 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric 1,8/13,2,x-M2C2B10 14-vertex metallacarboranes by direct electrophilic insertion reactions; the VCD and BHD methods in critical analysis of cage C atom positions†‡§
Amelia
McAnaw
,
Maria Elena
Lopez
,
David
Ellis
,
Georgina M.
Rosair
and
Alan J.
Welch
*
Institute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: a.j.welch@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 3217
First published on 8th October 2013
Abstract
The isolation of six isomeric, low-symmetry, dicobaltacarboranes with bicapped hexagonal antiprismatic cage structures, always in low yield, is described from reactions in which 13-vertex cobaltacarborane anions and sources of cobalt-containing cations were present. The vertex-to-centroid distance (VCD) and boron–H distance (BHD) methods are used to locate the correct C atom positions in the cages, thus allowing the compounds to be identified as 1,13-Cp2-1,13,2,10-closo-Co2C2B10H12 (1), 1,8-Cp2-3-OEt-1,8,2,10-closo-Co2C2B10H11 (2), 1,13-Cp2-1,13,2,9-closo-Co2C2B10H12 (3), 1,8-Cp2-1,8,2,4-closo-Co2C2B10H12 (4), 1,13-Cp2-1,13,2,4-closo-Co2C2B10H12 (5) and 1,8-Cp2-1,8,2,5-closo-Co2C2B10H12 (6). It is shown that a common alternative method of cage C atom identification, using refined (as B) Ueq values, does not work well, at least in these cases. Having identified the correct isomeric forms of the six dicobaltacarboranes, their syntheses are tentatively rationalised in terms of the direct electrophilic insertion of a {CpCo+} fragment into [CpCoC2B10]− anions and it is demonstrated that compounds 1, 4, 5 and 6 can be successfully prepared by deliberately performing such reactions.
Introduction
Although there are now hundreds of 13-vertex MC2B10 metallacarboranes whose origin can be traced to Hawthorne's original synthesis of 4-Cp-4,1,6-closo-CoC2B10H12,1 there are very few 14-vertex analogues.2 The expected shape3 of a closo 14-vertex heteroborane with 15 skeletal electron pairs is the bicapped hexagonal antiprism, shown together with its numbering scheme in Fig. 1a. It was Hawthorne, again, who prepared the first examples of 14-vertex metallacarboranes by reduction and subsequent metallation of 13-vertex MC2B10 precursors.4 Expansion of 4-Cp-4,1,12-closo-CoC2B10H12 afforded 1,14-Cp2-1,14,2,10-closo-Co2C2B10H12, whose spectroscopically-presumed structure was recently confirmed crystallographically.5 Similarly, reduction and metallation of 4-(p-cymene)-4,1,12-closo-RuC2B10H12 (p-cymene = η-C6H4MeiPr-1,4) afforded both homo- and heterobimetallic M2C2B10 species, again with 1,14,2,10-MC2B10 architectures.6 Hawthorne also expanded 4-Cp-4,1,8-closo-CoC2B10H12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and we recently established that the product here is the 1,14,2,9-closo-Co2C2B10 isomer.5 In an alternative approach, Grimes and co-workers reacted [Me4C4B8H8]2− with a source of {CpFe+} fragments to yield kinetic isomers of (CpFe)2Me4C4B8H8 which had irregular (non bicapped hexagonal antiprismatic) cage structures. However, progressive isomerisation to thermodynamically-preferred regular structures was achieved by heating, affording 1,14,2,4,10,12- and 1,14,2,5,10,12-Fe2C4B8 species.7 Note that in all the above examples of bicapped hexagonal antiprismatic M2C2B10 metallacarboranes both metal atoms occupy degree-6 vertices (vertices 1 and 14 of Fig. 1a) in which they have maximum interaction with other cage atoms, consistent with the relatively diffuse nature of the frontier orbitals of transition metals compared to those of boron and especially carbon.8
4 and we recently established that the product here is the 1,14,2,9-closo-Co2C2B10 isomer.5 In an alternative approach, Grimes and co-workers reacted [Me4C4B8H8]2− with a source of {CpFe+} fragments to yield kinetic isomers of (CpFe)2Me4C4B8H8 which had irregular (non bicapped hexagonal antiprismatic) cage structures. However, progressive isomerisation to thermodynamically-preferred regular structures was achieved by heating, affording 1,14,2,4,10,12- and 1,14,2,5,10,12-Fe2C4B8 species.7 Note that in all the above examples of bicapped hexagonal antiprismatic M2C2B10 metallacarboranes both metal atoms occupy degree-6 vertices (vertices 1 and 14 of Fig. 1a) in which they have maximum interaction with other cage atoms, consistent with the relatively diffuse nature of the frontier orbitals of transition metals compared to those of boron and especially carbon.8
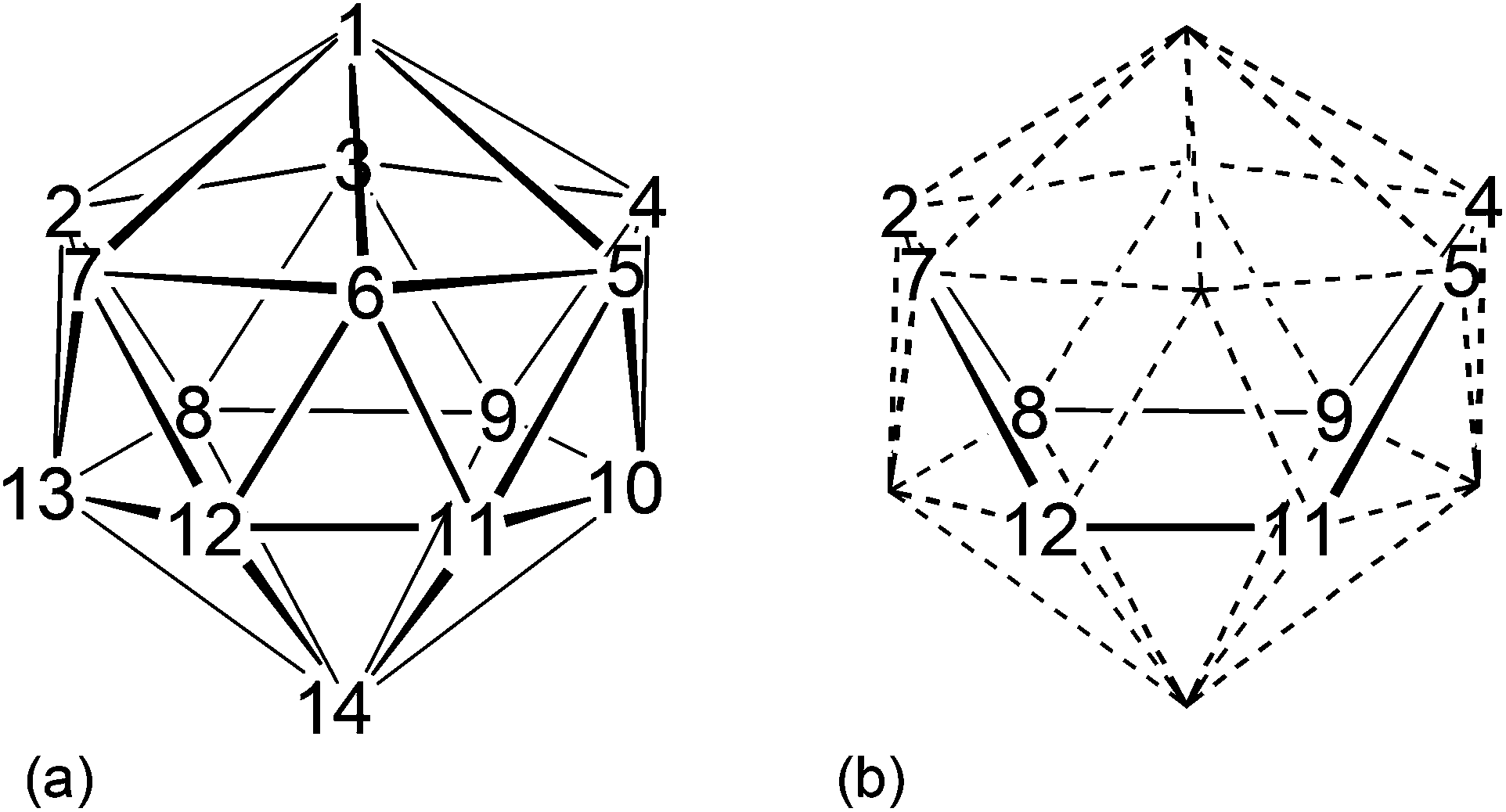 | ||
| Fig. 1 (a) The bicapped hexagonal antiprism and vertex numbering scheme; (b) the eight vertices used in VCD calculations. | ||
14-vertex MC2B11 species are also known. Reduction and subsequent metallation (with {(p-cymene)Ru2+}) of the 13-vertex tethered carborane 1,2-μ-(CH2)3-1,2-closo-C2B11H11 afforded the first such species in two isomeric forms, 1,2,3-RuC2B11 and 1,2,8-RuC2B11,9 whilst a 1,2,9-RuC2B11 species was later prepared by reduction and metallation of a tether-free carborane.10 Note that in these compounds the single metal atom is again located at a degree-6 vertex. The only known exceptions to this rule are two 8,2,3-NiC2B11 compounds afforded by treatment of [μ-(CH2)3-C2B11H11]2− with {nickel(chelating diphosphine)2+} fragments.11
In the present study we report the synthesis and structural characterisation of six bicapped hexagonal antiprismatic Co2C2B10 species in which one metal atom is in the degree-6 vertex 1 but, uniquely, the other is in a degree-5 site on the lower hexagonal belt (vertex 8 or 13, dependent on the C atom positions). We present evidence which suggests that these compounds are not formed by 2-e reduction and metallation of 13-vertex CoC2B10 species (although in some cases they were first isolated from reactions in which this was the intention) rather that they may arise as the result of direct electrophilic attack by a metal fragment cation on a [CoC2B10]− monoanion. Crucial to rationalising their synthesis is the identification of the correct positions of the cage C atoms in the crystallographically-determined structures (in no cases do the C atoms carry exo-polyhedral substituents other than H) and for this we have used both the recently reported vertex-to-centroid distance (VCD) method12 and a complementary approach, the boron–H distance (BHD) method which we first communicated in 200213 but for which we now provide more detail.
Results and discussion
Syntheses
Hawthorne's communication describing the original synthesis of the 14-vertex dicobaltacarborane 1,14-Cp2-1,14,2,10-closo-Co2C2B10H12, by reduction and subsequent metallation (Na[Cp]/CoCl2) of 4-Cp-4,1,12-closo-CoC2B10H12 followed by aerial oxidation (CoII → CoIII) noted “a mixture of products”.4 In repeating this experiment we found, by thin layer chromatography (TLC), evidence for at least eight products including the known compounds 3-Cp-3,1,2-closo-CoC2B9H11,14 1,14-Cp2-1,14,2,10-closo-Co2C2B10H124 and 4,5-Cp2-4,5,1,6-closo-Co2C2B9H11,15 these compounds being identified by a combination of 11B{1H} and 1H NMR spectroscopies. In addition we isolated in low yield a brown product 1 which by mass spectrometry appears to be (CpCo)2C2B10H12 but which, unlike 1,14-Cp2-1,14,2,10-closo-Co2C2B10H12, is clearly asymmetric with two Cp resonances and two CcageH resonances in the 1H NMR spectrum, and eight resonances, 1:2:2:1:1:1:1:1 from high frequency to low frequency, in the 11B{1H} spectrum.
Similarly, when we repeated the polyhedral expansion of 4-Cp-4,1,8-closo-CoC2B10H12 by reduction, metallation and oxidation,4,5 we isolated not only the target species 1,14-Cp2-1,14,2,9-closo-Co2C2B10H12 but also a small amount of a second brown compound, 3. Compound 3 also appears to be (CpCo)2C2B10H12 by mass spectrometry but by NMR spectroscopy it is clearly different to 1 although it again appears asymmetric in the 11B spectrum, with eight resonances, 1:1:1:1:1:2:2:1. Several of these, however, are unusually broad. Whilst there are two CcageH resonances in the 1H spectrum there is only one cyclopentadienyl resonance, appearing as a broad unresolved singlet. We believe that this may be evidence for a functional process in 3 in solution which equivalences the two metal fragments whilst keeping distinct the two cage C atoms, and which occurs very near to room temperature. Further studies are currently underway.16
We initially isolated two further brown solids, compounds 4 and 5, in trace amounts during the synthesis, first reported by Hawthorne,1b of 4-Cp-4,1,6-closo-CoC2B10H12 by the reduction and subsequent metallation then oxidation of 1,2-closo-C2B10H12. Similarly, during the synthesis17 of 4-Cp-4,1,10-closo-CoC2B10H12 starting from 1,12-closo-C2B10H12, yet another brown trace product, 6, was observed. Like 1 and 3, compounds 4–6 all appear by mass spectrometry to be (CpCo)2C2B10H12, but spectroscopically they are all different and clearly asymmetric. We therefore conclude that compounds 1, 3, 4, 5 and 6 are all related as positional isomers. Table 1 summarises the 1H and 11B NMR chemical shifts for 1 and 3–6. We have previously noted both the 11B range and the weighted average 11B chemical shift, 〈δ11B〉, for the compounds 1,14-Cp2-1,14,2,10-closo-Co2C2B10H12 and 1,14-Cp2-1,14,2,9-closo-Co2C2B10H12.5 In moving from the 1,14,2,10- to 1,14,2,9- isomer the chemical shift range increases from ca. 13 ppm to ca. 24 ppm, but 〈δ11B〉 is fairly constant, −14.3 and −13.4 ppm, respectively. In compounds 1 and 3–6 the chemical shift ranges are much greater, ca. 35 to 48 ppm, with the weighted average shift moving significantly to high frequency, lying in the range −3.6 to −8.1 ppm. Clearly 1 and 3–6 are structurally quite different to both the 14,2,10- and 1,14,2,9-isomers.
| Compound | 1H NMR | 11B NMR | |||||
|---|---|---|---|---|---|---|---|
| C5H5 | C5H5 | CcageH | CcageH | 11B patternb | 11B range | 〈δ11B〉 | |
| a Chemical shifts in ppm from CDCl3 solutions at room temperature. b 11B pattern from high frequency to low frequency. | |||||||
| 1 | 5.29 | 4.89 | 3.10 | 2.57 | 1:2:2:1:1:1:1:1 | 13.9 to −28.2 | −6.31 |
| 3 | 5.13(br) | 5.13(br) | 2.60 | 2.11 | 1:1:1:1:1:2:2:1 | 18.2 to −30.1 | −3.62 |
| 4 | 5.46 | 5.07 | 3.80 | 2.89 | 1:1:2:1:1:1:1:1:1 | 7.0 to −27.7 | −7.12 |
| 5 | 5.20 | 5.02 | 2.96 | 2.67 | 1:1:1:1:1:1:1:1:1:1 | 13.7 to −26.4 | −6.64 |
| 6 | 5.21 | 5.04 | 2.71 | 2.25 | 1:2:1:1:1:1:1:1:1 | 8.4 to −28.3 | −8.06 |
Finally, a related brown compound, 2, was isolated in trace amount from a complex mixture of products following an attempt to prepare a 14-vertex analogue of the 12- and 13-vertex cobaltacarborane sandwich compounds [3,3′-Co-(1,2-closo-C2B9H11)2]−,18 [3,3′-Co-(1,7-closo-C2B9H11)2]−,14 [3,3′-Co-(1,12-closo-C2B9H11)2]−,19 [4,4′-Co-(1,6-closo-C2B9H11)2]−1b and [4,4′-Co-(1,10-closo-C2B9H11)2]−20 part of which involved exposure of the reagents to EtOH. Although we never had sufficient amounts of 2 for NMR spectroscopy we were able to obtain a mass spectrum revealing a molecular ion consistent with the formula (CpCo)2C2B10H11(OEt) and we were fortunate to grow a few single crystals of the compound.
Crystallographic studies – identification of the cage C atoms
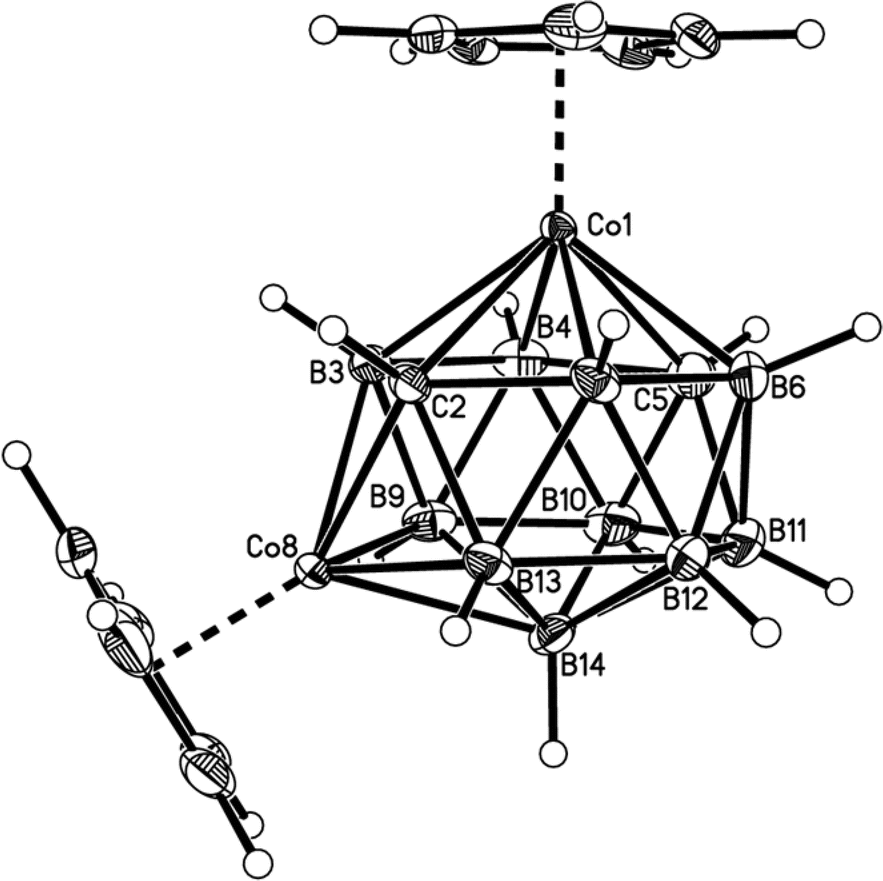
Compounds 1–6 were studied by single-crystal X-ray diffraction. These studies establish that in all cases the heteroborane cage has a (distorted) bicapped hexagonal antiprismatic shape with one Co atom in a degree-6 (capping) site and the other at a degree-5 vertex in the hexagonal belt distant from the degree-6 metal. Consistent with empirical electron counting rules3 (15 skeletal electron pairs for these 14-vertex closo clusters) and fully in agreement with the spectroscopic data described above, 1 and 3–6 are all formulated as (CpCo)2C2B10H12, whilst 2 is the compound (CpCo)2C2B10H11(OEt). However, to establish the precise identities of compounds 1–6 it is essential that the positions of the cage C atoms are correctly identified, and for this we first made use of the vertex-to-centroid distance (VCD) method that we recently described.12 Initially cage vertices were numbered according to Fig. 1a such that the Co atoms are at vertices 1 and 13. All other cage atoms were assumed to be B, and the structures were refined to convergence (including free refinement of cage H atoms). Using OLEX221 the cage centroid was calculated only from vertices 2,4,5,7,8,9,11 and 12, as shown in Fig. 1b. We omit the metal at vertex 1 and, for balance, the antipodal atom at vertex 14, to avoid compromising the centroid calculation. We also omit the metal at vertex 13 but, because the bicapped hexagonal antiprism does not contain a centre of inversion, we also omit vertex 10 (opposite 13 on the lower hexagonal belt) and, for balance, vertices 3 and 6 from the upper belt.Table 2 lists the VCDs for compounds 1–6. The shortest VCDs are those from vertex 14 but this is exceedingly unlikely to be the correct site of a cage C atom because the vertex is of degree-6.8 These VCDs are artificially short because vertex 14 is pulled up towards the cage centroid simply by virtue of it capping a six atom face.22 Notice that VCDs from the degree-6 Co atom at vertex 1 are consistently 0.3 Å shorter than those from the degree-5 Co atom at vertex 13 for the same reason. Ignoring, then, the VCDs from vertex 14, the two shortest VCDs are taken to be those from the cage C atoms, thus identifying the C atoms as being at vertices 2 & 10 (compound 1), 2 & 11 (2), 2 & 9 (3), 2 & 6 (4), 2 & 4 (5) and 2 & 5 (6). In all cases except for compound 1 the two VCDs from the C atoms are at least 0.025 Å shorter than all VCDs from B atoms. However the situation is less clear in the case of 1 with VCDs from vertices 9 and 11 being close to that from vertex 2. Hence we have sought additional structural evidence for the cage C atom locations.
| Vertex | 1 | 2 | 3 | 4 | 5 | 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
a Vertex numbers (left column) refer to the model before the C atoms were assigned; ![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) ![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) entries identify C vertices; atom labels to the right of each entry are the final atom identifiers, shown in Fig. 2–7. entries identify C vertices; atom labels to the right of each entry are the final atom identifiers, shown in Fig. 2–7.
|
||||||||||||
| 1 | 2.018(2) | Co1 | 2.020(3) | Co1 | 2.0047(7) | Co1 | 2.0304(18) | Co1 | 2.043(3) | Co1 | 2.0307(8) | Co1 |
| 2 | ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) . .![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) ( (![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) ) ) |
C2 | .![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) ( (![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) ) ) |
C2 | .(![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ) ) |
C2 | .![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ( (![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ) ) |
C2 | .() |
C2 | .() |
C2 |
| 3 | 1.895(6) | B3 | 1.865(9) | B7 | 1.907(3) | B3 | 1.907(5) | B7 | 1.884(6) | B3 | 1.9005(19) | B7 |
| 4 | 1.935(7) | B4 | 1.916(10) | B6 | 1.941(2) | B4 | 1.931(6) | B6 | .() |
C4 | 1.917(2) | B6 |
| 5 | 1.947(7) | B5 | 1.928(9) | B5 | 1.923(3) | B5 | 1.904(6) | B5 | 1.893(6) | B5 | .() |
C5 |
| 6 | 1.908(7) | B6 | 1.904(9) | B4 | 1.894(2) | B6 | .() |
C4 | 1.897(6) | B6 | 1.9021(18) | B4 |
| 7 | 1.917(6) | B7 | 1.940(9) | B3 | 1.905(2) | B7 | 1.896(5) | B3 | 1.883(8) | B7 | 1.9220(18) | B3 |
| 8 | 1.878(6) | B8 | 1.929(8) | B13 | 1.910(3) | B8 | 1.893(6) | B13 | 1.882(6) | B8 | 1.8809(18) | B13 |
| 9 | 1.829(7) | B9 | 1.870(10) | B12 | .() |
C9 | 1.866(5) | B12 | 1.871(7) | B9 | 1.859(2) | B12 |
| 10 | .() |
C10 | 1.876(10) | B11 | 1.880(3) | B10 | 1.882(5) | B11 | 1.892(6) | B10 | 1.900(2) | B11 |
| 11 | 1.830(7) | B11 | .() |
C10 | 1.868(3) | B11 | 1.865(5) | B10 | 1.861(7) | B11 | 1.877(2) | B10 |
| 12 | 1.895(7) | B12 | 1.923(10) | B9 | 1.908(2) | B12 | 1.915(5) | B9 | 1.876(6) | B12 | 1.909(2) | B9 |
| 13 | 2.434(2) | Co13 | 2.375(4) | Co8 | 2.3792(10) | Co13 | 2.396(2) | Co8 | 2.372(2) | Co13 | 2.3723(8) | Co8 |
| 14 | 1.596(6) | B14 | 1.601(9) | B14 | 1.6016(18) | B14 | 1.591(5) | B14 | 1.589(5) | B14 | 1.5851(18) | B14 |
In 2002 we described an early alternative method of distinguishing between cage B and cage C atoms in (hetero)carboranes, the B–H distance (BHD) method whereby we examined the vertex–H distances following refinement of all cage C or B atoms as B.13 Under crystallographic refinement an H atom bonded to a vertex at which insufficient electron density has been specified will compensate by moving towards that vertex, affording an artificially short vertex–H bond. Thus short distances identify where in the cage the C atoms are. In Table 3 are the BHDs for compounds 1–6 calculated from such all-boron models (left hand entries). Whilst the true B–H distances are all around 1.1 Å, two distances in each structure are between 0.17(3) and 0.48(5) Å, and these identify exactly the same C atom positions as found by the VCD method in all compounds, including compound 1. The right hand entries are the vertex–H distances once the cage C atoms have been identified as such and refinement completed; note that in all cases the artificially short “B”–H distances lengthen to sensible values when “B” is properly described as C. In some respects the BHD method might appear to be superior to the VCD method (at least in the case of compound 1) but a drawback of the former is that it requires successful free crystallographic refinement of H atom positions, something which is not always practicable. Overall, we have always advocated a multi-modal approach to the problem of distinguishing cage B and cage C atoms in crystallographic studies of (hetero)carboranes, and for compounds 1–6 we can have complete confidence in the results from the VCD and the BHD methods since they are in perfect agreement with each other.
| Vertex | 1 | 2 | 3 | 4 | 5 | 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a For each structure the left hand entry is the vertex–H distance for the “all-B” model (where all non-metal vertices are assigned as B atoms); entries identify C vertices. The right hand entry is the vertex–H distance following assignment of the cage C atoms.
|
||||||||||||
| 2 | .() |
0.99(6) | .() |
0.86(7) | .() |
0.95(2) | .() |
0.95(5) | .() |
0.97(5) | .() |
0.99(2) |
| 3 | 1.06(7) | 1.05(5) | 1.09(5) | 1.04(5) | 1.10(2) | 1.09(2) | 1.12(6) | 1.04(5) | 1.12(5) | 1.14(5) | 1.13(2) | 1.11(2) |
| 4 | 1.06(7) | 1.05(7) | 1.10(5) | 1.18(7) | 1.04(2) | 1.04(2) | 1.19(6) | 1.09(5) | .() |
1.04(5) | 1.13(2) | 1.11(2) |
| 5 | 1.07(7) | 1.08(6) | 1.10(5) | 1.10(6) | 1.07(2) | 1.06(2) | 1.18(6) | 1.19(5) | 0.98(5) | 1.18(5) | .() |
1.03(2) |
| 6 | 1.18(6) | 1.19(6) | 1.11(4) | 1.14(6) | 1.06(2) | 1.05(2) | .() |
0.96(5) | 1.12(5) | 1.09(5) | 1.06(2) | 1.06(2) |
| 7 | 1.14(6) | 1.13(5) | n/a | n/a | 1.05(2) | 1.09(2) | 1.05(6) | 1.09(5) | 1.06(5) | 1.04(5) | 1.12(2) | 1.12(2) |
| 8 | 1.08(6) | 1.08(6) | 1.07(5) | 0.92(6) | 1.06(2) | 1.06(2) | 1.14(6) | 1.15(5) | 1.07(5) | 1.04(5) | 1.07(2) | 1.07(2) |
| 9 | 1.07(7) | 1.04(7) | 1.10(3) | 1.07(6) | .() |
0.99(2) | 1.02(6) | 0.90(5) | 0.98(5) | 1.08(5) | 1.07(2) | 1.08(2) |
| 10 | .() |
0.86(8) | 1.09(5) | 1.02(7) | 1.10(3) | 1.10(2) | 1.06(6) | 1.13(5) | 1.12(5) | 1.18(5) | 1.08(3) | 1.07(2) |
| 11 | 1.05(7) | 1.05(7) | .() |
0.89(7) | 1.07(3) | 1.06(2) | 1.10(6) | 1.10(5) | 1.04(5) | 1.01(5) | 1.07(2) | 1.08(2) |
| 12 | 1.09(7) | 1.09(7) | 1.09(6) | 1.09(6) | 1.05(2) | 1.07(2) | 1.03(6) | 1.05(5) | 1.06(5) | 1.06(5) | 1.10(2) | 1.12(2) |
| 14 | 1.13(7) | 1.13(7) | 1.10(5) | 1.10(6) | 1.082(17) | 1.09(2) | 1.12(6) | 1.12(5) | 1.12(4) | 1.10(4) | 1.08(2) | 1.08(2) |
In this respect it is instructive to examine critically a third often-used method of C/B discrimination, that of using the refined (as B atoms) Ueq values. The argument here is that if the model describes insufficient electron density at a vertex (i.e. the vertex is really C not B) crystallographic refinement will compensate by Ueq being significantly smaller. In Table 4 we list the Ueq values for vertices 2–12 and 14 in compounds 1–6. Only in the case of compound 4 are the two smallest Ueq values correctly associated with the C atom positions. In compound 2 the Ueq of vertex 7 is as small as that of vertex 11, and in all the other structures there are at least two Ueq(B) smaller than one Ueq(C). We have previously noted12 the potential of adjacent heavy atoms to artificially suppress Ueq(B) and we see several examples of this in Table 4 (note the consistently low values of Ueq for B atoms at vertex 7, the other vertex in addition to vertex 2 that is bound to both metal vertices). Overall, we would argue strongly ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif)
![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif)
![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) using Ueq values to identify cage C atoms in (hetero)carboranes.
using Ueq values to identify cage C atoms in (hetero)carboranes.
| Vertex | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
|
a Vertex numbers (left column) refer to the model before the C atoms were assigned; entries indicate C vertices identified by the VCD and BHD methods. Italicised entries show Ueq values for genuine B atoms that are ≤ those of atoms which are actually C.
|
||||||
| 2 | .() |
.() |
.() |
.() |
.() |
.() |
| 3 | 0.0209(12) | 0.024(2) | 0.0168(4) | 0.0152(8) | 0.0240(13) | 0.0166(3) |
| 4 | 0.0311(16) | 0.035(2) | 0.0182(4) | 0.0162(9) | .() |
0.0206(3) |
| 5 | 0.0308(15) | 0.036(2) | 0.0179(4) | 0.0158(9) | 0.0294(14) | .() |
| 6 | 0.0237(13) | 0.029(2) | 0.0153(4) | .() |
0.0239(12) | 0.0192(3) |
| 7 | 0.0178(11) | 0.021(2) | 0.0135(4) | 0.0145(8) | 0.0186(13) | 0.0151(3) |
| 8 | 0.0163(10) | 0.027(2) | 0.0175(4) | 0.0143(8) | 0.0278(13) | 0.0155(3) |
| 9 | 0.0269(14) | 0.030(2) | .() |
0.0176(9) | 0.0283(13) | 0.0200(3) |
| 10 | .() |
0.040(2) | 0.0216(5) | 0.0172(9) | 0.0316(12) | 0.0241(4) |
| 11 | 0.0340(17) | .() |
0.0183(4) | 0.0170(9) | 0.0295(13) | 0.0223(3) |
| 12 | 0.0231(12) | 0.030(2) | 0.0158(4) | 0.0149(8) | 0.0249(12) | 0.0188(3) |
| 14 | 0.0210(12) | 0.034(2) | 0.0184(4) | 0.0137(8) | 0.0181(7) | 0.0169(3) |
Having identified the cage C atoms by both the VCD and BHD methods the cages were renumbered according to accepted convention,2 and this numbering is shown as the final column of Table 2. Thus compounds 1–6 are correctly described as 1,13-Cp2-1,13,2,10-closo-Co2C2B10H12 (1), 1,8-Cp2-3-OEt-1,8,2,10-closo-Co2C2B10H11 (2), 1,13-Cp2-1,13,2,9-closo-Co2C2B10H12 (3), 1,8-Cp2-1,8,2,4-closo-Co2C2B10H12 (4), 1,13-Cp2-1,13,2,4-closo-Co2C2B10H12 (5) and 1,8-Cp2-1,8,2,5-closo-Co2C2B10H12 (6).
Fig. 2–7 show perspective views of compounds 1–6, respectively, and Table 5 lists the lengths of the connectivities in the cobaltacarborane cages. The Co1–vertex distances span the range 2.08–2.19 Å, similar to that (2.13–2.19 Å) in a series of 1,14,2,9- and 1,14,2,10-MCoC2B10 species (M = Ru or Co) we recently studied (five compounds and nine crystallographically independent Co atoms).5 In contrast the Co–vertex distances from the degree-5 Co atom in 1–6 are more widely spread, spanning the range 1.96–2.28 Å. C–B and B–B distances involving only degree-5 atoms are in the ranges 1.64–1.72 and 1.71–1.81 Å which are perfectly normal.12 However, distances to the degree-6 atom B14 are considerably longer, as expected, with B–B in the range 1.85–1.98 Å and three C–B distances of 1.844(11), 1.904(3) and 2.015(9) Å.
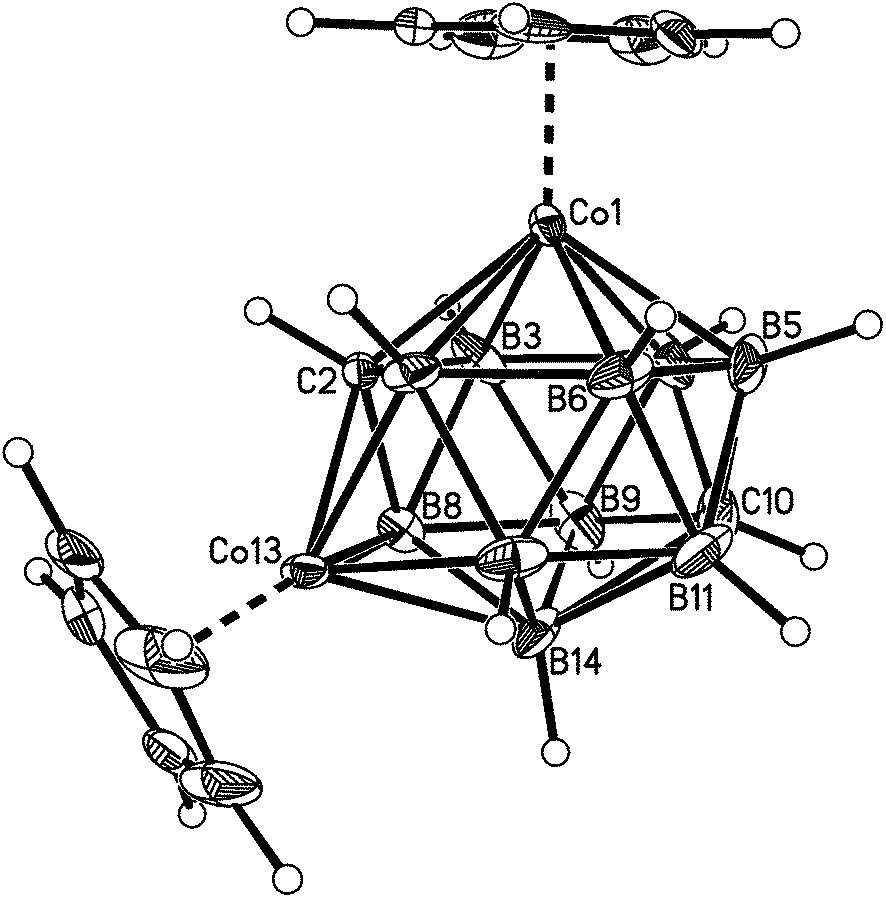 | ||
| Fig. 2 Perspective view of compound 1. Displacement ellipsoids are drawn at the 50% probability level except for hydrogen. | ||
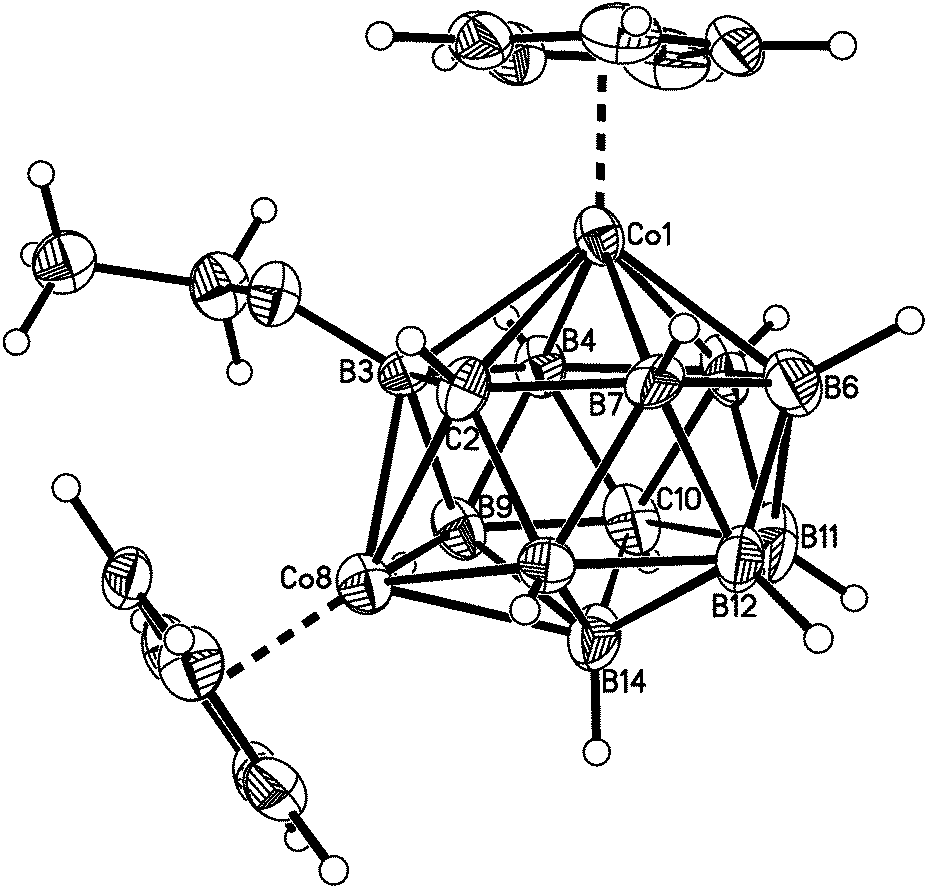 | ||
| Fig. 3 Perspective view of compound 2. Displacement ellipsoids as for 1. | ||
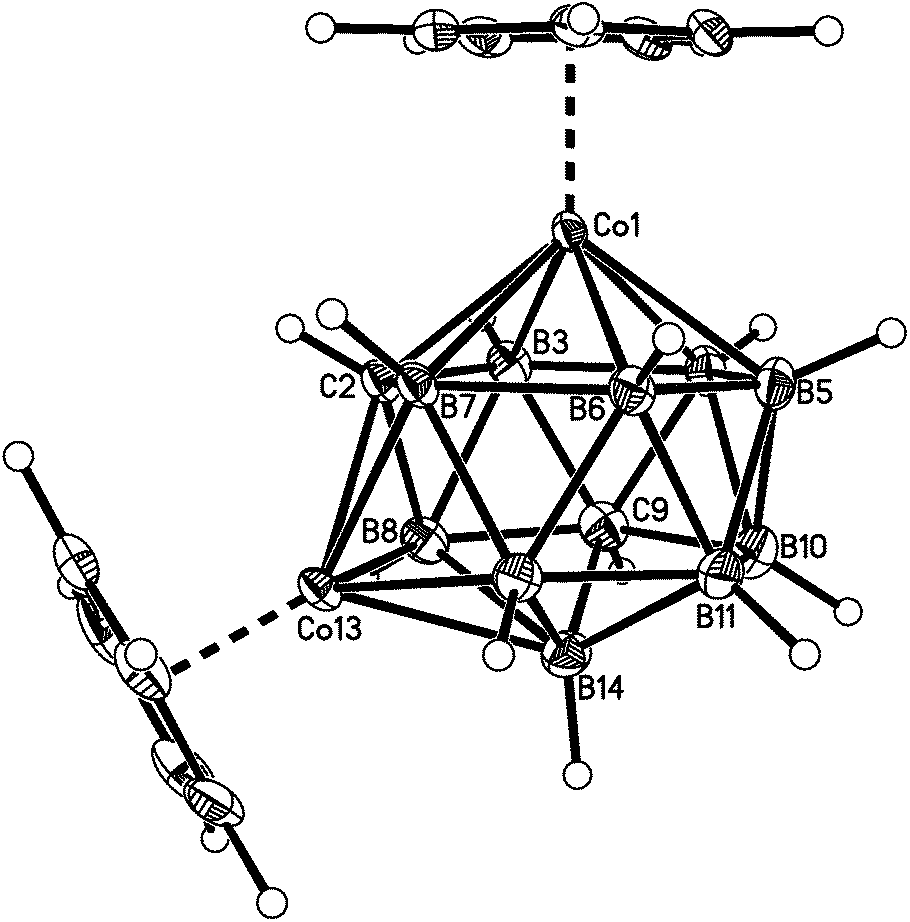 | ||
| Fig. 4 Perspective view of compound 3. Displacement ellipsoids as for 1. | ||
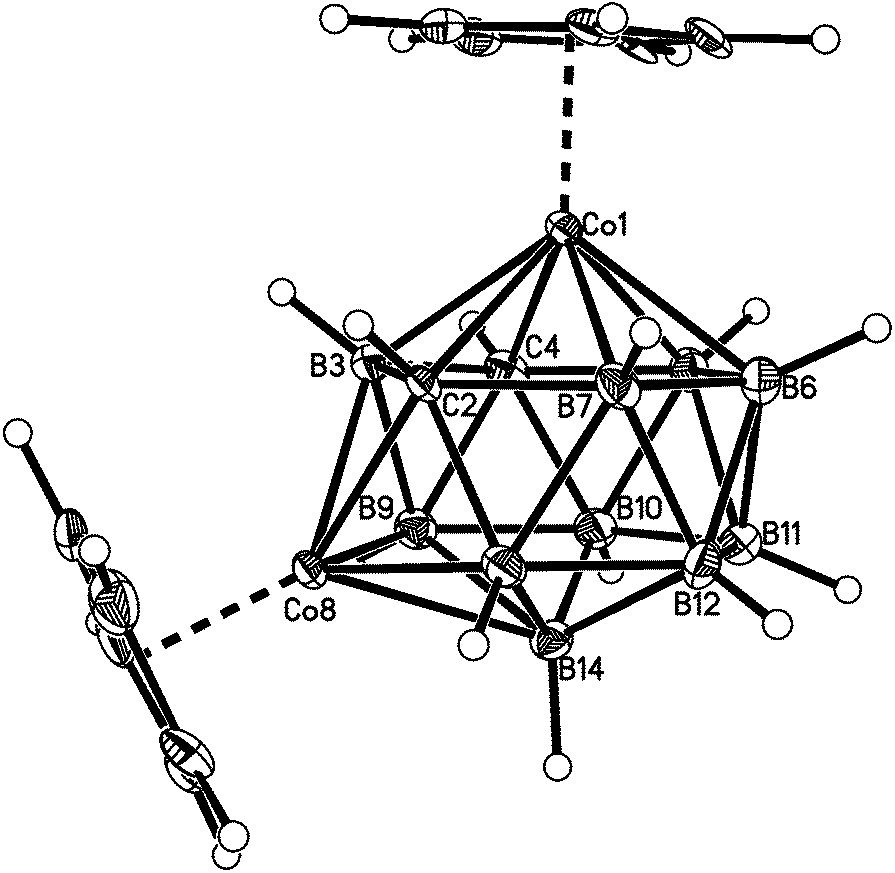 | ||
| Fig. 5 Perspective view of compound 4. Displacement ellipsoids as for 1. | ||
 | ||
| Fig. 6 Perspective view of compound 5. Displacement ellipsoids as for 1. | ||
 | ||
| Fig. 7 Perspective view of compound 6. Displacement ellipsoids as for 1. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
|
a
Bold entries, distances involving metal vertex; entries, distances involving C vertex.
|
||||||
| 1–2 |
![[2 with combining low line]](https://www.rsc.org/images/entities/b_char_0032_0332.gif) . .![[1 with combining low line]](https://www.rsc.org/images/entities/b_char_0031_0332.gif) ![[4 with combining low line]](https://www.rsc.org/images/entities/b_char_0034_0332.gif) ![[0 with combining low line]](https://www.rsc.org/images/entities/b_char_0030_0332.gif) ( (![[5 with combining low line]](https://www.rsc.org/images/entities/b_char_0035_0332.gif) ) )
|
.![[3 with combining low line]](https://www.rsc.org/images/entities/b_char_0033_0332.gif) ![[8 with combining low line]](https://www.rsc.org/images/entities/b_char_0038_0332.gif) ( (![[7 with combining low line]](https://www.rsc.org/images/entities/b_char_0037_0332.gif) ) )
|
.(![[6 with combining low line]](https://www.rsc.org/images/entities/b_char_0036_0332.gif) ) )
|
.()
|
.()
|
.()
|
| 1–3 | 2.128(5) | 2.199(7) | 2.0842(19) | 2.122(4) | 2.125(5) | 2.1569(16) |
| 1–4 | 2.153(6) | 2.127(8) | 2.151(2) |
.()
|
.![[9 with combining low line]](https://www.rsc.org/images/entities/b_char_0039_0332.gif) () ()
|
2.1269(17) |
| 1–5 | 2.153(6) | 2.146(8) | 2.1839(19) | 2.161(4) | 2.146(5) |
.()
|
| 1–6 | 2.148(6) | 2.185(8) | 2.1719(18) | 2.175(4) | 2.134(5) | 2.1484(17) |
| 1–7 | 2.153(5) | 2.108(8) | 2.1670(18) | 2.153(5) | 2.159(6) | 2.1238(15) |
| 2–7 | .() |
.() |
.() |
.() |
.() |
.() |
| 2–13 |
.()
|
.() |
.()
|
.() |
.()
|
.() |
| 2–8 | .() |
.()
|
.() |
.()
|
.() |
.()
|
| 2–3 | .() |
.() |
.() |
.() |
.() |
.() |
| 3–8 | 1.798(8) | 2.087(7) | 1.786(3) | 2.045(5) | 1.768(9) | .() |
| 3–9 | 1.784(9) | 1.797(10) | .() |
1.812(6) | 1.771(8) | 1.801(2) |
| 3–4 | 1.752(9) | 1.787(10) | 1.796(3) | .() |
.() |
1.804(2) |
| 4–9 | 1.768(9) | 1.750(11) | .() |
.() |
.() |
1.763(2) |
| 4–10 | .() |
.() |
1.762(3) | .() |
.() |
1.786(2) |
| 4–5 | 1.803(10) | 1.780(11) | 1.742(3) | .() |
.() |
.() |
| 5–10 | .() |
.() |
1.767(3) | 1.769(6) | 1.760(8) | .() |
| 5–11 | 1.755(11) | 1.777(13) | 1.773(3) | 1.756(7) | 1.733(9) | .() |
| 5–6 | 1.760(10) | 1.722(12) | 1.786(3) | 1.760(6) | 1.742(9) | .() |
| 6–11 | 1.805(9) | 1.733(12) | 1.774(3) | 1.757(7) | 1.779(8) | 1.771(3) |
| 6–12 | 1.778(9) | 1.760(12) | 1.760(3) | 1.774(7) | 1.767(9) | 1.765(2) |
| 6–7 | 1.776(9) | 1.780(11) | 1.793(3) | 1.791(7) | 1.769(9) | 1.789(2) |
| 7–12 | 1.793(8) | 1.775(11) | 1.793(3) | 1.757(6) | 1.782(9) | 1.769(2) |
| 7–13 | 2.058(6) | 1.799(10) | 2.0585(18) | 1.762(6) | 2.051(7) | 1.781(2) |
| 8–13 | 1.992(5) | 1.998(8) | 1.9597(19) | 2.003(4) | 1.983(7) | 1.9869(16) |
| 8–14 | 1.862(8) | 2.192(8) | 1.973(3) | 2.284(4) | 1.954(7) | 2.2412(16) |
| 8–9 | 1.730(8) | 2.005(8) | .() |
2.008(5) | 1.727(9) | 2.0247(18) |
| 9–14 | 1.932(9) | 1.979(12) | .() |
1.925(6) | 1.892(8) | 1.947(2) |
| 9–10 | .() |
.() |
.() |
1.721(7) | 1.726(9) | 1.741(3) |
| 10–14 | .() |
.() |
1.925(3) | 1.883(7) | 1.905(5) | 1.883(2) |
| 10–11 | .() |
.() |
1.725(3) | 1.728(7) | 1.699(10) | 1.718(3) |
| 11–14 | 1.893(9) | 1.890(12) | 1.858(3) | 1.931(6) | 1.897(8) | 1.913(2) |
| 11–12 | 1.758(10) | 1.714(12) | 1.744(3) | 1.735(7) | 1.744(9) | 1.719(3) |
| 12–14 | 1.858(8) | 1.903(11) | 1.893(3) | 1.921(6) | 1.930(8) | 1.937(3) |
| 12–13 | 2.029(6) | 1.747(11) | 2.0360(19) | 1.730(6) | 2.002(6) | 1.723(2) |
| 13–14 | 2.213(6) | 1.950(12) | 2.224(2) | 1.937(6) | 2.266(3) | 1.937(2) |
Mechanistic implications
It is rare to find transition metal atoms in degree-5 sites in bicapped hexagonal antiprismatic metallacarboranes. As already noted, the only currently known examples are the Ni atoms in two 8,1,2-NiC2B11 species.11 The Co8 and Co13 atoms in compounds 1–6 represent further examples. We believe that the unexpected finding of these degree-5 Co atoms, coupled with the unambiguous location of the cage C atoms, allows comment on the possible mechanisms of formation of 1–6.Compounds 1 and 3 were both isolated from reactions in which 4,1,8- and 4,1,12-CoC2B10 13-vertex cobaltacarboranes were treated firstly with large excess of Na and then with Na[Cp] and CoCl2. The sodium reduction would have been expected to open up the cobaltacarborane to generate a nido dianion with a 6-atom open face opposite the original metal atom which would then have been capitated by the second metal and, indeed, 1,14,2,9- and 1,14,2,10-Co2C2B10 species, respectively, were formed in these reactions in significantly greater yields than were 1 and 3. Nevertheless, it remains possible that 1 and 3 were produced via reduction to an alternative nido intermediate with a 5-atom open face which was subsequently capitated. However, compounds 2, 4, 5 and 6 were produced from reactions that did not involve 2-e reduction and subsequent metallation of a 13-vertex CoC2B10 precursor and we have ultimately also produced 1 not via 2-e reduction/metallation. We believe that the formation of compounds 1–6 may be rationalised instead by direct electrophilic insertion, and that the isomeric forms of the products are readily understood in terms of this process.
Direct electrophilic insertion, a term coined by Kudinov and co-workers,23 involves the polyhedral expansion of an anionic closo metallacarborane by its reaction with a cationic metal fragment. It is a complement to direct nucleophilic insertion of zerovalent metal fragments into neutral closo carboranes and metallacarboranes developed by Stone, Green and co-workers several decades ago,24 with both approaches offering interesting alternatives to the traditional method of polyhedral expansion via the two-stage approach of 2-e reduction followed by metallation.
We illustrate the possibility of direct electrophilic insertion as the mechanism by which the present compounds are afforded with respect to compounds 4 and 5. We first isolated 4 and 5 as trace co-products in the synthesis of 4-Cp-4,1,6-closo-CoC2B10H12. This neutral CoIII species is prepared by reaction between [7,9-nido-C2B10H12]2−, Na[Cp] and CoCl2. The initial product of the reaction is the anionic CoII species [4-Cp-4,1,6-closo-CoC2B10H12]−, subsequently oxidised to the final product by O2.1 However, Na[Cp] and CoCl2 (together a source of the {CoCp+} fragment) are used in excess in these reactions,1 so it is possible to envisage reaction between [4-Cp-4,1,6-closo-CoC2B10H12]− and {CoCp+} to afford (CpCo)2C2B10H12 products by direct electrophilic insertion. Fig. 8 shows the docosahedral shape and numbering system of 4-Cp-4,1,6-closo-CoC2B10H12 and we presume that essentially the same shape is preserved in the anion.
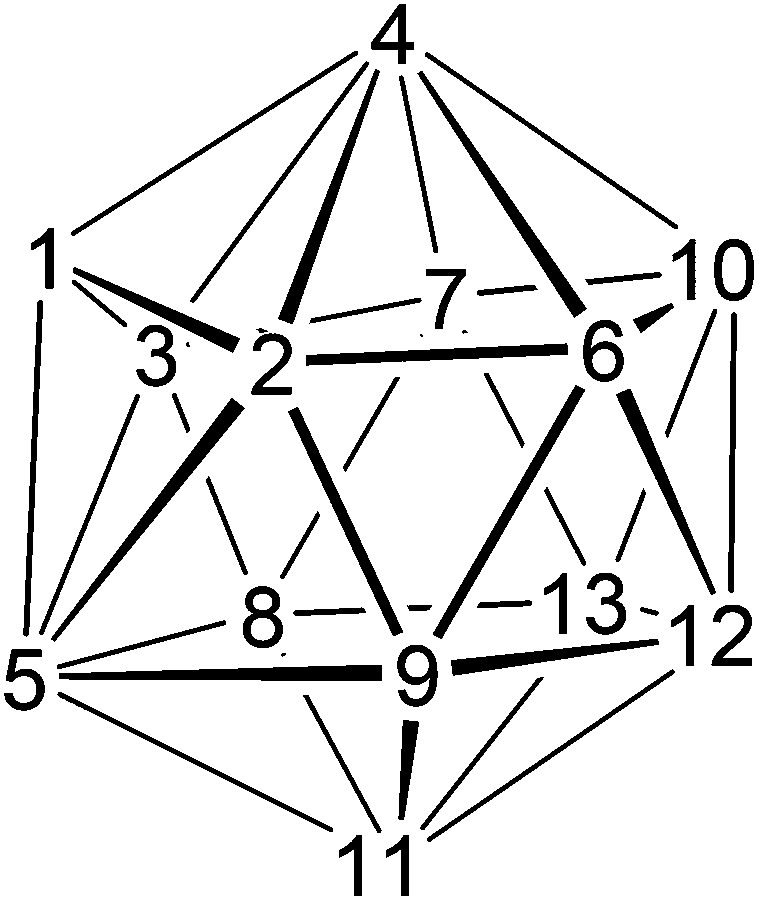 | ||
| Fig. 8 The docosahedron and numbering scheme. | ||
Docosahedral 4,1,x-MC2B10 metallacarboranes (x = 6, 8, 10, 11, 12) are known for a wide variety of metal types25 and structural determinations consistently shown long connectivities to the degree-6 atom B5, particularly the B2–B5, B3–B5, B/C8–B5 and B9–B5 connectivities.25b–f,26 It is therefore reasonable to imagine attack by the {CoCp+} fragment on both the forward (B2B5B9) and back (B3B5B8) triangles of [4-Cp-4,1,6-closo-CoC2B10H12]− with the new metal fragment breaking the presumably relatively weak B2–B527 and B9–B5 connectivities and bonding to the 1-2-9-11-5 open face so created, or breaking the B3–B5 and B8–B5 connectivities and bonding to the 1-3-8-11-5 face. The result of the former insertion is compound 4, and the result of the latter insertion is compound 5. The process of forming 4 from [4-Cp-4,1,6-closo-CoC2B10H12]− is perhaps best illustrated in the form of a Schlegel diagram, Fig. 9. In this the metal atoms are shown in red and the cage C atoms in blue, and squares, pentagons and hexagons are used to denote degree-4, -5 and -6 vertices, respectively. The process of breaking the B2–B5 and B9–B5 connectivities and inserting the new {CoCp+} fragment into the pentagonal face so created has the effect of increasing the degrees of vertices 1 and 11 by one unit and decreasing the degree of vertex 5 by one unit. The degrees of vertices 2 and 9 remain constant. The product, when labelled according to convention,2 would be 1,8-Cp2-1,8,2,4-closo-Co2C2B10H12, i.e. compound 4.
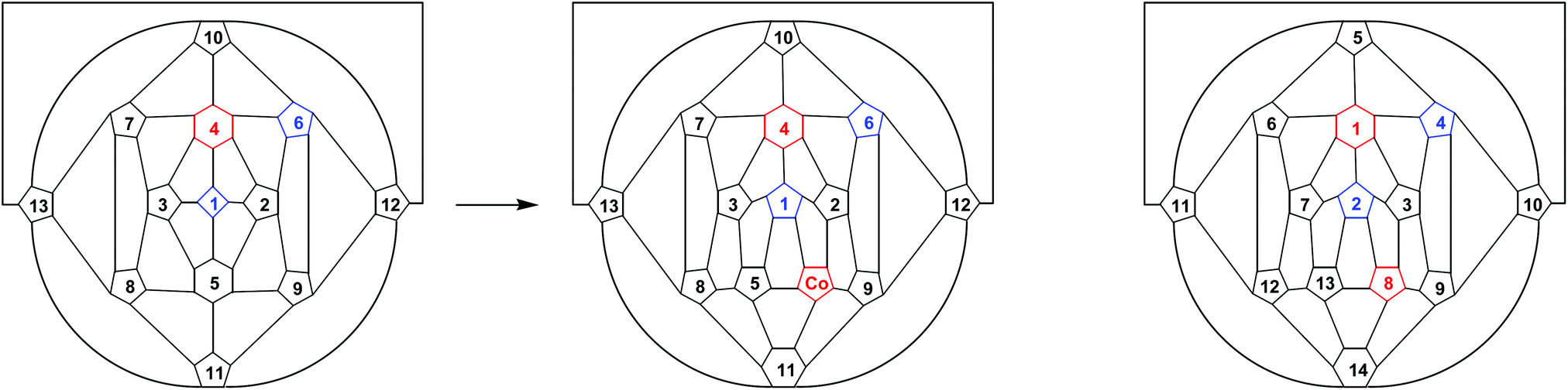 | ||
| Fig. 9 Schlegel diagrams showing schematically the transformation from a 4,1,6-MC2B10 docosahedron (left) to a bicapped hexagonal antiprism (centre) by the direct insertion of a new vertex (labelled Co) into the 1-2-9-11-5 face. Squares, pentagons and hexagons are used to represent degree-4, degree-5 and degree-6 vertices, respectively. To help follow the process atom numbering is maintained between left and centre diagrams, but on the right the product is renumbered according to convention, and is shown to be a 1,8,2,4-MCoC2B10 bicapped hexagonal antiprism (i.e. compound 4). | ||
In an attempt to support the possibility of direct electrophilic insertion, 4-Cp-4,1,6-closo-CoC2B10H12 was reduced with strictly one mole of electrons (sodium naphthalenide) and treated with Na[Cp] and CoCl2. Compounds 4 and 1,8-Cp2-1,8,2,6-closo-Co2C2B10H12, 5, were isolated (albeit in very low yields) following work-up.
Direct electrophilic attack of {CoCp+} on [4-Cp-4,1,10-closo-CoC2B10H12]− would be expected to afford only one product, 1,8-Cp2-1,8,2,5-closo-Co2C2B10H12, 6, because of the Cs symmetry of the precursor (the 1-2-9-11-5 and 1-3-8-11-5 pentagons are equivalent). Compound 6 was first isolated during the synthesis of 4-Cp-4,1,10-closo-CoC2B10H12,17 but again can be deliberately prepared by 1-e reduction of 4-Cp-4,1,10-closo-CoC2B10H12 followed by treatment with Na[Cp]/CoCl2. Similarly, 1,13-Cp2-1,13,2,10-closo-Co2C2B10H12, 1, can be deliberately prepared by 1-e reduction and subsequent metallation of 4-Cp-4,1,12-closo-CoC2B10H12, a result which is fully consistent with its formation by direct electrophilic insertion into the 1-3-8-11-5 pentagonal face of the 4,1,12-CoC2B10 precursor. Insertion into the 1-2-9-11-5 pentagon might also be expected to occur but we have not yet isolated the product of such an insertion, 1,8-Cp2-1,8,2,10-closo-Co2C2B10H12, from reduction and metallation of 4-Cp-4,1,12-closo-CoC2B10H12. However, a derivative of this “missing” product, 1,8-Cp2-3-OEt-1,8,2,10-closo-Co2C2B10H11, compound 2, was isolated in low yield from a reaction in which 4-Cp-4,1,12-closo-CoC2B10H12 was reduced, treated with CoCl2, and subjected to [K(18-crown-6)]Br in EtOH. The EtOH is clearly the source of the ethoxide substituent on B3 and we presume that the addition Cp ligand on Co8 was scavenged from another molecule of cobaltacarborane. Notwithstanding these complications it is possible that compound 2 was also formed by a direct electrophilic insertion reaction.
Similarly, direct electrophilic insertion of {CoCp+} into [4-Cp-4,1,8-closo-CoC2B10H12]− might be expected to lead to two products. Attack on the 1-2-9-11-5 pentagon of the 13-vertex precursor would result in a 1,13,2,9-Co2C2B10 14-vertex species, and indeed 1,13-Cp2-1,13,2,9-closo-Co2C2B10H12, 3, was recovered as a minor co-product during the attempted 2-e reduction then metallation of 4-Cp-4,1,8-closo-CoC2B10H12. Alternatively attack on the 1-3-8-11-5 pentagon would lead to 1,8-Cp2-1,8,2,9-closo-Co2C2B10H12, but this has not so far been isolated.
In Table 6 we summarise the expected products from direct electrophilic insertion of an {M′+} fragment into anionic 13-vertex metallacarboranes [4,1,x-MC2B10]− and list the examples of such insertions that are described herein. Although one example of a 4,1,11-MC2B10 metallacarborane is known26e we do not expect that direct electrophilic insertion into the anionic form of this will be very likely since the “product” would have a 1,8,2,14-MM′C2B10 architecture with a cage C atom in the very unfavoured8 degree-6 vertex 14.
| 13-Vertex precursor | 14-Vertex product | Example |
|---|---|---|
| [4,1,6-MC2B10]− | 1,8,2,4-MM′C2B10 | 4 |
| 1,13,2,4-MM′C2B10 | 5 | |
| [4,1,8-MC2B10]− | 1,8,2,9-MM′C2B10 | — |
| 1,13,2,9-MM′C2B10 | 3 | |
| [4,1,10-MC2B10]− | 1,8,2,5-MM′C2B10 | 6 |
| [4,1,12-MC2B10]− | 1,8,2,10-MM′C2B10 | 2 |
| 1,13,2,10-MM′C2B10 | 1 | |
| [4,1,11-MC2B10]− | 1,8,2,14-MM′C2B10 | Unlikely (see text) |
Conclusions
A series of six asymmetric, 14-vertex, (CpCo)2C2B10 dicobaltacarboranes with bicapped hexagonal antiprismatic cage structures in which one metal atom is at the capping vertex 1 and the other is at a degree-5 vertex (8 or 13) in the distant hexagonal belt, have been isolated. The VCD and BHD methods have been used to distinguish between cage B and cage C atoms in the crystallographically-determined structures, both leading to the same clear conclusions and thus allowing the identities of these species to be established unambiguously.28 The isomeric forms of the six compounds have been tentatively rationalised in terms of direct electrophilic insertion of a {CoCp+} fragment cation into a [CpCoC2B10]− monoanion.Experimental
Synthesis
Experiments were performed under dry, oxygen free N2, using standard Schlenk techniques, although subsequent manipulations were sometimes performed in the open laboratory. All solvents were freshly distilled under nitrogen from the appropriate drying agents immediately before use (CH2Cl2; CaH2:THF and 40–60 petroleum ether; sodium wire) or were stored over 4 Å molecular sieves and were degassed (3 × freeze–pump–thaw cycles) before use. Preparative TLC employed 20 × 20 cm Kieselgel F254 glass plates. NMR spectra at 400.1 MHz (1H) and 128.4 MHz (11B) or 300.1 MHz (1H) and 96.3 MHz (11B) were recorded on Bruker AVIII-400 or AVIII-300 spectrometers, respectively, from CDCl3 solutions at room temperature. Electron ionisation mass spectrometry (EIMS) was carried out using a Finnigan (Thermo) LCQ Classic ion trap mass spectrometer at the University of Edinburgh. The starting materials 4-Cp-4,1,6-closo-CoC2B10H12,1b 4-Cp-4,1,10-closo-CoC2B10H1217 and 4-Cp-4,1,12-closo-CoC2B10H121b,17 were prepared by literature methods or slight variations thereof. All other reagents were supplied commercially. Low yields of all the compounds isolated precluded elemental analyses.
:2 CH2Cl2:40–60 petroleum ether eluent. A complex mixture of at least nine mobile bands were observed including, in order of elution, the following compounds; 4-Cp-4,1,12-closo-CoC2B10H12 (starting material), 1,13-Cp2-1,13,2,10-closo-Co2C2B10H12 (Rf 0.68, brown, 0.012 g, 3.6%, 1), 3-Cp-3,1,2-closo-CoC2B9H11 (identified spectroscopically14), 1,14-Cp2-1,14,2,10-closo-Co2C2B10H12 (identified spectroscopically4) and 4,5-Cp2-4,5,1,6-closo-Co2C2B9H11 (identified spectroscopically15). For 1: 1H NMR: δ 5.29 (s, 5H, C5H5), 4.89 (s, 5H, C5H5), 3.10 (br s, 1H, CcageH), 2.57 (br s, 1H, CcageH). 11B{1H} NMR: δ 13.9 (1B), 5.2 (2B), −2.1 (2B), −5.7 (1B), −6.7 (1B), −15.8 (1B), −26.8 (1B), −28.2 (1B). EIMS: envelopes centred on m/z 392 (M+), 329 (M+ − Cp).
:2 CH2Cl2:40–60 petroleum ether). A complex mixture of six mobile bands was observed including 4-Cp-4,1,12-closo-CoC2B10H12 (Rf 0.86, starting material), 1,13-Cp2-1,13,2,10-closo-Co2C2B10H12 (Rf 0.75, identified spectroscopically, 1), 2-OEt-4-Cp-4,1,12-closo-CoC2B10H11 (Rf 0.39, identified crystallographically29) and 1,8-Cp2-3-OEt-1,8,2,10-closo-Co2C2B10H11 (Rf 0.21, brown, 0.006 g, ca. 1%, 2). For 2: EIMS: envelope centred on m/z 437 (M+).
:1 CH2Cl2:40–60 petroleum ether) to afford 1,13-Cp2-1,13,2,4-closo-Co2C2B10H12 (Rf 0.65, 5) and 1,8-Cp2-1,8,2,4-closo-Co2C2B10H12 (Rf 0.58, 4) as brown solids (on removal of solvent) in low yields (ca. 2 mg, 0.5%). For 4: 1H NMR: δ 5.46 (s, 5H, C5H5), 5.07 (s, 5H, C5H5), 3.80 (br s, 1H, CcageH), 2.89 (br s, 1H, CcageH). 11B{1H} NMR: δ 7.0 (1B), 3.6 (1B), 2.0 (2B), −4.2 (1B), −5.5 (1B), −10.2 (1B), −18.3 (1B), −19.9 (1B), −27.7 (1B). EIMS: envelopes centred on m/z 392 (M+), 268 (M+ − CpCo). For 5: 1H NMR: δ 5.20 (s, 5H, C5H5), 5.02 (s, 5H, C5H5), 2.96 (br s, 1H, CcageH), 2.67 (br s, 1H, CcageH). 11B{1H} NMR: δ 13.7 (1B), 11.5 (1B), 2.0 (1B), −0.7 (1B), −5.3 (1B), −8.2 (1B), −13.8 (1B), −18.0 (1B), −21.2 (1B), −26.4 (1B). EIMS: envelopes centred on m/z 392 (M+), 268 (M+ − CpCo).
:2 CH2Cl2:40–60 petroleum ether, Rf 0.51) in low yield (ca. 10 mg, 2%). 1H NMR: δ 5.21 (s, 5H, C5H5), 5.04 (s, 5H, C5H5), 2.71 (br s, 1H, CcageH), 2.25 (br s, 1H, CcageH). 11B{1H} NMR: δ 8.4 (1B), 3.0 (2B), −0.7 (1B), −2.4 (1B), −9.9 (1B), −14.8 (1B), −17.7 (1B), −21.2 (1B), −28.3 (1B). EIMS: envelopes centred on m/z 392 (M+), 268 (M+ − CpCo).
:2 CH2Cl2:40–60 petroleum ether, Rf 0.51) in low yield (ca. 5 mg, 1%) and identified spectroscopically.
Crystallography
Diffraction-quality crystals of compounds 1–6 were grown by slow diffusion of a CH2Cl2 solution of the appropriate compound and 40–60 petroleum ether at −30 °C. Intensity data were collected on a Bruker X8 APEX2 diffractometer using Mo-Kα X-radiation, with crystals mounted in inert oil on a cryoloop and cooled to 100 K by an Oxford Cryosystems Cryostream. Indexing, data collection and absorption correction were performed using the APEXII suite of programs.30 The structures were solved by direct methods (SHELXS-97) and refined by full-matrix least-squares (SHELXL-97).31Cage vertices were numbered as in Fig. 1a with the two Co atoms at positions 1 and 13. Initially all non-metal cage vertices were treated a B atoms. An ethoxide group as identified attached to vertex 7 in compound 2. With free (positional) refinement of cage H atoms all six structures were refined to convergence and the structures analysed by the VCD and BHD methods to locate the cage C atoms, as described in Results and discussion. Once this was done it was necessary to renumber some of the structures to concur with accepted convention.2 Finally, all structures were refined to full convergence.
The refinements of structures 3, 4 and 5 were as two component twins, whilst all other structures were refined conventionally. Non-cage H atoms were set in idealised positions and allowed to ride on their bound C atom, with C–H = 1.00 Å (Cp), 0.99 Å (CH2) or 0.98 Å (CH2). All H displacement parameters, Uiso, were constrained to be 1.2 × Ueq (bound B or C) except Me H atoms [Uiso(H) = 1.5 × Ueq C(Me)]. Table 7 contains further experimental details. Compound 2 has an OEt group bound to one B atom and compound 4 co-crystallises with one molecule of CH2Cl2 solvent, but 1, 3, 5 and 6 only differ in having the cage C atoms in different cage vertices. In that respect it is perhaps surprising that only two of these compounds, 1 and 6, are crystallographically isomorphous. Intermolecular contacts of possible significance are listed in the ESI,§ but in essence all six compounds crystallise as individual molecules.
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Formula | C12H22B10Co2 | C14H26B10Co2O | C12H22B10Co2 | C12H22B10Co2·CH2Cl2 | C12H22B10Co2 | C12H22B10Co2 |
| M | 392.26 | 436.31 | 392.26 | 477.18 | 392.26 | 392.26 |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | Triclinic | Monoclinic | Orthorhombic |
| Space group | Pbca | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21 | Pbca |
| a/Å | 14.9436(12) | 10.813(4) | 7.4419(4) | 7.6842(8) | 7.7083(6) | 14.8157(18) |
| b/Å | 13.3925(10) | 13.146(6) | 10.1756(6) | 11.0372(11) | 11.3413(9) | 13.3179(18) |
| c/Å | 16.7499(13) | 13.159(5) | 12.2035(7) | 11.7319(12) | 9.9968(8) | 16.818(2) |
| α (°) | 90.288(3) | 93.130(5) | ||||
| β (°) | 96.620(16) | 103.174(3) | 95.145(5) | 109.900(4) | ||
| γ (°) | 111.022(3) | 98.582(5) | ||||
| U/Å3 | 3352.2(5) | 1858.1(13) | 836.07(8) | 977.56(17) | 821.76(11) | 3318.4(8) |
| Z | 8 | 4 | 2 | 2 | 2 | 8 |
| F(000)/e | 1584 | 888 | 396 | 480 | 396 | 1584 |
| D calc/Mg m−3 | 1.554 | 1.560 | 1.558 | 1.621 | 1.585 | 1.570 |
| μ(Mo-Kα)/mm−1 | 1.966 | 1.786 | 1.971 | 1.965 | 2.005 | 1.986 |
| θ max (°) | 28.26 | 23.25 | 29.97 | 31.29 | 30.98 | 35.17 |
| Data measured | 93508 |
20203 |
16116 |
31755 |
16215 |
98827 |
| Unique data | 4134 | 2651 | 4740 | 6087 | 4694 | 7274 |
| R int | 0.0959 | 0.1138 | 0.0280 | 0.0847 | 0.0337 | 0.0424 |
| R, wR2 (obs. data) | 0.0611, 0.1447 | 0.0450, 0.0898 | 0.0291, 0.0669 | 0.0392, 0.1029 | 0.0469, 0.1026 | 0.0323, 0.0791 |
| S | 1.351 | 0.988 | 1.020 | 1.192 | 1.029 | 1.024 |
| Variables | 253 | 278 | 254 | 281 | 254 | 253 |
| Flack parameter | 0.21(3) | |||||
| E max, Emin/e Å−3 | 0.637, −0.857 | 0.480, −0.514 | 0.558, −0.475 | 1.211, −0.856 | 1.122, −1.202 | 0.906, −1.190 |
Acknowledgements
We thank the Carnegie Trust for the Universities of Scotland (AMcA) and the EPSRC (MEL, DE) for support.References
- (a) G. B. Dunks, M. M. McKown and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 2541 CrossRef; (b) D. F. Dustin, G. B. Dunks and M. F. Hawthorne, J. Am. Chem. Soc., 1973, 95, 1109 CrossRef CAS.
- R. N. Grimes, Carboranes, Academic Press, Oxford, UK, 2nd edn, 2011 Search PubMed.
- (a) K. Wade, J. Chem. Soc. D, 1971, 792 RSC; (b) K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1 CrossRef CAS.
- W. J. Evans and M. F. Hawthorne, J. Chem. Soc., Chem. Commun., 1974, 38 RSC.
- A. McAnaw, M. E. Lopez, D. Ellis, G. M. Rosair and A. J. Welch, Dalton Trans., 2013, 42, 671 RSC.
- D. Ellis, M. E. Lopez, R. McIntosh, G. M. Rosair and A. J. Welch, Chem. Commun., 2005, 1917 RSC.
- (a) W. M. Maxwell, R. F. Bryan and R. N. Grimes, J. Am. Chem. Soc., 1977, 99, 4008 CrossRef CAS; (b) W. M. Maxwell, R. Weiss, E. Sinn and R. N. Grimes, J. Am. Chem. Soc., 1977, 99, 4016 CrossRef CAS; (c) J. R. Pipal and R. N. Grimes, Inorg. Chem., 1978, 17, 6 CrossRef CAS; (d) R. N. Grimes, Acc. Chem. Res., 1978, 11, 420 CrossRef CAS; (e) R. N. Grimes, Adv. Inorg. Chem. Radiochem., 1983, 26, 55 CAS.
- (a) E. D. Jemmis, J. Am. Chem. Soc., 1982, 104, 7017 CrossRef CAS; (b) R. B. King, J. Organomet. Chem., 2007, 692, 1773 CrossRef CAS and references therein.
- R. D. McIntosh, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2006, 45, 4313 CrossRef CAS PubMed.
- J. Zhang, L. Deng, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2007, 129, 18 CrossRef CAS PubMed.
- L. Deng, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2006, 128, 5219 CrossRef CAS PubMed.
- A. McAnaw, G. Scott, L. Elrick, G. M. Rosair and A. J. Welch, Dalton Trans., 2013, 42, 645 RSC.
- A. Burke, R. McIntosh, D. Ellis, G. M. Rosair and A. J. Welch, Collect. Czech. Chem. Commun., 2002, 67, 991 CrossRef CAS.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, J. F. Warren and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879 CrossRef CAS.
- (a) D. F. Dustin and M. F. Hawthorne, J. Am. Chem. Soc., 1974, 96, 3462 CrossRef CAS; (b) M. E. Lopez, M. J. Edie, D. Ellis, A. Horneber, S. A. Macgregor, G. M. Rosair and A. J. Welch, Chem. Commun., 2007, 2243 RSC.
- W. Y. Man, A. McAnaw, D. Ellis, G. M. Rosair and A. J. Welch, work in progress.
- D. Ellis, M. E. Lopez, R. McIntosh, G. M. Rosair, A. J. Welch and R. Quenardelle, Chem. Commun., 2005, 1348 RSC.
- Reviews: (a) J. Plešek, Chem. Rev., 1992, 92, 269 CrossRef; (b) I. B. Sivaev and V. I. Bregadze, Collect. Czech. Chem. Commun., 1999, 64, 783 CrossRef CAS.
- M. E. Lopez, D. Ellis, P. R. Murray, G. M. Rosair, A. J. Welch and L. J. Yellowlees, Collect. Czech. Chem. Commun., 2010, 75, 853 CrossRef CAS.
- D. Ellis, R. D. McIntosh, S. Esquirolea, C. Viñas, G. M. Rosair, F. Teixidor and A. J. Welch, Dalton Trans., 2008, 1009 RSC.
- OLEX2, version 1.2.2 CrossRef CAS; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- As the size of the face being capped increases the perpendicular distance to it from a common capping atom necessarily decreases.
- A. R. Kudinov, D. S. Perekalin, S. S. Rynin, K. A. Lyssenko, G. V. Grintselev-Knyazev and P. V. Petrovskii, Angew. Chem., Int. Ed., 2002, 41, 4112 CrossRef CAS.
- (a) J. L. Spencer, M. Green and F. G. A. Stone, J. Chem. Soc., Chem. Commun., 1972, 1178 RSC; (b) M. Green, J. L. Spencer, F. G. A. Stone and A. J. Welch, J. Chem. Soc., Dalton Trans., 1975, 179 RSC.
- (a) First transition metal: ref. 1; (b) First group 1 metal: K. Chui, H.-W. Li and Z. Xie, Organometallics, 2000, 19, 5447 CrossRef CAS; (c) First group 2 metal: R. Khattar, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1990, 112, 4962 CrossRef CAS; (d) First p-block metal: N. M. M. Wilson, D. Ellis, A. S. F. Boyd, B. T. Giles, S. A. Macgregor, G. M. Rosair and A. J. Welch, Chem. Commun., 2002, 464 RSC; (e) First lanthanide: R. Khattar, C. B. Knobler, S. E. Johnson and M. F. Hawthorne, Inorg. Chem., 1991, 30, 1970 CrossRef CAS; (f) First actinide: Z. Xie, C. Yan, Q. Yang and T. C. W. Mak, Angew. Chem., Int. Ed., 1999, 38, 1761 CrossRef CAS.
- E.g. (a) M. R. Churchill and B. G. DeBoer, Inorg. Chem., 1974, 13, 1411 CrossRef CAS; (b) M. A. Laguna, D. Ellis, G. M. Rosair and A. J. Welch, Inorg. Chim. Acta, 2003, 347, 161 CrossRef CAS; (c) A. Burke, D. Ellis, D. Ferrer, D. L. Ormsby, G. M. Rosair and A. J. Welch, Dalton Trans., 2005, 1716 RSC; (d) K. J. Dalby, D. Ellis, S. Erhardt, R. D. McIntosh, S. A. Macgregor, K. Rae, G. M. Rosair, V. Settles, A. J. Welch, B. E. Hodson, T. D. McGrath and F. G. A. Stone, J. Am. Chem. Soc., 2007, 129, 3302 CrossRef CAS PubMed; (e) S. Zlatogorsky, M. J. Edie, D. Ellis, S. Erhardt, M. E. Lopez, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2007, 46, 6706 CrossRef CAS PubMed; (f) P. D. Abram, D. McKay, D. Ellis, S. A. Macgregor, G. M. Rosair, R. Sancho and A. J. Welch, Dalton Trans., 2009, 2345 RSC; (g) P. D. Abram, D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2009, 5403 RSC; (h) G. Scott, A. McAnaw, D. McKay, A. S. F. Boyd, D. Ellis, G. M. Rosair, S. A. Macgregor, A. J. Welch, F. Laschi, F. Rossi and P. Zanello, Dalton Trans., 2010, 39, 5286 RSC; (i) D. Ellis, D. McKay, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2010, 49, 4943 CrossRef CAS PubMed; (j) D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2010, 46, 7394 RSC; (k) J. S. Ward, H. Tricas, G. Scott, D. Ellis, G. M. Rosair and A. J. Welch, Organometallics, 2012, 31, 2523 CrossRef CAS; (l) A. McAnaw, M. E. Lopez, G. Scott, D. Ellis, D. McKay, G. M. Rosair and A. J. Welch, Dalton Trans., 2012, 41, 10957 RSC; (m) G. Scott, D. Ellis, G. M. Rosair and A. J. Welch, J. Organomet. Chem., 2012, 721–722, 78 CrossRef CAS and references therein.
- Supporting evidence for the relative weakness of B2–B5 in 4,1,6-MC2B10 species is the well-known double diamond-square-diamond process (involving breaking this connectivity) which accounts for the functionality of these species in solution at room temperature. See ref. 1, 25d, 26b, c, f, h.
- One referee commented on the relatively imprecise original structural determination of compound 6 and concluded “for this structure we cannot distinguish between B and C in the [metalla]carborane cage”. In response we recollected crystallographic data from a second crystal of better quality (all data in the paper relating to compound 6 are from this second determination). However, it is apparent from analysis of the VCD, BHD and Ueq data that
![[x with combining low line]](https://www.rsc.org/images/entities/char_0078_0332.gif)
![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif)
![[y with combining low line]](https://www.rsc.org/images/entities/char_0079_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) conclusions regarding C atom positions in compound 6 are reached using both the relatively imprecise and the relatively precise data. See ESI§ for full details.
conclusions regarding C atom positions in compound 6 are reached using both the relatively imprecise and the relatively precise data. See ESI§ for full details. - A. McAnaw, PhD thesis, Heriot-Watt University, 2013.
- Bruker AXS APEX2, version 2009-5, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Professor María Pilar García Clemente. |
| ‡ Celebrating 300 Years of Chemistry at Edinburgh. |
| § Electronic supplementary information (ESI) available. CCDC 939752–939756 and 939818. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52101j |
| This journal is © The Royal Society of Chemistry 2014 |