DOI:
10.1039/C3DT52234B
(Paper)
Dalton Trans., 2014,
43, 423-431
2-(1-(2-Benzhydrylnaphthylimino)ethyl)pyridylnickel halides: synthesis, characterization, and ethylene polymerization behavior†
Received
16th August 2013
, Accepted 25th September 2013
First published on 25th September 2013
Abstract
A series of 2-(1-(2-benzhydrylnaphthylimino)ethyl)pyridine derivatives (L1–L3) was synthesized and fully characterized. The organic compounds acted as bi-dentate ligands on reacting with nickel halides to afford two kinds of nickel complexes, either mononuclear bis-ligated L2NiBr2 (Ni1–Ni3) or chloro-bridged dinuclear L2Ni2Cl4 (Ni4–Ni6) complexes. The nickel complexes were fully characterized, and the single crystal X-ray diffraction revealed for Ni2, a distorted square pyramidal geometry at nickel comprising four nitrogens of two ligands and one bromide; whereas for Ni4, a centrosymmetric dimer possessing a distorted octahedral geometry at nickel was formed by two nitrogens of one ligand, two bridging chlorides and one terminal chloride along with oxygen from methanol (solvent). When activated with diethylaluminium chloride (Et2AlCl), all nickel complexes performed with high activities (up to 1.22 × 107 g (PE) mol−1 (Ni) h−1) towards ethylene polymerization; the obtained polyethylene possessed high branching, low molecular weight and narrow polydispersity, suggestive of a single-site active species. The effect of the polymerization parameters, including the nature of the ligands/halides on the catalytic performance is discussed.
Introduction
Studies of late-transition metal complexes, including those of iron, cobalt, nickel, and palladium, have seen a significant rise, particularly when applied to ethylene oligomerization and polymerization since the pioneering work on α-diiminometal (nickel or palladium) complexes (A, Scheme 1)1 and bis(imino)pyridyl metal (iron or cobalt) complexes which appeared in the middle to late 1990s.2 Interestingly, branched polyethylenes were obtained using nickel and palladium complex pre-catalysts,1,3 for which the proposed mechanism involving β-hydride elimination explained the formation of methyl branches. Other more attractive polyethylenes possessed longer branches (such as propyl, amyl and longer branches),4 and better properties are anticipated from other branched products. This has stimulated both academics and industrialists to further study new nickel-based complex pre-catalysts. In particular, the modification of α-diimino derivatives was extensively investigated,5 and in addition, various related ligand sets were explored such as 2-iminopyridines (B, Scheme 1),6 2-arylimino-5,6,7-trihydro-quinolines (C, Scheme 1),7 as well as bicyclic compounds.8 Furthermore, tridentate ligand sets have also been developed such as those based on conjugated 1,10-phenanthroline derivatives9 and quinoline derivatives10 as well as a number of non-conjugated derivatives.11 This progress has been illustrated in a number of review articles.12 Although the nickel catalytic system has been commercialized in the form of the SHOP process,13 the utilization of the newly developed nickel complex pre-catalysts is still problematic due to a number of critical issues concerning both the nature of the catalytic systems and the usefulness of the resulting products.
 |
| | Scheme 1 Representative N,N-bidentate nickel complex pre-catalysts. | |
To overcome some of these shortcomings (such as deactivation and production of polymers of low molecular weight at elevated reaction temperatures), we note that a number of iron complex pre-catalysts have been successfully modified with improved thermo-stability and have produced useful products,14 and now are considered promising catalytic systems for both ethylene oligomerization15 and polymerization.16 Ligands bearing bulky benzhydryl-substituents were recognized to exert a significant influence on improving the thermo-stabilities of their metal complexes and for producing polyethylene of narrow polydispersity.16,17 As a consequence, nickel complex pre-catalysts bearing benzhydryl-substituted ligands have also been explored, and better performances in terms of both thermo-stability and catalytic activities have been reported.18 In general, benzhydryl-substituents have been introduced through the use of benzhydryl-substituted anilines.16–18 Following on from this, the next challenge is to design benzhydryl-substituted 1-aminonaphthalene derivatives, and subsequently a new series of 2-iminopyridine derivatives. With this in mind, we have prepared new 2-iminopyridine derivatives, from which their nickel halide complexes were synthesized. These nickel complexes performed with high activities toward ethylene polymerization. During the preparation of this manuscript, 2-bulky-substituted 1-aminonaphthalene derivatives (D, Scheme 1) have been used for preparing α-diimino nickel complex pre-catalysts in ethylene polymerization.19 Herein, the synthesis and characterization of the 2-(1-(2-benzhydrylnaphthylimino)ethyl)pyridine derivatives and their nickel halides are reported as well as the catalytic performance of the nickel complexes in ethylene polymerization.
Results and discussion
Synthesis and characterization of 2-(1-(2-benzhydrylnaphthyl imino)ethyl)pyridine derivatives (L1–L3) and their nickel complexes (Ni1–Ni6)
The 2-benzhydrylnaphthylamine, 2,4-dibenzhydrylnaphthylamine, and 2,4,7-tribenzhydrylnaphthylamine compounds were prepared in acceptable yields using modified synthetic procedures according to the literature.20 The routine condensation reaction of 2-acetylpyridine with the above benzhydryl-substituted naphthylamine derivatives formed the respective 2-(1-(2-benzhydrylnaphthylimino)ethyl)pyridine derivatives (L1–L3) in moderate yields (Scheme 2). All organic compounds were characterized by 1H/13C NMR and FT-IR spectroscopy, and were further confirmed by elemental analysis data.
 |
| | Scheme 2 Synthetic procedure for the 2-(1-(2-benzhydrylnaphthylimino) ethyl)pyridines and their nickel complexes. | |
The 2-(1-(2-benzhydrylnaphthylimino)ethyl)pyridine derivatives (L1–L3) readily reacted with the nickel halides NiCl2·6H2O or (DME)NiBr2 in a mixture of ethanol and dichloromethane to form the corresponding complexes (Scheme 2). From these stoichiometric reactions, the nickel bromide complexes were commonly isolated in low yield, whereas the nickel chloride complexes were obtained in high yields. The FT-IR spectroscopy indicated effective coordination of the cationic nickel with the ligands as evidenced by the vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations of the complexes Ni1–Ni6 shifting to lower frequencies and with weaker intensities in the region 1619–1628 cm−1versus the free organic compounds in the region 1642–1643 cm−1. Moreover, elemental analysis data were consistent with the bromide complexes having the formula L2NiBr2 (Ni1–Ni3) and the chloride complexes as LNiCl2 (Ni4–Ni6). Given this, the reaction of (DME)NiBr2 was conducted with two equivalents of 2-(1-(2-benzhydrylnaphthyl-imino)ethyl)pyridine derivatives to form the corresponding nickel bromide complexes L2NiBr2 (Ni1–Ni3) in much higher yield. To confirm their molecular structures, single crystals of the nickel complexes Ni2 and Ni4 were obtained and were subjected to single crystal X-ray crystallographic studies.
N stretching vibrations of the complexes Ni1–Ni6 shifting to lower frequencies and with weaker intensities in the region 1619–1628 cm−1versus the free organic compounds in the region 1642–1643 cm−1. Moreover, elemental analysis data were consistent with the bromide complexes having the formula L2NiBr2 (Ni1–Ni3) and the chloride complexes as LNiCl2 (Ni4–Ni6). Given this, the reaction of (DME)NiBr2 was conducted with two equivalents of 2-(1-(2-benzhydrylnaphthyl-imino)ethyl)pyridine derivatives to form the corresponding nickel bromide complexes L2NiBr2 (Ni1–Ni3) in much higher yield. To confirm their molecular structures, single crystals of the nickel complexes Ni2 and Ni4 were obtained and were subjected to single crystal X-ray crystallographic studies.
Single-crystal X-ray diffraction study
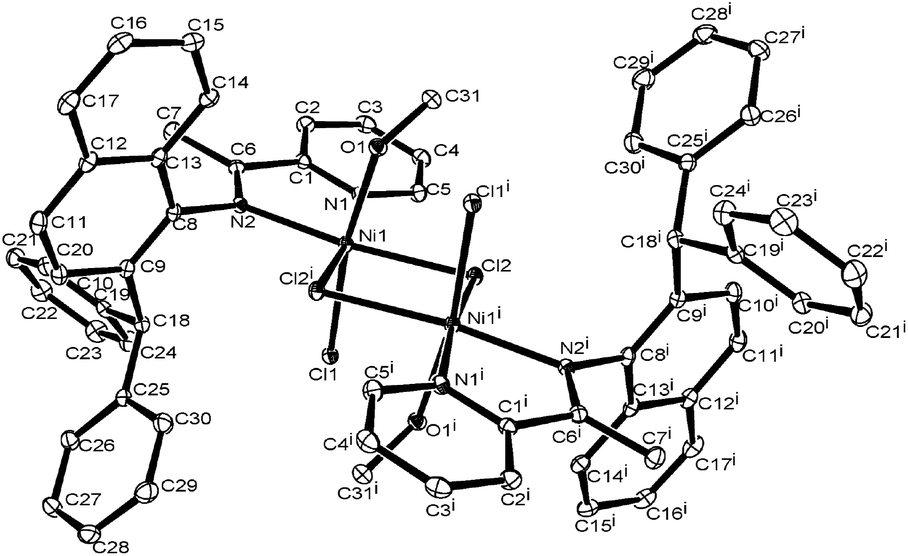
A single crystal of the nickel bromide complex Ni2 suitable for X-ray diffraction analysis was obtained by slow diffusion of heptane into an ethanol solution at room temperature. The molecular structure is shown in Fig. 1, and the selected bond lengths and angles are tabulated in Table 1.
 |
| | Fig. 1 ORTEP drawing of Ni2. Thermal ellipsoids are shown at 30% probability level. Hydrogen atoms have been omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for Ni2 and Ni4·2CH3OH
|
Ni2
|
Ni4·2CH3OH |
| Bond lengths (Å) |
| Ni(1)–N(1) |
2.056(4) |
Ni(1)–N(1) |
2.0410(15) |
| Ni(1)–N(2) |
2.059(4) |
Ni(1)–N(2) |
2.1097(15) |
| Ni(1)–N(3) |
2.058(4) |
Ni(1)–Cl(1) |
2.4162(6) |
| Ni(1)–N(4) |
2.062(4) |
Ni(1)–Cl(2) |
2.4358(6) |
| Ni(1)–Br(1) |
2.4012(10) |
Ni(1)–O(1) |
2.1635(14) |
| Bond angles (°) |
| N(1)–Ni(1)–N(2) |
79.04(16) |
N(1)–Ni(1)–N(2) |
79.21(6) |
| N(1)–Ni(1)–N(3) |
165.93(16) |
N(1)–Ni(1)–Cl(1) |
93.71(5) |
| N(3)–Ni(1)–N(2) |
92.67(15) |
N(2)–Ni(1)–Cl(1) |
97.95(5) |
| N(1)–Ni(1)–N(4) |
92.62(16) |
N(1)–Ni(1)–O(1) |
89.20(6) |
| N(3)–Ni(1)–N(4) |
78.48(16) |
N(2)–Ni(1)–O(1) |
91.20(6) |
| N(2)–Ni(1)–N(4) |
105.15(15) |
N(1)–Ni(1)–Cl(2) |
96.24(5) |
| N(1)–Ni(1)–Br(1) |
95.96(12) |
N(2)–Ni(1)–Cl(2) |
173.21(4) |
| N(3)–Ni(1)–Br(1) |
98.11(11) |
O(1)–Ni(1)–Cl(1) |
170.76(4) |
| N(2)–Ni(1)–Br(1) |
125.67(10) |
O(1)–Ni(1)–Cl(2) |
83.65(4) |
| N(4)–Ni(1)–Br(1) |
129.17(11) |
Cl(1)–Ni(1)–Cl(2) |
87.31(2) |
As shown in Fig. 1, complex Ni2 possesses a distorted square-based pyramidal geometry at the nickel, comprising four nitrogen atoms (N1, N2, N3 and N4) of two ligands and one bromide (Br1), and another bromide (Br2) as a free-anion, which is similar to bis(2-((2,4-dibenzhydryl-6-ethylphenylimino)ethyl)pyridyl) nickel dibromide.18e In this structure, there is a five-membered heteronickel-cycle constructed from Ni1, N1, C5, C6 and N2, in which the C5 atom deviates by 1.222 Å from the co-plane of the atoms N1, N2 and Ni1, whilst the C6 atom deviates by 1.506 Å. The Ni1–N1 bond length is similar to the Ni1–N2 at 2.056(4) Å and 2.059(4) Å, respectively. The dihedral angle between the pyridyl and imino-naphthalene planes is 46.86°, which is consistent with the structures of the 2-iminopyridylnickel halide analogs reported previously.18e
Single crystal of the nickel chloride complex Ni4 suitable for X-ray diffraction analysis was obtained by laying diethyl ether on to their dichloromethane–methanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solutions at room temperature. The nickel chloride complex Ni4 (Fig. 2) was found to be a centrosymmetric dimer with a slightly distorted octahedral geometry at the nickel center, in which the nickel is symmetrically bridged by the two chloride atoms (Cl2 and Cl2i), and the two nickel atoms, as evidenced by the intramolecular distance of 3.404 Å, which is slightly shorter than 3.475 Å observed in the analogous 2-iminopyridine-Ni(II) dimers,6a but slightly longer than 3.363 Å found in 2-((2,4-dibenzhydryl-6-methylphenylimino)methyl)pyridylnickel dichloride.18e For the Ni–N bond lengths of different types, the length of the Ni–Npyridine is slightly shorter than the Ni–Nimino, (Ni1–N1, 2.0410(15) and Ni1–N2, 2.1097(15)). The pyridyl and imino-naphthalene planes are near perpendicular with a dihedral angle of 85.55° consistent with the dihedral angles between the pyridyl and imino-phenyl for the reported 2-aryliminopyridylnickel analogues.6a,c,7,18c
:1) solutions at room temperature. The nickel chloride complex Ni4 (Fig. 2) was found to be a centrosymmetric dimer with a slightly distorted octahedral geometry at the nickel center, in which the nickel is symmetrically bridged by the two chloride atoms (Cl2 and Cl2i), and the two nickel atoms, as evidenced by the intramolecular distance of 3.404 Å, which is slightly shorter than 3.475 Å observed in the analogous 2-iminopyridine-Ni(II) dimers,6a but slightly longer than 3.363 Å found in 2-((2,4-dibenzhydryl-6-methylphenylimino)methyl)pyridylnickel dichloride.18e For the Ni–N bond lengths of different types, the length of the Ni–Npyridine is slightly shorter than the Ni–Nimino, (Ni1–N1, 2.0410(15) and Ni1–N2, 2.1097(15)). The pyridyl and imino-naphthalene planes are near perpendicular with a dihedral angle of 85.55° consistent with the dihedral angles between the pyridyl and imino-phenyl for the reported 2-aryliminopyridylnickel analogues.6a,c,7,18c
 |
| | Fig. 2 ORTEP drawing of Ni4·2CH3OH. Thermal ellipsoids are shown at 30% probability level. Hydrogen atoms have been omitted for clarity. | |
Catalytic behavior toward ethylene polymerization
The complex Ni2 was used to optimize the polymerization parameters. Various alkylaluminium reagents such as MAO, MMAO, and Et2AlCl were explored as activators under 10 atm of ethylene pressure and all catalytic systems indicated high activities toward ethylene polymerization (Table 2). With potential industrial applications in mind, the cheaper and more readily available Et2AlCl was used for further systematically exploring the catalysis.
Table 2 Ethylene polymerization by Ni2 using various co-catalystsa
| Run |
Co-catalyst |
Al/Ni |
Yield/g |
Activityb |
M
wc/g mol−1 |
M
w/Mnc |
T
md/°C |
|
General conditions: 2 μmol of Ni; 30 min; 30 °C; 100 mL of toluene for 10 atm of ethylene.
106 g (PE) mol−1 (Ni) h−1.
Determined by GPC.
Determined by DSC.
|
| 1 |
MAO |
1500 |
8.58 |
8.58 |
2653 |
2.19 |
89.0 |
| 2 |
MMAO |
1500 |
10.9 |
10.9 |
2272 |
2.37 |
101.3 |
| 3 |
Et2AlCl |
400 |
9.89 |
9.89 |
1676 |
1.99 |
72.3 |
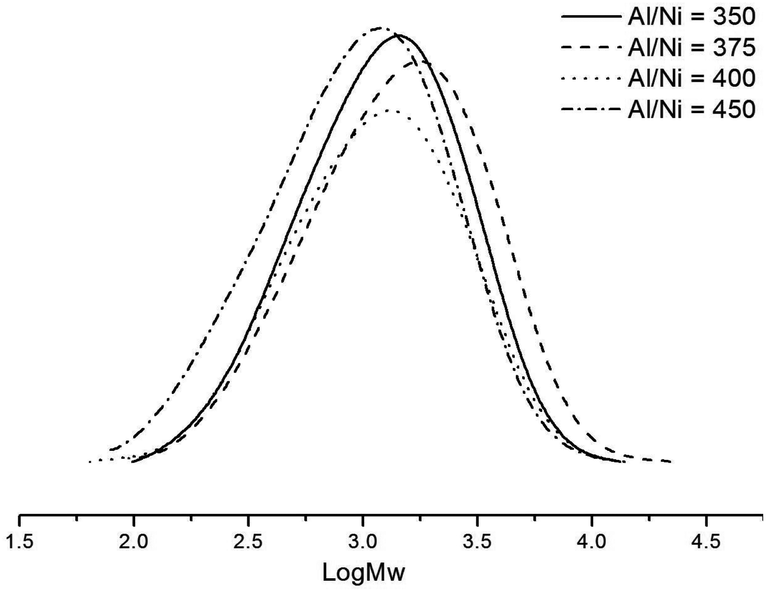
Variations of the Al/Ni ratio from 300 to 450 (runs 1–5 in Table 3) were conducted at 30 °C, and the highest activity was observed at 9.89 × 106 g (PE) mol−1 (Ni) h−1 using the Al/Ni ratio of 400 (run 4 in Table 3). Catalytic activities increased with higher molar ratios of Al/Ni (runs 1–4 in Table 3), however, on further increasing the Al/Ni molar ratio to 450, a dramatic decrease of the catalytic activity 5.19 × 106 g (PE) mol−1 (Ni) h−1 (run 5 in Table 3) was observed. As indicated by the data and the curves of the GPC for the obtained polyethylene (Fig. 3), these catalytic systems exhibited single-site active species; the molecular weights of the obtained polyethylenes slightly decreased on increasing the molar ratio of Al/Ni, indicative of more chain transfers from the nickel species to aluminium and chain termination.6f,21 The molecular weight/polydispersity values herein are at the limits of what can be determined accurately using such columns, and so we will limit our discussions to general trends.
 |
| | Fig. 3 GPC traces of polyethylenes by the Ni2–Et2AlCl system with various Al/Ni ratios (runs 2–5 in Table 3). | |
Table 3 Ethylene polymerization by Ni2–Et2AlCla
| Run |
Al/Ni |
T/°C |
t/min |
Yield/g |
Activityb |
M
wc/g mol−1 |
M
w/Mnc |
T
md/°C |
|
General conditions: 2 μmol of Ni; 100 mL of toluene.
106 g (PE) mol−1 (Ni) h−1.
Determined by GPC.
Determined by DSC.
|
| 1 |
300 |
30 |
30 |
Trace |
Trace |
nd |
nd |
nd |
| 2 |
350 |
30 |
30 |
1.54 |
1.54 |
1833 |
2.13 |
80.8 |
| 3 |
375 |
30 |
30 |
8.59 |
8.59 |
1809 |
2.08 |
79.9 |
| 4 |
400 |
30 |
30 |
9.89 |
9.89 |
1676 |
1.99 |
72.3 |
| 5 |
450 |
30 |
30 |
5.19 |
5.19 |
1445 |
1.89 |
68.6 |
| 6 |
400 |
20 |
30 |
8.08 |
8.08 |
2529 |
2.15 |
88.2 |
| 7 |
400 |
40 |
30 |
4.22 |
4.22 |
1117 |
1.71 |
62.6 |
| 8 |
400 |
50 |
30 |
1.54 |
1.54 |
981 |
1.58 |
56.0 |
| 9 |
400 |
20 |
10 |
3.40 |
10.2 |
2270 |
2.05 |
85.5 |
| 10 |
400 |
20 |
20 |
6.57 |
9.86 |
2378 |
2.11 |
87.5 |
| 11 |
400 |
20 |
40 |
8.49 |
6.37 |
2898 |
2.49 |
90.1 |
| 12 |
400 |
20 |
60 |
9.37 |
4.69 |
3388 |
2.53 |
105.7 |
To understand the influence of the reaction temperature, the ethylene polymerization was conducted over 30 min at the Al/Ni ratio of 400 and at 10 atm of ethylene using temperatures from 20 to 50 °C (runs 4 and 6–8 in Table 3). The data confirmed that the optimized temperature was 30 °C (run 4 in Table 3), and that the higher the reaction temperature, the lower the molecular weight of the obtained polyethylene (Fig. 4), which is consistent with observations on nickel pre-catalysts bearing α-diimino5a or 2-iminopyridine ligands.18 This is likely to arise from faster chain termination versus chain propagation at the elevated temperature.12a
 |
| | Fig. 4 GPC curves of polyethylenes by the Ni2–Et2AlCl system at different temperature (runs 4, 6–8 in Table 3). | |
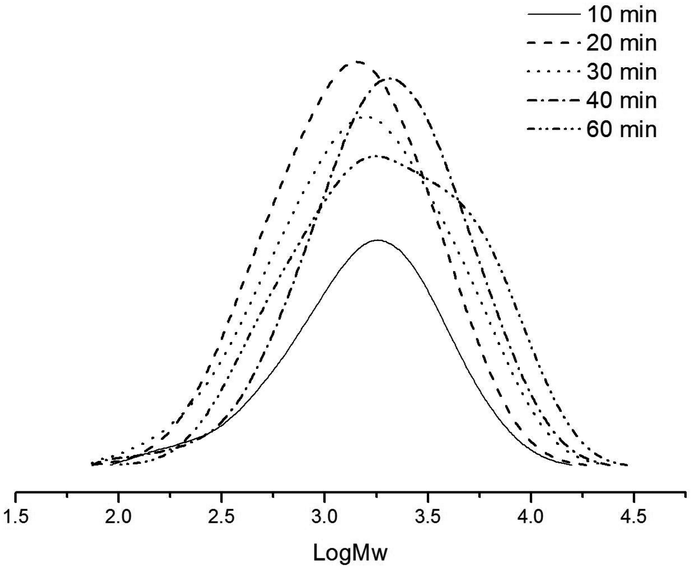
With regard to the life time of the catalytic system, the ethylene polymerization by Ni2–Et2AlCl was quenched over different time periods, typically 10, 20, 30, 40 and 60 min (runs 6 and 9–12 in Table 3). On extending the reaction time, more polyethylene was obtained and with higher molecular weights, but the catalytic activities gradually decreased (Fig. 5). A similar catalytic behavior was observed for the nickel analogs bearing 2-iminopyridine.18a,e After 30 min, the catalytic system exhibited low activity towards polymerizing ethylene, but the isolation of polyethylene of increased molecular weight indicated that the catalytic species still remained active. In general, all the polyethylene products possessed narrow polydispersity in the range of 1.58–2.49, indicative of a single site active species.
 |
| | Fig. 5 GPC curves of polyethylenes by the Ni2–Et2AlCl system with different time (runs 6, 9–12 in Table 3). | |
The melting points of all resultant polyethylenes exhibited lower Tm values, not only reflecting the lower molecular weights but also indicating that branched polyethylene had formed. To understand the polyethylene obtained using Ni2–Et2AlCl (run 8 in Table 3) at 50 °C, 13C NMR spectroscopic measurements (Fig. 6) were conducted which indicated the presence of 199 branches/1000 carbons as measured according to the literature.22
 |
| | Fig. 6
13C NMR spectrum of the polyethylene by Ni2–Et2AlCl at 50 °C (run 8 in Table 3). | |
The signals were assigned and are listed in Table 4, which indicated that the main branches were methyl (32%), propyl (13%) and long chains (33%) as well as amyl chains (14%).
Table 4 Percentage of branching for the polyethylene (run 8 in Table 3)22
| Peak no. |
Chem. shifts/ppm |
Integral exp. |
Branch content |
Percentage over total branching |
| 1 |
11.22 |
0.13 |
Nm |
0.76 |
26.00% |
| 1 |
11.38 |
0.08 |
Nm(1,4) |
0 |
0.00% |
| 2 |
14.08 |
1 |
Nm(1,5) |
0 |
0.00% |
| 3 |
14.58 |
0.14 |
Nm(1,6) |
0.17 |
5.82% |
| 4 |
19.98 |
1.76 |
Ne |
0.17 |
5.82% |
| 7 |
22.87 |
0.84 |
Np |
0.37 |
12.67% |
| 8 |
23.24 |
0.14 |
Nb |
0.07 |
2.40% |
| 10 |
26.72 |
0.28 |
Na |
0.41 |
14.04% |
| 11 |
27.3 |
0.74 |
Nl |
0.97 |
33.22% |
| 12 |
27.44 |
2.51 |
Nl(1,4) |
0 |
0.00% |
| 13 |
27.81 |
0.34 |
δδCH2 |
23.54 |
|
| 14 |
29.6 |
2.4 |
[E] |
11.77 |
|
| 16 |
30 |
24.51 |
[R] |
2.92 |
100.00% |
| 17 |
30.38 |
3.56 |
Total branching = 199 |
|
| 20 |
32.19 |
0.97 |
Branches/1000C |
|
| 21 |
32.79 |
0.41 |
|
|
|
| 22 |
33.23 |
1.1 |
Methyl branches |
31.82% |
| 24 |
34.05 |
0.44 |
Ethyl branches |
5.82% |
| 26 |
34.52 |
0.43 |
Propyl branches |
12.67% |
| 26 |
34.85 |
0.38 |
Butyl branches |
2.40% |
| 28 |
37.06 |
0.37 |
Amyl branches |
14.04% |
| 29 |
37.54 |
2.24 |
Long branches |
33.22% |
In comparison, the polyethylene obtained using the Ni2–Et2AlCl (run 6 in Table 3) at 20 °C was also measured by 13C NMR spectroscopy (Fig. 7), which indicated 91 branches/1000 carbons; the main branches were methyl (46%) and long chains (37%) as well as amyl chains (11%). Highly branched PEs are commonly obtained using nickel catalytic systems5d,23 due to β-hydride migrations.
 |
| | Fig. 7
13C NMR spectrum of the polyethylene by Ni2–Et2AlCl at 20 °C (run 6 in Table 3). | |
To understand the influence of the ligands and halides present on the catalytic behavior of these nickel complex pre-catalysts, the complexes Ni1–Ni6 were investigated under optimum conditions (Al/Ni molar ratio of 400:1 at 30 °C) under 10 atm of ethylene, and the catalytic results are tabulated in Table 5. Previous bis-ligated complexes were reported to be relatively inactive for olefin polymerization due to detrimental olefin insertions.24 However, the current bis-ligated nickel bromide pre-catalysts exhibited higher activities than did the corresponding chloride analogs. Besides the better solubility of the bromide complexes, the bis-ligated nickel bromide pre-catalysts probably transformed into mono-ligated complexes during catalysis.25 Overall, catalytic activities of these nickel complex pre-catalysts gradually decreased when more benzhydryl-substituted groups were incorporated (Fig. 8): Ni1 > Ni2 > Ni3 and Ni4 > Ni5 > Ni6, illustrating that the bulky groups retard the insertion of ethylene.
 |
| | Fig. 8 The catalytic performances by Ni1–Ni6/Et2AlCl (Table 5). | |
Table 5 Ethylene polymerization by Ni1–Ni6/Et2AlCla
| Run |
Pre-catalyst |
Yield/g |
Activityb |
M
wc/g mol−1 |
M
w/Mnc |
T
md/°C |
|
General conditions: 2 μmol of Ni; Al/Ni = 400/1; 30 min; 30 °C; 100 mL toluene; 10 atm of ethylene.
106 g (PE) mol−1 (Ni) h−1.
Determined by GPC.
Determined by DSC.
|
| 1 |
Ni1 |
12.2 |
12.2 |
1309 |
2.11 |
59.2 |
| 2 |
Ni2 |
9.89 |
9.89 |
1676 |
1.99 |
72.3 |
| 3 |
Ni3 |
8.18 |
8.18 |
515 |
1.78 |
56.7 |
| 4 |
Ni4 |
4.73 |
4.73 |
864 |
1.98 |
44.2 |
| 5 |
Ni5 |
4.44 |
4.44 |
978 |
2.02 |
55.9 |
| 6 |
Ni6 |
2.27 |
2.27 |
541 |
1.45 |
47.0 |
Conclusion
2-((2-Benzhydrylnaphthylimino)ethyl)pyridine derivatives and their nickel complexes were synthesized and fully characterized. The anionic halides caused a different coordination and a different structural geometry at nickel, viz distorted square pyramidal at nickel for the bromides versus a centrosymmetric dimer with distorted octahedral nickel for the chlorides. All nickel complexes exhibited high activities (up to 1.22 × 107 g (PE) mol−1 (Ni) h−1) for ethylene polymerization. The obtained polyethylene was found to be of low molecular weight waxes with narrow PDI and high branching. Such nickel complex pre-catalysts are of potential industrial interest for the production of polyethylene waxes and lubricants.
Experimental section
General consideration
All manipulations involving air- and moisture-sensitive compounds were performed using standard Schlenk techniques under a nitrogen atmosphere. Toluene was refluxed over sodium and distilled under nitrogen prior to use. Methylaluminoxane (MAO, 1.46 M solution in toluene) and modified methylaluminoxane (MMAO, 1.93 M in heptane) were purchased from Akzo Nobel Corp. Diethylaluminium chloride (Et2AlCl, 0.5 M in toluene) was purchased from Acros Chemicals. High-purity ethylene was purchased from Beijing Yansan Petrochemical Co. and used as received. Other reagents were purchased from Aldrich, Acros, or local suppliers. NMR spectra were recorded on a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard; IR spectra were recorded using a Perkin-Elmer System 2000 FT-IR spectrometer. Elemental analysis was carried out using a Flash EA 1112 micro-analyzer. Molecular weights and molecular weight distribution (MWD) of polyethylene were determined by PL-GPC220 at 150 °C, with 1,2,4-trichlorobenzene as the solvent. The melting points of polyethylene were measured from the second scanning run using a Perkin-Elmer TA-Q2000 differential scanning calorimetry (DSC) analyzer under a nitrogen atmosphere. In the procedure, a sample of about 4.0 mg was heated to 140 °C at the rate of 20 °C min−1 and kept for 2 min at 140 °C to remove the thermal history and then cooled at the rate of 20 °C min−1 to −40 °C. 13C NMR spectra of the polyethylenes were recorded using a Bruker DMX 300 MHz instrument at 135 °C in deuterated 1,2-dichlorobenzene with TMS as an internal standard.
Synthesis of various bulky naphthylamines
A variety of benzhydryl-substituted naphthylamines were successfully synthesized according to modified literature procedures.20 2-Benzhydrylnaphenamine was obtained as a red solid, Mp: 136–137 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.84–7.80 (m, 2H, Ar–H), 7.49–7.47 (m, 2H, Ar–H), 7.39–7.22 (m, 11H, Ar–H), 6.96 (d, 1H, J = 8.4 Hz, Ar–H), 5.79 (s, 1H, –CHPh2), 3.98 (s, 2H, –NH2). 13C NMR (100 MHz, CDCl3, TMS): 142.8, 139.2, 133.3, 129.7, 128.7, 128.6, 128.1, 126.8, 125.5, 125.1, 123.7, 122.8, 120.5, 118.2, 52.5. FT-IR (KBr, disk, cm−1): 3382, 3057, 3023, 1620, 1597, 1563, 1511, 1491, 1396, 1243, 1075, 1027, 917, 858, 801, 747, 697. Anal. Calcd for C23H19N (309.15): C, 89.28; H, 6.19; N, 4.53%; found: C, 88.89; H, 6.26; N, 4.38%.
2,4-Dibenzhydrylnaphthylamine was obtained as a red solid, Mp: 145–146 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.84–7.80 (m, 2H, Ar–H), 7.49–7.47 (m, 2H, Ar–H), 7.39–7.22 (m, 11H, Ar–H), 6.96 (d, 1H, J = 8.4 Hz, Ar–H), 5.79 (s, 1H, –CHPh2), 3.98 (s, 2H, –NH2). 13C NMR (100 MHz, CDCl3, TMS): 144.2, 142.6, 137.9, 131.5, 130.6, 129.7, 129.5, 129.4, 128.6, 128.3, 126.6, 126.1, 125.7, 125.1, 124.8, 124.3, 122.2, 121.1, 52.9, 52.5. FT-IR (KBr, disk, cm−1): 3397, 3057, 3024, 1624, 1598, 1513, 1490, 1448, 1379, 1322, 1249, 1178, 1075, 1028, 917, 815, 743, 695. Anal. Calcd for C36H29N (475.23): C, 90.91; H, 6.15; N, 2.94%; found: C, 90.65; H, 6.13; N, 3.19%.
2,4,7-Tribenzhydrylnaphthylamine was obtained as a light yellow solid, Mp: 161–162 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.86 (d, 1H, J = 8.8, Ar–H), 7.47 (s, 1H, Ar–H), 7.31–7.11 (m, 23H, Ar–H), 7.01–6.96 (m, 8H, Ar–H), 6.35 (s, 1H, Ar–H), 6.06 (s, 1H, –CHPh2), 5.69 (s, 1H, –CHPh2), 5.57 (s, 1H, –CHPh2), 3.90 (s, 2H, –NH2). 13C NMR (100 MHz, CDCl3, TMS): 144.1, 143.7, 142.5, 140.2, 137.9, 130.3, 130.1, 129.6, 129.5, 129.4, 129.3, 128.5, 128.4, 128.2, 127.8, 126.6, 126.5, 126.0, 125.0, 124.0, 122.4, 121.1, 57.2, 52.9, 52.5. FT-IR (KBr, disk, cm−1): 3412, 3353, 3057, 3024, 1627, 1598, 1563, 1514, 1492, 1447, 1378, 1251, 1179, 1151, 1075, 1029, 918, 824, 784, 747, 697. Anal. Calcd for C49H39N (641.31): C, 91.69; H, 6.12; N, 2.18%; found: C, 91.38; H, 6.28; N, 2.09%.
Synthesis of ligands
2-((2-Benzhydrylnaphthylimino)ethyl)pyridine (L1).
A mixture of 2-acetylpyridine (0.459 g, 3.79 mmol), 2-benzhydrylnaphthylamine (1.23 g, 3.98 mmol) and a catalytic amount of p-toluenesulfonic acid (0.144 g, 0.76 mmol) in toluene (80 mL) was refluxed for 8 h. After solvent evaporation at reduced pressure, the residue was purified by column chromatography on basic alumina with the eluent of petroleum ether–ethyl acetate (v:v = 20:1) to afford a yellow solid in 71% isolated yield. Mp: 114–115 °C. 1H NMR (400 MHz, CDCl3, TMS): δH 8.68 (d, 1H, J = 4.4 Hz, Py–H), 8.47 (d, 1H, J = 8.0 Hz, Py–H), 7.87–7.82 (m, 2H, Py–H), 7.51–7.07 (m, 16H, Ar–H), 5.72 (s, 1H, –CHPh2), 1.55 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3, TMS): 170.4, 156.2, 148.8, 145.4, 144.0, 142.7, 136.6, 133.0, 129.9, 129.6, 128.5, 128.3, 128.2, 127.5, 126.3, 125.9, 125.1, 123.2, 123.0, 121.6, 52.2, 17.4. FT-IR (KBr, disk, cm−1): 3051, 3003, 1643 (νCN), 1565, 1464, 1302, 1275, 1098, 810, 748, 725, 696. Anal. Calcd for C30H24N2 (412.19): C, 87.35; H, 5.86; N, 6.79%; found: C, 87.06; H, 6.11; N, 6.59%.
2-((2,4-Dibenzhydrylnaphthylimino)ethyl)pyridine (L2).
In a manner similar to that described for L1, L2 was prepared as a chartreuse solid in 61% yield. Mp: 111–112 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 8.67 (d, 1H, J = 4.0 Hz, Py–H), 8.45 (d, 1H, J = 8.0 Hz, Py–H), 7.96 (d, 1H, J = 8.4 Hz, Py–H), 7.85 (t, 1H, J = 7.4 Hz, Py–H), 7.43–6.88 (m, 24H, Ar–H), 6.67 (s, 1H, Ar–H), 6.18 (s, 1H, –CHPh2), 5.58 (s, 1H, –CHPh2), 1.59 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3, TMS): 170.5, 156.2, 148.8, 144.2, 144.0, 143.8, 142.7, 136.6, 134.6, 131.2, 130.1, 129.7, 129.6, 129.2, 128.3, 128.1, 127.0, 126.3, 126.2, 125.9, 125.4, 125.1, 123.8, 121.6, 53.0, 52.2, 17.6. FT-IR (KBr, disk, cm−1): 3058, 3024, 1643 (νCN), 1566, 1448, 1363, 1301, 1242, 1099, 1029, 787, 737, 696. Anal. Calcd for C43H34N2 (578.27): C, 89.24; H, 5.92; N, 4.84%; found: C, 88.60; H, 6.24; N, 4.73%.
2-((2,4,7-Tribenzhydrylnaphthylimino)ethyl)pyridine (L3).
In a manner similar to that described for L1, L3 was prepared as a yellow solid in 72% yield. Mp: 106–107 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 8.67 (d, 1H, J = 4.8 Hz, Py–H), 8.26 (d, 1H, J = 8.0 Hz, Py–H), 7.88 (d, 1H, J = 8.8 Hz, Py–H), 7.81 (t, 1H, J = 7.6 Hz, Py–H), 7.43–6.86 (m, 33H, Ar–H), 6.65 (s, 1H, Ar–H), 6.14 (s, 1H, –CHPh2), 5.55 (s, 1H, –CHPh2), 5.54 (s, 1H, –CHPh2), 1.42 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3, TMS): 170.5, 156.2, 148.5, 144.0, 143.9, 143.8, 143.6, 142.9, 141.0, 136.5, 134.6, 129.9, 129.7, 129.6, 129.2, 128.4, 128.3, 128.1, 127.8, 126.4, 126.3, 126.2, 126.1, 125.0, 124.6, 124.3, 124.2, 121.6, 56.8, 53.0, 52.2, 17.7. FT-IR (KBr, disk, cm−1): 3057, 3024, 1642 (νCN), 1596, 1492, 1447, 1362, 1101, 1075, 1030, 785, 743, 697. Anal. Calcd for C56H44N2 (744.35): C, 90.29; H, 5.95; N, 3.76%; found: C, 89.95; H, 6.03; N, 3.70%.
Synthesis of nickel complexes
Bis(2-((2-benzhydrylnaphthylimino)ethyl)pyridyl)nickel bromide (Ni1).
(DME)NiBr2 (0.25 mmol) and the ligand L1 (0.5 mmol) were dissolved in a mixture of 10 mL ethanol–CH2Cl2 (v/v = 1:1). The mixture was stirred for 12 h, and then diethyl ether was added to the mixture to precipitate the complex. The precipitant was collected by filtration, washed with diethyl ether (3 × 5 mL), and dried in vacuum to obtain a yellow powder in 84% yield. FT-IR (KBr, disk, cm−1): 3058, 3026, 1619 (νCN), 1594, 1493, 1446, 1373, 1318, 1261, 1119, 813, 776, 750, 700. Anal. Calcd for C60H48Br2N4Ni (1040.16): C, 69.06; H, 4.64; N, 5.37%. Found: C, 68.95; H, 4.82; N, 5.16%.
Bis(2-((2,4-dibenzhydrylnaphthylimino)ethyl)pyridyl)nickel bromide (Ni2).
In a manner similar to that described for Ni1, Ni2 was prepared as a yellow powder in 88% yield. FT-IR (KBr, disk, cm−1): 3060, 3024, 1622 (νCN), 1596, 1493, 1446, 1375, 1322, 1261, 1076, 1027, 782, 741, 700. Anal. Calcd for C86H68Br2N4Ni (1372.32): C, 75.07; H, 4.98; N, 4.07%. Found: C, 74.91; H, 4.75; N, 4.16%.
Bis(2-((2,4,7-tribenzhydrylnaphthylimino)ethyl)pyridyl)nickel bromide (Ni3).
In a manner similar to that described for Ni1, Ni3 was prepared as a yellow powder in 84% yield. FT-IR (KBr, disk, cm−1): 3058, 3025, 1624 (νCN), 1596, 1494, 1446, 1369, 1319, 1257, 1076, 1029, 837, 742, 699. Anal. Calcd for C112H88Br2N4Ni (1704.47): C, 78.74; H, 5.19; N, 3.28%. Found: C, 78.48; H, 5.14; N, 3.28%.
2-((2-Benzhydrylnaphthylimino)ethyl)pyridylnickel chloride (Ni4).
NiCl2·6H2O (0.5 mmol) and the ligand L1 (0.5 mmol) were dissolved in a mixture of 10 mL ethanol–CH2Cl2 (v/v = 1:1). The mixture was stirred for 12 h, and then diethyl ether was added to the mixture to precipitate the complex. The precipitant was collected by filtration, washed with diethyl ether (3 × 5 mL), and dried in vacuum to obtain a yellow powder in 96% yield. FT-IR (KBr, disk, cm−1): 3057, 2972, 2902, 1625 (νCN), 1596, 1493, 1446, 1374, 1318, 1259, 1161, 1074, 1046, 814, 787, 752, 727, 703. Anal. Calcd for C30H24Cl2N2Ni (540.07): C, 66.46; H, 4.46; N, 5.17%. Found: C, 66.28; H, 4.48; N, 4.88%.
2-((2,4-Dibenzhydrylnaphthylimino)ethyl)pyridylnickel chloride (Ni5).
In a manner similar to that described for Ni4, Ni5 was prepared as a yellow powder in 92% yield. FT-IR (KBr, disk, cm−1): 3061, 3025, 1624 (νCN), 1598, 1493, 1446, 1370, 1321, 1256, 1164, 1125, 1029, 821, 743, 701. Anal. Calcd for C43H34Cl2N2Ni (706.15): C, 72.91; H, 4.84; N, 3.95%. Found: C, 72.80; H, 5.02; N, 3.99%.
2-((2,4,7-Tribenzhydrylnaphthylimino)ethyl)pyridylnickel chloride (Ni6).
In a manner similar to that described for Ni4, Ni6 was prepared as a yellow powder in 82% yield. FT-IR (KBr, disk, cm−1): 3058, 3025, 1628 (νCN), 1597, 1493, 1447, 1368, 1319, 1256, 1076, 1029, 838, 743, 698. Anal. Calcd for C56H44Cl2N2Ni (872.22): C, 76.91; H, 5.07; N, 3.20%. Found: C, 76.68; H, 5.30; N, 3.00%.
X-ray crystallographic studies
A single crystal of Ni2 suitable for X-ray diffraction analyses was obtained by laying heptane on ethanol solution at room temperature. A single crystal of the nickel chloride complex Ni4 suitable for X-ray diffraction analysis was obtained by laying diethyl ether on their dichloromethane–methanol (v/v = 1:1) solutions at room temperature. X-ray studies were carried out using a Rigaku Saturn724+CCD diffractometer with MoKα radiation (λ = 0.71073 Å) at 173(2) K (Ni2) and 100 K (Ni4), cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least squares on F2. All hydrogen atoms were placed in calculated positions. Structure solution and refinement were performed by using the SHELXL-97 package.26 During structure refinement of Ni2 and Ni4·2CH3OH, there were free solvent molecules which have no influence on the geometry of the main compounds. Therefore, the SQUEEZE option of the crystallographic program PLATON27 was used to remove these free solvents from the structure. Details of the X-ray structure determinations and refinements are provided in Table 6.
Table 6 Crystal data and structure refinement for Ni2 and Ni4·2CH3OH
| Identification code |
Ni2
|
Ni4·2CH3OH |
| Crystal color |
Brown |
Yellow |
| Empirical formula |
C86H68Br2N4Ni |
C62H56Cl4N4Ni2O2 |
| Formula weight |
1375.97 |
1148.33 |
| Temperature/K |
173 (2) |
100 (2) |
| Wavelength/Å |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/c |
|
a/Å |
11.391(2) |
15.786(3) |
|
b/Å |
18.201(4) |
10.024(2) |
|
c/Å |
18.612(4) |
18.193(4) |
| Alpha/° |
91.61(3) |
90 |
| Beta/° |
104.33(3) |
98.30(3) |
| Gamma/° |
95.61(3) |
90 |
| Volume/Å3 |
3715.4(13) |
2848.6(10) |
|
Z
|
2 |
2 |
|
D
calcd (g cm−3) |
1.230 |
1.339 |
|
μ/mm−1 |
1.382 |
0.895 |
|
F(000) |
1420 |
1192 |
| Crystal size/mm |
0.175 × 0.122 × 0.056 |
0.248 × 0.127 × 0.109 |
|
θ range (°C) |
1.13–27.43 |
2.41–27.48 |
| Limiting indices |
−14 ≤ h ≤ 14 |
−20 ≤ h ≤ 20 |
| −23 ≤ k ≤ 23 |
−12 ≤ k ≤ 13 |
| −22 ≤ l ≤ 24 |
−23 ≤ l ≤ 23 |
| No. of rflns collected |
33431 |
18670 |
| No. unique rflns |
16797 |
6483 |
|
R(int) |
0.0624 |
0.0326 |
| No. of params |
838 |
336 |
| Completeness to θ |
99.1% |
99.5% |
| Goodness of fit on F2 |
1.024 |
1.129 |
| Final R indices [I > 2∑(I)] |
R
1 = 0.0825 |
R
1 = 0.0368 |
| wR2 = 0.2210 |
wR2 = 0.0866 |
|
R indices (all data) |
R
1 = 0.1150 |
R
1 = 0.0399 |
| wR2 = 0.2529 |
wR2 = 0.0885 |
| Largest diff. peak, and hole (e Å−3) |
0.728 and −0.783 |
0.479 and −0.284 |
General procedure for ethylene polymerization
Ethylene polymerizations were carried out in a 250 mL stainless steel autoclave equipped with a mechanical stirrer and a temperature controller. The autoclave was evacuated by a vacuum pump and back-filled three times with N2 and once with ethylene. When the desired reaction temperature was reached, 30 mL toluene was added under ethylene atmosphere, and the nickel pre-catalyst in 20 mL toluene was injected. The required amount of co-catalysts (MAO, MMAO or Et2AlCl) and additional toluene (maintaining total volume at 100 mL in the reactor) were added by syringe. The reaction mixture was intensively stirred for the desired time under 10 atm of ethylene and maintained at this level by constant feeding of ethylene. The reaction was quenched by addition of acidic ethanol. The precipitated polymer was washed with ethanol several times and dried in vacuo.
Acknowledgements
This work was supported by NSFC no. 20874105 and 21374123. The EPSRC is thanked for the awarded of a travel grant (to CR).
Notes and references
-
(a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS;
(b) C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11665 CrossRef CAS.
-
(a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS;
(b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC.
-
(a) S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634–10635 CrossRef CAS;
(b) D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS;
(c) L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS PubMed;
(d) M. D. Leatherman, S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 2003, 125, 3068–3081 CrossRef CAS PubMed.
-
(a) X. Tang, W.-H. Sun, T. Gao, J. Hou, J. Chen and W. Chen, J. Organomet. Chem., 2005, 69, 1570–1580 CrossRef;
(b) X. Tang, Y. Cui, W.-H. Sun, Z. Miao and S. Yan, Polym. Int., 2004, 53, 2155–2161 CrossRef CAS.
-
(a) D. P. Gates, S. A. Svejda, E. Onate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS;
(b) R. J. Maldanis, J. S. Wood, A. Chandrasekaran, M. D. Rausch and J. C. W. Chien, J. Organomet. Chem., 2002, 645, 158–167 CrossRef CAS;
(c) B. K. Bahuleyan, G. W. Son, D.-W. Park, C.-S. Ha and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1066–1082 CrossRef CAS;
(d) M. M. Wegner, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 3624–3633 CrossRef CAS;
(e) C. S. Popeney and Z. Guan, Macromolecules, 2010, 43, 4091–4097 CrossRef CAS;
(f) F.-S. Liu, H.-B. Hu, Y. Xu, L.-H. Guo, S.-B. Zai, K.-M. Song, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789–7796 CrossRef CAS;
(g) S. A. Svejda and M. Brookhart, Organometallics, 1999, 18, 65–74 CrossRef CAS.
-
(a) T. V. Laine, M. Klinga and M. Leskelä, Eur. J. Inorg. Chem., 1999, 959–964 CrossRef CAS;
(b) T. V. Laine, K. Lappalainen, J. Liimatta, E. Aitola, B. Löfgren and M. Leskelä, Macromol. Rapid Commun., 1999, 20, 487–491 CrossRef CAS;
(c) T. V. Laine, U. Piironen, K. Lappalainen, M. Klinga, E. Aitola and M. Leskel, J. Organomet. Chem., 2000, 606, 112–124 CrossRef CAS;
(d) B. Y. Lee, X. Bu and G. C. Bazan, Organometallics, 2001, 20, 5425–5431 CrossRef CAS;
(e) J. M. Benito, E. De Jesús, F. J. de la Mata, J. C. Flores, R. Gómez and P. Gómez-Sal, Organometallics, 2006, 25, 3876–3887 CrossRef CAS;
(f) Z. Huang, K. Song, F. Liu, J. Long, H. Hu, H. Gao and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1618–1628 CrossRef CAS;
(g) S. Jie, D. Zhang, T. Zhang, W.-H. Sun, J. Chen, Q. Ren, D. Liu, G. Zheng and W. Chen, J. Organomet. Chem., 2005, 690, 1739–1749 CrossRef CAS.
-
(a) J. Yu, X. Hu, Y. Zeng, L. Zhang, C. Ni, X. Hao and W.-H. Sun, New J. Chem., 2011, 35, 178–183 RSC;
(b) J. Yu, Y. Zeng, W. Huang, X. Hao and W.-H. Sun, Dalton Trans., 2011, 40, 8436–8443 RSC;
(c) L. Zhang, X. Hao, W.-H. Sun and C. Redshaw, ACS Catal., 2011, 1, 1213–1220 CrossRef CAS.
- C. Shao, W.-H. Sun, Z. Li, Y. Hu and L. Han, Catal. Commun., 2002, 3, 405–410 CrossRef CAS.
-
(a) L. Wang, W.-H. Sun, L. Han, H. Yang, Y. Hu and X. Jin, J. Organomet. Chem., 2002, 658, 62–70 CrossRef CAS;
(b) W.-H. Sun, S. Zhang, S. Jie, W. Zhang, Y. Li, H. Ma, J. Chen, K. Wedeking and R. Föhlich, J. Organomet. Chem., 2006, 691, 4196–4203 CrossRef CAS;
(c) M. Zhang, S. Zhang, P. Hao, S. Jie, W.-H. Sun, P. Li and X. Lu, Eur. J. Inorg. Chem., 2007, 3816–3826 CrossRef CAS;
(d) W.-H. Sun, K. Wang, K. Wedeking, D. Zhang, S. Zhang, J. Cai and Y. Li, Organometallics, 2007, 26, 4781–4790 CrossRef CAS;
(e) P. Hao, S. Zhang, W.-H. Sun, Q. Shi, S. Adewuyi, X. Lu and P. Li, Organometallics, 2007, 26, 2439–2446 CrossRef CAS;
(f) Y. Chen, P. Hao, W. Zuo, K. Gao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1829–1840 CrossRef CAS;
(g) S. Adewuyi, G. Li, S. Zhang, W. Wang, P. Hao, W.-H. Sun, N. Tang and J. Yi, J. Organomet. Chem., 2007, 692, 3532–3541 CrossRef CAS;
(h) A. P. Armitage, Y. D. M. Champouret, H. Grigoli, J. D. A. Pelletier, K. Singh and G. A. Solan, Eur. J. Inorg. Chem., 2008, 4597–4607 CrossRef CAS;
(i) Y. Yang, P. Yang, C. Zhang, G. Li, X.-J. Yang, B. Wu and C. Janiak, J. Mol. Catal. A: Chem., 2008, 296, 9–17 CrossRef CAS;
(j) L. Xiao, M. Zhang, R. Gao, X. Cao and W.-H. Sun, Aust. J. Chem., 2010, 63, 109–115 CrossRef CAS;
(k) X. Chen, L. Zhang, J. Yu, X. Hao, H. Liu and W.-H. Sun, Inorg. Chim. Acta, 2011, 370, 156–163 CrossRef CAS;
(l) L. Xiao, M. Zhang and W.-H. Sun, Polyhedron, 2010, 29, 142–147 CrossRef CAS;
(m) M. Zhang, P. Hao, W. Zuo, S. Jie and W.-H. Sun, J. Organomet. Chem., 2008, 693, 483–491 CrossRef CAS;
(n) G. J. P. Britovsek, S. P. D. Baugh, O. Hoarau, V. C. Gibson, D. F. Wass, A. J. P. White and D. Williams, Inorg. Chim. Acta, 2003, 345, 279–291 CrossRef CAS.
- F. He, X. Hao, X. Cao, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 712, 46–51 CrossRef CAS.
-
(a) F. A. Kunrath, R. F. Souza, O. L. Casagrande, N. R. Brooks and V. G. Young, Organometallics, 2003, 22, 4739–4743 CrossRef CAS;
(b) N. Ajellal, M. C. A. Kuhn, A. D. G. Boff, M. Hörner, C. M. Thomas, J.-F. Carpentier and O. L. Casagrande, Organometallics, 2006, 25, 1213–1216 CrossRef CAS;
(c) S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, Organometallics, 2009, 28, 2127–2133 CrossRef CAS.
-
(a) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS PubMed;
(b) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed;
(c) C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS;
(d) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS PubMed;
(e) C. Bianchini, G. Giambastiani, L. Luconi and A. Meli, Coord. Chem. Rev., 2010, 254, 431–455 CrossRef CAS;
(f) F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 38, 784–793 CrossRef CAS PubMed;
(g) R. Gao, W.-H. Sun and C. Redshaw, Catal. Sci. Technol., 2013, 3, 1172–1179 RSC;
(h) S. Wang, W.-H. Sun and C. Redshaw, J. Organomet. Chem. DOI:10.1016/j.jorganchem.2013.08.021.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235–244 CrossRef.
- W. Zhang, W.-H. Sun and C. Redshaw, Dalton Trans., 2013, 42, 8988–8997 RSC.
-
(a) W.-H. Sun, S. Jie, S. Zhang, W. Zhang, Y. Song and H. Ma, Organometallics, 2006, 25, 666–677 CrossRef CAS;
(b) S. Jie, S. Zhang, W.-H. Sun, X. Kuang, T. Liu and J. Guo, J. Mol. Catal. A: Chem., 2007, 269, 85–96 CrossRef CAS;
(c) M. Zhang, P. Hao, W. Zuo, S. Jie and W.-H. Sun, J. Organomet. Chem., 2008, 693, 483–491 CrossRef CAS;
(d) W.-H. Sun, P. Hao, S. Zhang, Q. Shi, W. Zuo, X. Tang and X. Lu, Organometallics, 2007, 26, 2720–2734 CrossRef CAS;
(e) Y. Chen, P. Hao, W. Zuo, K. Gao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1829–1840 CrossRef CAS;
(f) L. Xiao, R. Gao, M. Zhang, Y. Li, X. Cao and W.-H. Sun, Organometallics, 2009, 28, 2225–2233 CrossRef CAS;
(g) J. Lai, W. Zhao, W. Yang, C. Redshaw, T. Liang, Y. Liu and W.-H. Sun, Polym. Chem., 2012, 3, 787–793 RSC;
(h) F. He, W. Zhao, X.-P. Cao, T. Liang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 713, 209–216 CrossRef CAS;
(i) S. Wang, W. Zhao, X. Hao, B. Li, C. Redshaw, Y. Li and W.-H. Sun, J. Organomet. Chem., 2013, 731, 78–84 CrossRef CAS.
-
(a) J. Yu, H. Liu, W. Zhang, X. Hao and W.-H. Sun, Chem. Commun., 2011, 47, 3257–3259 RSC;
(b) W. Zhao, J. Yu, S. Song, W. Yang, H. Liu, X. Hao, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 130–137 CrossRef CAS;
(c) X. Cao, F. He, W. Zhao, Z. Cai, X. Hao, T. Shiono, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 1870–1880 CrossRef CAS;
(d) S. Wang, B. Li, T. Liang, C. Redshaw, Y. Li and W.-H. Sun, Dalton Trans., 2013, 42, 9188–9197 RSC.
-
(a) J. Lai, W. Zhao, W. Yang, C. Redshaw, T. Liang, Y. Liu and W.-H. Sun, Polym. Chem., 2012, 3, 787–793 RSC;
(b) F. He, W. Zhao, X.-P. Cao, T. Liang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 713, 209–216 CrossRef CAS;
(c) S. Wang, W. Zhao, X. Hao, B. Li, C. Redshaw, Y. Li and W.-H. Sun, J. Organomet. Chem., 2013, 731, 78–84 CrossRef CAS.
-
(a) H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418–2424 CrossRef CAS;
(b) H. Liu, W. Zhao, J. Yu, W. Yang, X. Hao, C. Redshaw, L. Chen and W.-H. Sun, Catal. Sci. Technol., 2012, 2, 415–422 RSC;
(c) X. Hou, Z. Cai, X. Chen, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2012, 41, 1617–1623 RSC;
(d) J. Yu, W. Huang, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2011, 40, 10209–10214 RSC;
(e) W.-H. Sun, S. Song, B. Li, C. Redshaw, X. Hao, Y. Li and F. Wang, Dalton Trans., 2012, 41, 11999–12010 RSC.
- J. Yuan, F. Wang, W. Xu, T. Mei, J. Li, B. Yuan, F. Song and Z. Jia, Organometallics, 2013, 32, 3960–3968 CrossRef CAS.
- H. A. Iddles and W. L. Hartop Jr., J. Am. Chem. Soc., 1950, 72, 4589–4591 CrossRef CAS.
- K. Tomov, V. C. Gibson, G. J. P. Britovsek, R. J. Long, M. van Meurs, D. J. Jones, K. P. Tellmann and J. J. Chirinos, Organometallics, 2009, 28, 7033–7040 CrossRef.
- G. B. Galland, R. F. de Souza, R. S. Mauler and F. F. Nunes, Macromolecules, 1999, 32, 1620–1625 CrossRef CAS.
- R. Chen and S. F. Mapolie, J. Mol. Catal. A: Chem., 2003, 193, 33–40 CrossRef CAS.
-
(a) T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS PubMed;
(b) E. F. Connor, T. R. Younkin, J. I. Henderson, A. W. Waltman and R. H. Grubbs, Chem. Commun., 2003, 2272–2273 RSC;
(c) V. C. Gibson, C. K. A. Gregson, C. M. Halliwell, N. J. Long, P. J. Oxford, A. J. P. White and D. J. Williams, J. Organomet. Chem., 2005, 690, 6271–6283 CrossRef CAS.
-
(a) J. M. Benito, E. De Jesús, F. J. de la Mata, J. C. Flores, R. Gómez and P. Gómez-Sal, Organometallics, 2006, 25, 3876–3887 CrossRef CAS;
(b) Z. Huang, K. Song, F. Liu, J. Long, H. Hu, H. Gao and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1618–1628 CrossRef CAS.
-
G. M. Sheldrick, SHELXTL-97 Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a