 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning ligand electronics and peripheral substitution on cobalt salen complexes: structure and polymerisation activity†‡
Linus
Chiang
a,
Laura E. N.
Allan
b,
Juan
Alcantara
a,
Michael C. P.
Wang
a,
Tim
Storr
*a and
Michael P.
Shaver
*b
aDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada. E-mail: tim_storr@sfu.ca
bSchool of Chemistry, University of Edinburgh, Joseph Black Building, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: michael.shaver@ed.ac.uk; Tel: +44 (0) 131 650 4726
First published on 9th September 2013
Abstract
A series of cobalt salen complexes, where salen represents an N2O2 bis-Schiff-base bis-phenolate framework, are prepared, characterised and investigated for reversible-termination organometallic mediated radical polymerisation (RT-OMRP). The salen ligands contain a cyclohexane diimine bridge and systematically altered para-substituted phenoxide moieties as a method to examine the electronic impact of the ligand on complex structure and reactivity. The complexes are characterised by single crystal X-ray diffraction, cyclic voltammetry, X-ray photoelectron spectroscopy, electron paramagnetic resonance spectroscopy and computational methods. Structural studies all support a tailorable metal centre reactivity altered by the electron-donating ability of the salen ligand. RT-OMRP of styrene, methyl methacrylate and vinyl acetate is reported and suggests that cobalt–carbon bond strength varies with the ligand substitution. Competing β-hydrogen abstraction affords long-chain olefin-terminated polymer chains and well controlled vinyl acetate polymerisations, contrasting with the lower temperature associative exchange mechanism of degenerative transfer OMRP.
Introduction
Metal complexes of tetradentate salen (salen is a common abbreviation for N2O2 bis-Schiff-base bis-phenolates) ligands have received considerable attention in transition metal chemistry due to their relative ease of synthesis, ability to form stable complexes with many metals in a variety of oxidation states and versatility as catalysts for important organic transformations.1–9 Notable examples of catalysis include Mn salen olefin epoxidation,1,2 Co salen hydrolytic kinetic resolution of epoxides,10 Co salen enantioselective polymerisation of epoxides,11,12 and Cr/Co salen coupling of CO2 and epoxides.5,9,13 The modular synthesis of the salen framework allows easy tuning of both sterics and electronics, providing a means to study how subtle changes to the ligand framework influence reactivity.In many cases the steric and electronic properties of the salen ligand exerts a profound effect on the reaction outcome. For example, enantiomeric excess (ee) values for the Mn salen – catalysed asymmetric epoxidation of pro-chiral olefins depends on the nature of the chiral diimine bridge and the identity of the ortho- and para-ring substituents.2,14–16 While the ortho-ring substituents primarily provide steric bulk to influence substrate approach, the electron-donating ability of the para-ring substituents has a significant effect on the reaction outcome by altering the position of the transition state along the reaction coordinate.14,15 In the case of Mn salen complexes, electron-donating para-ring substituents lead to a late transition state and afford the highest ee values. A smaller effect of the para-ring substituents (Br, H, tBu) is observed for Co salen copolymerisation of propylene oxide and CO2, with the tBu derivative showing the highest regioselectivity.17 Similar to the Mn salen results, electron-donating substituents increased the rate of copolymer formation for Cr salen catalysed cyclohexene oxide/CO2 copolymerisation.5,18 These results highlight the influence of the electron-donating ability of the para-ring substituents on the catalytic activity of the metal centre.
We were interested in studying the polymerisation activity of a series of Co salen complexes wherein we have varied the electron-donating ability of the para-ring substituents (NMe2 > OMe > tBu > NO2) (Chart 1). In particular, the role these complexes could play in controlling organometallic mediated radical polymerisation (OMRP) is important.19 In this variant of controlled radical polymerisation, the fast and reversible homolytic cleavage of a metal–carbon bond is exploited to lower radical concentrations and control the properties of the polymer products. OMRP may proceed through reversible termination (RT), where a transition metal complex acts as a reversible spin-trap to deactivate the propagating chains, or through degenerative transfer (DT) whereby associative exchange between an active radical and a metal-bound radical of the dormant species takes place. Cobalt complexes dominate the OMRP literature,20 imparting the best control, although the development of other metals as OMRP mediators is a recent topic of interest.19,21–25
 | ||
| Chart 1 Structures of the Co Schiff-base complexes CoSalR1,R2. | ||
Early cobalt OMRP systems were based on nitrogen donors, such as porphyrins,26–29 which were particularly efficient for the OMRP of acrylates. A more recent report examined a series of 1,3-bis(2-pyridylimino)isoindolate ligands, structurally related to porphyrins, but lacking a fourth N-donor.30 These complexes were active for acrylate polymerisation but did not show the anticipated structure–activity relationship, leading the authors to conclude that the cobalt–carbon bond was decoupled from the ligand substituents and so inclusion of electron-withdrawing or electron-donating groups had no effect on the polymerisation behaviour. Cobalt species incorporating oxygen donors, such as bis-acetylacetonate Co(II) and related complexes,19–22 are very efficient for the OMRP of vinyl acetate31–36 and careful choice of reaction conditions has allowed expansion of the monomer scope to include acrylates,37–39 acrylonitrile,40–43 other vinyl ester monomers44,45 and N-vinylpyrrolidone.44,46 The use of mixed N,O-donor systems in cobalt-mediated OMRP is limited to reports on the β-ketoiminates,47,48 which exert reasonable control over methyl acrylate and vinyl acetate polymerisation, and a very recent report on the highly efficient degenerative transfer OMRP of vinyl acetate and methyl acrylate mediated by cobalt(II) [N,N-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine],49 (CoSaltBu,tBu), which was published during the course of our study.
To complement these recent efforts by Liao et al. we have focused our efforts on the more challenging reversible-termination (RT) OMRP methodology. In this work we have investigated the electronic structure of a series of Co(II) salen complexes where we have varied the para-ring substituents (Chart 1). We have shown that the para-ring substituents influence both the electron density at the metal centre as well as the overall reactivity via increasing stability of the Co(II) forms in the order of NMe2 > OMe > tBu > NO2. The more electron-donating substituents should thus increase the concentration of propagating radicals in the system and the use of the Co(II) salen complexes in RT-OMRP is reported.
Results and discussion
Synthesis and characterisation
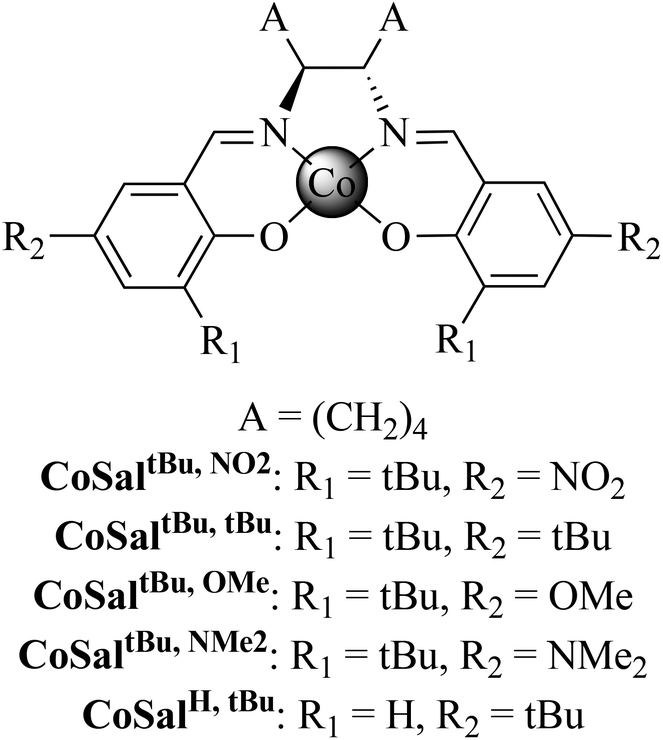
The Co complexes CoSalR1,R2 were synthesised in good yields from the metallation of the corresponding ligands H2SalR1,R2 with Co(OAc)2·4H2O.50–55 CoSaltBu,NO2 yielded crystals suitable for X-ray analysis, using THF–pentane as the recrystallisation solvents. The molecular structure of CoSaltBu,NO2·THF is shown in Fig. 1 and selected crystallographic data for the complex are presented in Table 1. CoSaltBu,NO2 exhibits an essentially square planar geometry at the Co centre with a molecule of THF weakly coordinated in the axial position (Co–OTHF ∼ 2.21 Å). The dihedral angle between the N–Co–O planes is 9°, likely due to the steric interaction between the two ortho t-butyl groups.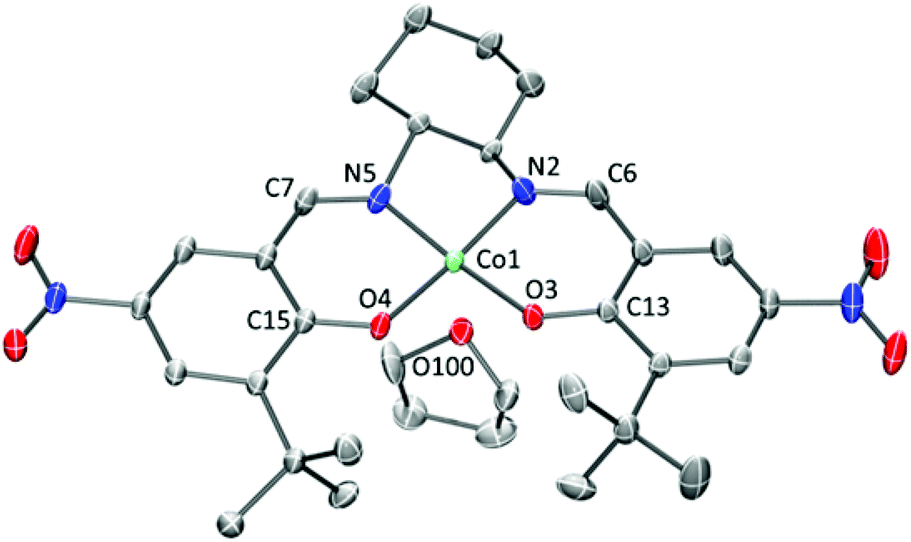 | ||
| Fig. 1 POV-ray representation (50% probability) of CoSaltBu,NO2·THF, excluding hydrogen atoms. Selected interatomic distances (Å) and angles (°): Co(1)–O(4), 1.901; Co(1)–O(3), 1.895; Co(1)–N(5), 1.876; Co(1)–N(2), 1.874; N(5)–C(7), 1.292; N(2)–C(6), 1.288; O(4)–C(15), 1.296; O(3)–C(13), 1.291; Co(1)–O(100), 2.211; O(3)–Co(1)–O(4), 86.9(4); O(4)–Co(1)–N(5), 93.5(7); O(3)–Co(1)–N(2), 93.4(8); N(2)–Co(1)–N(5), 85.2(7). | ||
| Bond/distance | CoSaltBu,NO2·THF (Å) experimental | CoSaltBu,NO2·THF (Å) calculated |
|---|---|---|
| a See Experimental section for calculation details. | ||
| Co(1)–O(4) | 1.901 | 1.901 |
| Co(1)–O(3) | 1.895 | 1.900 |
| Co(1)–N(5) | 1.876 | 1.884 |
| Co(1)–N(2) | 1.874 | 1.893 |
| Co(1)–O(100) | 2.211 | 2.282 |
| N(5)–C(7) | 1.292 | 1.296 |
| N(2)–C(6) | 1.288 | 1.297 |
| O(4)–C(15) | 1.296 | 1.295 |
| O(3)–C(13) | 1.291 | 1.293 |
Electrochemistry
Redox processes for CoSaltBu,R2 were probed by cyclic voltammetry (CV) in CH2Cl2 using tetra-n-butylammonium perchlorate (nBu4NClO4) as the supporting electrolyte. CoSaltBu,NO2 was not soluble in CH2Cl2 and thus the CV experiments were completed in THF. Three quasi-reversible, one-electron redox processes were observed for R2 = tBu, OMe and NMe2, where only a single redox process was observed for R2 = NO2 (Fig. 2). The reversibility of these processes was evaluated via comparison of the peak-to-peak difference (|Epa − Epc|) for a specific redox process to that of the Fc+/Fc couple under identical conditions (Table 2). The redox potentials versus ferrocenium/ferrocene (Fc+/Fc) are reported in Table 2. The three redox couples can be assigned to the metal centre Co(II)/Co(III) and the two redox-active phenolate moieties. The redox chemistry of the tBu, and OMe derivatives have been reported previously with the first redox process assigned as a one-electron Co(II)/Co(III) couple (Fig. S1‡).53,56 The more positive redox potential for the NO2 derivative is due to the presence of electron-withdrawing groups in the para position. The positive shift in the potential for the NO2 derivative (and the use of THF as the solvent) results in only one of the redox processes being visible by CV in this work. Overall there is a clear shift in the redox potentials towards more negative values as the electron-donating ability of the para-substituents is increased (NMe2 > OMe > tBu > NO2). Modulation of the electron-donating ability of the salen ligands is subsequently shown to have an effect on the polymerisation activity of the Co complexes (vide infra).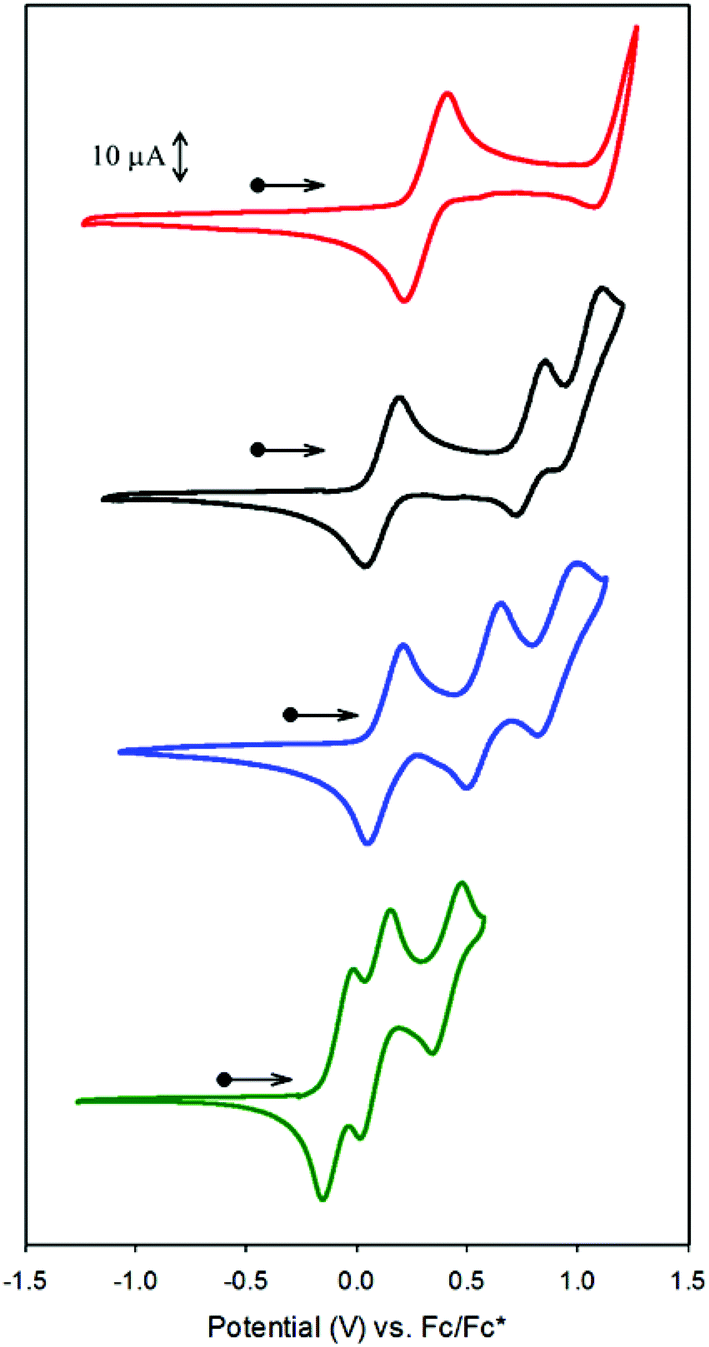 | ||
| Fig. 2 Cyclic voltammograms of CoSaltBu,R2 (red: R2 = NO2; black: R2 = tBu; blue: R2 = OMe; green: R2 = NMe2) versus Fc+/Fc. | ||
| CoSaltBu,R2 | E 11/2 (mV) | E 21/2 (mV) | E 31/2 (mV) |
|---|---|---|---|
| a Peak-to-peak differences in brackets (|Epa − Epc| in mV). Peak-to-peak difference for the Fc+/Fc couple at 233 K is 80 mV. b Solvent = THF. | |||
R2 = NO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
310 (190) | — | — |
| R2 = tBu | 110 (160) | 780 (130) | 1020 (190) |
| R2 = OMe | 130 (160) | 570 (160) | 910 (180) |
| R2 = NMe2 | −90 (140) | 80 (140) | 400 (130) |
X-ray photoelectron spectroscopy
The electronic structures of the Co complexes CoSaltBu,R2 were investigated by XPS (Fig. 3). Referenced to the C 1s binding energy, the Co 2p3/2 and Co 2p1/2 binding energies (Table 3) for all four CoSaltBu,R2 complexes indicate a common 2+ oxidation state for Co.57–59 Comparing the binding energies a general trend is observed: as the electron-donating ability of the para-ring substituents is increased, there is a decrease in the Co 2p3/2 and 2p1/2 binding energies. These data are further evidence for the influence of the para-ring substituents on the electronics at the metal centre.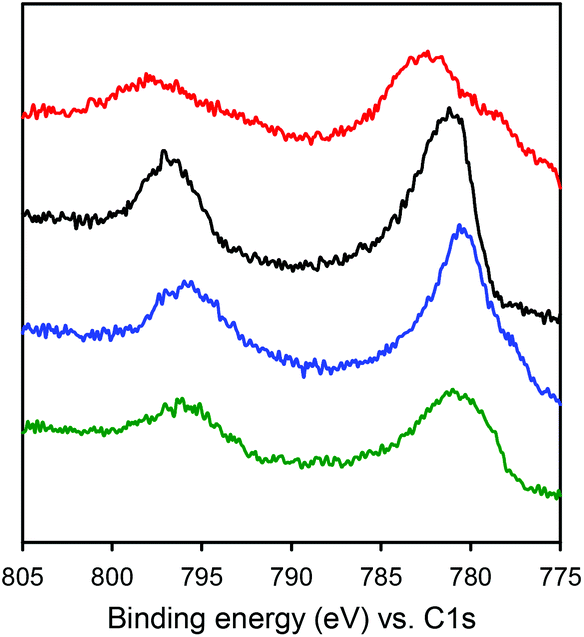 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) spectra of CoSaltBu,R2 compounds showing the Co 2p3/2 and 2p1/2 binding energies (red: R2 = NO2; black: R2 = tBu; blue: R2 = OMe; green: R2 = NMe2). Full spectra shown in Fig. S2.‡ | ||
| CoSaltBu,R2 | Binding energy (eV) | |
|---|---|---|
| Co (2p3/2) | Co (2p1/2) | |
| R2 = NO2 | 797.6 | 782.8 |
| R2 = tBu | 796.8 | 781.1 |
| R2 = OMe | 795.7 | 780.4 |
| R2 = NMe2 | 795.6 | 780.6 |
Electron paramagnetic resonance
The X-band EPR spectra of the complexes CoSaltBu,R2 were studied at 20 K and are consistent with a low spin Co(II) (S = 1/2) ground state (Fig. 4).60,61 Based on previous work of Daul et al., and the fitting of the experimental EPR data, the CoSaltBu,tBu, CoSaltBu,OMe and CoSaltBu,NMe2 derivatives exhibit a |yz, 2A2〉 ground state in frozen PhMe (Table 4). This result is consistent with other reported 4-coordinate Co(II) salen complexes.60,61 The CoSaltBu,NO2 derivative was not soluble in PhMe, and thus the isolated THF adduct was dissolved in a 2:1 mixture of PhMe–CH2Cl2 and subsequently frozen for EPR analysis. The observed spectrum displays a different pattern in comparison to the tBu, OMe, and NMe2 analogues (Fig. 4), suggesting a different ground state for this complex. EPR fitting analysis provides evidence for a |z2, 2A1〉 ground state for CoSaltBu,NO2 under these conditions, consistent with a 5-coordinate structure (likely THF adduct). This analysis is further corroborated by theoretical calculations (vide infra).
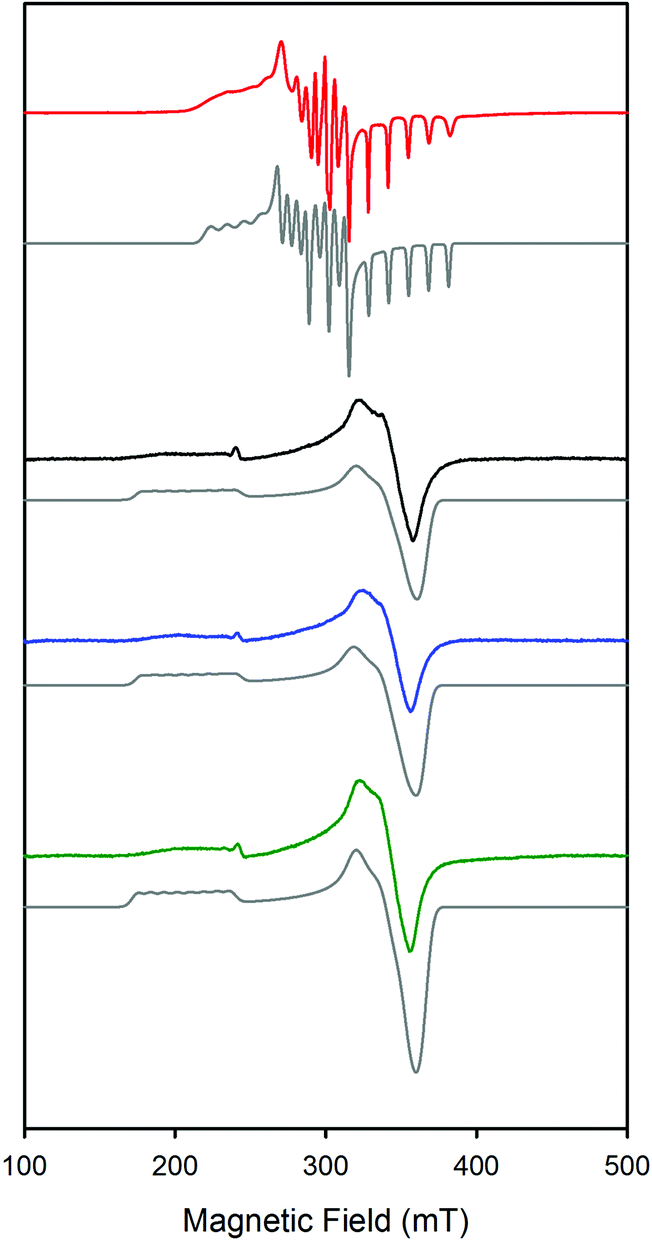 | ||
| Fig. 4 X-band EPR spectra of CoSaltBu,R2. Red: R2 = NO2, ν = 9.384 GHz; black: R2 = tBu, ν = 9.383 GHz; blue: R2 = OMe, ν = 9.383 GHz; green: R2 = NMe2, ν = 9.385 GHz. Grey spectra are respective simulations. Conditions: power = 2.0 mW; modulation frequency = 100 kHz; modulation amplitude = 0.4 mT; T = 20 K. | ||
| g x | g y | g z | A x (Co)a | A y (Co)a | A z (Co)a | |
|---|---|---|---|---|---|---|
|
a Estimated values due to large g and A strain (A in MHz).
b See ref. 59.
c The fits for tBu, OMe and NMe2 are identical within experimental error.
d Solvent 2:1 CH2Cl2–PhMe.
|
||||||
| CoSaltBu,tBub,c |
3.21 | 1.88 | 1.98 | 400 | 80 | 125 |
| CoSaltBu,OMec |
3.21 | 1.89 | 1.99 | 400 | 80 | 125 |
| CoSaltBu,NMe2c |
3.19 | 1.89 | 1.99 | 400 | 80 | 125 |
| CoSaltBu NO2d |
2.56 | 2.30 | 2.00 | 410 | 196 | 346 |
Theoretical analysis
Density functional theory (DFT) calculations of the CoSaltBu,R2 complexes provided further insight into the geometric and electronic structure of these complexes. We first compared the optimised geometry for the CoSaltBu,NO2·THF complex with the experimental X-ray metrical data (Table 1). The calculations reproduce the coordination sphere bond lengths to within ±0.02 Å. In the absence of the THF molecule, the predicted coordination sphere bond lengths differ significantly from the experimental data. The calculations of the neutral square planar Co(II) complexes provided further details of the effect of the peripheral substituents on the metal centre. Mulliken population analysis62,63 predicts the lowest partial charge at Co (0.78) for CoSaltBu,NMe2 and the highest partial charge at Co (0.80) for CoSaltBu,NO2 consistent with the expected electronic effects of the para-ring substituents.We further investigated the electronic structure of the CoSaltBu,R2 complexes and in particular the nature of the singularly occupied molecular orbital (SOMO). Based on the EPR data and fitting, the NMe2, OMe, and tBu derivatives display a common |yz, 2A2〉 ground state.60,61 The DFT calculations predict correctly a dyz-containing SOMO for these three derivatives (Fig. 5). In addition, without the axial THF molecule the NO2 derivative also displays a dyz SOMO (Fig. S3‡). In contrast, the 5-coordinate THF adduct CoSaltBu,NO2·THF exhibits a |z2, 2A1〉 ground state based on the EPR analysis. The DFT calculation for the THF adduct predicts correctly a dz2 SOMO for this analogue (Fig. 5). Axial ligand binding in this case raises the energy of the dz2 orbital in comparison to dyz, resulting in the change in electronic ground state.
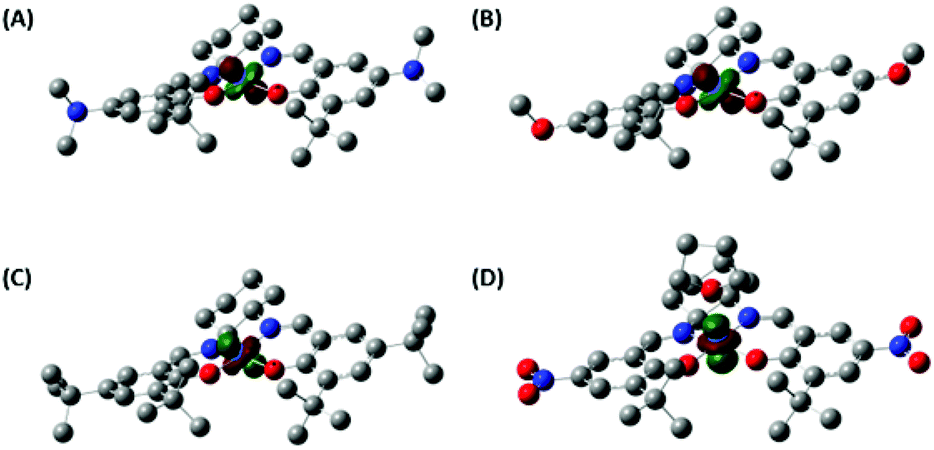 | ||
| Fig. 5 Predicted singularly occupied molecular orbital (dyz) for the Co(II) salen complexes, except the THF adduct of CoSaltBu,NO2 (dz2): (A) CoSaltBu,NMe2; (B) CoSaltBu,OMe; (C) CoSaltBu,tBu; (D) CoSaltBu,NO2·THF. See the Experimental section for calculation details. | ||
The 5-coordinate X–Co(III)Sal complexes (X = 1-phenylethane) were investigated via computations to better understand the influence of the para-ring substituents on the axial Co–C bond strength. The Co–C bond dissociation energies (BDE) were calculated by subtracting the energy of the X–Co(III)Sal reactant from the sum of the energies of the products as shown in eqn (1).
| X–Co(III)SaltBu,R → Co(II)SaltBu,R + X˙ | (1) |
For this series of calculations the BP86 functional was used,64 based on prior work by Kozlowski et al. on alkyl-Co(III) macrocycles.65–67 The Co–C BDE for the NO2 derivative (15.4 kcal mol−1) was predicted to be at least 1.3 kcal mol−1 higher than the values for the tBu (14.1 kcal mol−1), OMe (13.7 kcal mol−1) and NMe2 (13.3 kcal mol−1) derivatives. The calculations predict the highest Co–C BDE for the NO2 derivative, consistent with the electron-withdrawing para-substituents and resultant increased Lewis acidity at the metal centre. The predicted BDE values are lower than those previously reported for alkyl cobalt salen complexes (methyl, ethyl, etc.), likely due to the increased steric bulk of 1-phenylethane and presence of an additional axial ligand in the previous work.68 Overall, the calculated Co–C BDE decrease as the electron-donating ability of the para-substituents is increased in the series.
Organometallic mediated radical polymerisation
Although recent research has significantly expanded the number of active systems in OMRP,19,21,22 ligand design has an important role to play in increasing both the monomer scope and efficacy of OMRP catalysts. While the effect of carbon–halogen bond strengths on atom transfer radical polymerisation (ATRP) has been studied both experimentally and computationally, no similar data have been compiled for OMRP systems. Few investigations of systematic variation of ligand substituents have been reported, but a better understanding of the role of the metal–carbon bond strengths will facilitate improved control and tunability of polymerisations. To clarify the impact of bond strength, we wished to investigate our series of CoSaltBu,R2 complexes for reversible termination OMRP, with the strong electronic variation anticipated to significantly alter the cobalt–carbon bond strengths, potentially allowing us to tune our system for different monomers. This complements recently reported degenerative transfer OMRP of vinyl acetate by a CoSaltBu,tBu complex.49 Of note, DT-OMRP is outside the scope of this initial report on electronic effects as we wished to examine the strength of the cobalt–carbon bond directly, not through associative exchange.a
| CoSaltBu,R2 | % Conv. | M n,th | M n | PDI |
|---|---|---|---|---|
|
a Bulk styrene polymerisations, 1 h at 120 °C, initiated with AIBN with complex:initiator:monomer ratio of 1:0.6:100. Mn,th = [M]0/2[I]0 × MW(monomer) × conversion + MW(catalyst).
|
||||
| R2 = NO2 | 55 | 5340 | 4630 | 2.23 |
| R2 = tBu | 61 | 5880 | 5800 | 1.78 |
| R2 = OMe | 72 | 6770 | 14310 |
2.38 |
| R2 = NMe2 | 85 | 7920 | 9110 | 1.65 |
To investigate whether the steric bulk of the tBu group at the ortho position of the aromatic ring was hindering the deactivation equilibrium and causing the broadened PDIs, we synthesised CoSalH,tBu and screened it for styrene OMRP. 58% conversion in 1 h was obtained, with molecular weights which were slightly lower than theoretical values (Mn,th = 5500, Mn = 4320) and an improved, but still broad, PDI of 1.65. This suggested that the bulk at the ortho position played, at most, a minor role in controlling the OMRP equilibrium.
To study whether the high polymerisation temperatures of 120 °C were favouring side reactions and thus broadening polydispersities, we investigated styrene polymerisation using V-70 as the initiator (Table S1, ESI‡). As expected, conversions were lower at 65 °C, as the dormant species is favoured and propagation rates are reduced. PDIs were actually broader, with more deviation between theoretical and experimental molecular weights indicating that the lower temperatures were not preventing termination reactions. The productive, if uncontrolled, polymerisation at this lower temperature is also suggestive that the monomer scope of DT-OMRP by CoSaltBu,tBu may be limited to monomers that form stronger cobalt–carbon bonds at these temperatures.
Despite reasonable agreement of experimental molecular weights with the theoretical values, the broad PDIs of 1.6–2.4 indicated that styrene polymerisation was inadequately controlled by Co complexes CoSalR1,R2. To further investigate this, we examined kinetic data for the polymerisation of styrene by CoSaltBu,tBu. Although the plot of ln([M]0/[M]t) was linear for the first 2 hours (Fig. 6a), with kobs. = 0.40 h−1, deviations after this time indicated that the radical concentration did not remain constant. Interestingly, after just 10 minutes the conversion was 36%, indicating an initial rapid period of polymerisation where molecular weights quickly reached ca. 5500 Da. Molecular weights then increased in a linear fashion for the first hour, up to ca. 7200 Da at 56%. However, after this point the molecular weights stagnated, remaining at around 7000 Da for the rest of the polymerisation (Fig. 6b). This behaviour suggested the occurrence of catalytic chain transfer, which was confirmed by the presence of olefin end-groups at δ 6.2 ppm in the 1H NMR spectra of the polymer samples (Fig. S4, ESI‡). Many cobalt(II) complexes are excellent CCT catalysts, particularly the cobaloximes and cobalt porphyrins,69,70 and it is likely that a low metal–carbon bond dissociation energy in the polymerisation of styrene results in high radical concentrations which, coupled with a high concentration of Co(II), favours β-hydrogen abstraction. Reinitiation from the metal-hydride species gives new propagating chains, resulting in the broad PDIs which increase from 1.62 at early stages of the polymerisation to 2.04 at 80% conversion. Importantly, by altering the monomer concentration we can tune the ‘top-out’ molecular weight of our olefin-terminated polymer chains. For instance, with 500 eq. of monomer the molecular weights are increased to ca. 20000 Da, achieved from 25% conversion onwards (Fig. S5, ESI‡). These moderate-length, olefin-terminated poly(styrene) chains could potentially be used as building blocks for extended macromolecular structures and offer an alternative to the short-chain oligomers traditionally synthesised through efficient CCT polymerisations. Attempted controlled radical polymerisations mediated by molybdenum71,72 and iron73–75 catalysts have previously been reported to yield olefin-terminated poly(styrene) through CCT processes, with the molecular weights of the polymers obtained typically ca. 1000–5000 Da.
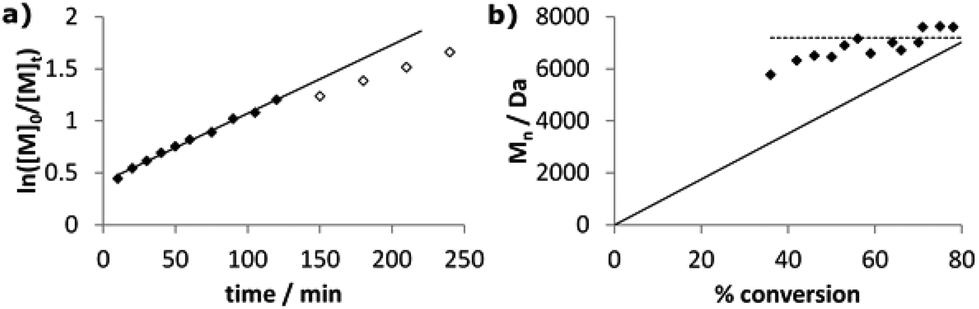 | ||
| Fig. 6 Plots of (a) ln([M]0/[M]t) vs. time and (b) molecular weight vs. conversion for bulk styrene polymerisation at 120 °C using CoSaltBu,tBu and AIBN. Monomer:catalyst:initiator ratio of 100:1:0.6 used. Solid data points in (a) used to calculate least-squares fit. Solid line in (b) represents Mn,th, while dashed line indicates ‘top-out’ molecular weight. | ||
Building from the recently published work on the DT-OMRP of vinyl acetate by a cobalt salen complex (CoSaltBu,tBu),49 we also examined the four CoSaltBu,R2 complexes for the RT-OMRP of vinyl acetate (Table 6). Although a notoriously difficult monomer to control due to the difficulty in activating the monomer and then controlling the equilibrium between the unstabilised radical and the dormant species, vinyl acetate has been successfully polymerised using Co(acac)2 and other Co-systems using both V-70 and AIBN initiators.19,20,22 We anticipated that the formation of a stronger metal–carbon bond between the vinyl acetate radical and the Co complex would either hinder CCT and favour controlled radical polymerisation or, as expected for a system operating solely by DT-OMRP, form no polymeric products.
a
| Complex | % Conv. | M n,th | M n | PDI |
|---|---|---|---|---|
|
a Bulk vinyl acetate polymerisations, 3 h at 120 °C, initiated with AIBN with complex:initiator:monomer ratio of 1:0.6:100. Mn,th = [M]0/2[I]0 × MW(monomer) × conversion + MW(catalyst).
|
||||
| R2 = NO2 | 17 | 1850 | 338300 |
1.81 |
| R2 = tBu | 49 | 4260 | 1280 | 1.61 |
| R2 = OMe | 48 | 4130 | 4720 | 1.29 |
| R2 = NMe2 | 50 | 4310 | 4240 | 1.60 |
Complex CoSaltBu,NO2 was an unsuccessful mediator of VAc OMRP, with only small amounts of very high molecular weight polymer isolated. The strong electron-withdrawing substituent makes the Co centre electron-poor and irreversible binding of the vinyl acetate monomer to the complex is likely to occur, with a small amount of thermal polymerisation yielding the observed high molecular weight polymer. As observed with the styrene system, as more electron-donating substituents were incorporated into the CoSaltBu,R2 complexes, monomer conversion increased. However, the substituent effect on polymerisation rate was less profound for vinyl acetate, with complexes where R2 = tBu, OMe and NMe2 all yielding ca. 50% conversion in 3 hours. More interesting was the effect on the molecular weight data, with complex CoSaltBu,tBu yielding molecular weights which were significantly lower than theoretical values. Complexes CoSaltBu,OMe and CoSaltBu,NMe2 both gave poly(vinyl acetate) with molecular weights which were in good agreement with the theoretical values, although the PDI of 1.29 obtained with complex CoSaltBu,OMe suggests that the OMe substituent yields the optimal CoSaltBu,R2 electronic structure out of this series. However, the reported molecular weight represented only 80% of the sample, with a higher molecular weight peak at ca. 97000 Da making up the other 20%. We thought this was likely to be due to an inefficient deactivation process, allowing some chains to propagate in an uncontrolled manner, and so reduced the radical concentration. Using 0.5 equivalents of AIBN produced the same bimodal distribution (Table S2, ESI‡), with PDIs of 1.30 and 1.69 for the low and high molecular weight peaks, but reducing the amount of initiator further did yield a monomodal distribution. With 0.4 eq. of AIBN, conversion was 39% in 3 h (lower radical concentrations resulting in slower polymerisation) and the observed molecular weight of 4300 was in good agreement with the Mn,th of 4765, with the PDI of 1.33 illustrating the reasonable control exerted over VAc OMRP by CoSaltBu,OMe.
Removing the steric bulk of the tBu group at the ortho position of the aromatic ring did not improve the control over VAc polymerisation. Complex CoSalH,tBu behaved similarly to complex CoSaltBu,tBu, reaching 44% conversion in 3 h and yielding PVAc of lower molecular weight than the theoretical value (Mn = 1010, Mn,th = 3780), but the increased PDI of 2.65 (cf. PDI of 1.61 for CoSaltBu,tBu) indicated less control over the polymerisation.
Conclusions
A series of cobalt salen complexes have been prepared and studied by single crystal X-ray diffraction, cyclic voltammetry, X-ray photoelectron spectroscopy, electron paramagnetic resonance spectroscopy and computational methods. Characterisation supports a reactive metal centre tailored by altering ligand electronics. Electrochemistry showed an inverse correlation between redox potentials and electron-donating ability, corroborated by a decrease in the Co 2p3/2 and 2p1/2 binding energies measured by XPS. Tuning the framework was further evident in the theoretical calculations, in particular, the NO2 derivative exhibited the highest Co(III)–carbon bond dissociation energy.These initial results from RT-OMRP using CoSalR1,R2 illustrate the potential of these complexes in controlled radical polymerisation. The data show that the cobalt–carbon bond strength varies with the ligand substitution and, while we could not achieve well-controlled styrene polymerisation under the conditions studied, our current work focuses on degenerative transfer OMRP. Monomers which are susceptible to β-hydrogen abstraction, including styrene, may be successfully polymerised at the lower temperatures used in DT-OMRP and we are particularly interested in studying the effects of our electron-donating and electron-withdrawing ligand substituents on the rate of VAc polymerisation with complexes CoSalR1,R2 under DT-OMRP conditions. The RT-OMRP regime will be used for the synthesis of specific molecular weight, olefin-terminated polymer chains with a wide scope of monomers.
Experimental
Materials and methods
All chemicals used were of the highest grade available and were further purified whenever necessary.76 Literature methods were followed to prepare Schiff-base ligands50–54 H2SaltBu,NO2, H2SaltBu,tBu, H2SaltBu,OMe, H2SaltBu,NMe2, and H2SalH,tBu and Co complexes50,53,55 CoSaltBu,tBu, CoSaltBu,OMe, and CoSalH,tBu Monomers styrene, methyl methacrylate, methyl acrylate and vinyl acetate were purchased from Aldrich Chemical Co. and dried by stirring over calcium hydride for 24 hours, before being vacuum transferred, degassed and stored at −35 °C under inert atmosphere. Azobis(isobutyronitrile), AIBN, was purchased from Aldrich, recrystallised from methanol prior to use and then stored at −35 °C under inert atmosphere. V-70 was purchased from Wako and used as received. 1H NMR and 2-D spectra were recorded at 298 K with a Bruker Avance Spectrometer (300 MHz) in CDCl3. Cyclic voltammetry (CV) was performed on a PAR-263A potentiometer, equipped with a Ag wire reference electrode, a platinum disk working electrode, and a Pt counter electrode with 0.1 M NBu4ClO4 solutions in CH2Cl2. Decamethylferrocene was used as an external standard, and redox processes assigned to the Co complexes were directly referenced to the standard. The redox potential for decamethylferrocene is reported as −0.59 V vs. ferrocene.77 Mass spectra (positive ion) were obtained on an Agilent 6210 TOF ESI-MS instrument. All EPR spectra were collected using a Bruker EMXplus spectrometer operating with a premiumX X-band (∼9.5 GHz) microwave bridge. Low temperature measurements of frozen solutions used a Bruker helium temperature-control system and a continuous flow cryostat. Samples for X-band measurements were placed in 4 mm outer-diameter sample tubes with sample volumes of ∼300 μL. EPR spectra were simulated with EasySpin 4.0.0 software.78 X-ray photoelectron spectra were obtained using a Kratos Analytical Axis ULTRA spectrometer containing a DLD detector. Gel permeation chromatography (GPC) was carried out in THF (flow rate: 1 mL min−1) at 50 °C with a Polymer Labs PL-GPC 50 Plus integrated GPC system with two 300 × 7.8 mm Jordi gel DVB mixed bed columns, utilising a refractive index detector coupled with a Wyatt Technology miniDAWN™ TREOS® multiple angle light scattering (MALS) detector operating at 658 nm. Literature dn/dc values of 0.185, 0.088, 0.063 and 0.052 for poly(styrene),79 poly(methyl methacrylate),80 poly(methyl acrylate)81 and poly(vinyl acetate),79 respectively, were used.Synthesis
:1) to afford long dark red crystals of CoSaltBu,NO2·THF (63 mg, 46% yield). MS-ESI m/z (%): 582.2 (100) [CoSaltBu,NO2]+. Anal. calcd (found) for C28H34N4O6Co·THF: C 58.80 (58.77), H 6.48 (6.61), N 8.57 (8.65).
X-ray analysis
Single crystal X-ray crystallographic analysis of CoSaltBu,NO2 was performed on a Bruker X8 APEX II diffractometer with graphite monochromated Mo-Kα radiation. A dark red needle crystal of C28H34N4O6Co·THF, isolated from the slow evaporation of a THF–pentane solution was mounted on a glass fibre. The data were collected at a temperature of −100.0 ± 0.1 °C to a maximum 2θ value of 55.0°. Data were collected in a series of ϕ and ω scans in 0.50° oscillations with 10.0-second exposures. The crystal-to-detector distance was 36.00 mm. The structure was solved by direct methods.82 The material crystallises with 1 molecules of THF in the asymmetric unit. All non-hydrogen atoms were refined anisotropically. All C–H hydrogen atoms were placed in calculated positions but were not refined. All crystal structure plots were produced using ORTEP-3. A summary of the crystal data and experimental parameters for structure determinations are given in Table S4.‡Calculations
Geometry optimisations were performed using the Gaussian 09 program (Revision A.02),83 the B3LYP functional,84,85 and the 6-31g* basis set on all atoms. Frequency calculations at the same level of theory confirmed that the optimised structures were located at a minimum on the potential energy surface. Single point calculations were performed using the same functional and the TZVP basis set of Ahlrichs86,87 on all atoms. The corresponding orbital transformation (COT) was used to determine the singularly occupied molecular orbital for each of the Co complexes.88,89 AOMix90–92 was used for determining atomic orbital compositions employing Mulliken Population Analysis. Bond dissociation energies were calculated using the BP86 functional,64 and the TZVP basis set following a published procedure.65–67 The XYZ coordinates of the optimised structures are provided in the ESI.‡Polymerisations
:1:0.6 were placed in an ampoule under inert atmosphere. The ampoule was stirred in a preheated oil-bath at the desired temperature for the required length of time, then removed from the heat and cooled quickly under running water. Work-up procedures were dependent on the monomer: poly(styrene), poly(methyl methacrylate) and poly(methacrylate) samples were dissolved in 5 mL of THF and precipitated into 150 mL of acidified methanol (1% HCl). Monomer conversion for these reactions was determined by 1H NMR spectroscopic analysis of crude samples, by comparing the integration of the polymer versus monomer resonances. For poly(vinyl acetate), excess monomer was removed under reduced pressure, the samples were dried to constant mass and then weighed to determine monomer conversion gravimetrically.
Acknowledgements
This work is supported by an NSERC Discovery Grant and the University of Edinburgh (M. S.) and an NSERC Discovery Grant (T. S.). Compute Canada and Westgrid are thanked for access to computational resources.Notes and references
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS.
- L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–93 RSC.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS PubMed.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- D. J. Darensbourg, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, 2012, vol. 245, pp. 1–27 Search PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS PubMed.
- R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- E. N. Jacobsen, W. Zhang and M. L. Guler, J. Am. Chem. Soc., 1991, 113, 6703–6704 CrossRef CAS.
- M. Palucki, N. S. Finney, P. J. Pospisil, M. L. Guler, T. Ishida and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 948–954 CrossRef CAS.
- H. Jacobsen and L. Cavallo, Chem.–Eur. J., 2001, 7, 800–807 CrossRef CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 CrossRef CAS PubMed.
- L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127–156 CrossRef CAS.
- A. Debuigne, R. Poli, C. Jérôme, R. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2009, 34, 211–239 CrossRef CAS.
- F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS.
- M. Hurtgen, C. Detrembleur, C. Jérôme and A. Debuigne, Polym. Rev., 2011, 51, 188–213 CrossRef CAS.
- M. P. Shaver, M. E. Hanhan and M. R. Jones, Chem. Commun., 2010, 46, 2127–2129 RSC.
- M. R. Perry, L. E. N. Allan, A. Decken and M. P. Shaver, Dalton Trans., 2013, 42, 9157–9165 RSC.
- Z. Xue and R. Poli, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3494–3504 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943–7944 CrossRef CAS.
- B. B. Wayland, L. Basickes, S. Mukerjee, M. Wei and M. Fryd, Macromolecules, 1997, 30, 8109–8112 CrossRef CAS.
- Z. Lu, M. Fryd and B. B. Wayland, Macromolecules, 2004, 37, 2686–2687 CrossRef CAS.
- B. B. Wayland, C.-H. Peng, X. Fu, Z. Lu and M. Fryd, Macromolecules, 2006, 39, 8219–8222 CrossRef CAS.
- B. Langlotz, J. Lloret Fillol, J. Gross, H. Wadepohl and L. Gade, Chem.–Eur. J., 2008, 14, 10267–10279 CrossRef CAS PubMed.
- A. Debuigne, J. R. Caille, C. Detrembleur and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 3439–3442 CrossRef CAS PubMed.
- A. Debuigne, J. R. Caille and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 1101–1104 CrossRef CAS PubMed.
- C. Detrembleur, A. Debuigne, R. Bryaskova, B. Charleux and R. Jérôme, Macromol. Rapid Commun., 2006, 27, 37–41 CrossRef CAS.
- V. Sciannamea, A. Debuigne, Y. Piette, R. Jérôme and C. Detrembleur, Chem. Commun., 2006, 4180–4182 RSC.
- S. Maria, H. Kaneyoshi, K. Matyjaszewski and R. Poli, Chem.–Eur. J., 2007, 13, 2480–2492 CrossRef CAS PubMed.
- A. Debuigne, Y. Champouret, R. Jérôme, R. Poli and C. Detrembleur, Chem.–Eur. J., 2008, 14, 4046–4059 CrossRef CAS PubMed.
- H. Kaneyoshi and K. Matyjaszewski, Macromolecules, 2005, 38, 8163–8169 CrossRef CAS.
- M. Hurtgen, A. Debuigne, C. Jérôme and C. Detrembleur, Macromolecules, 2010, 43, 886–894 CrossRef CAS.
- C. Detrembleur, D.-L. Versace, Y. Piette, M. Hurtgen, C. Jerome, J. Lalevee and A. Debuigne, Polym. Chem., 2012, 3, 1856–1866 RSC.
- A. Debuigne, C. Michaux, C. Jérôme, R. Jérôme, R. Poli and C. Detrembleur, Chem.–Eur. J., 2008, 14, 7623–7637 CrossRef CAS PubMed.
- A. Debuigne, J. Warnant, R. Jérôme, I. Voets, A. de Keizer, M. A. Cohen Stuart and C. Detrembleur, Macromolecules, 2008, 41, 2353–2360 CrossRef CAS.
- M. R. Buchmeiser and M. G. Marino, Macromol. Mater. Eng., 2012, 297, 894–901 CrossRef CAS.
- C. Detrembleur, A. Debuigne, O. Altintas, M. Conradi, E. H. H. Wong, C. Jerome, C. Barner-Kowollik and T. Junkers, Polym. Chem., 2012, 3, 135–147 RSC.
- H. Kaneyoshi and K. Matyjaszewski, Macromolecules, 2006, 39, 2757–2763 CrossRef CAS.
- D. N. Bunck, G. P. Sorenson and M. K. Mahanthappa, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 242–249 CrossRef CAS.
- A. Debuigne, N. Willet, R. Jérôme and C. Detrembleur, Macromolecules, 2007, 40, 7111–7118 CrossRef CAS.
- K. S. S. Kumar, Y. Li, Y. Gnanou, U. Baisch, Y. Champouret, R. Poli, K. C. D. Robson and W. S. McNeil, Chem.–Asian J., 2009, 4, 1257–1265 CrossRef PubMed.
- R. K. Sherwood, C. L. Kent, B. O. Patrick and W. S. McNeil, Chem. Commun., 2010, 46, 2456–2458 RSC.
- C.-M. Liao, C.-C. Hsu, F.-S. Wang, B. B. Wayland and C.-H. Peng, Polym. Chem., 2013, 4, 3098–3104 RSC.
- W. H. Leung, E. Y. Y. Chan, E. K. F. Chow, I. D. Williams and S. M. Peng, Dalton Trans., 1996, 1229–1236 RSC.
- I. Sylvestre, J. Wolowska, C. A. Kilner, E. J. L. McInnes and M. A. Halcrow, Dalton Trans., 2005, 3241–3249 RSC.
- Y. N. Belokon, W. Clegg, R. W. Harrington, M. North and C. Young, Inorg. Chem., 2008, 47, 3801–3814 CrossRef CAS PubMed.
- A. Kochem, H. Kanso, B. Baptiste, H. Arora, C. Philouze, O. Jarjayes, H. Vezin, D. Luneau, M. Orio and F. Thomas, Inorg. Chem., 2012, 51, 10557–10571 CrossRef CAS PubMed.
- L. Chiang, A. Kochem, O. Jarjayes, T. J. Dunn, H. Vezin, M. Sakaguchi, T. Ogura, M. Orio, Y. Shimazaki, F. Thomas and T. Storr, Chem.–Eur. J., 2012, 18, 14117–14127 CrossRef CAS PubMed.
- R. I. Kureshy, K. J. Prathap, S. Agrawal, M. Kumar, N. U. H. Khan, S. H. R. Abdi and H. C. Bajaj, Eur. J. Org. Chem., 2009, 2863–2871 CrossRef CAS.
- T. Kurahashi and H. Fujii, Inorg. Chem., 2013, 52, 3908–3919 CrossRef CAS PubMed.
- C. A. Bessel and D. R. Rolison, J. Phys. Chem. B, 1997, 101, 1148–1157 CrossRef CAS.
- V. D. Chaube, S. Shylesh and A. P. Singh, J. Mol. Catal. A: Chem., 2005, 241, 79–87 CrossRef CAS.
- Y.-S. Kim, C.-Y. Lee and G.-J. Kim, Bull. Korean Chem. Soc., 2010, 31, 2973–2979 CrossRef CAS.
- C. Daul, C. W. Schläpfer and A. von Zelewsky, Struct. Bonding, 1979, 36, 129–171 CrossRef CAS.
- E. Vinck, S. Van Doorslaer, D. M. Murphy and I. A. Fallis, Chem. Phys. Lett., 2008, 464, 31–37 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- I. G. Csizmadia, Int. J. Quantum Chem., 1978, 13, 159–159 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- J. Kuta, S. Patchkovskii, M. Z. Zgierski and P. M. Kozlowski, J. Comput. Chem., 2006, 27, 1429–1437 CrossRef CAS PubMed.
- P. M. Kozlowski, J. Kuta and W. Galezowski, J. Phys. Chem. B, 2007, 111, 7638–7645 CrossRef CAS PubMed.
- W. Galezowski, J. Kuta and P. M. Kozlowski, J. Phys. Chem. B, 2008, 112, 3177–3183 CrossRef CAS PubMed.
- G. Li, F. F. Zhang, H. Chen, H. F. Yin, H. L. Chen and S. Y. Zhang, Dalton Trans., 2002, 105–110 RSC.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3660 CrossRef CAS PubMed.
- J. P. A. Heuts and N. M. B. Smeets, Polym. Chem., 2011, 2, 2407–2423 RSC.
- E. Le Grognec, J. Claverie and R. Poli, J. Am. Chem. Soc., 2001, 123, 9513–9524 CrossRef CAS PubMed.
- S. Maria, F. Stoffelbach, J. Mata, J.-C. Daran, P. Richard and R. Poli, J. Am. Chem. Soc., 2005, 127, 5946–5956 CrossRef CAS PubMed.
- M. P. Shaver, L. E. N. Allan, H. S. Rzepa and V. C. Gibson, Angew. Chem., Int. Ed., 2006, 45, 1241–1244 CrossRef CAS PubMed.
- L. E. N. Allan, M. P. Shaver, A. J. P. White and V. C. Gibson, Inorg. Chem., 2007, 46, 8963–8970 CrossRef CAS PubMed.
- R. K. O'Reilly, M. P. Shaver, V. C. Gibson and A. J. P. White, Macromolecules, 2007, 40, 7441–7452 CrossRef.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, New York, 1st edn, 1988 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. Grcev, P. Schoenmakers and P. Iedema, Polymer, 2004, 45, 39–48 CrossRef CAS.
- K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597–1603 CrossRef CAS.
- B. Metin and F. D. Blum, J. Chem. Phys., 2006, 124, 054908–054910 CrossRef PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- D. Herebian, K. E. Wieghardt and F. Neese, J. Am. Chem. Soc., 2003, 125, 10997–11005 CrossRef CAS PubMed.
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis, University of Ottawa, Canada, 2007, http://www.sg-chem.net/ Search PubMed.
- S. I. Gorelsky and E. I. Solomon, Theor. Chem. Acc., 2008, 119, 57–65 CrossRef CAS.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic supplementary information (ESI) available: Full XPS spectra for CoSalR1,R2, DFT metrical parameters, further polymerisation data and summary of crystallographic data. CCDC 948259. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51846a |
| This journal is © The Royal Society of Chemistry 2014 |