
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An integrated strategy for the conversion of cellulosic biomass into γ-valerolactone†
Valerio
Molinari
,
Markus
Antonietti
and
Davide
Esposito
*
Max-Planck-Institute of Colloids and Interfaces, Department of Colloid Chemistry, 14424 Potsdam, Germany. E-mail: davide.esposito@mpikg.mpg.de; Tel: (+49) 0331 567 9538
First published on 11th August 2014
Abstract
An integrated method for the production of γ-valerolactone from cellulosic biomass is presented here. A combination of acidic water hydrolysis of the biomass followed by extraction with methyltetrahydrofuran is used to generate a levulinic acid feed, which is further hydrogenated into the platform chemical γ-valerolactone using RANEY® nickel as a catalyst.
Fuels and chemicals produced in a sustainable fashion from non-edible lignocellulosic biomass are currently considered as a promising alternative to fossil feedstocks.1 A variety of different molecules have been identified as possible high value products of the catalytic upgrade of biomass. One of the illustrative examples in this direction is represented by γ-valerolactone (GVL),2 a cyclic ester with optimal properties as a fuel additive3 and solvent4 and as a precursor for different platform chemicals and fuels.3,5 GVL can be produced by hydrogenation of levulinic acid (LA), obtained in turn by hydrolysis of the cellulosic portion of raw biomass.6 Several hydrogenation schemes relying on the use of different heterogeneous catalysts have been proposed for the conversion of LA into GVL.7–11 In general, Ru-based catalysts have shown the highest efficiency, followed by other transition metal-based catalysts containing Pd, Pt, Re or RANEY® nickel.2,12–16 Since LA is usually produced by acidic hydrolysis of biomass with mineral acids (sulfuric acid in particular), novel preparation of catalysts with increased tolerance towards acids and poisoning agents like sulfur has been attempted5 in order to enable the direct processing of biomass-derived LA. Despite the success, the stability of such catalysts still requires additional improvements to become compatible with high-throughput conversion schemes. Alternative solutions have been proposed to bypass this problem; for instance, reactive extraction has been used to convert LA into different aliphatic esters,17,18 with the drawback of adding a synthetic step to the process. Alternatively, extraction with alkylphenol solvents has been proposed to selectively separate LA from the water phase,19 although in this case, the use of an environmentally questionable solvent was required. Ideally, the use of a sustainable and green solvent to replace the alkylphenol could tremendously improve such an approach. In addition, in order to favor the industrial application of these methodologies, the exploitation of rare and expensive transition metals for the preparation of the hydrogenation catalysts should be avoided, while the development of catalytic schemes based on cheap and widely available metals should be encouraged.
In this work we propose an efficient integrated strategy to convert biomass into GVL, which is based on the use of the biomass-derived solvent 2-methyltetrahydrofuran (2-MeTHF) to extract LA from aqueous feeds obtained by acidic hydrolysis of lignocellulosic materials. The so-obtained organic feed is thus subjected to a continuous-flow hydrogenation catalyzed by the commercial and relatively economical20 RANEY® nickel.
The suggested integrated scheme for the conversion of lignocellulosic carbohydrates into GVL is depicted in Fig. 1.
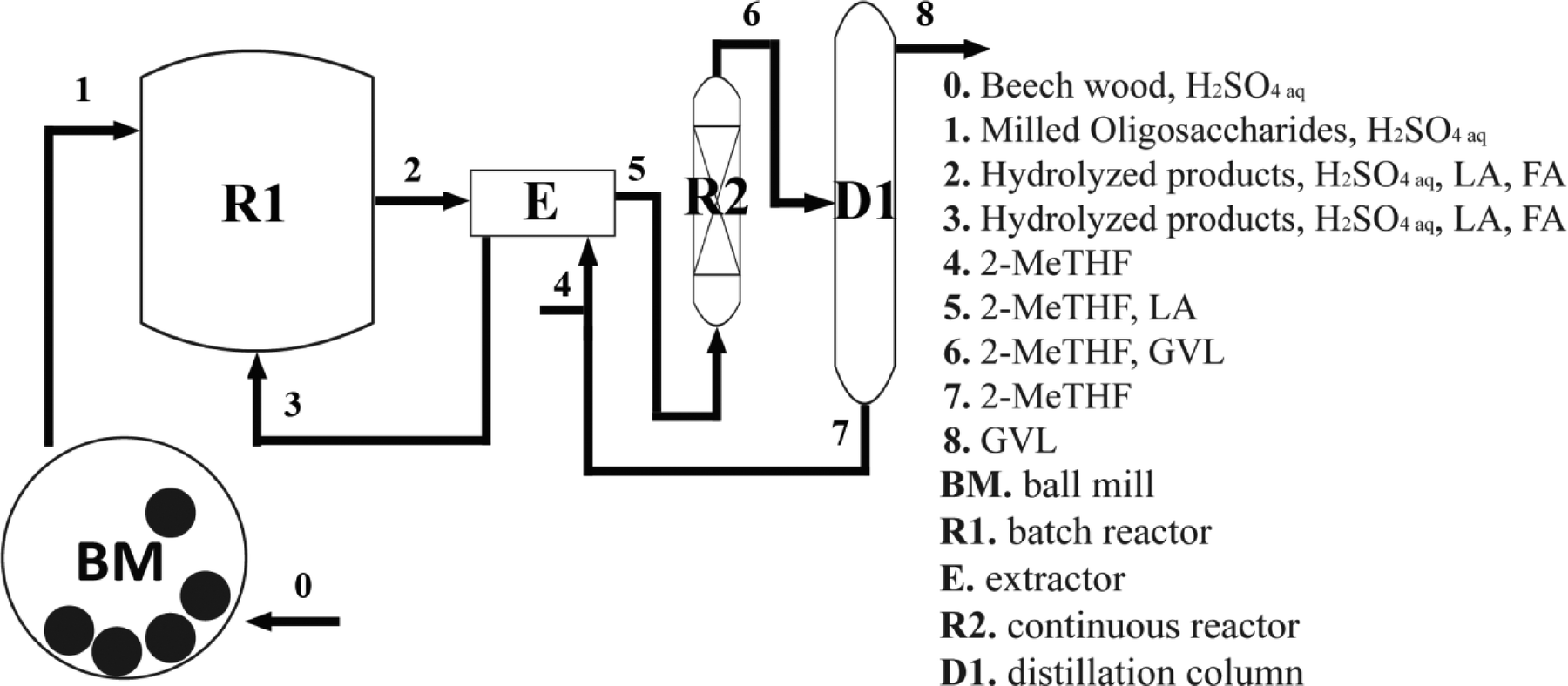 | ||
| Fig. 1 Schematic representation of the integrated strategy for the multistep synthesis of GVL from lignocellulosic biomass. | ||
Initially, following a previously published route,21 biomass is conveniently pretreated by ball milling to obtain water-soluble partially depolymerized lignocellulosic fragments. Then, a batch reactor is employed to perform carbohydrate hydrolysis using sulfuric acid as the catalyst. The water phase can be introduced into an extractor after separation of the by-products derived from the decomposition of carbohydrates. Extraction of LA using 2-MeTHF regenerates the water phase, which can be recycled for iterative hydrolysis steps, while producing a LA-containing organic feed ready to be introduced in the continuous reactor R2. Here, hydrogenolysis with RANEY® Ni yields GVL that can be conveniently separated from 2-MeTHF by distillation. 2-MeTHF is distilled as a stable azeotrope with water,22 which is still suitable for further reactions.
We commenced our studies by evaluating the possible hydrogenation of LA using commercial RANEY® nickel. Interestingly, the successful use of RANEY® nickel for the hydrogenation of ethyl levulinate to GVL has been recently reported on simulated feeds.14 In contrast, impurities and poisoning agents contained in biomass-derived LA were not tolerated. It is worth noting that the use of RANEY® Ni for the direct hydrogenation of LA in batch has also been proposed,12,13 although the authors reported very low efficiency under moderate conditions, whereas adequate preparative conditions were attained at high temperatures (175–200 °C) and pressure (35 bar).
We believe that such findings could be due to a non-optimized reaction setup, while milder reaction conditions could be obtained by careful engineering of the reaction parameters in a continuous reactor. In this direction, a recent work proposed the continuous synthesis of γ-valerolactone exploiting RANEY® nickel as a catalyst and water as solvent, obtaining interesting results.23 We therefore investigated the use of a continuous system but focused on the use of organic solvents, which integrate better in our strategy. In particular, we screened the effect of temperature and solvent on the reactions using a fixed-bed reactor equipped with a packed catalyst column (0.9 g of wet RANEY® Ni) and supplied with LA and H2 feeds (for details see ESI†). The results are summarized in Table 1.
| Entry | Solvent | T (°C) | Conversion (%) |
|---|---|---|---|
| a Reaction conditions: 0.9 g RaNi (wet), 12 bar, 0.3 mL min−1, 50 mM LA in EtOH, EtOH/water and 2-MeTHF. | |||
| 1 | EtOH | 115 | 57 |
| 2 | EtOH | 125 | >99 |
| 3 | EtOH | 135 | >99 |
| 4 | EtOH/H2O | 125 | >99 |
| 5 | 2-MeTHF | 125 | >99 |
LA was already fully converted into GVL at moderate conditions (entries 1–4). We continued to explore the possible use of other solvents characterized by a low environmental footprint and we focused our attention on 2-MeTHF, a biomass-derived solvent,22 which can be in fact obtained by reduction of GVL.24,25 Interestingly, during hydrogenation of LA in ethanol and ethanol–water, we did not observe the formation of 2-MeTHF, which probably requires more severe conditions. The use of 2-MeTHF as the reaction solvent (entry 5) yielded GVL with full conversion and selectivity under the same conditions applied in the case of ethanol and ethanol–water. Having demonstrated the viability of reducing LA in flow using RANEY® Ni under moderate conditions, we proceeded to evaluate a possible strategy to extend this approach to LA feeds extracted from an acidic hydrolysate of the lignocellulosic material. As mentioned above, one of the major problems associated with the hydrogenation of LA in biomass-derived feeds is the presence of mineral acids and impurities which result in the deactivation of the catalyst.
The results obtained with 2-MeTHF (Table 1, entry 5) therefore appeared particularly attractive since this solvent could be used in principle to extract LA from acidic aqueous solutions. For instance, exploiting a continuous extraction system would enable accumulation of LA in the organic phase and potentially lead to the recycling of the aqueous sulfuric acid for iterative production of LA. Interestingly, 2-MeTHF has been recently exploited for biomass processing in water-containing biphasic mixtures to extract apolar lignin fragments from the aqueous phase.26
To assess the feasibility of such an approach, we performed experiments to determine the partition ratio (Kd) for LA in the system 2-MeTHF/H2SO4 (0.5 M) (Fig. S4†). The estimated Kd resulted in a value of ca. 1.57, anticipating the possible extraction of LA using 2-MeTHF.
We performed the reduction of LA using a simulated feed obtained by partitioning a water solution of LA in 0.5 M sulfuric acid with 2-MeTHF (Table 2, entries 1–3). The efficiency of the resulting reaction was unaffected after the extraction (>99% conversion, corresponding to a GVL yield of 96%, based on GC analysis) independent of the temperature, indicating that no significant amounts of sulfuric acid were carried into the organic feed. At this point, the robustness of the reduction step was challenged by processing real levulinic acid feeds obtained by acidic hydrolysis of biomass models and lignocellulosic biomass in order to take into account the presence of possible additional poisoning agents that originated during sugar depolymerization. While glucose was used initially as a model substrate, we later focused our attention on real biomass. The direct use of biomass is nevertheless complicated by the relative hydrolytic stability and low solubility of cellulose and lignin. To overcome this problem, several techniques have been recently applied to process biomass into water-soluble partially depolymerized cellulosic fragments. Among others, the mechano-catalytic depolymerization of lignocellulose has been successfully exploited as an entry process for the preparation of oligosaccharides, alcohols and furfurals.21,27,28 The main benefits of this technique reside in the efficient comminution of the substrate, which makes it compatible with various reaction setups, and the simultaneous partial depolymerization of polysaccharides and lignin, which facilitates the successive hydrolytic treatments. We performed our experiments using water-soluble oligosaccharides prepared by mechano-catalytic processing of cellulose and beechwood. LA was synthesized following known procedures6,29 treating the desired precursor in 0.5 M aqueous sulfuric acid at 220 °C in autoclaves for 8 hours. Reaction time was not optimized further, while a range of concentrations between 30 and 50 mg mL−1 was explored without observing significant changes in the yield. The obtained water phase was thus filtered to remove the humins and the insoluble products generated during the hydrolysis.30 LA was thus extracted with 2-MeTHF, and the resulting concentration, depending on the initial concentration of biomass used for the hydrolysis, was measured by chromatographic methods. Formic acid is a known co-product of the hydrolysis of sugars into levulinic acid.25 Although the formic acid potentially extracted in the organic phase can undergo transfer hydrogenation processes,31,32 its presence was neglected due to the excess of hydrogen applied during hydrogenation. Three different standardized feeds obtained from glucose, milled cellulose, and milled wood were supplied to the continuous reactor, and the effect of temperature and flow rate on the conversion was analyzed.
| Hydrolysis | Hydrogenation | |||||
|---|---|---|---|---|---|---|
| Hydrolysis conditions: substrate in 0.5 M H2SO4, 220 °C, 8 h. Hydrogenation conditions: LA in 2-MeTHF, RANEY® nickel, H2, 12 bar.a Obtained by partitioning a LA solution in 0.5 M H2SO4 with 2-MeTHF.b Pretreated via ball milling with catalytic H2SO4.c Determined by HPLC analysis; reactions were carried out multiple times and yields were averaged.d Determined by HPLC analysis measuring the difference in the concentration of LA in the water phase before and after extraction with 2-MeTHF.e Determined by GC-MS analysis. | ||||||
| Entry | Substrate | LA yield (w/w %)c | Feed conc. (mM) | Flow (mL min−1) | T (°C) | Conversion (%)e |
| 1 | Simulated feeda | 52d | 0.3 | 125 | >99 | |
| 2 | Simulated feed | 52 | 0.5 | 135 | 98 | |
| 3 | Simulated feed | 52 | 1.0 | 150 | >99 | |
| 4 | Glucose | 32c | 52d | 0.3 | 125 | >99 |
| 5 | Glucose | — | 52 | 0.4 | 125 | 87 |
| 6 | Glucose | — | 52 | 0.5 | 125 | 64 |
| 7 | Glucose | — | 52 | 1.0 | 125 | 54 |
| 8 | Glucose | — | 52 | 0.3 | 135 | >99 |
| 9 | Glucose | — | 52 | 0.4 | 135 | 98 |
| 10 | Glucose | — | 52 | 0.5 | 135 | 85 |
| 11 | Glucose | — | 52 | 1.0 | 135 | 64 |
| 12 | Celluloseb | 23c | 36d | 0.5 | 125 | >99 |
| 13 | Cellulose | — | 36 | 0.7 | 125 | 66 |
| 14 | Cellulose | — | 36 | 1.0 | 125 | 36 |
| 15 | Beechwoodb | 13c | 40d | 0.5 | 125 | 93 |
| 16 | Beechwood | — | 40 | 0.7 | 125 | 81 |
| 17 | Beechwood | — | 40 | 1.0 | 125 | 49 |
| 18 | Beechwood | — | 40 | 0.5 | 135 | >99 |
| 19 | Beechwood | — | 40 | 1.0 | 135 | 88 |
Hydrolysis of glucose afforded LA in 32% weight yield, ca. 50% on a molar basis (Table 2, entry 4). Hydrogenation of a 2-MeTHF extract using RANEY® nickel proceeded smoothly, already yielding to full conversion at 125 °C (Table 2, entry 4). Increasing the flow rate (entries 5, 6 and 7) resulted in a progressive decrease in the conversion. This effect could be compensated for by increasing the reaction temperature (entries 8–11). Interestingly, these experiments showed that the use of a feed obtained from real biomass does not affect the efficiency of the conversion of LA into GVL.
Performing hydrolysis of water-soluble oligosaccharides (WSO) obtained by mechano-catalytic depolymerization of cellulose with sulfuric acid33 (entry 12) produced LA with yields that are similar to the ones obtained from glucose, confirming the efficiency of ball milling for biomass pretreatment.
Hydrogenation of 32 mM LA extracted in 2-MeTHF afforded GVL quantitatively at 125 °C with a flow rate of 0.5 mL min−1.
In this case, the advantage of working with dilute LA feeds was clearly visible, since in comparison, the use of a 48 mM solution of LA and a volumetric flow of 0.5 mL min−1 resulted in only 64% conversion at 125 °C (entry 6). Direct hydrolysis of milled beechwood as an exemplary real biomass afforded LA in 13% yield (w/w). Considering the composition of beechwood, which contains ca. 41 wt% glucans (see the ESI†), this corresponds to a LA yield of 44% on a molar basis.
HPLC analysis of the water phase revealed the presence of LA, FA and acetic acid together with additional minor impurities. Insoluble products containing humins and lignin fragments were isolated at the end of the reaction by filtration. Extraction of a concentrated beechwood hydrolysate with 2-MeTHF was used to prepare a 40 mM LA feed. Interestingly, GC analysis of this solution revealed the presence of the sole LA in 2-MeTHF, indicating that the extraction proceeds in a very selective manner. Hydrogenation was performed at different temperatures and full conversion could be achieved at 135 °C with a flow of 0.5 mL min−1 (entry 18), in line with the trend observed for glucose- and cellulose-derived LA feeds. It is worth noting that the efficiency of LA hydrogenation proved to be independent of the nature of the precursor exploited, whereas the feed concentration played a determining role. The stability of RANEY® Ni as a function of time was evaluated showing that the catalyst retains its activity up to 130 hours on stream with the values for the conversion of LA into γ-valerolactone remaining stable (Fig. S6†). The efficiency of the proposed integrated scheme relies on the possibility of recycling the acidic aqueous phase for iterative hydrolytic steps. In order to verify this condition, we recycled the acidic water phase for the hydrolysis of glucose after extraction with 2-MeTHF over 4 cycles without observing a substantial decrease in the yield of LA (Fig. 2, red bars). Similarly, recycling of the water phase after hydrolysis of beechwood over four cycles did not compromise LA formation (green bars). It is worth noting that performing recycling experiments after extraction with 2-MeTHF prevented the formation of insoluble precipitates. In fact, at the end of the reaction we observed the formation of a biphasic system composed of water and a less dense organic layer. We attributed this to the presence of 2-MeTHF (ca. 7–8%) dissolved in the water phase after extraction. A control experiment performed by treating a 7% solution of 2-MeTHF in 0.5 M H2SO4 at 220 °C for 8 hours resulted in the formation of a similar bio-oil. GC-MS analysis revealed a complicated mixture of different aliphatic and aromatic molecules, probably originating through non-selective decomposition pathways involving 2-MeTHF. In addition, gel permeation chromatography and ESI mass analysis of a bio-oil obtained after hydrothermal treatment of beechwood during recycling experiments revealed the presence of molecular species with mass in the range of 500–100 g mol−1. We surmised that such a “bio-oil” could be responsible for the solubilization of the solid by-products otherwise observed during the acidic hydrothermal treatment of the biomass, including lignin fragments corresponding to different degrees of depolymerization.
 | ||
| Fig. 2 Levulinic acid yield per cycle during recycling experiments of the acidic aqueous phase for the hydrolysis of glucose and beechwood. | ||
Conclusions
In conclusion, we described a convenient integrated strategy for the conversion of biomass into GVL. Ball milling is used as an efficient entry step to generate water-soluble partially depolymerized lignocellulosic feeds. Thus, an acidic hydrothermal treatment is used to produce LA. Following extraction with 2-MeTHF to prepare a LA-containing feed, a final RANEY® nickel-catalyzed hydrogenation affords GVL. Ball milling was performed with catalytic amounts of sulfuric acid in line with the following sulfuric acid-catalyzed hydrolysis. The use of 2-MeTHF resulted in a particularly convenient strategy providing LA in acid/sulfur-free organic feeds which do not compromise the activity of RANEY® nickel. Furthermore, 2-MeTHF proved to be an optimal solvent for hydrogenation reactions.One of the main advantages of the proposed scheme is the possibility of recycling both the water and the organic streams for iterative processing. It is worth noting that blending of the water phase with little amounts of 2-MeTHF during hydrolysis prevented the formation of insoluble precipitates and in turn resulted in the formation of an enriched “bio-oil”, which in the case of beechwood contained different lignin fragments. This renders the proposed refining schemes particularly attractive due to the possible simultaneous upgrading of both the cellulosic and the lignin fractions of the biomass. Since 2-MeTHF is considered a green/ecofriendly solvent22,34,35 and RANEY® Ni is prepared from an alloy of two widely available metals, Al and Ni, the strategy disclosed in this paper represents a very sustainable alternative to methods based on the use of rare transition metals or non-green reaction media and could facilitate the preparation of GVL from biomass on an industrial scale. The possible extension of the proposed integrated strategy also to the preparation of 2-MeTHF, which would render the proposed approach even more sustainable, will be the subject of future studies.
Acknowledgements
We gratefully thank Dr. Roberto Rinaldi and his team at the Max Planck for Coal Research for providing the milled cellulose and wood samples as well as for fruitful discussion. The authors also thank the Max Planck–Fraunhofer cooperation scheme for financial support.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- I. T. Horvath, Green Chem., 2008, 10, 1024–1028 RSC.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- W. Fitzpatrick Stephen, in Feedstocks for the Future, American Chemical Society, 2006, vol. 921, pp. 271–287 Search PubMed.
- J. Yuan, S.-S. Li, L. Yu, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Energy Environ. Sci., 2013, 6, 3308–3313 CAS.
- V. Mohan, V. Venkateshwarlu, C. V. Pramod, B. D. Raju and K. S. R. Rao, Catal. Sci. Technol., 2014, 4, 1253–1259 CAS.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- P. A. Son, S. Nishimura and K. Ebitani, RSC Adv., 2014, 4, 10525–10530 RSC.
- K. Yan, T. Lafleur, G. Wu, J. Liao, C. Ceng and X. Xie, Appl. Catal., A, 2013, 468, 52–58 CrossRef CAS PubMed.
- Z.-P. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- R. V. Christian, H. D. Brown and R. M. Hixon, J. Am. Chem. Soc., 1947, 69, 1961–1963 CrossRef CAS.
- Z. Yang, Y.-B. Huang, Q.-X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330 RSC.
- O. A. Abdelrahman, A. Heyden and J. Q. Bond, ACS Catal., 2014, 4, 1171–1181 CrossRef CAS.
- M. G. Al-Shaal, W. R. H. Wright and R. Palkovits, Green Chem., 2012, 14, 1260–1263 RSC.
- L. Peng, L. Lin, H. Li and Q. Yang, Appl. Energy, 2011, 88, 4590–4596 CrossRef CAS PubMed.
- J.-P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437–441 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081 CrossRef CAS PubMed.
- T. E. Graedel, Annu. Rev. Mater. Res., 2011, 41, 323–335 CrossRef CAS.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- J. M. Tukacs, R. V. Jones, F. Darvas, G. Dibo, G. Lezsak and L. T. Mika, RSC Adv., 2013, 3, 16283–16287 RSC.
- R. Palkovits, Angew. Chem., Int. Ed., 2010, 49, 4336–4338 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- T. vom Stein, P. M. Grande, H. Kayser, F. Sibilla, W. Leitner and P. Dominguez de Maria, Green Chem., 2011, 13, 1772–1777 RSC.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS PubMed.
- J. J. Verendel, T. L. Church and P. G. Andersson, Synthesis, 2011, 2011, 1649–1677 CrossRef PubMed.
- M. Shalom, V. Molinari, D. Esposito, G. Clavel, D. Ressnig, C. Giordano and M. Antonietti, Adv. Mater., 2014, 26, 1272–1276 CrossRef CAS PubMed.
- F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS PubMed.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schuth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- S.-J. Tenne, J. Kinzel, M. Arlt, F. Sibilla, M. Bocola and U. Schwaneberg, J. Chromatogr., B, 2013, 937, 13–17 CrossRef CAS PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2006, 11, 156–159 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00717d |
| This journal is © The Royal Society of Chemistry 2014 |