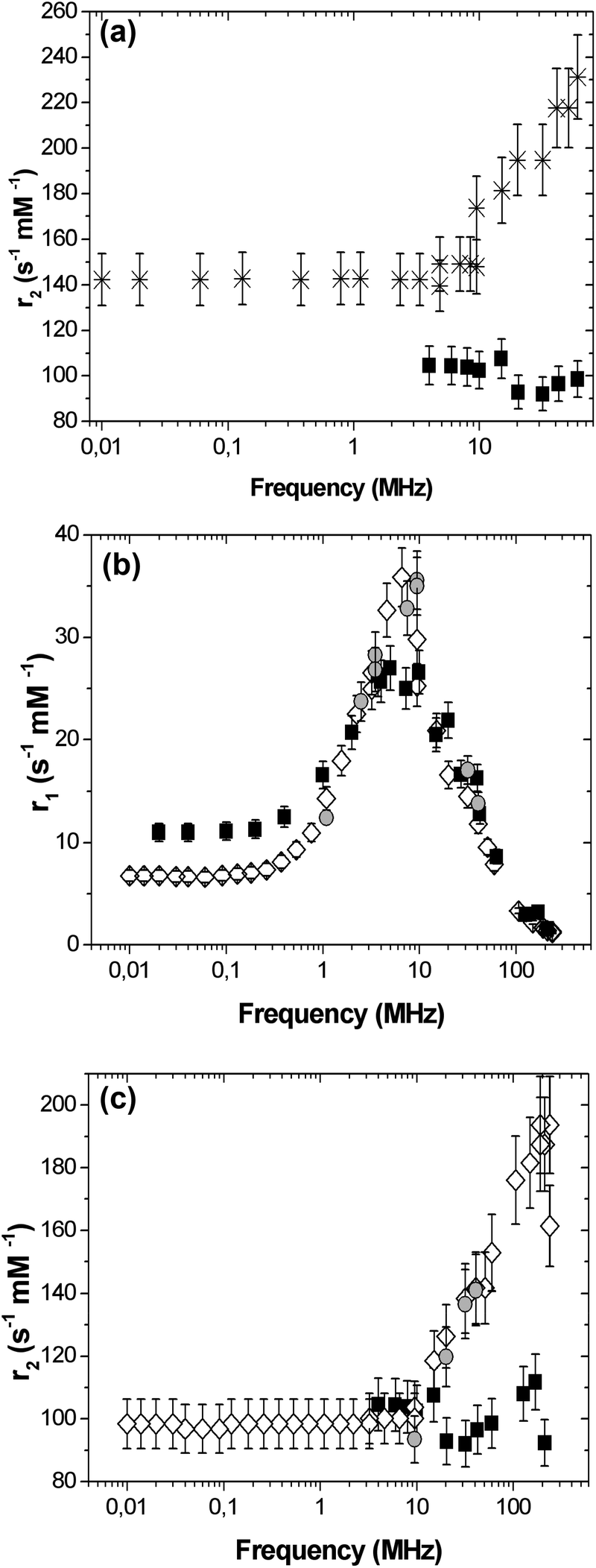 | ||
| Fig. 6 (a) Transverse r2 relaxivity in SPIO@PAA-Re(1) (star symbols), dispersed in ultrapure water. Data are compared with the ones measured for the commercial compound Endorem® (black squares). (b) Longitudinal r1 relaxivity and (c) transverse r2 relaxivity of SPIO@PAA(2) (open diamonds) and SPIO@PAA-Re(2) (grey circles) dispersed in water. Data are compared with the ones measured for the commercial compound Endorem® (black squares). | ||
To get a confirmation of the contrast enhancement ability of our SPIO@PAA(2) samples, MRI in vitro experiments have been performed on a phantom composed of four vials containing: a suspension of the SPIO@PAA(2) sample, Magnevist® (positive or T1 relaxing commercial CA), Endorem® (negative or T2 relaxing commercial CA) and ultrapure water. A standard high resolution spin echo sequence was used to verify the MRI efficiency of SPIO@PAA(2) both as T1 and T2 relaxing agents. In Fig. 7a the T1-weighted image is shown, where a mild ability of the sample to contrast the image as a positive CA is evident. The T2-weighted image, reported in Fig. 7b, is in good agreement with the measured r2 relaxivities at approximately 8 MHz, corresponding to our MRI tomograph operating field (0.2 T). As can be seen from the image, the solution containing SPIO@PAA(2) contrasts with the image at the same quality level of the commercial CA, confirming the efficacy of our material.
 | ||
| Fig. 7 MRI images of vials containing SPIO@PAA(2), Magnevist® and Endorem® at the same concentration (∼0.85 mM), and ultrapure water, obtained by an Artoscan (by Esaote SpA) imager at 8.5 MHz by means of a high resolution spin echo sequence: (a) T1 weighted and (b) T2 weighted images. | ||
3.4 Preliminary stability test of SPIO@PAA-Re(2) under physiological conditions
A preliminary stability assay mimicking physiological conditions was performed on the most promising sample, i.e.SPIO@PAA-Re(2), to test possible aggregation phenomena and the stability of the binding of the rhenium moiety to the nanocomposite, in the presence of possible competitors. A suspension of the dialyzed nanocomposite was added with NaCl (up to 0.15 M) and 20 equivalents (with respect to the rhenium moieties) of a ubiquitous amino acid like cysteine, which should be able to compete with the chelating pendant of the polymer in the coordination of the metal centre. After 24 h at 37 °C no aggregation was observed; moreover, after ultracentrifugation no IR bands ascribable to carbonyl ligands were observed in the filtrate, while the IR spectrum of the retained portions was unchanged, indicating no detachment of rhenium from the nanocomposite. The stability test was repeated in the presence of 10% (v/v) fetal bovine serum. The NP suspension remained clear over 24 h and DLS monitoring (Fig. S8†) showed that the intensity peak remained centred at about 100 nm, as before FBS addition.4. Conclusions
An amphoteric PAA named ISA23SH (containing carboxyl and ter-amine groups in each repeating unit and 10% units also carrying one mercapto group) has been found to effectively stabilize iron oxide NPs, acting either post synthesis (peptization) or simultaneously with the NP formation. In both cases, very stable hybrid organic–inorganic nanocomposites have been produced, in which small SPIO nanoparticles are coated with a large excess of polymer coils decorated with a “Re(CO)3” fragment. The one-pot synthetic route gave better results from the point of view of size and shape homogeneity of the iron oxide cores. Moreover, this procedure is more convenient for rhenium conjugation, since it strongly reduces the manipulation times of the reactants containing hot rhenium isotopes.The synthesized hybrid organic–inorganic nanocomposites show superparamagnetic behaviour at room temperature. Remarkably, the samples have demonstrated the capacity to behave as MRI contrast agents similar or superior to a well-known commercial product, i.e. Endorem®, as demonstrated both by transverse relaxation measurements and in vitro MRI experiments. The presented nanocomposites have thus proven to be very interesting systems, since the capability to encapsulate rhenium might allow the NPs to behave as bifunctional imaging probes, useful for both MRI and SPECT, if hot 186/188Re isotopes were used. On the other hand, the potential of the nanocomposites to behave as theranostic agents must be noted, since Re-containing hybrid materials could be employed for radiotherapy, owing to the β-emission of the hot rhenium isotopes.45 This makes fundamental the achievement of selectivity in cancer tissues addressing. The spontaneous tendency of NPs to accumulate in the tumour tissues (by enhanced permeability and retention effects)63 and the magnetic transport64 could be exploited, but functionalization of the nanocomposites with actively targeting moieties would also be highly advisable.65 Further investigation in this direction is under way.
In the future, efforts will be devoted also to the preparation of SPIO NPs with a suitable size for hyperthermia treatments,22,66,67 thus enhancing the multifunctionality of the hybrid organic–inorganic nanocomposite. A further possible development towards multifunctional magnetic/luminescent/γ-emitter materials could be provided by using, for SPIO NPs stabilization, the recently reported ISA23-based copolymer bearing a phenanthroline pendant in its minority part, able to rapidly bind to Re(CO)3 fragments, affording luminescent polymeric complexes.68
Acknowledgements
A. L. and D. M. thank the INSTM-Regione Lombardia (Italy) MAG-NANO project for partly supporting the research. A. L., P. A. and F. O. thank Fondazione Cariplo project no. 2010-0612. A. M. F. gratefully acknowledges financial support by Fondazione Cariplo (Milano, Italy) under grant no. 2011-2114, and by the Italian MIUR under the grant FIRB RBAP115AYN “Oxides at the nanoscale: multifunctionality and applications”. The authors warmly thank Dr Vladimiro Dal Santo for ICP-OES measurements.Notes and references
- D. L. J. Thorek, A. Chen, J. Czupryna and A. Tsourkas, Ann. Biomed. Eng., 2006, 34, 23–38 CrossRef PubMed.
- Y. Gossuin, P. Gillis, A. Hocq, Q. L. Vuong and A. Roch, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 299–310 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- C. Xu and S. Sun, Dalton Trans., 2009, 5583–5591 RSC.
- A. K. Gupta and M. Gupta, Biomaterials, 2006, 26, 3995–4021 CrossRef PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- L. Harivardhan Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef PubMed.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- C. C. Berry, J. Phys. D: Appl. Phys., 2009, 42, 224003 CrossRef (9pp).
- T. D. Schladt, K. Schneider, H. Schild and W. Tremel, Dalton Trans., 2011, 40, 6315–6343 RSC.
- W.-Y. Huang and J. J. Davis, Dalton Trans., 2011, 40, 6087–6103 RSC.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- L. Zhang, Y. Wang, Y. Tang, Z. Jiao, C. Xie, H. Zhang, P. Gu, X. Wei, G.-Y. Yang, H. Gu and C. Zhang, Nanoscale, 2013, 5, 4506–4516 RSC.
- M. Mahmoudi, V. Serpooshan and S. Laurent, Nanoscale, 2011, 3, 3007–3026 RSC.
- Z. Liu, B. Li, B. Wang, Z. Yang, Q. Wang, T. Li, D. Qin, Y. Li, M. Wang and M. Yan, Dalton Trans., 2012, 41, 8723–8728 RSC.
- F. Hu and Y. S. Zhao, Nanoscale, 2012, 4, 6235–6243 RSC.
- S. Sahu and S. Mohapatra, Dalton Trans., 2013, 42, 2224–2231 RSC.
- A. G. Roca, R. Costo, A. F. Rebolledo, S. Veintemillas-Verdaguer, P. Tartaj, T. González-Carreño, M. P. Morales and C. J. Serna, J. Phys. D: Appl. Phys., 2009, 42, 224002 CrossRef.
- S. Laurent, S. Dutz, U. O. Häfeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8–23 CAS.
- S. Monret, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161–2175 RSC.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. von Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neurooncol., 2007, 81, 53–60 CrossRef CAS PubMed.
- F. Goya, V. Grazu and M. R. Ibarra, Curr. Nanosci., 2008, 4, 1–16 CrossRef.
- Q. Wan, L. Xie, L. Gao, Z. Wang, X. Nan, H. Lei, X. Long, Z.-Y. Chen, C.-Y. He, G. Liu, X. Liu and B. Qiu, Nanoscale, 2013, 5, 744–752 RSC.
- D. Liu, W. Wu, X. Chen, S. Wen, X. Zhang, Q. Ding, G. Teng and N. Gu, Nanoscale, 2012, 4, 2306–2310 RSC.
- V. M. Khot, A. B. Salunkhe, N. D. Thorat, R. S. Ningthoujamb and S. H. Pawar, Dalton Trans., 2013, 42, 1249–1258 RSC.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- E. Pöselt, H. Kloust, U. Tromsdorf, M. Janschel, C. Hahn, C. Maßlo and H. Weller, ACS Nano, 2012, 6, 1619–1624 CrossRef PubMed.
- P. Arosio, J. Thévenot, T. Orlando, F. Orsini, M. Corti, M. Mariani, L. Bordonali, C. Innocenti, C. Sangregorio, H. Oliveira, S. Lecommandoux, A. Lascialfari and O. Sandre, J. Mater. Chem. B, 2013, 1, 5317–5328 RSC.
- T. Togashi, T. Naka, S. Asahina, K. Sato, S. Takamia and T. Adschiri, Dalton Trans., 2011, 40, 1073–1078 RSC.
- J. R. Rockenberger, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 1999, 121, 11595–11596 CrossRef CAS; S. Sun and H. J. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef PubMed; T. Hyeon, Chem. Commun., 2003, 927–934 RSC; K. Woo, J. Hong, S. Choi, H.-W. Lee, J.-P. Ahn, C. S. Kim and S. W. Lee, Chem. Mater., 2004, 16, 2814–2818 CrossRef; J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nature Mater., 2004, 3, 891–895 CrossRef PubMed.
- V. Amendola, M. Meneghetti, O. M. Bakr, P. Riello, S. Polizzi, S. Fiameni, D. H. Anjum, P. Arosio, T. Orlando, C. de Julian Fernandez, F. Pineider, C. Sangregorio and A. Lascialfari, Nanoscale, 2013, 5, 5611–5619 RSC.
- L.-H. Shen, J.-F. Bao, D. Wang, Y.-X. Wang, Z.-W. Chen, L. Ren, X. Zhou, X.-B. Ke, M. Chen and A.-Q. Yang, Nanoscale, 2013, 5, 2133–2141 RSC.
- T. Krasia-Christoforou and T. K. Georgiou, J. Mater. Chem. B, 2013, 1, 3002–3025 RSC.
- G. D. Moeser, K. A. Roach, W. H. Green, P. E. Laibinis and T. A. Hatton, Ind. Eng. Chem. Res., 2002, 41, 4739–4749 CrossRef CAS; G. D. Moeser, W. H. Green, P. E. Laibinis, P. Linse and T. A. Hatton, Langmuir, 2004, 20, 5223–5234 CrossRef; H. Lee, E. Lee, D. K. Kim, N. K. Jang, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2006, 128, 7383–7389 CrossRef PubMed; M. Kim, J. Jung, J. Lee, K. Na, S. Park and J. Hyun, Colloids surf., B, 2010, 76, 236–240 CrossRef PubMed; Q. Liang, D. Zhao, T. Qian, K. Freeland and Y. Feng, Ind. Eng. Chem. Res., 2012, 51, 2407–2418 CrossRef.
- P. Ferruti, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2319–2353 CrossRef CAS.
- S. Richardson, P. Ferruti and R. Duncan, J. Drug Targeting, 1999, 6, 391–404 CrossRef CAS PubMed.
- A.-H. An-Hui, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef PubMed.
- T. Tierui Zhang, J. Ge, Y. Hu and Y. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef PubMed.
- R. T. Ragheb and J. S. Riffle, Polymer, 2008, 49, 5397–5404 CrossRef CAS PubMed.
- U. O. Häfeli, J. S. Riffle, L. Harris-Shekhawat, A. Carmichael-Baranauskas, F. Mark, J. P. Dailey and D. Bardenstein, Mol. Pharm., 2009, 6, 1417–1428 CrossRef PubMed.
- D. Yang, J. Hu and S. Fu, J. Phys. Chem. C, 2009, 113, 7646–7651 CAS.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- M. Garnett, P. Ferruti and E. Ranucci (University of Nottingham, Università degli Studi di Milano). PatentWO/2008/038038, 2008 Search PubMed.
- D. Donghi, D. Maggioni, G. D'Alfonso, F. Amigoni, E. Ranucci, P. Ferruti, A. Manfredi, F. Fenili, A. Bisazza and R. Cavalli, Biomacromolecules, 2009, 10, 3273–3282 CrossRef CAS PubMed.
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC; W. A. Volkert and T. J. Hoffman, Chem. Rev., 1999, 99, 2269–2292 CrossRef CAS PubMed; S. Liu, Chem. Soc. Rev., 2004, 33, 445–461 RSC; U. Mazzi and M. Riondato, Radiometal Labelled Complexes for Diagnostic Imaging and Radiotherapy, in Recent Development in Bioinorganic Chemistry, ed. M. Saviano, Transword Research Network, Trivandrum, India, 2006, pp. 1–33 CrossRef; T. Mindt, H. Struthers, E. Garcia-Garayoa, D. Desbouis and R. Schibli, Chimia, 2007, 61, 725–731 CrossRef.
- P. W. Causey, T. R. Besanger, P. Schaffer and J. F. Vaillant, Inorg. Chem., 2008, 47, 8213–8221 CrossRef CAS PubMed.
- R. Alberto, R. Schibli, R. Waibel, U. Abram and A. P. Schubiger, Coord. Chem. Rev., 1999, 190–192, 901–919 CrossRef CAS.
- R. Schibli, R. Schwarzbach, R. Alberto, K. Ortner, H. Schmalle, C. Dumas, A. Egli and A. Schubiger, Bioconjugate Chem., 2002, 13, 750–756 CrossRef CAS PubMed.
- R. Sharma, Y. Xu, S. W. Kim, M. J. Schueller, D. Alexoff, S. D. Smith, W. Wang and D. Schlyer, Nanoscale, 2013, 5, 7476–7483 RSC.
- H. He, M. Lipowska, X. Xu, A. T. Taylor, M. Carlone and L. Marzilli, Inorg. Chem., 2005, 44, 5437–5446 CrossRef CAS PubMed ; slightly modified by the addition of a small amount of CF3SO3H before refluxing, see ref. 44.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- S. Freddi, L. D'Alfonso, M. Collini, M. Caccia, L. Sironi, G. Tallarida, S. Caprioli and G. Chirico, J. Phys. Chem. C, 2009, 113, 2722–2730 CAS.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- J.-C. Bacri, R. Perzynski, D. Salin, V. Cabuil and R. Massart, J. Magn. Magn. Mater., 1990, 85, 27–32 CrossRef CAS.
- E. Ranucci, P. Ferruti, E. Lattanzio, A. Manfredi, M. Rossi, P. R. Mussini, F. Chiellini and C. Bartoli, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6977–6991 CrossRef CAS.
- C. Oliver Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef PubMed.
- G. Ferro-Flores and C. Arteaga de Murphy, Adv. Drug Delivery Rev., 2008, 60, 1389–1401 CrossRef CAS PubMed.
- See for instance the claims for Feridex I.V.® dosage in the following patents: U.S.P. 4,770,183, U.S.P. 4,827,945, U.S.P. 4,951,675, U.S.P. 5,055,288, U.S.P. 5,102,652, U.S.P. 5,219,554, U.S.P. 5,248,492 .
- N. Hondow, R. Brydson, P. Wang, M. D. Holton, M. Rowan Brown, P. Rees, H. D. Summers and A. Brown, J. Nanopart. Res., 2012, 14, 977 CrossRef.
- L. Bordonali, T. Kalaivani, K. P. V. Sabareesh, C. Innocenti, E. Fantechi, C. Sangregorio, M. F. Casula, L. Lartigue, J. Larionova, Y. Guari, M. Corti, P. Arosio and A. Lascialfari, J. Phys.: Condens. Matter, 2013, 25, 066008 CrossRef CAS PubMed.
- L. Lartigue, C. Innocenti, T. Kalaivani, A. Awwad, M. M. Sanchez, Y. Guari, J. Larionova, C. Guérin, J. L. Montero, V. Barragan, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, J. Am. Chem. Soc., 2011, 133, 10459–10472 CrossRef CAS PubMed.
- Q. L. Vuong, J.-F. Berret, J. Fresnais, Y. Gossuin and O. Sandre, Adv. Health. Mater., 2012, 1, 502–512 CrossRef CAS PubMed.
- Y. Noguchi, J. Wu, R. Duncan, J. Strohalm, K. Ulbrich, T. Akaike and H. Maeda, Jpn. J. Cancer Res., 1998, 89, 307–314 CrossRef CAS; H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef; M. Fox, S. Szoka and JM. Frechet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef PubMed; F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef PubMed.
- E. Kim, K. Lee, Y.-M. Huh and S. Haam, J. Mater. Chem. B, 2013, 1, 729–739 RSC.
- S. J. Shin, J. R. Beech and K. A. Kelly, Integr. Biol., 2013, 5, 29–42 RSC.
- J.-P. Fortin, C. Wilhelm, J. Servais, C. Menager, J.-C. Bacri and F. Gazeau, J. Am. Chem. Soc., 2007, 129, 2628–2635 CrossRef CAS PubMed.
- K. Okawa, M. Sekine, M. Maeda, M. Tada, M. Abe, N. Matsushita, K. Nishio and H. Handa, J. Appl. Phys., 2006, 99, 08H102 CrossRef.
- D. Maggioni, F. Fenili, L. D'Alfonso, D. Donghi, M. Panigati, I. Zanoni, R. Marzi, A. Manfredi, P. Ferruti, G. D'Alfonso and E. Ranucci, Inorg. Chem., 2012, 51, 12776–12788 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3dt52377b |
| This journal is © The Royal Society of Chemistry 2014 |