DOI:
10.1039/C3DT52132J
(Paper)
Dalton Trans., 2014,
43, 1736-1743
Oxidative addition of disulfide/diselenide to group 10 metal(0) and in situ functionalization to form neutral thiasalen/selenasalen group 10 metal(II) complexes†
Received 5th August 2013, Accepted 14th October 2013
First published on 15th October 2013
Abstract
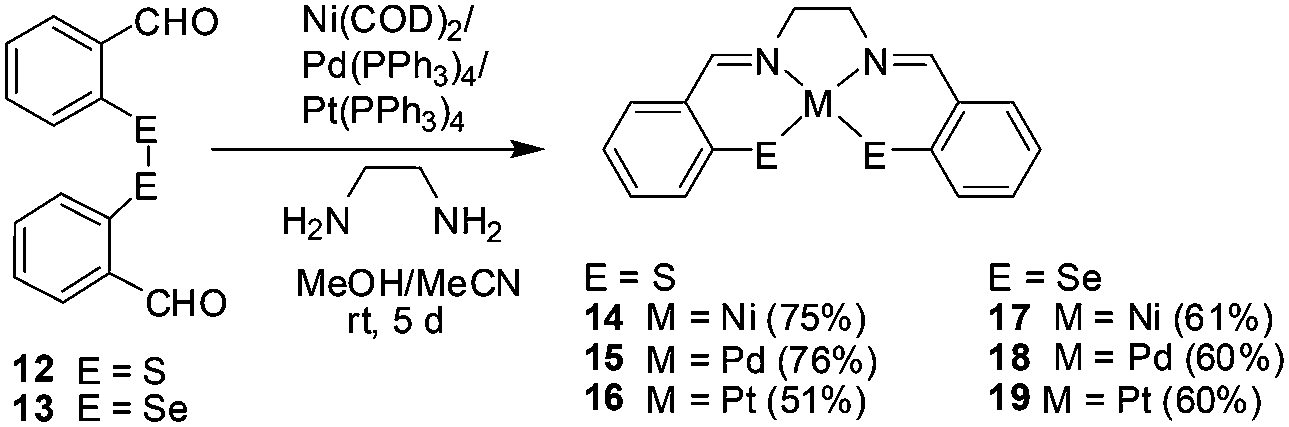
Three components, one pot synthesis of thiasalen/selenasalen Ni(II), Pd(II) and Pt(II) complexes, 14–19, by the oxidative addition of S–S/Se–Se bond of bis(o-formylphenyl)disulfide/-diselenide to Ni(0), Pd(0) and Pt(0) followed by in situ Schiff base formation with ethylenediamine is reported. S–S or Se–Se bonds were cleaved and coordinated to the metal center as thiolate (ArS−) or selenolate (ArSe−) while the formal oxidation state of metal centers was changed from ‘0’ to ‘+2’. The disulfide/diselenide reacted with zero-valent metals at room temperature to give only the monometallic complexes. All complexes (except Pd–thiolate complex 15) were studied by single crystal X-ray crystallography and revealed the square planar geometry around metal centers.
Introduction
Schiff base synthesized by the reaction of two equivalents of salicylaldehyde with one equivalent of ethylenediamine is known as ‘salen’ which provides potentially tetradentate chelating systems with a N2O2 donor set. Metal complexes based on salen and its derivatives have drawn considerable attention in last few decades1 due to their applications in catalysis, biological studies, material science, molecular magnetism and sensory materials.2 As catalysts, such complexes have been successfully applied in many reactions such as oxidation, olefin epoxidation, cycloaddition and in asymmetric synthesis.3 Recently, salen-complexes were utilized to develop metal organic frameworks (MOFs) for their novel applications such as hydrogen gas storage and highly efficient catalysis.4 Sulfur and selenium analogues of salen (thiasalen and selenasalen with N2S2 and N2Se2 ligating sites) have attracted scant attention due to instability of thiol and selenol groups compared to the hydroxyl group of salen.Oxidative addition of E–E bonds (E = S and Se) of diorganodisulfides and -diselenides to low-valent transition metal complexes is a mild and efficient method to synthesize chalcogenolato-metal complexes.5 The oxidative addition of diorganyl disulfides/diselenides to Pd(0) complexes, such as [Pd(PPh3)4], was investigated for their addition to alkynes, which is an efficient single step method for the formation of two C–S/Se bonds in a stereoselective manner.6 Mechanistic studies on these reactions have been carried out using various types of disulfides/diselenides and alkynes from both the experimental and theoretical point of views.7 Yamamoto and Sekine have reported the oxidative addition of diaryl disulfides, ArS-SAr, with [Ni(COD)2] (COD = 1,5-cyclooctadiene) in the presence of basic ligands such as 2,2′-bipyridine, triethylphosphine.8 Oxidative addition of diaryl disulfides (ArS)2 with [Pd(PPh3)4] and [Pt(PPh3)4] formed dimeric and monomeric complexes, respectively.9 The tendency of formation of dimeric complexes can be minimized by using electron withdrawing substituents on the aromatic rings of diaryl disulfide. This study also revealed that Pt(0) complexes have less tendency to form dimeric complexes compared to Pd(0) precursors. The oxidative addition of S–S bond of the ring systems to Ni(0), Pd(0) and Pt(0) to form S,S-dithiolate chelate complexes were also studied.10
Morley et al.11 have shown the formation of selenolate complexes via oxidative addition of Ph2Se2 and Fc2Se2 [Fc = ferrocenyl] to Pd(0) and Pt(0). Laitinen et al. reported the oxidative addition of Th2Se2 and Ph2Se2 to Pd(0) and Pt(0).12 Reversible oxidative addition of Se–Se bonds to Pt(0) and Pt(II) precursors was observed by changing the ligand environment.13 However, oxidative addition of Se–Se bond of diorganodiselenide to Ni(0) is not reported till the date. In addition to oxidative addition of E–E bonds to group 10 M(0) precursors, the cleavage of E–E bonds also have also been reported with Group 10 metals in higher oxidation states.14
Synthesis of ligands and complexes simultaneously by the formation of both carbon–heteroatom and heteroatom–metal bonds is a powerful technique in the preparation of metallo-organic self assemblies.15 The introduction of thiolates/selenolates into metal complexes directly by the oxidative addition of dichalcogenides are often complicated by the presence of dimeric/polynuclear and non-stoichiometric compounds, however, it can be avoided by using the chelating diselenide.5a Here we have used this approach to synthesize thiasalen and selenasalen complexes.
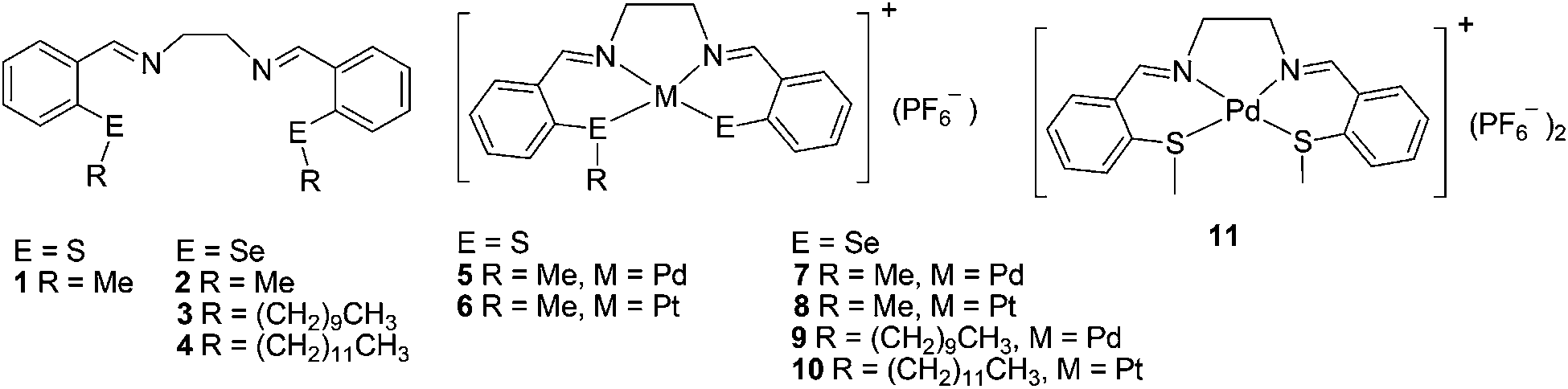
Earlier we reported the Pd(II) and Pt(II) complexes (5–11) which were prepared by the reactions of bis(alkyl)thiasalen (1) and bis(alkyl)selenasalen (2–4) ligands with Pd(II) and Pt(II) metal precursors.16 We have observed dealkylative complexation via nucleophilic substitution at C–E bond (E = S, Se) where monocationic complexes act as a leaving groups. However, the attempts to prepare the analogous Ni(II) complexes were unsuccessful. In the synthesis of complexes 5–11, the two step process was applied; (i) synthesis and isolation of the ligand and then (ii) complexation with the metal precursors. Here we report the complete series of six neutral group 10 d8 metal complexes of thia/selenasalen ligands via oxidative addition of E–E bond of bis(orthoformyl)phenyl disulfide (12) and bis(orthoformyl)phenyl diselenide (13) to Ni(0), Pd(0) and Pt(0) and subsequent in situ reaction of ethylene diamine. Thus it is a three component one pot reaction for facile synthesis of sulfur and selenium analogues of salen group 10 metal complexes. It is the first report on the synthesis of neutral thiasalen complexes of Pd and Pt and selenosalen complexes of Ni, Pd and Pt.
Result and discussion
Synthesis of ligands
The compounds 1217 and 1318 were synthesized according to the reported procedures. 1H NMR spectrum of the ligand 12 showed a singlet at δ 10.23 ppm for aldehyde protons whereas for ligand 13 showed a singlet at δ 10.17 ppm.Complexation of compound 12 and 13 with Ni(0), Pd(0) and Pt(0)
Reactions of ligands 12 and 13 with Ni(0) ([Ni(COD)2]), Pd(0) ([Pd(PPh3)4]) and Pt(0) ([Pt(PPh3)4]) afforded six neutral thiolate and selenolate complexes 14–19via oxidative addition of E–E bond (E = S, Se) to Ni(0), Pd(0) and Pt(0) at room temperature (Scheme 1). As a result, S–S or Se–Se bonds were cleaved and coordinated to the metal center as thiolate (ArS−) or selenolate (ArSe−) while the formal oxidation state of metal center was changed from ‘0’ to ‘+2’. The intermediate aldehyde complexes were used in situ as template for the preparation of desired Schiff base derivatives. Thus, it provides a one pot three component method for synthesis of the thiasalen and selenasalen d8 metal complexes by template synthesis using oxidative addition of bis(o-formylphenyl)disulfide/diselenide to M(0) (M = Ni, Pd, Pt) precursors and imine bond formation by reaction with ethylenediamine. Template synthesis is a well established method for the one pot synthesis of Schiff-base complexes. In the template synthesis method a metal ion first coordinates with aldehyde to create the template for the reaction with amine to form metal ion coordinated Schiff-base.19 Thus, the concept of simultaneous use of oxidative addition and template synthesis is used here for one pot synthesis of thiasalen/selenasalen based metal complexes. |
| | Scheme 1 Synthesis of complexes 14–19. | |
When one equivalent of ligand 12 was treated with one equivalent of [Ni(COD)2], [Pd(PPh3)4] and [Pt(PPh3)4] and one equivalent of 1,2-ethelenediamine, complexes 14, 15 and 16 were formed, respectively. All the three complexes are red in colour and stable in solid as well as in solution phase at ambient conditions. Complex 14 is soluble in DCM, CHCl3, DMF and DMSO while complexes 15 and 16 are soluble in DMF and DMSO. Previously, Yamamura et al.20 reported the synthesis of complex 14 by the reaction of bis(2-(tert-butylthio)benzylidene)ethylenediamine with NiCl2·6H2O with in situ cleavage of tert-butyl group. Goswami and Eichhorn15 reported the synthesis of 14 by the cleavage of disulfide bond in bis(o-formylphenyl)disulfide using [Ni(en)3]Cl2. In both the cases, Ni(II) ion was taken as metal source. Elemental analysis data of the compounds 14, 15 and 16 are consistent with the proposed formula and structures. 1H/13C NMR spectra of the complexes 14, 15 and 16 are in good agreement with the proposed structures. 1H NMR spectra of the complexes 14, 15 and 16 showed a singlet at δ 8.59, 8.72 and 9.02 ppm for azomethine protons, respectively. Complexes 14, 15 and 16 showed eight signals in the 13C NMR spectra for eight different types of carbons indicating symmetrical nature of the complexes in solution. FT-IR spectra of the complexes 14, 15 and 16 displayed the characteristic ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching frequencies at 1610, 1626 and 1617 cm−1, respectively, which were shifted to lower energy values compared to the ν(CO) stretching frequency (1691 cm−1) of precursor 12.
N) stretching frequencies at 1610, 1626 and 1617 cm−1, respectively, which were shifted to lower energy values compared to the ν(CO) stretching frequency (1691 cm−1) of precursor 12.
The selenium analogues 17, 18 and 19 were prepared by similar procedure using precursor bis(orthoformylphenyl) diselenide 13. Formulations of complexes 17–19 were supported by elemental analyses, however, elemental analysis for 18 was found to be slightly deviated from the calculated value of percentage of carbon. Color, stability and solubility of these complexes are quite similar to that of their sulfur analogues. 1H NMR and 13C NMR spectra of 17, 18 and 19 showed symmetrical nature of these complexes. The characteristic imine protons were observed at δ 8.66, 8.76 and 9.09 ppm, respectively, in 1H NMR spectra of 17, 18 and 19. 77Se NMR of complexes 17, 18, 19 showed the single resonance at δ 354.9, 393.7, 318.9 ppm, respectively. In the FT-IR spectra of complexes 17, 18 and 19, peaks at 1609, 1622 and 1611 cm−1, respectively, were assigned to ν(CN) stretching frequency. ESI-MS spectra of complexes 14 and 17 displayed the molecular ion peak at 356.37 and 452.1934, respectively as [M + H]+ moieties, while the Pd(II) and Pt(II) complexes did not ionize in ESI mass spectrometer.
Crystal structure study
The molecular structures and supramolecular assemblies of 14, 16, 17, 18 and 19 have been determined by X-ray crystallography.Crystal structure of Ni–thiolate complex 14
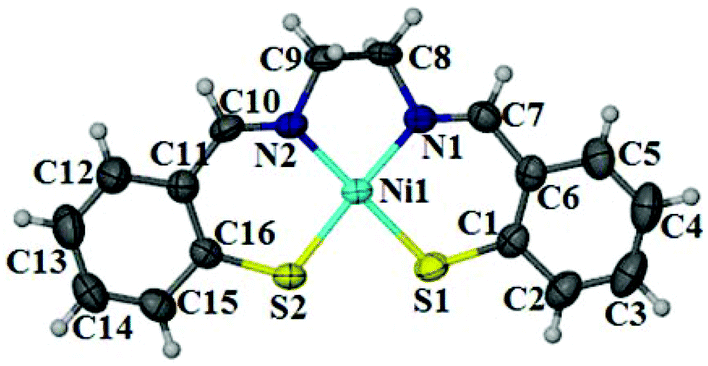
Complex 14 crystallizes in orthorhombic space group Pna21 with the square planar geometry around metal center (Fig. 1). Space group, unit cell and bond lengths/angles are quite similar to those reported by Yamamura et al.,20 while Goswami and Eichhorn15 reported the monoclinic space group P21/c with asymmetric unit containing two independent molecules and a dichloromethane molecule. |
| | Fig. 1 ORTEP representation of nickel complex 14; thermal ellipsoids are drawn at 50% probability level. | |
Crystal structure of Pt–thiolate complex 16
Complex 16 crystallizes in the monoclinic space group P21/c with a square planar geometry around the Pt(II) center (Fig. 2). The two Pt–S bonds are nearly equal and slightly shorter than that in thioether–thiolate complex 12 (thioether Pt–S, 2.2614(7) Å and thiolate Pt–S, 2.2539(7) Å), respectively.16b Selected bond lengths and bond angles are shown in Table 1. Crystal packing of complex 16 shows the formation of 2D sheet along bc plane via C–H⋯π (H1A⋯C1–C6), H5⋯H14 and H1A⋯S1 interactions (Fig. S1†). Intermolecular S1⋯H2B and H5⋯H14 interactions present in the crystal lattice construct the two dimensional sheet along ac plane (Fig. S2†). |
| | Fig. 2 ORTEP representation of platinum complex 16; thermal ellipsoids are drawn at 50% probability level. | |
Table 1 Selected bond lengths (Å) and angles (°) for 16
| S. no | Bond lengths (Å) | Bond angles (°) |
|---|
| 1 | Pt1–S1 | 2.246(4) | S1–Pt1–S2 | 89.53(16) |
| 2 | Pt1–S2 | 2.225(4) | N1–Pt1–N2 | 82.4(6) |
| 3 | Pt1–N1 | 2.025(14) | S1–Pt1–N2 | 176.6(4) |
| 4 | Pt1–N2 | 2.039(13) | S2–Pt1–N1 | 173.2(4) |
| 5 | S1–C1 | 1.760(16) | S1–Pt1–N1 | 95.7(4) |
| 6 | S2–C16 | 1.83(2) | S2–Pt1–N2 | 92.6(4) |
Crystal structure of Ni–selenolate complex 17
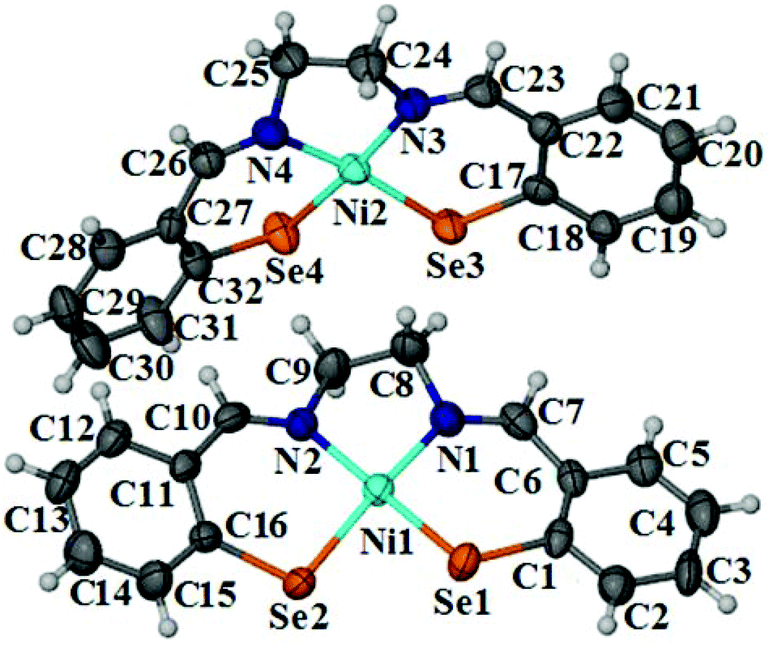
The square planar geometry around Ni(II) center in complex 17 was confirmed by single crystal X-ray crystallography. Complex 17 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two asymmetric molecules in the unit cell (Fig. 3). The four Ni–Se bonds are nearly equal (average 2.263 Å) and comparable to those reported for selenocarbamoyl benzamidine base Ni(II) complex (2.278 and 2.293 Å)21 and 2-aminophenyl diselenolate based Ni(II) complex (2.295 Å)22 which also have square planar geometry around the Ni(II) center with N2Se2 donor set. The average Ni–N bond distance is 1.894 Å. The various intermolecular nonbonding interactions including CH⋯π (aromatic) interaction between the two molecules led to the formation of 1-D chain along b axis (Fig. S3†). Selected bond lengths and bond angles are shown in Table 2.
with two asymmetric molecules in the unit cell (Fig. 3). The four Ni–Se bonds are nearly equal (average 2.263 Å) and comparable to those reported for selenocarbamoyl benzamidine base Ni(II) complex (2.278 and 2.293 Å)21 and 2-aminophenyl diselenolate based Ni(II) complex (2.295 Å)22 which also have square planar geometry around the Ni(II) center with N2Se2 donor set. The average Ni–N bond distance is 1.894 Å. The various intermolecular nonbonding interactions including CH⋯π (aromatic) interaction between the two molecules led to the formation of 1-D chain along b axis (Fig. S3†). Selected bond lengths and bond angles are shown in Table 2. |
| | Fig. 3 ORTEP representation of platinum complex 17; thermal ellipsoids are drawn at 50% probability level. | |
Table 2 Selected bond lengths (Å) and bond angles (°) for complex 17
| S. no | Bond lengths (Å) | Bond angles (°) |
|---|
| 1 | Ni1–Se1 | 2.2576(13) | Se1–Ni1–Se2 | 83.60(5) |
| 2 | Ni1–Se2 | 2.2578(13) | Se3–Ni2–Se4 | 84.03(4) |
| 3 | Ni2–Se3 | 2.2860(13) | N1–Ni1–N2 | 86.9(2) |
| 4 | Ni2–Se4 | 2.2518(13) | N3–Ni2–N4 | 86.52(19) |
| 5 | Ni1–N1 | 1.897(4) | Se1–Ni1–N2 | 171.68(14) |
| 6 | Ni1–N2 | 1.887(5) | Se2–Ni1–N1 | 171.53(14) |
| 7 | Ni2–N3 | 1.896(5) | Se2–Ni1–N2 | 95.99(15) |
| 8 | Ni2–N4 | 1.894(4) | Se1–Ni1–N1 | 94.73(15) |
| 9 | Se1–C1 | 1.902(3) | Se3–Ni2–N3 | 93.10(13) |
| 10 | Se2–C16 | 1.887(3) | Se3–Ni2–N4 | 172.87(15) |
| 11 | Se3–C17 | 1.890(3) | Se4–Ni2–N3 | 172.33(16) |
| 12 | Se4–C32 | 1.898(3) | Se4–Ni2–N4 | 97.20(14) |
Crystal structure of Pd–selenolate complex 18
Similar to complex 17, the crystal structure of complex 18 crystallizes in the triclinic space group P with two molecules in the asymmetric unit (Z = 4, Z′ = 2) and a square planar geometry around the Pd(II) centers (Fig. 4). The average distance of four Pd–Se bonds is 2.366 Å which is slightly shorter than Pd–Se bonds of nicotinoyl selenide based square planar Pd(II) complex (2.444 and 2.428 Å).23 The average of four Pd–N bond distances is 2.036 Å. Selected bond lengths and bond angles are shown in Table 3. Crystal packing of 18 shows the intermolecular C–H⋯π, π⋯π and Se⋯H short contacts (Fig. S4†). |
| | Fig. 4 ORTEP representation of palladium complex 18; thermal ellipsoids are drawn at 50% probability level. | |
Table 3 Selected bond lengths (Å) and angles (°) for 18
| S. no | Bond lengths (Å) | Bond angles (°) |
|---|
| 1 | Pd1–Se1 | 2.3587(3) | Se1–Pd1–Se2 | 87.281(10) |
| 2 | Pd1–Se2 | 2.3639(3) | Se3–Pd2–Se4 | 87.796(10) |
| 3 | Pd2–Se3 | 2.3809(3) | N1–Pd1–N2 | 84.44(7) |
| 4 | Pd2–Se4 | 2.3588(3) | N3–Pd2–N4 | 83.60(7) |
| 5 | Pd1–N1 | 2.0328(18) | Se1–Pd1–N2 | 173.09(5) |
| 6 | Pd1–N2 | 2.0358(18) | Se2–Pd1–N1 | 173.30(5) |
| 7 | Pd2–N3 | 2.0344(19) | Se2–Pd1–N2 | 94.02(5) |
| 8 | Pd2–N4 | 2.0390(17) | Se1–Pd1–N1 | 95.02(5) |
| 9 | Se1–C1 | 1.893(2) | Se3–Pd2–N3 | 92.60(5) |
| 10 | Se2–C16 | 1.899(2) | Se3–Pd2–N4 | 174.89(5) |
| 11 | Se3–C17 | 1.898(2) | Se4–Pd2–N3 | 175.76(5) |
| 12 | Se4–C32 | 1.895(2) | Se4–Pd2–N4 | 96.24(5) |
Crystal structure of Pt–selenolate complex 19
Complex 19 crystallizes in the triclinic space group P with two molecules in the asymmetric unit (Z = 4, Z′ = 2) and a square planar geometry around the Pt(II) centre (Fig. 5). The four Pt–Se bonds are nearly equal and comparable to those in selenoether–selenolate complex 8 (selenoether Pt–Se, 2.3636(6) Å and selenolate Pt–Se, 2.3583(6) Å).16a The average Pt–N bond distance is 2.019 Å. Selected bond lengths and bond angles are shown in Table 4. Molecular packing of the complex is similar to that of nickel and palladium analogues. |
| | Fig. 5 ORTEP representation of palladium complex 19; thermal ellipsoids are drawn at 50% probability level. | |
Table 4 Selected bond lengths (Å) and angles (°) for 19
| S. no | Bond lengths (Å) | Bond angles (°) |
|---|
| 1 | Pt1–Se1 | 2.3641(7) | Se1–Pt1–Se2 | 87.08(3) |
| 2 | Pt1–Se2 | 2.3731(7) | Se3–Pt2–Se4 | 87.58(2) |
| 3 | Pt2–Se3 | 2.3866(7) | N1–Pt1–N2 | 83.3(2) |
| 4 | Pt2–Se4 | 2.3672(7) | N3–Pt2–N4 | 83.2(2) |
| 5 | Pt1–N1 | 2.017(5) | Se1–Pt1–N2 | 174.44(15) |
| 6 | Pt1–N2 | 2.015(6) | Se2–Pt1–N1 | 174.43(15) |
| 7 | Pt2–N3 | 2.027(5) | Se2–Pt1–N2 | 94.55(16) |
| 8 | Pt2–N4 | 2.020(5) | Se1–Pt1–N1 | 95.53(17) |
| 9 | Se1–C1 | 1.883(7) | Se3–Pt2–N3 | 93.28(15) |
| 10 | Se2–C16 | 1.900(7) | Se3–Pt2–N4 | 174.95(15) |
| 11 | Se3–C17 | 1.895(6) | Se4–Pt2–N3 | 175.96(15) |
| 12 | Se4–C32 | 1.882(6) | Se4–Pt2–N4 | 96.20(15) |
Complete crystallographic data for complex 14, 16–19 are provided in Table 5.
Table 5 Crystallographic information of complex 14, 16, 17, 18 and 19
| Formula | C16H14N2NiS2 | C16H14N2PtS2 | C16H14N2NiSe2 | C16H14N2PdSe2 | C16H14N2PtSe2 |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | Pna2(1) | P21/c | P | P | P |
| a [Å] | 8.9500(7) | 7.5651(9) | 10.398(6) | 10.3810(10) | 10.4940(12) |
| b [Å] | 22.688(2) | 7.9224(9) | 12.088(7) | 12.0674(11) | 12.1603(15) |
| c [Å] | 7.5123(7) | 26.820(3) | 12.955(7) | 12.7544(11) | 12.8887(16) |
| α [°] | 90 | 90 | 71.269(13) | 72.987(2) | 72.482(2) |
| β [°] | 90 | 97.961(2) | 86.197(18) | 85.737(2) | 85.863(2) |
| γ [°] | 90 | 90 | 82.699(14) | 83.075(2) | 83.337(2) |
| V [Å3] | 1525.4(2) | 1591.9(3) | 1529.0(15) | 1515.4(2) | 1556.7(3) |
| Z | 4 | 4 | 4 | 4 | 4 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρcalcd [g cm−3] | 1.555 | 2.059 | 1.959 | 2.185 | 2.506 |
| F [000] | 736 | 936 | 880 | 952 | 1080 |
| μ [mm−1] | 1.538 | 9.070 | 6.023 | 6.021 | 13.684 |
| θ [°] | 2.86–24.96 | 2.57–24.97 | 2.51–23.95 | 2.54–25.04 | 2.61–31.94 |
| Index ranges | −10 ≤ h ≤ 10 | −8 ≤ h ≤ 9 | −11 ≤ h ≤ 12 | −12 ≤ h ≤ 12 | −7 ≤ h ≤ 13 |
| −26 ≤ k ≤ 26 | −5 ≤ k ≤ 9 | −14 ≤ k ≤ 14 | −14 ≤ k ≤ 14 | −15 ≤ k ≤ 15 |
| −8 ≤ l ≤ 8 | −31 ≤ l ≤ 31 | −15 ≤ l ≤ 14 | −15 ≤ l ≤ 13 | −16 ≤ l ≤ 16 |
| T [K] | 100(2) | 298(2) | 298(2) | 298(2) | 298(2) |
| R1 | 0.0215 | 0.0644 | 0.0438 | 0.0166 | 0.0320 |
| wR2 | 0.0481 | 0.1849 | 0.1065 | 0.0394 | 0.0820 |
| Rmerge | 0.0257 | 0.0751 | 0.0731 | 0.0183 | 0.0410 |
| Parameters | 190 | 160 | 331 | 379 | 379 |
| GOF | 1.024 | 1.085 | 1.025 | 1.050 | 1.185 |
| Total reflns | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 553 553 | 11915 | 21036 | 29289 | 24568 |
| Unique reflns | 2681 | 2806 | 5597 | 5407 | 6805 |
| Obsd reflns | 2445 | 2295 | 3920 | 5049 | 5752 |
| CCDC no. | 946622 | 946625 | 946624 | 946623 | 946626 |
Conclusion
In summary, the reactions of ligands bis(o-formylphenyl)disulfide/-diselenide (12 and 13) with [Ni(COD)2], [Pd(PPh3)4] and [Pt(PPh3)4] afforded six neutral square planar thiolate and selenolate complexes 14–19via oxidative addition of E–E bond (E = S, Se) to Ni(0), Pd(0) and Pt(0) followed by imine bond formation by reaction with ethylene diamine. Thus we developed three-component (group 10 M(0) precursor, bis(o-formylphenyl)disulfide/-diselenide, and ethylene diamine), one-pot reaction for the synthesis of thiasalen and selenasalen group 10 metal complexes. We believe that the reaction can be extended to the various derivatives of complexes 14–19 by using substituted bis(o-formylphenyl)disulfide/-diselenide and various diamines. It is the first report on the synthesis of neutral thiasalen complexes of Pd and Pt and selenosalen complexes of Ni, Pd and Pt.Experimental section
All reagents were purchased from Aldrich/Merck and used without further purification. Acetonitrile was distilled from P2O5 and kept over molecular sieves. 1H and 13C NMR spectra were recorded either on Bruker 500 MHz or on JEOL-FT NMR-AL 400 MHz spectrometer using DMSO-d6 as solvent and tetramethylsilane (SiMe4) as internal standards. 77Se NMR spectra were recorded on Bruker 500 MHz spectrometer using DMSO-d6 as solvent. 77Se NMR chemical shifts are reported using Ph2Se2 as external standard with chemical shifts of 470 ppm with respect to Me2Se, thus the values are reported with respect to Me2Se. IR spectra of the compounds have been recorded on a Perkin-Elmer spectrophotometer as KBr pellets. The mass spectra were recorded on a MICROMAX Q-TOF-MICRO instrument. Melting points were measured using a digital melting point apparatus, SECOR INDIA.Crystal structure determination
Single crystals of the compounds suitable for X-ray diffraction were grown from dimethyl formamide/dimethyl sulfoxide solution by diffusing diethyl ether vapors in a closed beaker. The crystals were carefully chosen using a stereomicroscope supported by a rotatable polarizing stage. The data were collected on Bruker's KAPPA APEX II CCD Duo with graphite monochromated Mo-Kα radiation (0.71073 Å). The crystals were glued to a thin glass fibre using FOMBLIN immersion oil and mounted on the diffractometer. The intensity data were processed using Bruker's suite of data processing programs (SAINT), and absorption corrections were applied using SADABS.24 The crystal structure was solved by direct methods using SHELXS-97 and the data were refined by full matrix least-squares refinement on F2 with anisotropic displacement parameters for non-H atoms, using SHELXL-97.25 ORTEP diagrams are drawn from X-seed version 2.0.26Thiasalen Ni(II) complex 14
To a solution of bis(o-formylphenyl)disulfide 12 (74 mg, 0.27 mmol) in 10 mL dry methanol, [Ni(COD)2] (74 mg, 0.27 mmol) and ethylenediamine (16 mg, 0.27 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with methanol and dried in vacuum (yield: 72 mg, 75%). Mp 272 °C (d). 1H NMR (400 MHz, DMSO-d6): δ 8.59 (s, 2H); 7.49 (m, 4H); 7.22 (m, 2H); 7.03 (m, 2H), 3.72 (s, 4H) ppm. 13C NMR (125 MHz, DMSO-d6): δ 163.77, 144.70, 134.96, 130.41, 129.69, 129.36, 121.65, 61.26 ppm. IR νmax (KBr, cm−1): 1610 (CN). Anal. Calcd (%) for C16H14N2 Ni S2: C, 53.81; H, 3.95; N, 7.84. Found: C, 54.65; H, 4.02; N, 7.91%. ESI-MS calcd for {(C16H14N2NiS2 + H)+}, 357.0030, found, 356.3700.Thiasalen Pd(II) complex 15
To a solution of bis(o-formylphenyl)disulfide 12 (100 mg, 0.36 mmol) in 10 mL dry acetonitrile, [Pd(PPh3)4] (410 mg, 0.36 mmol) and ethylenediamine (26 mg, 0.44 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with acetonitrile and dried in vacuum (yield: 117 mg, 76%). Mp 290 °C (d). 1H NMR (400 MHz, DMSO-d6): δ 8.72 (1H), 7.61–7.06 (4H, Ar–H), 3.99 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 161.80, 144.06, 136.93, 131.82, 130.87, 130.62, 121.91, 61.76 ppm. IR νmax (KBr, cm−1): 1626 (CN). Anal. Calcd (%) for C16H14N2PdS2: C, 47.47; H, 3.49; N, 6.92. Found: C, 47.03; H, 3.29; N, 6.63%.Thiasalen Pt(II) complex 16
To a solution of bis(o-formylphenyl)disulfide 12 (100 mg, 0.36 mmol) in 10 mL dry acetonitrile, [Pt(PPh3)4] (450 mg, 0.36 mmol) and ethylenediamine (26 mg, 0.44 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with acetonitrile and dried in vacuum (yield: 92 mg, 51%). Mp 285 °C (d). 1H NMR (400 MHz, DMSO-d6): δ 9.02 (1H), 7.67–7.04 (4H, Ar–H), 3.94 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.20, 139.84, 135.89, 131.95, 131.08, 130.63, 122.03, 62.76 ppm. IR νmax (KBr, cm−1): 1617 (CN). Anal. Calcd (%) for C16H14N2PtS2: C, 38.94; H, 2.86; N, 5.68. Found: C, 38.76; H, 2.74; N, 5.31%.Selenasalen Ni(II) complex 17
To a solution of bis(o-formylphenyl) diselenide 13 (100 mg, 0.27 mmol) in 10 mL dry methanol, [Ni(COD)2] (74 mg, 0.27 mmol) and ethylene diamine (17 mg, 0.27 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with methanol and dried in vacuum (yield: 78 mg, 61%). Mp 285 °C (d). 1H NMR (500 MHz, DMSO-d6): δ 8.66 (s, 1H); 7.68 (m, 2H), 7.56 (m, 2H), 7.19 (m, 4H), 3.74 (s, 4H) ppm. 13C NMR (125 MHz, DMSO-d6): δ 164.76, 136.40, 136.00, 132.72, 131.78, 130.22, 123.45, 62.04 ppm. 77Se NMR (95 MHz, DMSO-d6): δ 354.9 ppm. IR νmax (KBr, cm−1): 1609 (CN). Anal. Calcd (%) for C16H14N2Se2Ni: C, 42.62; H, 3.13; N, 6.21. Found: C, 41.98; H, 2.95; N, 6.41%. ESI-MS calcd for {(C16H14N2NiSe2 + H)+}, 452.8919, found, 452.1934.Selenasalen Pd(II) complex 18
To a solution of bis(o-formylphenyl) diselenide 13 (100 mg, 0.27 mmol) in 10 mL dry methanol, [Pd(PPh3)4] (310 mg, 0.27 mmol) and ethylenediamine (16 mg, 0.27 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with methanol and dried in vacuum (yield: 81 mg, 60%). Mp 283 °C (d). 1H NMR (400 MHz, DMSO-d6): δ 8.76 (1H), 7.80–7.18 (4H, Ar–H), 3.97 (s, 2H) ppm. 13C NMR (125 MHz, DMSO-d6): δ 162.69, 137.70, 134.70, 133.66, 132.84, 131.46, 123.39, 62.45 ppm. 77Se NMR (95 MHz, DMSO-d6): δ 393.7 ppm. IR νmax (KBr, cm−1): 1622 (CN). Anal. Calcd (%) for C16H14N2PdSe2: C, 38.54; H, 2.83; N, 5.62. Found: C, 40.79; H, 2.74; N, 5.67%.Selenasalen Pt(II) complex 19
To a solution of bis (o-formylphenyl)diselenide 13 (100 mg, 0.27 mmol) in 10 mL dry methanol, [Pt(PPh3)4] (340 mg, 0.27 mmol) and ethylenediamine (16 g, 0.27 mmol) were added at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 5 days. Precipitate was filtered, washed thoroughly with methanol and dried in vacuum (yield: 96 mg, 60%). Mp 297 °C (d). 1H NMR (500 MHz, DMSO-d6): δ 9.09 (1H), 7.80–7.16 (4H, Ar–H), 3.91 (s, 2H) ppm. 13C NMR (125 MHz, DMSO-d6): δ 158.26, 137.01, 134.59, 132.71, 131.06, 123.67, 63.77 ppm. 77Se NMR (95 MHz, DMSO-d6): δ 318.9 ppm. IR νmax (KBr, cm−1): 1611 (CN). Anal. Calcd (%) for C16H14N2PtSe2: C, 32.72; H, 2.40; N, 4.77. Found: C, 33.98; H, 2.26; N, 5.05%.Acknowledgements
SP is thankful to Department of Science and Technology (DST), India for funding this work through Fast-Track project. PD is thankful to UGC for fellowship.References
-
(a) S. Yamada, Coord. Chem. Rev., 1999, 190–192, 537–555 CrossRef CAS;
(b) C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622–631 RSC;
(c) J. Lewinski, J. Zachara, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185–1199 CrossRef CAS PubMed;
(d) M. Kojima, H. Taguchi, M. Tsuchimoto and K. Nakajima, Chem. Soc. Rev., 2003, 237, 183–196 CAS.
-
(a) K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS PubMed;
(b) A. Tzubery and E. Y. Tshuva, Inorg. Chem., 2011, 50, 7946–7948 CrossRef CAS PubMed;
(c) S. J. Wezenberg and A. W. Kleij, Angew. Chem., Int. Ed., 2008, 47, 2354–2364 CrossRef CAS PubMed;
(d) H. Miyasaka, A. Saitoh and S. Abe, Coord. Chem. Rev., 2007, 251, 2622–2664 CrossRef CAS PubMed;
(e) T. Glaser, Chem. Commun., 2011, 47, 116–130 RSC;
(f) L. D. Chen, D. Mandal, G. Pozzi, J. A. Gladysz and P. Bühlmann, J. Am. Chem. Soc., 2011, 133, 20869–20877 CrossRef CAS PubMed.
-
(a) K. C. Gupta, A. K. Sutar and C.-C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS PubMed;
(b) R. Drozdzak, B. Allaert, N. Ledoux, I. Dragutan, V. Dragutan and F. Verpoort, Coord. Chem. Rev., 2005, 249, 3055–3074 CrossRef CAS PubMed;
(c) C.-M. Che and J.-S. Huang, Coord. Chem. Rev., 2003, 242, 97–113 CrossRef CAS.
-
(a) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS PubMed;
(b) R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS PubMed;
(c) F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed;
(d) Y.-M. Jeon, G. S. Armatas, J. Heo, M. G. Kanatzidis and C. A. Mirkin, Adv. Mater., 2008, 20, 2105–2110 CrossRef CAS.
-
(a) T. Chakraborty, K. Srivastava, H. B. Singh and R. J. Butcher, J. Organomet. Chem., 2011, 696, 2782–2788 CrossRef CAS PubMed;
(b) R. Kaur, S. C. Menon, S. Panda, H. B. Singh, R. P. Patel and R. J. Butcher, Organometallics, 2009, 28, 2363–2371 CrossRef CAS;
(c) E. Becker, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 2004, 23, 2876–2883 CrossRef CAS;
(d) S. H. Park, H. J. Gwon and K. B. Park, Chem. Lett., 2004, 33, 1278–1279 CrossRef CAS;
(e) W.-F. Liaw, C.-H. Hsieh, S.-M. Peng and G.-H. Lee, Inorg. Chim. Acta, 2002, 332, 153–159 CrossRef CAS;
(f) R. Oilunkaniemi, R. S. Laitinen and M. Ahlgren, J. Organomet. Chem., 2001, 623, 168–175 CrossRef CAS;
(g) C.-M. Lee, G.-Y. Lin, C.-H. Hsieh, C.-H. Hu, G.-H. Lee, S.-M. Peng and W.-F. Liaw, J. Chem. Soc., Dalton Trans., 1999, 2393–2398 RSC;
(h) W.-F. Liaw, C.-H. Chen, G.-H. Lee and S.-M. Peng, Organometallics, 1998, 17, 2370–2372 CrossRef CAS;
(i) M. A. Aubart and R. G. Bergmann, J. Am. Chem. Soc., 1998, 120, 8755–8766 CrossRef CAS;
(j) L.-Y. Chia and W. R. McWhinnie, J. Organomet. Chem., 1978, 148, 165–170 CrossRef CAS;
(k) R. J. Haines, J. A. de Beer and R. Greatrex, J. Organomet. Chem., 1975, 85, 89–99 CrossRef CAS.
-
(a) I. P. Beletskaya and V. P. Ananikov, Pure Appl. Chem., 2007, 79, 1041–1056 CrossRef CAS;
(b) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320–2354 CrossRef CAS PubMed;
(c) H. Kuniyasu, A. Ogawa, S.-I. Miyazaki, I. Ryu, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1991, 113, 9796–9803 CrossRef CAS;
(d) V. P. Ananikov, I. P. Beletskaya, G. G. Aleksandrov and I. L. Eremenko, Organometallics, 2003, 22, 1414–1421 CrossRef CAS;
(e) V. P. Ananikov, K. A. Gayduk, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Eur. J. Inorg. Chem., 2009, 1149–1161 CrossRef CAS;
(f) V. P. Ananikov, M. A. Kabeshov, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Organometallics, 2005, 24, 1275–1283 CrossRef CAS.
-
(a) V. P. Ananikov and I. P. Beletskaya, Russ. Chem. Bull., 2004, 53, 561–565 CrossRef CAS;
(b) V. P. Ananikov, M. A. Kabeshov, I. P. Beletskaya, G. G. Aleksandrov and I. L. Eremenko, J. Organomet. Chem., 2003, 687, 451–461 CrossRef CAS;
(c) V. P. Ananikov, I. P. Beletskaya, G. G. Aleksandrov and I. L. Eremenko, Organometallics, 2003, 22, 1414–1421 CrossRef CAS;
(d) A. Ogawa, J. Organomet. Chem., 2000, 611, 463–474 CrossRef CAS;
(e) J. M. Gonzales, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 4908–4914 CrossRef CAS.
- T. Yamamoto and Y. Sekine, Inorg. Chim. Acta, 1984, 83, 47–53 CrossRef CAS.
- R. Zanella, R. Ros and M. Graziani, Inorg. Chem., 1973, 12, 2736–2738 CrossRef CAS.
-
(a) K. Kudoh, T. Okamoto and S. Yamaguchi, Organometallics, 2006, 25, 2374–2377 CrossRef CAS;
(b) H.-D. Stachel, B. Zimmer, E. Eckl, K. Semmlinger, W. Weigand, R. Wuensch and P. Mayer, Helv. Chim. Acta, 2005, 88, 1208–1220 CrossRef CAS;
(c) N. Kano, S. Kusaka and T. Kawashima, Dalton Trans., 2010, 39, 456–460 RSC;
(d) U. Siemeling, F. Bretthauer and C. Bruhn, J. Organomet. Chem., 2010, 695, 626–629 CrossRef CAS PubMed;
(e) H. Petzold, T. Weisheit, S. Braeutigam, H. Goerls, G. Mloston and W. Weigand, Eur. J. Inorg. Chem., 2010, 3636–3641 CrossRef CAS;
(f) B. Khalili Najafabadi, M. Hesari, M. S. Workentin and J. F. Corrigan, J. Organomet. Chem., 2012, 703, 16–24 CrossRef CAS PubMed;
(g) T. Weisheit, A. Kriltz, H. Goerls, G. Mloston, W. Imhof and W. Weigand, Chem.–Asian J., 2012, 7, 1383–1393 CrossRef CAS PubMed;
(h) A. Ishii, S. Kashiura, Y. Hayashi and W. Weigand, Chem.–Eur. J., 2007, 13, 4326–4333 CrossRef CAS PubMed.
- C. P. Morley, C. A. Webster and M. D. Vaira, J. Organomet. Chem., 2006, 691, 4244–4249 CrossRef CAS PubMed.
-
(a) R. Oilunkaniemi, R. S. Laitinen and M. Ahlgrén, J. Organomet. Chem., 1999, 587, 200–206 CrossRef CAS;
(b) R. Oilunkaniemi, R. S. Laitinen and M. Ahlgrén, J. Organomet. Chem., 2001, 623, 168–175 CrossRef CAS.
-
(a) A. Panunzi, G. Roviello and F. Ruffo, Organometallics, 2002, 21, 3503–3505 CrossRef CAS;
(b) V. G. Albano, M. Monari, I. Orabona, A. Panunzi and F. Ruffo, J. Am. Chem. Soc., 2001, 123, 4352–4353 CrossRef CAS.
-
(a) K. J. Bonnington, M. C. Jennings and R. J. Puddephatt, Organometallics, 2008, 27, 6521–6530 CrossRef CAS;
(b) M. S. McCready and R. J. Puddephatt, Inorg. Chem. Commun., 2011, 14, 210–212 CrossRef CAS PubMed;
(c) A. J. Canty, H. Jin, B. W. Skelton and A. H. White, Inorg. Chem., 1998, 37, 3975–3981 CrossRef CAS PubMed;
(d) T. Schaub, M. Backes, O. Plietzsch and U. Radius, Dalton Trans., 2009, 7071–7079 RSC;
(e) J. R. Zimmerman, B. W. Smucker, R. P. Dain, M. J. Van Stipdonk and D. M. Eichhorn, Inorg. Chim. Acta, 2011, 373, 54–61 CrossRef CAS PubMed;
(f) A. J. Canty, M. C. Denney, J. Patel, H. Sun, B. W. Skelton and A. H. White, J. Organomet. Chem., 2004, 689, 672–677 CrossRef CAS PubMed;
(g) C.-H. Hsieh, I.-J. Hsu, C.-M. Lee, S.-C. Ke, T.-Y. Wang, G.-H. Lee, Y. Wang, J.-M. Chen, J.-F. Lee and W.-F. Liaw, Inorg. Chem., 2003, 42, 3925–3933 CrossRef CAS PubMed;
(h) R. Kaur, S. C. Menon, S. Panda, H. B. Singh, R. P. Patel and R. J. Butcher, Organometallics, 2009, 28, 2363–2371 CrossRef CAS;
(i) S. Dey, V. K. Jain, B. Varghese, T. Schurr, M. Niemeyer, W. Kaim and R. J. Butcher, Inorg. Chim. Acta, 2006, 359, 1449–1457 CrossRef CAS PubMed;
(j) S. Dey, V. K. Jain, S. Chaudhury, A. Knoedler, F. Lissner and W. Kaim, J. Chem. Soc., Dalton Trans., 2001, 723–728 RSC.
- N. Goswami and D. M. Eichhorn, Inorg. Chim. Acta, 2000, 303, 271–276 CrossRef CAS.
-
(a) S. Panda, G. R. Ramakrishna, C. M. Reddy and S. S. Zade, Dalton Trans., 2011, 40, 6684–6690 RSC;
(b) P. K. Dutta, S. Panda, G. R. Ramakrishna, C. M. Reddy and S. S. Zade, Dalton Trans., 2013, 42, 476–483 RSC.
- M. Martinek, M. Korf and J. Srogl, Chem. Commun., 2010, 46, 4387–4389 RSC.
- S. Panda, S. S. Zade, H. B. Singh and G. Wolmershauser, J. Organomet. Chem., 2005, 690, 3142–3148 CrossRef CAS PubMed.
-
(a) P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2012, 256, 953–1114 CrossRef CAS PubMed;
(b) A. D. Garnovskii, A. L. Nivorozhkin and V. I. Minkin, Coord. Chem. Rev., 1993, 126, 1–69 CrossRef CAS;
(c) D. J. Sheeran and K. B. Mertes, J. Am. Chem. Soc., 1990, 112, 1055–1061 CrossRef CAS.
- T. Yamamura, M. Tadokoro, K. Tanaka and R. Kuroda, Bull. Chem. Soc. Jpn., 1993, 66, 1984–1990 CrossRef CAS.
- C.-H. Hsieh, I-.J. Hsu, C.-M. Lee, S.-C. Ke, T.-Y. Wang, G.-H. Lee, Y. Wang, J.-M. Chen, J.-F. Lee and W.-F. Liaw, Inorg. Chem., 2003, 42, 3925–3933 CrossRef CAS PubMed.
- A. Bredenkamp, X. Zeng and F. Mohr, Polyhedron, 2012, 33, 107–113 CrossRef CAS PubMed.
- R. S. Chauhan, C. P. Prabhu, P. P. Phadnis, G. Kedarnath, J. A. Golen, A. L. Rheingold and V. K. Jain, J. Organomet. Chem., 2013, 723, 163–170 CrossRef CAS PubMed.
- Bruker, SADABS V2008-1, Bruker AXS, Madison, WI, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, X-Seed, Graphical Interface to SHELX-97 and POV-Ray, University of Missouri, Columbia, MO, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and 77Se spectra of complexes 14–19. CCDC 946622–996626. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52132j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a