 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopentadienyl molybdenum alkyl ester complexes as catalyst precursors for olefin epoxidation†
Nidhi
Grover
,
Alexander
Pöthig
and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center and Department of Chemistry, Technische Universität München, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
First published on 11th July 2014
Abstract
New molybdenum complexes of the type [CpMo(CO)3X] containing ligands of the formula X = CHR2CO(OR1) where R1 = ethyl (1), menthyl (4), and bornyl (5) and R2 = H; R1 = ethyl and R2 = methyl (2) and phenyl (3) have been synthesized and characterized by NMR and IR spectroscopy and X-ray crystallography. These compounds have been applied as catalyst precursors for achiral and chiral epoxidation of unfunctionalized olefins with tert-butyl hydroperoxide (TBHP) as the oxidant at 22 °C (in CH2Cl2) and 55 °C (in CHCl3). The substrates cis-cyclooctene, 1-octene, cis- and trans-stilbene, and trans-β-methylstyrene were selectively and quantitatively converted into their epoxides using a catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate:oxidant ratio of 1:100:200 within 4 h at room temperature in CH2Cl2 and within 15 min at 55 °C in CHCl3. Complexes 1–5 are precursors of active epoxidation catalysts and turnover frequencies (TOFs) of ca. 1200 h−1 are obtained with cis-cyclooctene as the substrate. No enantioselectivity is observed with trans-β-methylstyrene as the substrate despite the application of enantiomerically pure precatalysts. In situ monitoring of catalytic epoxidation of cis-cyclooctene with complex 5 by 1H and 13C NMR spectroscopy suggests that the chiral alkyl ester side chain is retained during oxidation with TBHP. During epoxidation, the primary catalytic species is the dioxo complex [CpMoO2X]. After near complete conversion of cis-cyclooctene to its epoxide, further oxidation of the dioxo complex to oxo–peroxo complex [CpMo(η2-O2)(O)X] takes place. The oxo–peroxo complex is also an active epoxidation catalyst.
:substrate:oxidant ratio of 1:100:200 within 4 h at room temperature in CH2Cl2 and within 15 min at 55 °C in CHCl3. Complexes 1–5 are precursors of active epoxidation catalysts and turnover frequencies (TOFs) of ca. 1200 h−1 are obtained with cis-cyclooctene as the substrate. No enantioselectivity is observed with trans-β-methylstyrene as the substrate despite the application of enantiomerically pure precatalysts. In situ monitoring of catalytic epoxidation of cis-cyclooctene with complex 5 by 1H and 13C NMR spectroscopy suggests that the chiral alkyl ester side chain is retained during oxidation with TBHP. During epoxidation, the primary catalytic species is the dioxo complex [CpMoO2X]. After near complete conversion of cis-cyclooctene to its epoxide, further oxidation of the dioxo complex to oxo–peroxo complex [CpMo(η2-O2)(O)X] takes place. The oxo–peroxo complex is also an active epoxidation catalyst.
Introduction
Since the development of the Halcon–ARCO process for industrial propylene oxide production using molecular Mo(VI) catalysts and organic hydroperoxides in the homogeneous phase, improvements in stability, selectivity and applicability of specialized molybdenum complexes for homogeneous and heterogeneous epoxidation catalysis have been focal points of studies.1 Mononuclear complexes such as [MoO2X2Ln] (X = halide, alkyl, siloxy; L = mono- or bidentate ligand),2–6 [η5-(C5R5)Mo(CO)3X] (R = H, CH3, CH2Ph; X = halide, Me, Et, ansa-bridge, CF3)7–11 and binuclear complexes of the type [(η5-(C5R5)2M2O5] (M = Mo, W)12,13 have been used as epoxidation catalysts with tert-butyl hydroperoxide (TBHP) or H2O2 as the oxidant.13–15 Cyclopentadienyl (Cp) Mo complexes sometimes display activities similar to or even higher than that of the well-examined methyltrioxorhenium (MTO)-H2O2 system.16–20 During the past decade, monomeric CpMo tricarbonyl complexes have been established as suitable precursors for catalytically active molybdenum dioxo or oxo–peroxo complexes, which are formed in situ with organic hydroperoxides after oxidative decarbonylation.21–23 Homogeneous epoxidation activities of CpMo complexes as catalysts have been compared in a recent review10 and other reviews have discussed heterogenization and catalytic applications of the aforementioned category of molybdenum catalysts.24–26Enantiopure epoxides are valuable in organic synthesis and ubiquitous in pharmaceutical, agrochemical and other fine chemical industrial applications.27 Numerous molybdenum-based complexes have been utilized in enantioselective catalysis.28,29 Specifically for epoxidation of unfunctionalized prochiral alkenes, chiral dioxo-molybdenum-based complexes have been extensively studied in both homogeneous and heterogeneous catalysis.28,30 However, the limited enantioselectivity achieved with such complexes is, in general, a consequence of either weakly coordinating chiral ligands or transition states that are symmetrical during oxygen transfer from the oxo–bisperoxo species.31 Although stereoselective epoxidation in the homogeneous phase with readily available Mo catalysts is a lucrative target, only very few examples are reported in the literature and the enantiomeric excess (ee) does not exceed ca. 20% (for trans-β-methylstyrene as the substrate).32 Efforts towards the synthesis of chiral CpMo catalysts mostly involve the introduction of chiral substituents on the Cp ring.32,33 However, as a consequence of the fast rotation of the Cp ring in solution, chiral information is lost, and hence, the ee obtained is very poor. The rotation of the chiral Cp ligand can be suppressed by an ansa-bridge from the Cp ligand to the Mo centre that is coordinated either in a heteroatomic fashion14 or may be σ-C bound.34 In these cases, the chiral centres are located either at the ansa-bridge directly or at substituents at the bridge, which is apparently too far away from the metal to be able to effectively transfer chiral information to the substrate. Royo et al. have investigated a chiral oxazoline-substituted Cp molybdenum complex, which forms a heteroatomic ansa-bridge in order to introduce chiral centres in close proximity to the metal centre.14 However, the oxazoline moiety decoordinates and loss of the Cp ligand during catalysis occurs. Hence, the efficiency of stereoselectivity in catalytic epoxidation with [CpMo(CO)3X] precatalysts also depends on the strength of the Mo–X bond.
In view of the strategic importance of enantiopure epoxides in many industrial endeavors and the efficiency of CpMo complexes in achiral epoxidation,10 complexes 1–5 have been synthesized (Scheme 1). The ligands utilized are alkyl moieties of the type –CHR2–COOR1 (R1 = ethyl (1), menthyl (4), and bornyl (5), R2 = H; R1 = ethyl, R2 = methyl (2) and phenyl (3)) where the chiral information is located at the R1 group in complexes 4 and 5. Compared to the inductive effect of the chloro (electron-withdrawing) and the methyl (electron-donating) group in complexes [CpMo(CO)3Cl] and [CpMo(CO)3Me], respectively, the alkyl ester group renders the metal centre more electron poor than the methyl substituent but less than the chloro substituent since the electron-withdrawing ester group is not directly bound to the metal. This is useful for verifying ligand effects on the Lewis acidity of the metal centre in the precatalyst and establishing a correlation with catalytic activity in the epoxidation reaction. Furthermore, complexes 1–5 are designed to be less active than the chloro and methyl analogues and thus may allow for better thermodynamic and kinetic control during enantioselective epoxidation as well as in situ monitoring of the catalyzed reaction. The presence of the alkyl ester moiety also eliminates the possibility of β-hydrogen elimination decomposition processes, which are possible for complexes where molybdenum is attached to a large alkyl group.35
 | ||
| Scheme 1 Synthesis of cyclopentadienyl molybdenum tricarbonyl alkyl ester complexes 1–5. | ||
In this work, the synthesis, characterization and applications of CpMo complexes 1–5 for epoxidation of unfunctionalized olefins such as cis-cyclooctene, 1-octene, trans- and cis-stilbene and trans-β-methylstyrene with TBHP as the oxidant are reported. In addition, the progress of catalytic epoxidation of cis-cyclooctene has been monitored by 1H and 13C NMR spectroscopy in order to confirm that the chiral side chain remains coordinated during catalysis.
Experimental
Methods and materials
[Mo(CO)6], NaH (60% dispersion in mineral oil), ethyl chloroacetate, ethyl lactate, ethyl mandelate, (–)-borneol, (–)-menthol, tert-butyl hydroperoxide (TBHP, ~5.5 M solution in n-decane), and trans-β-methylstyrene were purchased from Sigma Aldrich. All manipulations involving air-sensitive materials were performed under an argon atmosphere using standard Schlenk techniques and dry solvents. Menthyl and bornyl chloroacetate ligands were synthesized by the reaction of L-(–)-menthol and (–)-borneol with chloroacetyl chloride and N,N-dimethylamine at 0–30 °C. Ethyl lactate mesylate and ethyl mandelate mesylate were also obtained by reaction with mesyl chloride in toluene.36 Ethyl chloroacetate was degassed and dried by the freeze–pump–thaw method and ethyl lactate mesylate and ethyl mandelate mesylate were distilled at low pressures prior to use for the synthesis of complexes 1–5. High-resolution NMR spectra were recorded using a Bruker Avance DPX-400 spectrometer. 1H and 13C spectra are referenced to solvent residual signals37 and 95Mo spectra to an internal standard of 2 M Na2MoO4 in D2O set at 0 ppm. Solid-state NMR spectra were recorded using a Bruker Avance 300 spectrometer at room temperature in a 4-mm ZrO2 rotor at 10 kHz or 12 kHz. Adamantane was used as an external (secondary) standard for referencing TMS (1H: 2.00 ppm, 13C: 29.472 ppm). IR spectra were recorded using a Varian ATR-FTIR instrument. Thermogravimetric analyses were performed with a Netzsch TG 209 system at a heating rate of 10 °C min−1 under argon. Microanalyses were performed in the Mikroanalytisches Labor of the Technische Universität München, Garching. Mass spectra were recorded with Finnigan MAT 311 A and MAT 90 spectrometers. Catalysis was performed under an ambient atmosphere and catalytic runs were monitored using a Varian CP-3800 instrument equipped with an FID, an Optima 5 Amine column (cis-cyclooctene, 1-octene, trans- and cis-stilbene) and an Optima Delta Amine column (trans-β-methylstyrene).X-ray crystallography
Data were collected using a single-crystal X-ray diffractometer equipped with a CCD detector (APEX II, κ-CCD), a rotating anode (Bruker AXS, FR591) with MoKα radiation (λ = 0.71073 Å) and a graphite monochromator. Depending on the complex, either a Montel-type focusing optic (compound 2) or a fine-focus sealed tube (compounds 1 and 5) was used. Collected data were analyzed by using the SMART software package.38 The measurements were performed on single crystals coated with perfluorinated ether. The crystals were fixed on top of a glass fiber and transferred to the diffractometer. The crystals were frozen under a stream of cold nitrogen. A matrix scan was used to determine the initial lattice parameters. Reflections were merged and corrected for Lorenz and polarization effects, scan speed, and background using SAINT.39 Absorption corrections including odd- and even-ordered spherical harmonics were performed using SADABS.39 Space group assignments were based on systematic absences, E statistics, and successful refinement of the structures. Structures were solved by direct methods with the aid of successive difference Fourier maps and refined against all data using the APEX 2 software38,40 in conjunction with SHELXL-9741 and SHELXLE.42 Unless otherwise stated, methyl hydrogen atoms were refined as a part of rigid rotating groups with a C–H distance of 0.98 Å and Uiso(H) = 1.5 × Ueq(C). Other H atoms were placed in calculated positions and refined using a riding model with methylene and aromatic C–H distances of 0.99 and 0.95 Å, respectively, and Uiso(H) = 1.2 × Ueq(C). If not mentioned otherwise, non-hydrogen atoms were refined with anisotropic displacement parameters. Full-matrix least-squares refinements were carried out by minimizing ∑w(Fo2 − Fc2)2 with the SHELXL-9741 weighting scheme. Neutral atom scattering factors for all atoms and anomalous dispersion corrections for non-hydrogen atoms were taken from the International Tables for Crystallography.43 Images of the crystal structures were generated by PLATON.44 Full refinement was straightforward for complex 1, while the ethyl moieties in 2 were refined using split-layer positions. Due to physically meaningless ADPs, the following restraints were applied: SIMU for C1 > C5 (Cp moiety) and ISOR for C10 of complex 5.Crystallographic data (excluding structure factors) for the structures have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-934898 (1), CCDC-934899 (2) and CCDC-934900 (5).
General procedure for synthesis of 1–5
Na[CpMo(CO)3] (1 equiv.) was prepared by refluxing Mo(CO)6 and NaCp in freshly distilled, dry THF overnight. The yellow-orange oily residue obtained after removing THF in vacuo was purified by washing with cold, dry Et2O (3 × 10 mL). 40 mL of dry THF was then added followed by dropwise addition of a degassed solution of a chloro or a mesylate reagent (1.05 equiv.) in 10 mL of THF under steady argon flow in the dark at −40 °C. The reaction flask was stirred at r.t. in the dark for a suitable time followed by evaporation of the solvent to dryness. The obtained residues were extracted with dry pentane or hexane and concentrated under vacuum. The red-yellow products were then purified by column chromatography (SiO2). Complexes 1–5 eluted as yellow bands after a deep red band of Cp2Mo2(CO)6 using n-pentane:diethyl ether = 9:1. The fractions were concentrated under vacuum and 1–5 were obtained as yellow solids/oil in yields 54–85%.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 93.44 (Cp), 59.37 (COOCH2CH3), 14.74 (COOCH2CH3), −3.77 (Mo–CH2). 95Mo (C6D6) = δ −1546. Elemental analysis calcd. (%): C 43.39, H 3.64; found: C 43.56, H 3.70. IR (cm−1, C6D6) 2026 s (Mo–CO), 1926 vs. (Mo–CO), 1682 w (ester CO).
O), 93.90 (Cp), 59.33 (COOCH2CH3), 23.61 (Mo–CH), 14.68 (COOCH2CH3), 11.32 (Mo–CH–CH3). 95Mo (C6D6) = δ −1484. Elemental analysis calcd. (%): C 45.10, H 4.08, Mo 27.71; found: C 45.72, H 4.23, Mo 25.65. IR (cm−1, C6D6) 2010 s (Mo–CO), 1914 vs. (Mo–CO), 1666 vs. (ester CO).
O), 147.97 (MoCH(COOEt)C (phenyl)), 128.15, 124.74 (phenyl ring C, one signal not clearly observed due to C6D6 solvent residual peaks in the same region), 94.82 (Cp), 59.53 (COOCH2CH3), 21.39 (Mo–CH), 14.53 (COOCH2CH3). 95Mo (C6D6) = δ −1515. Elemental analysis calcd. (%): C 52.96, H 3.95, Mo 23.50; found: C 54.10, H 4.06, Mo 22.43. IR (cm−1, C6D6) 2023 s (Mo–CO), 1930 vs. (Mo–CO), 1689 w (ester CO).
O), 93.44 (Cp), 73.00 (MoCH2(CO)OC*H(menthyl)), 47.78 (CH2C(iPr)C*HO–(CO)–CH2Mo), 41.89 (CH(Me)CH2C*HO–(CO)–CH2Mo), 34.65 (C(iPr)CH2CH2CH(Me)), 31.75 (CH2–CH(Me)–CH2–), 26.65 (CH(CH3)2), 23.82 (C(iPr)CH2CH2CH(Me)), 22.43, 21.16, 16.76 (CH3 groups (menthyl)), −3.36 (Mo–CH2). 95Mo (C6D6) = δ −1553. Elemental analysis calcd. (%): C 54.30, H 5.92, Mo 21.69; found: C 55.36, H 6.28, Mo 21.85. IR (cm−1, C6D6) 2028 s (Mo–CO), 1934 vs. (Mo–CO), 1679 w (ester CO).
O), 93.53 (Cp), 79.27 (Mo–CH2(CO)OC*H(bornyl)), 48.91, 48.04, 45.40 (bridgehead quaternary C of the bornyl moiety), 37.54, 28.56, 27.74 (–CH2– (bornyl)), 19.92, 18.99, 13.98 (CH3 groups (bornyl)), −3.35 (Mo–CH2). 95Mo (C6D6) δ −1555. Elemental analysis calcd. (%): C 54.55, H 5.49, Mo 21.79; found: C 54.81, H 5.62, Mo 21.91. IR (cm−1, C6D6) 2023 s (Mo–CO), 1931 vs. (Mo–CO), 1669 s (ester CO).
O), 93.44 (Cp), 59.37 (COOCH2CH3), 14.74 (COOCH2CH3), −3.77 (Mo–CH2). 95Mo (C6D6) = δ −1546. Elemental analysis calcd. (%): C 43.39, H 3.64; found: C 43.56, H 3.70. IR (cm−1, C6D6) 2026 s (Mo–CO), 1926 vs. (Mo–CO), 1682 w (ester CO).
O), 93.90 (Cp), 59.33 (COOCH2CH3), 23.61 (Mo–CH), 14.68 (COOCH2CH3), 11.32 (Mo–CH–CH3). 95Mo (C6D6) = δ −1484. Elemental analysis calcd. (%): C 45.10, H 4.08, Mo 27.71; found: C 45.72, H 4.23, Mo 25.65. IR (cm−1, C6D6) 2010 s (Mo–CO), 1914 vs. (Mo–CO), 1666 vs. (ester CO).
O), 147.97 (MoCH(COOEt)C (phenyl)), 128.15, 124.74 (phenyl ring C, one signal not clearly observed due to C6D6 solvent residual peaks in the same region), 94.82 (Cp), 59.53 (COOCH2CH3), 21.39 (Mo–CH), 14.53 (COOCH2CH3). 95Mo (C6D6) = δ −1515. Elemental analysis calcd. (%): C 52.96, H 3.95, Mo 23.50; found: C 54.10, H 4.06, Mo 22.43. IR (cm−1, C6D6) 2023 s (Mo–CO), 1930 vs. (Mo–CO), 1689 w (ester CO).
O), 93.44 (Cp), 73.00 (MoCH2(CO)OC*H(menthyl)), 47.78 (CH2C(iPr)C*HO–(CO)–CH2Mo), 41.89 (CH(Me)CH2C*HO–(CO)–CH2Mo), 34.65 (C(iPr)CH2CH2CH(Me)), 31.75 (CH2–CH(Me)–CH2–), 26.65 (CH(CH3)2), 23.82 (C(iPr)CH2CH2CH(Me)), 22.43, 21.16, 16.76 (CH3 groups (menthyl)), −3.36 (Mo–CH2). 95Mo (C6D6) = δ −1553. Elemental analysis calcd. (%): C 54.30, H 5.92, Mo 21.69; found: C 55.36, H 6.28, Mo 21.85. IR (cm−1, C6D6) 2028 s (Mo–CO), 1934 vs. (Mo–CO), 1679 w (ester CO).
O), 93.53 (Cp), 79.27 (Mo–CH2(CO)OC*H(bornyl)), 48.91, 48.04, 45.40 (bridgehead quaternary C of the bornyl moiety), 37.54, 28.56, 27.74 (–CH2– (bornyl)), 19.92, 18.99, 13.98 (CH3 groups (bornyl)), −3.35 (Mo–CH2). 95Mo (C6D6) δ −1555. Elemental analysis calcd. (%): C 54.55, H 5.49, Mo 21.79; found: C 54.81, H 5.62, Mo 21.91. IR (cm−1, C6D6) 2023 s (Mo–CO), 1931 vs. (Mo–CO), 1669 s (ester CO).
Epoxidation catalysis
All catalytic investigations were carried out under air either in CH2Cl2, under solvent-free conditions (22 °C) or in CHCl3 (55 °C). Under standardized conditions, 1 mol% or 0.1 mol% catalyst and the substrate were dissolved in 5 mL of solvent. Catalysis was started with the addition of TBHP to the catalyst and substrate reaction mixture. Aliquots were obtained from the reaction flask at suitable time intervals and treated with activated MnO2 to destroy the oxidant followed by filtration through a MgSO4 plug to remove traces of water. Appropriate amounts of external standard solutions (indane and p-xylene for cis-cyclooctene, toluene and mesitylene for 1-octene, hexadecane and octadecane for cis- and trans-stilbene, 1,2,4-trimethylbenzene and tetraline for trans-β-methylstyrene in isopropanol) were then added to the aliquot. The sample was injected into a GC-MS instrument with a column pre-calibrated with r2 = 0.999 to the chosen substrate. Enantiomeric excess (ee) was calculated from the integration of the two peaks relative to the calibration method optimized with pure sample injections of (2S,3S)-2-methyl-3-phenyloxirane and (2R,3R)-2-methyl-3-phenyloxirane.Typical reaction conditions and data acquisition for NMR study of catalytic epoxidation
For catalytic epoxidation of cis-cyclooctene, a mixture of ca. 0.1 mmol of 5 and a known amount of mesitylene was dissolved in 0.4 mL of CDCl3 in an NMR tube and its 1H and 13C spectra were recorded. 10 equiv. of cis-cyclooctene was then added to the NMR tube and after mixing properly, 1H and 13C NMR were measured. Subsequently, 20 equiv. of TBHP (5.5 M n-decane solution) was added at 22 °C and mixed with the precatalyst and substrate solution. It was necessary to shim the magnet again after addition of TBHP for field locking. Using the multizg acquisition program of a Bruker© TopSpin spectrometer, data collection could be automated. The 1H spectrum (16 scans (~1.5 min)) was first recorded at 5 min after addition of TBHP followed by the 13C spectrum (164 scans (~8.5 min)). Thus, in an alternating manner, 1H and 13C spectra were collected at 10-minute intervals for a total time of 4 h.Data analysis for 1H NMR experiments: using NMR software MestReNova©, the characteristic signal of the internal standard mesitylene centred at 6.65 ppm was integrated to 3 H in all 1H NMR spectra. The concentration of oxidized species in each spectrum was then determined by the integral of the Cp signal with correlation for 5 H.
Results and discussion
Synthesis and characterization of complexes 1–5
Complexes 1–5 were synthesized in yields of 54–85% by procedures analogous to previous reports for the synthesis of complex 145,46 using Na[CpMo(CO)3] as the metal precursor and either α-chloroesters (complexes 1, 4 and 5) or α-methanesulfonoxyesters (for 2 and 3) as alkylating agents (Scheme 1).47Complexes 1–3 and 5 are yellow solids and complex 4 is obtained as a bright yellow oil after column purification. All complexes are soluble in benzene, toluene, tetrahydrofuran and dichloromethane. In the solid state, they are stable in air and moisture for several hours, but in solution, they are significantly more sensitive and show visible decomposition associated with a colour change from yellow to blue-green and formation of blue residues overnight. Compounds 1–5 can be handled briefly under ambient conditions without any apparent decomposition, but similar complexes are known to be susceptible to either slow photochemical transformation in solution to μ-CO-bridged species48 or rearrangement to the η3-coordinated side chain on the loss of a carbonyl ligand.49 Complexes similar to 2 are known to be susceptible to β-hydrogen elimination50 but in the case described here, decomposition of 2 to an α-alkenyl type of complex was not observed. These complexes are stable for over a year when stored in the dark under argon at −30 °C. While complexes 2 and 3 decompose at 150 °C and 110 °C, respectively, Mo complexes 4 and 5 bearing menthyl and bornyl moieties decompose at 210 °C and 205 °C, respectively, as shown by TGA-MS measurements. Although elemental analyses of complexes 2, 3 and 4 give poor results, solution 1H, 13C and 95Mo NMR and mass spectra (see the ESI†) do not indicate any impurities. Formation of Mo oxides is a possibility under combustion analysis conditions, which is a probable cause for deviation from calculated results.
NMR spectroscopy
1H NMR spectra of compounds 1–5 (in C6D6) show the C5H5 ligand in the range 4.48–4.70 ppm and 93.44–94.82 ppm in 13C NMR (Table 1). These chemical shift values for the Cp ligand are in the range observed for other structurally similar [CpMo(CO)3X] complexes (where X = Cl and CH3). The deshielding effect of the ester group on Mo–CH2 protons is partly offset by the metal centre, which is coordinated to three backbonding CO ligands, and therefore, these protons appear upfield (1.88–2.16 ppm) for 1, 4 and 5. On the other hand, the Mo–αCH signals of 2 and 3 are highly deshielded, appearing downfield at 2.90 and 4.15 ppm, respectively. This deshielding is also reflected in the respective 13C NMR shifts of the α-carbon observed at 21.39 and 23.61 ppm for complexes 2 and 3, respectively, in contrast to the highly upfield Mo–αCH2 signals ranging from −3.35 to −3.78 ppm for 1, 4 and 5.| Complex | 1Ha | 13C{1H}a | 95Moc | ||||
|---|---|---|---|---|---|---|---|
| Cp | Mo–αCH | Cp | Mo–αCH |
C(O)O |
Mo–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O O |
||
| a All signals are referenced to deuterated solvent C6D6δ 7.16 (for 1H) and δ 128.06 (for 13C{1H}). b Ref. 45. c All signals are referenced to 2 M Na2MoO4 in D2O set at 0 ppm. | |||||||
| 1 | 4.68 | 1.84 (s, 2H) | — | −3.94 | — | 227.04, 240.63 | — |
| 1 | 4.62 | 1.88 (s, 2H) | 93.44 | −3.78 | 181.05 | 226.99, 240.63 | −1546 |
| 2 | 4.63 | 2.87–2.93 (q, 1H) | 93.90 | 23.61 | 182.63 | 227.63, 228.28, 241.00 | −1484 |
| 3 | 4.48 | 4.15 (s, 1H) | 94.82 | 21.39 | 178.80 | 228.72, 228.81, 241.35 | −1349 |
| 4 | 4.70 | 2.16 (m, 2H) | 93.44 | −3.36 | 180.71 | 226.77, 226.91, 240.72 | −1553 |
| 5 | 4.66 | 1.93 (m, 2H) | 93.53 | −3.35 | 181.48 | 226.85, 227.00, 240.58 | −1555 |
| [CpMo(CO)3Cl] | 4.62 | — | 95.58 | — | — | 225.21, 242.99 | −887 |
| [CpMo(CO)3CH3] | 4.42 | 0.39 | 92.41 | 1.44 | — | 227.37, 240.49 | −1736 |
In 13C NMR spectra, the carbonyl resonances of complexes 2–5 (in C6D6) appear as three well-resolved peaks, one each for the two syn carbonyls and one for the third trans carbonyl known to appear downfield to the two cis CO.51 Similar to [CpMo(CO)3X] complexes (X = Me and Cl), compound 1 shows no such inequivalence in the chemical shift for the terminal carbonyl groups and only two signals are observed.
Variable temperature 13C NMR studies of 5 in C6D6 demonstrate that the stereoelectronic asymmetry observed in the form of two suitably resolved peaks for the electronically inequivalent ‘cis-CO’ is present even until 70 °C. Coalescence of the two carbonyl signals does not occur even at this high temperature (Fig. S6, ESI†). Solid-state 1H MAS and 13C CPMAS NMR spectra of 5 have been compared to those of [CpMo(CO)3Me] (Fig. S7, ESI†). For complex 5, all three Mo-bound carbonyl groups show chemical shift anisotropy in the solid state and appear at 226.59, 230.16 and 242.25 ppm, in contrast to [CpMo(CO)3Me] where the two cis-CO are equivalent at 230.3 ppm and the third appears at 242.3 ppm. This suggests that the possible fluxional processes, namely, rotation of Cp about the Mo-(η5-Cp) C5 axis, rapid interchangeability equivalence of the square pyramidal basal cis-CO ligands, rotation about the Mo–αC σ bond, and Berry-type pseudorotation52–56 might be slower or restricted probably due to the presence of bulky substituents in compounds 2–5. However, for the [CpMo(CO)3Me] complex, no such inequivalence is apparent and only two carbonyl peaks can be observed in solution and solid-state 13C NMR spectra. Complexes 2–5 are thus examples of monomeric cyclopentadienyl tricarbonyl molybdenum piano-stool complexes where the barriers to various fluxional processes involving Cp and basal ligands are significant even at high temperatures. It is worth noting that the solution NMR spectra of complexes 4 and 5 and the X-ray structure of 5 (Flack parameter 0.02(7), see the ESI†) confirm the enantiopurity of the prepared complexes.
95Mo NMR chemical shifts are regarded as a suitable indicator of the electronic situation or the Lewis acidity of the metal centre.57 The 95Mo chemical shifts of complexes 1, 4 and 5 are similar, seen at ca. −1550 ppm. For complexes 2 and 3, the chemical shifts are observed at −1484 ppm and −1349 ppm, respectively. These lie in between the 95Mo shift of known tricarbonyl complexes [CpMo(CO)3Cl] (at −887 ppm) and [CpMo(CO)3CH3] (at −1736 ppm). The trend in chemical shifts (–Cl compared to the –CH3 complex) can be interpreted to indicate that an electron-withdrawing substituent at the Mo centre shows a downfield shift in comparison with an electron-donating group.57 This implies that the Mo centre is more electron-deficient in compound 3 compared to that in complex 2, which is in agreement with the expected substituent effects (–CH3vs. –C6H5 at α-C in conjunction with the ester functional group).
Vibrational spectroscopy
The carbonyl ester group in 1–5 absorbs in the range of 1666–1690 cm−1, which is typical for complexes of this type.46 The absorption frequencies for the terminal carbonyl groups differ only about 15 cm−1 in 1–5. The chemical shift anisotropy, which is observed in solution and solid-state NMR spectra of metal-bound carbonyl groups, is not clearly evident in the absorption pattern and only two bands are seen in the range of 2023–2028 cm−1 and 1926–1931 cm−1.58 This is not altogether unusual as deviation from the original D4h geometry to the slightly perturbed C3v geometry in piano-stool complexes results in experimental observation of only two bands, A′ and A′′, out of the three possible νCO.Thermogravimetry and mass spectrometry
Thermogravimetric analysis combined with mass spectrometry data for complexes 2–5 indicate that the loss of a Mo-bound carbonyl group (as CO+) initiates their decomposition. This is followed by a nearly simultaneous loss of Cp, the other two carbonyls or the alkyl ester side chain. These transformations are responsible for the complete decomposition of the precatalysts. The propensity of both processes is accentuated when one CO is lost. Mass spectrometry and decomposition points determined from TGA-MS are given in Table 2.| Complex | MIa,b | Base peak | Methodd | Decomposition point (°C)e |
|---|---|---|---|---|
| a % Relative abundance in parenthesis. b CI-MS (+) m/z are M + 1 peaks. c Ref. 46. d CI refers to chemical ionization; FAB refers to fast atom bombardment method. e Determined by TGA-MS under an inert argon atmosphere with Al2O3 correction; temperature gradient 10 K min−1. f Gradual decomposition, triggered by the loss of one CO+. | ||||
| 1 | 333 | 89 | CI | 32.5–33.5c |
| 2 | 348.6 (14.8) | 182.8 | CI (+) | 150 |
| 3 | 410.5 (13.7) | 164.9 | CI (+) | 110f |
| 4 | 444.6 (4.6) | 146.9 | FAB (+) | 210–220f |
| 5 | 442.5 (19.9) | 246.6 | FAB (+) | 205 |
Single-crystal X-ray diffraction
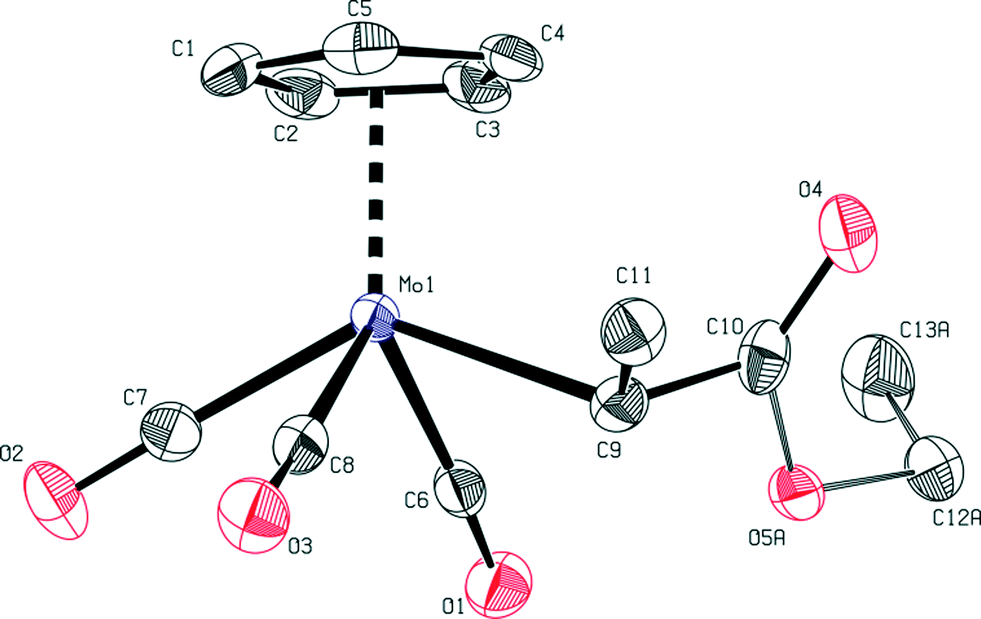
Crystals of complexes 1, 2 and 5 were obtained from a pentane–diethyl ether solvent mixture by slow vapor diffusion and were suitable for single-crystal X-ray diffraction experiments. The crystal structures of these complexes prove indisputably the η1-coordination of the ester side chain (Fig. 1–3). | ||
| Fig. 1 ORTEP view of the single-crystal X-ray structure of compound 1. Thermal ellipsoids are drawn at a 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances [Å], angles [°] and torsion angles [°]: Mo1–C9 2.325(2), Mo1⋯Cp 1.9956(2), Mo1–C6 2.002(3), Mo1–C7 1.990(3), Mo1–C8 2.000(3), C6–O1 1.148(4), C7–O2 1.143(3), C8–O3 1.138(4), Cp–Mo1–C9 109.01(6), Mo1–C9–C10 116.0(2), C6–Mo1–C9 77.7(2), C7–Mo1–C9 136.2(1), C8–Mo1–C9 78.4 (2), Mo1–C9–C10–O4 88.1(3), Cp–Mo1–C9–C10 −177.8(2). | ||
 | ||
| Fig. 2 ORTEP view of the single-crystal X-ray structure of compound 2. Thermal ellipsoids are drawn at a 50% probability level. The hydrogen atoms and the disorder in the ester moiety are omitted for clarity. Selected bond distances [Å], angles [°] and torsion angles [°]: Mo1–C9 2.377(2), Mo1⋯Cp 2.0118(3), Mo1–C6 1.997(2), Mo1–C7 1.997(3), Mo1–C8 2.004(2), C6–O1 1.145(3), C7–O2 1.145(3), C8–O3 1.141(3), Cp–Mo1–C9 110.71(6), Mo1–C9–C10 108.3(2), C6–Mo1–C9 78.9(1), C7–Mo1–C9 134.4(1), C8–Mo1–C9 73.6(1), Mo1–C9–C10–O4 96.3(3), Cp–Mo1–C9–C10 −59.8(2). | ||
 | ||
| Fig. 3 ORTEP view of the single-crystal X-ray structure of compound 5. Thermal ellipsoids are drawn at a 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances [Å], angles [°] and torsion angles [°]: Mo1–C9 2.349(5), Mo1⋯Cp 1.9917(5), Mo1–C6 2.008(5), Mo1–C7 1.983(7), Mo1–C8 1.978(7), C6–O1 1.141(6), C7–O2 1.170(8), C8–O3 1.161(7), Cp–Mo1–C9 111.0(2), Mo1–C9–C10 111.0(4), C6–Mo1–C9 77.6(2), C7–Mo1–C9 132.7(3), C8–Mo1–C9 73.5(2), Mo1–C9–C10–O4 91.4(6), Cp–Mo1–C9–C10 −51.2(4). | ||
The bond length Mo1–C9 (Mo–αC) in the crystal structures of complexes 1, 2 and 5 differs significantly. This bond distance is the shortest for complex 1 (2.325(2) Å) due to the least steric demand in the absence of any α-C substituent, while in complex 2 (2.377(2) Å), the presence of the methyl substituent exerts a higher steric demand and thus the bond length increases. For complex 5, the bond length Mo1–C9 is 2.349(5) Å, which is in between those for 1 and 2. Here, the steric influence is a result of the bulky bornyl ester group even though there is no additional α-C substituent. The Mo–αC bond length in complex [CpMo(CO)3CH3] is 2.326(3) Å,59 which is almost identical to the bond length in complex 1.
All terminal Mo–CO bonds and C–O bond lengths for 1, 2 and 5 are equal (within statistical error) and lie in the expected range. Furthermore, the bond angle Cp–Mo1–C9 is smaller for complex 1 (109.01(6)°) when compared to those for complexes 2 and 5 (110.71(6)° and 111.0(2)°, respectively). The torsion angle Cp–Mo1–C9–C10 is −177.8(2)° in 1, −59.8(2)° in 2 and −51.2(4)° in 5, which indicates that the alkyl ester moiety can rotate freely in 2 and 5 but only a staggered conformation of Cp–Mo1–C9–C10 is possible in complex 1. In addition, the torsion angle C7–Mo1–C9 differs in the three complexes significantly. It has the highest value for 1 (136.2(1)°), which is possibly a consequence of conformation or packing effects, while in complexes 2 (134.4(1)°) and 5 (132.7(3)°), the higher steric demand and the gauche conformation to Mo1–Cp make a close proximity between C9 and CO ligands possible, making the torsion angle smaller than that for 1.
Epoxidation catalysis
All complexes were applied as catalyst precursors for the epoxidation of cis-cyclooctene and 1-octene as well as the prochiral olefins trans- and cis-stilbene and trans-β-methylstyrene with TBHP (5.5 M in n-decane) as the oxidant. Different molar ratios of the catalysts, 1 mol% and 0.1 mol%, were investigated and the ratio of substrate:TBHP = 1:2 was utilized in all reactions, which were carried out at 22 and 55 °C. Catalytic reactions were investigated under air in 5 mL of solvent dichloromethane, chloroform or in the absence of a solvent. For all catalytic reactions, <1% conversion of all substrates to their epoxides was observed in the absence of the molybdenum precatalyst, and similarly, the oxidant alone without the catalyst was ineffective in any appreciable epoxidation of the chosen substrates. Yield and TOF for each catalytic experiment are given in Tables S1 and S2 in the ESI.†
During catalytic epoxidation of cis-cyclooctene with 1–5 and TBHP, an induction period is observed that lasts for 30 min to 2 h depending on the catalyst. This initial time period is attributed to oxidative decarbonylation of the Mo(II) precatalyst to give the catalytically active oxomolybdenum(VI) species (see Fig. 4 and Scheme 2).21,22,60 The concentration of active species present in the reaction mixture is very small in the beginning of the reaction, and therefore, conversion of the substrate to its epoxide is originally also small. Once a critical amount of oxidized species is formed, epoxidation of the substrate becomes quite fast, as indicated by the steep part of the plots in Fig. 4.
 | ||
| Fig. 4 Conversion vs. time plot of different substrates with 1 mol% complexes 1–5 and TBHP oxidant at room temperature in dichloromethane unless otherwise stated: (a) cis-cyclooctene, (b) cis-cyclooctene + 0.1 mol% catalyst, (c) cis-cyclooctene, no co-solvent, (d) cis-cyclooctene, 55 °C, CHCl3 solvent, (e) cis-stilbene, (f) trans-stilbene, (g) 1-octene, (h) trans-β-methylstyrene. Ratio of catalyst:substrate:oxidant = 1:100:200. | ||
 | ||
| Scheme 2 The oxidation of tricarbonyl precatalyst 5 with 10 equiv. of TBHP (in n-decane) results in the formation of both dioxo (I) and oxo–peroxo (II) species at room temperature in CDCl3. | ||
1–5 are active catalysts for the epoxidation of cis-cyclooctene, forming cyclooctene oxide selectively and quantitatively within 2–4 h (1 mol% catalyst) in CH2Cl2 (Fig. 4(a)). Activities in the range of 120–190 cycles per hour are observed for cyclooctene oxide formation, which increase to 230–360 h−1 when 0.1 mol% of the precatalysts is used (Fig. 4(b)). In the absence of a cosolvent, catalytic epoxidation of cis-cyclooctene was accompanied by evolution of heat after the addition of TBHP, indicating that oxidative decarbonylation is exothermic. This is, at least in part, responsible for faster conversions to cyclooctene oxide along with the smaller dilution factor and thus increased TOFs (210–500 h−1) (Fig. 4(c)). At a higher reaction temperature of 55 °C, conversion of cis-cyclooctene to its epoxide is very fast and quantitative yields are obtained within 10 min after addition of the oxidant (Fig. 4(d)). There is no clearly discernible induction period for these catalysis experiments and TOFs are 780 h−1 (1, 4, 5), 1020 h−1 (2) and 1190 h−1 (3). TOFs for catalytic epoxidation of cis-cyclooctene in the case of complexes [CpMo(CO)3Cl]21 and [CpMo(CO)3CH3]11 are 1300 h−1 and 820 h−1, respectively, using a catalyst:substrate:TBHP ratio of 1:100:200 at 55 °C. For catalysis with complexes 2 and 3, within 5 min of addition of TBHP, rapid evolution of gases is observed with a simultaneous increase in temperature over 55 °C. This temperature increase is a result of oxidative decarbonylation of the tricarbonyl complexes and is responsible for the high activity (as indicated by TOFs) of these complexes. During catalysis with complexes 1, 4 and 5, such violent exothermic reactions are not observed; however, due to the high reaction temperature, the conversion of cis-cyclooctene is very fast, and for all complexes, quantitative yield of the epoxide is obtained within 15 min.
The stilbene substrates are selectively transformed to their respective epoxides in yields of up to 50% within 4 h and these yields only marginally increase up to 65% after 24 h (Fig. 4(e) and (f)). A more challenging substrate, 1-octene, is converted to its epoxide slowly, and yields of about 40% are obtained after 24 h with 1 mol% of the catalysts. The conversion of the terminal alkene (1-octene) and aromatic substrates (stilbene, methylstyrene) is both poor and slow relative to cis-cyclooctene. This can be due to deactivation of the primary catalyst before complete epoxidation of these substrates. There is little influence of the increasing steric bulk of the ester alkyl group from ethyl (1, 2 and 3) to menthyl (4) or bornyl (5) on catalytic activity, which is not surprising as the electronic situation at the metal centre is similar for the three α-carbon-unsubstituted precatalysts. In addition, the reaction site is farther from the ethyl group or the sterically encumbered menthyl or bornyl groups located at the end of the oxoalkyl side chain.
Although epoxidation of trans-β-methylstyrene is selective towards the epoxide product, there is negligible (within the experimental error) stereo-differentiation during catalysis and only equimolar amounts of (2S,3S)-2-methyl-3-phenyloxirane and (2R,3R)-2-methyl-3-phenyloxirane are obtained. Poor ee obtained with these complexes can be reasoned to be due to the location of chiral information being still too far away from the reactive metal centre.
Complexes 2 and 3 with a methyl and a phenyl substituent on Mo–αC, respectively, are generally more active than complexes 1, 4, and 5, which are unsubstituted at this position. Furthermore, catalysis with 3 gives a slightly higher yield of epoxides for nearly all substrates tested compared to that with complex 2. This trend may be accounted for by the observation that the molybdenum centre appears slightly more electron-deficient in 2 and 3 compared to those in precatalysts 1, 4 and 5.
NMR study of catalytic epoxidation of cis-cyclooctene
In order to confirm the stability of the Mo–R bond in the chiral catalyst during epoxidation, we followed the progress of catalytic epoxidation of cis-cyclooctene with TBHP using 1H and 13C NMR. Complex 5 was chosen as the precatalyst since its oxidative transformation is slow enough to be studied on a suitable time scale in NMR at room temperature especially in comparison with precatalysts 2, 3, [CpMo(CO)3Cl] and [CpMo(CO)3CH3] for which the reaction was observed to be exothermic. In analogy with previous reports,9,23 it is proposed that the reaction of 5 with TBHP proceeds as illustrated in Scheme 2, and the oxidized complexes I and II are the catalytically active species.A mixture of ca. 0.1 mmol of 5 in CDCl3 and 10 equiv. of cis-cyclooctene was treated with 20 equiv. of TBHP at 22 °C and the reaction progress was monitored by 1H (Fig. 5) and 13C NMR (Fig. 6). Quantitative epoxidation of cis-cyclooctene to its epoxide takes place within 3.5 h, as indicated by the disappearance of the cis-cyclooctene multiplet at 5.68–5.85 ppm. However, complex 5 does not undergo complete oxidative decarbonylation and all three terminal CO signals can be observed even after 4 h in 13C NMR (Fig. 6(a)). This indicates that although only a part of the precatalyst is converted to the active species, the rate of epoxidation is quite high. The signal for Cp of precatalyst 5 at 5.22 ppm and a new signal for the oxidized complex at 6.28 ppm can both be observed after 4 h of monitoring the catalysis reaction, confirming that the Cp ligand is retained after the oxidative transformation.
 | ||
| Fig. 5 1H NMR profile for the reaction of 5 with 10 equiv. of cis-cyclooctene and 20 equiv. of TBHP (in decane) in CDCl3 at 22 °C (with mesitylene as the internal standard). | ||
 | ||
| Fig. 6
13C NMR profile for the reaction of 5 with 10 equiv. of cis-cyclooctene and 20 equiv. of TBHP (in n-decane) in CDCl3 at 22 °C showing spectral regions for (a) terminal CO, (b) alkylester CO, (c) mesitylene (internal standard), (d) Cp ligand, and (e) solvent CDCl3, tert-butanol, epoxycyclooctane, and alkyl moieties CH2 and CH3 of R1 group in complex 5. | ||
In 13C NMR spectra, a prominent signal from the Cp ligand of the oxidized complex (Cp(ox)) is observed at 111.3 ppm, evolving from the Cp signal at 93.4 ppm of the tricarbonyl precatalyst 5 and has been assigned to the dioxo complex I. The proposal outlined in Scheme 2 asserts that in situ oxidation of 5 with TBHP forms complexes I (dioxo) and (later) II (oxo–peroxo) and both are catalytically active for olefin epoxidation. The work presented here supports the possibility of the oxidation shown in Scheme 2 but does not unambiguously support the structures of I and II. A comparison with NMR data of similar complexes21 suggests, however, that this assignment is most likely correct. A small signal at 111.7 ppm can be seen (Fig. 6(d)) when the amount of cyclooctene decreases appreciably at later stages of the reaction and has been assigned to the Cp ligand of the oxo–peroxo complex II based on the 95Mo NMR chemical shift (see discussion below, Fig. S11 in the ESI†) and crystallographic evidence (Table S3 in the ESI†). Since cyclooctene is converted to its epoxide before the amount of the oxo–peroxo complex is significant, it is evident that the rate of oxidation of olefin with the dioxo complex is quite high. Alternatively, this observation suggests that the presence of the olefin (in its role as a reductant) affects the oxidation of the precatalyst, i.e. by suppressing the conversion of I to II.
In Fig. 6(b), the signals of the ester carbonyl carbon are not clearly observed in the period following oxidative decarbonylation (between 15 and 45 min) but they reappear at about 55 min. It is unclear why this might occur since the signals of other quaternary carbon (CO group) can still be observed. The chiral side chain does not dissociate during the catalysis reaction, as evident from a persistent multiplet in the 1H NMR spectrum from 4.6 to 4.76 ppm after 4 h (corresponding to the hydrogen at the bornyl chiral centre from the dioxo complex I) and a signal at 170.8 ppm (for the ester carbonyl) in the 13C NMR spectrum. tert-Butanol is evolved as a side product, appearing as a broad signal in 1H spectra from 2.9 to 3.3 ppm. The complete assignment of observed 1H and 13C NMR chemical shifts is given in Table 3.
:CyOc:TBHP = 1:10:20). R1 = Bornyl, CyOc = cis-cyclooctene, EpCy = epoxycyclooctane
| Complex | 1H NMR, δ (ppm) | 13C NMR, δ (ppm) | Time | ||
|---|---|---|---|---|---|
|
a See ‘Bornyl-HC–O–C(O)’ in Fig. 5 for changes in the observed multiplet of this proton.
b Chemical shift changes due to the change in polarity.
c Observed after 3 h when epoxidation of cyclooctene is complete.
|
|||||
| 5 | Cp | 5.22 | Cp | 93.4 | 0–4 h |
| C(O)OCH | 4.93–4.99 | Mo–CO | 226.14, 226.36, 239.67 | ||
| (OC)OR1 |
182.3 → 184.0b | ||||
| I | Cp | 6.28 | Cp | 111.3 | 35 min–4 h |
| C(O)OCH | 4.6–4.76a | (OC)OR1 |
176.16 | ||
| II | Cp | 6.27 | Cp | 111.7c | 175 min–4 h |
| C(O)OCH | (OC)OR1 |
171.4c | |||
| CyOc | –HCCH– |
5.68–5.85 | –HCCH– |
130.0 | 0–165 min |
| EpCy | –HC–(O)–CH– | 2.64–2.75 | –HC–(O)–CH– | 55.5 | 15 min–4 h |
The concentration vs. time plots illustrated in Fig. 7 follow the progress of catalytic epoxidation and the changes in the concentrations of the substrate, precatalyst 5 and dioxo complex I. From a starting concentration of [5] = 0.249 M, the amount of 5 present in solution after 3.5 h left unreacted is 0.079 M (31.7%) and the concentration of the catalytically active dioxo species is 0.083 M (~33.3%). This suggests that in situ generation of catalytically active complexes is not very efficient. Alternatively, since about one-third of the precatalyst is left unreacted during catalytic epoxidation, these results suggest that the conversion of the cyclooctene substrate to its epoxide takes precedence over a complete oxidative decarbonylation of the precatalyst.
 | ||
| Fig. 7 Concentration vs. time plots for (a) catalytic epoxidation of cis-cyclooctene with TBHP and (b) the concentration of precatalyst 5 and the catalytically active oxidized complex (I) during epoxidation. | ||
In an attempt to evaluate how such homogeneous catalysts perform in subsequent catalytic runs without isolating the active oxo complex, we added the substrate after treating the precatalyst with TBHP. The 95Mo NMR spectrum of the reaction mixture on treating 5 with 50 equiv. of TBHP in CDCl3 shows a broad signal at −628 ppm (Fig. S11 in the ESI†). This chemical shift is similar to 95Mo signals of other [CpMo(O)(O2)R] complexes (R = CH3, −609 ppm; R = CF3, −709 ppm).9,23 For this reason, the persistent Cp signal at 6.27 ppm in the 1H NMR spectrum and 111.7 ppm in the 13C NMR spectrum is assigned to the oxidized species (II). After 48 h, cis-cyclooctene (10 equiv.) was added into the NMR tube containing the pre-oxidized complex (II). The concentration of the oxidized complex available for epoxidation of the substrate was determined to be ca. 0.054 M. This amount is ~50% less than that present after 4 h of oxidation of the precatalyst (ca. 0.11 M), indicating that either II is slowly transformed into another species or it undergoes decomposition. Complex II also catalyses the transformation of cis-cyclooctene to its epoxide, although conversion occurs gradually (Fig. S12 in the ESI†). This may be attributed to the auto-retardation effect of tert-butanol, which is present in the reaction mixture after in situ oxidation of the precatalyst and/or lower activity of the oxo–peroxo species relative to the dioxo complex.
Accordingly, it can be confirmed that complex II (oxo–peroxo) is also an active catalyst for olefin epoxidation.
Conclusion
Cyclopentadienyl molybdenum complexes with different oxoalkyl side chains have been synthesized and investigated for achiral and chiral epoxidation catalysis. The chiral ligands for the synthesized complexes are derived from cheap and readily available chiral pool compounds. A comparison between substituted and unsubstituted complexes at the α-carbon to molybdenum is presented. The former are more active for the epoxidation of olefins on account of a more electron-deficient metal centre and 3 is a better catalyst for nearly all substrates. 1–5 display moderate activities in epoxidation catalysis at room temperature compared to many other half-sandwich tricarbonyl Mo(II) complexes previously reported, and no stereoselectivity is achieved with prochiral substrates, probably due to the pronounced distance between the chiral centres and the catalytic sites. Nevertheless, due to the somewhat reduced catalytic activity, insight into the progress of the precatalyst oxidation and the epoxide formation is gained. Cyclooctene epoxidation progress with precatalyst 5 and TBHP monitored by NMR indicates that in situ oxidation of 5 with TBHP forms the highly active dioxo complex, and thus, both the Cp ligand and the chiral side chain are retained. However, ca. 32% of precatalyst 5 remain unreacted and catalytic epoxidation takes precedence over complete oxidation of the tricarbonyl precursor. The oxo–peroxo complex is also formed on oxidation of the dioxo complex, albeit only near-complete conversion of the substrate in the epoxidation reaction. The quantitative 1H NMR study also confirms that the oxo–peroxo complex [CpMo(O)(O2)R] obtained through oxidation of the precursor with TBHP is catalytically active for olefin epoxidation.Acknowledgements
N.G. is grateful to the TUM Graduate School of Chemistry for financial support.References
- Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, 2011 Search PubMed.
- S. M. Bruno, B. Monteiro, M. S. Balula, C. Lourenço, A. A. Valente, M. Pillinger, P. Ribeiro-Claro and I. S. Gonçalves, Molecules, 2006, 11, 298–308 CrossRef CAS PubMed.
- A. Günyar and F. E. Kühn, J. Mol. Catal. A: Chem., 2010, 319, 108–113 CrossRef.
- A. M. Al-Ajlouni, A. Günyar, M. Zhou, P. N. W. Baxter and F. E. Kühn, Eur. J. Inorg. Chem., 2009, 2009, 1019–1026 CrossRef.
- F. E. Kühn, A. M. Santos and M. Abrantes, Chem. Rev., 2006, 106, 2455–2475 CrossRef PubMed.
- A. Günyar, M.-D. Zhou, M. Drees, P. N. W. Baxter, G. Bassioni, E. Herdtweck and F. E. Kühn, Dalton Trans., 2009, 8746–8754 RSC.
- A. Capapé, A. Raith and F. E. Kühn, Adv. Synth. Catal., 2009, 351, 66–70 CrossRef.
- C. Freund, W. A. Herrmann and F. E. Kühn, Top. Organomet. Chem., 2007, 22, 39–77 CrossRef CAS.
- S. A. Hauser, M. Cokoja, M. Drees and F. E. Kühn, J. Mol. Catal. A: Chem., 2012, 363–364, 237–244 CrossRef CAS.
- N. Grover and F. E. Kühn, Curr. Org. Chem., 2012, 16, 16–32 CrossRef CAS.
- J. Zhao, A. M. Santos, E. Herdtweck and F. E. Kühn, J. Mol. Catal. A: Chem., 2004, 222, 265–271 CrossRef CAS.
- A. M. Martins, C. C. Romão, M. Abrantes, M. C. Azevedo, J. Cui, A. R. Dias, M. T. Duarte, M. A. Lemos, T. Lourenço and R. Poli, Organometallics, 2005, 24, 2582–2589 CrossRef CAS.
- C. Dinoi, M. Ciclosi, E. Manoury, L. Maron, L. Perrin and R. Poli, Chem. – Eur. J., 2010, 16, 9572–9584 CrossRef CAS PubMed.
- P. M. Reis, C. A. Gamelas, J. A. Brito, N. Saffon, M. Gómez and B. Royo, Eur. J. Inorg. Chem., 2011, 2011, 666–673 CrossRef.
- V. V. Krishna Mohan Kandepi, J. M. S. Cardoso and B. Royo, Catal. Lett., 2010, 136, 222–227 CrossRef.
- T. Michel, M. Cokoja, V. Sieber and F. E. Kühn, J. Mol. Catal. A: Chem., 2012, 358, 159–165 CrossRef CAS.
- P. Altmann, M. Cokoja and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 2012, 3235–3239 CrossRef CAS.
- T. Michel, D. Betz, M. Cokoja, V. Sieber and F. E. Kühn, J. Mol. Catal. A: Chem., 2011, 340, 9–14 CrossRef CAS.
- F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef.
- F. E. Kühn, A. M. Santos and W. A. Herrmann, Dalton Trans., 2005, 2483–2491 RSC.
- M. Abrantes, A. M. Santos, J. Mink, F. E. Kühn and C. C. Romão, Organometallics, 2003, 22, 2112–2118 CrossRef CAS.
- A. A. Valente, J. D. Seixas, I. S. Gonçalves, M. Abrantes, M. Pillinger and C. C. Romão, Catal. Lett., 2005, 101, 127–130 CrossRef CAS.
- A. M. Al-Ajlouni, D. Veljanovski, A. Capapé, J. Zhao, E. Herdtweck, M. J. Calhorda and F. E. Kühn, Organometallics, 2009, 28, 639–645 CrossRef CAS.
- D. Betz, A. Raith, M. Cokoja and F. E. Kühn, ChemSusChem, 2010, 3, 559–562 CrossRef CAS PubMed.
- D. Betz, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2009, 694, 3320–3324 CrossRef CAS.
- K. R. Jain and F. E. Kühn, Dalton Trans., 2008, 2221–2227 RSC.
- Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH Weinheim, 2006 Search PubMed.
- K. R. Jain, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2008, 252, 556–568 CrossRef CAS.
- J. A. Brito, B. Royo and M. Gómez, Catal. Sci. Technol., 2011, 1, 1109–1118 CAS.
- F. E. Kühn, J. Zhao and W. A. Herrmann, Tetrahedron: Asymmetry, 2005, 16, 3469–3479 CrossRef.
- A. J. Burke, Coord. Chem. Rev., 2008, 252, 170–175 CrossRef CAS.
- M. Abrantes, A. Sakthivel, C. C. Romão and F. E. Kühn, J. Organomet. Chem., 2006, 691, 3137–3145 CrossRef CAS.
- M. Abrantes, F. A. A. Paz, A. A. Valente, C. C. L. Pereira, S. Gago, A. E. Rodrigues, J. Klinowski, M. Pillinger and I. S. Gonçalves, J. Organomet. Chem., 2009, 694, 1826–1833 CrossRef CAS.
- J. Zhao, E. Herdtweck and F. E. Kühn, J. Organomet. Chem., 2006, 691, 2199–2206 CrossRef CAS.
- J. K. P. Ariyaratne, A. M. Bierrum, M. L. H. Green, M. Ishaq, C. K. Prout and M. G. Swanwick, J. Chem. Soc. A, 1969, 1969, 1309–1321 RSC.
- L. R. Hillis and R. C. Ronald, J. Org. Chem., 1981, 46, 3348–3349 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- APEX suite of crystallographic software. APEX 2 Version 2008.4, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- SAINT, Version 7.56a and SADABS Version 2008/1, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
- C. B. Huebschle, G. M. Sheldrick and B. Dittrich, SHELXLE, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef CAS PubMed.
- International Tables for Crystallography, Vol. C, Tables 6.1.1.4 (pp. 500–502), 4.2.6.8 (pp. 219–222), and 4.2.4.2 (pp. 193–199), ed. A. J. C. Wilson, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1992 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed.
- E. R. Burkhardt, J. J. Doney, R. G. Bergman and C. H. Heathcock, J. Am. Chem. Soc., 1987, 109, 2022–2039 CrossRef CAS.
- R. B. King, M. B. Bisnette and A. Fronzaglia, J. Organomet. Chem., 1966, 5, 341–356 CrossRef CAS.
- T. S. Piper and G. Wilkinson, J. Inorg. Nucl. Chem., 1956, 3, 104–124 CrossRef CAS.
- R. B. King, J. Organomet. Chem., 1975, 100, 111–125 CrossRef CAS.
- R. B. King and A. Fronzaglia, J. Am. Chem. Soc., 1966, 88, 709–712 CrossRef CAS.
- J. K. P. Ariyaratne and M. L. H. Green, J. Chem. Soc., 1964, 1–5 RSC.
- L. J. Todd, J. R. Wilkinson, J. P. Hickey, D. L. Beach and K. W. Barnett, J. Organomet. Chem., 1978, 154, 151–157 CrossRef CAS.
- R. Mynott, H. Lehmkuhl, E.-M. Kreuzer and E. Joussen, Angew. Chem., Int. Ed. Engl., 1990, 29, 289–290 CrossRef.
- R. F. Jordan, E. Tsang and J. R. Norton, J. Organomet. Chem., 1978, 149, 53–56 CrossRef.
- J. W. Faller, A. S. Anderson and C.-C. Chen, J. Chem. Soc. D, 1969, 719–720 RSC.
- R. Poli, Organometallics, 1990, 9, 1892–1900 CrossRef CAS.
- J. M. Smith and N. J. Coville, Organometallics, 1996, 15, 3388–3392 CrossRef CAS.
- C. G. Young, M. Minelli, J. H. Enemark, G. Miessler, N. Janietz, H. Kauermann and J. Wachter, Polyhedron, 1986, 5, 407–413 CrossRef CAS.
- R. B. King and L. W. Houk, Can. J. Chem., 1969, 47, 2959–2964 CrossRef CAS.
- M. Abrantes, P. Neves, M. M. Antunes, S. Gago, F. A. Almeida Paz, A. E. Rodrigues, M. Pillinger, I. S. Gonçalves, C. M. Silva and A. A. Valente, J. Mol. Catal. A: Chem., 2010, 320, 19–26 CrossRef CAS.
- M. K. Trost and R. G. Bergman, Organometallics, 1991, 10, 1172–1178 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 934898–934900. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cy00738g |
| This journal is © The Royal Society of Chemistry 2014 |