 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct evidence for a coordination–insertion mechanism of ethylene oligomerization catalysed by neutral 2,6-bisiminopyridine iron monoalkyl complexes†
M.
Ángeles Cartes
,
Antonio
Rodríguez-Delgado
,
Pilar
Palma
,
Luis J.
Sánchez
and
Juan
Cámpora
*
Instituto de Investigaciones Químicas, CSIC – Universidad de Sevilla, c/Américo Vespucio, 41092, Sevilla, Spain. E-mail: campora@iiq.csic.es; Fax: +34 954460565; Tel: +34 954489555
First published on 29th May 2014
Abstract
1H NMR studies on ethylene oligomerization catalysed by the neutral monoalkyl complex [Fe(Me)(iPrBIP)] allow direct observation of alkyl iron intermediates as well as reversible ethylene coordination to the metal center, providing for the first time experimental evidence for a coordination–insertion mechanism of iron-catalysed ethylene upgrade reactions.
Iron complexes with 2,6-bisiminopyridine (BIP) ligands have continued to attract the interest of many research groups more than 15 years after the discovery of their catalytic activity in olefin polymerization.1 Today, the uses of these compounds in catalysis have extended to many other important reactions.2 One of the key factors for the success of BIP ligands in catalysis is their redox non-innocence, which enables reversible redox processes that are not accessible for complexes containing uniquely spectator ancillary ligands.3 Even though olefin polymerization does not require redox changes during the catalytic cycle, iron catalysts somehow benefit from the special properties of BIP ligands since few other types of iron complexes exhibit comparable catalytic performance in such reactions.4 In fact, the non-innocent behaviour of BIP ligands casts uncertainty on the identity of the catalytic species involved in typical iron polymerization systems generated from halide precursors [FeXn(BIP)] (n = 2, 3) and organoaluminum co-catalysts. It is known that the reaction of these iron halocomplexes with main group organometallics frequently leads to neutral or even anionic alkyl complexes [Fe(R)(BIP)]0/− that are best described as arising from the reduction of the BIP ligand rather than the metal center.5 On the other hand, although Chirik demonstrated that cationic monoalkyls [Fe(R)(iPrBIP)]+ containing a Fe(II) center and an innocent, electroneutral iPrBIP ligand (iPrBIP = 2,6-bis-N-(2,6-diisopropylphenyl)acetimidoylpyridine) are very active single-component ethylene polymerization catalysts,6 this does not rule out the possibility that different alkyl iron complex species could be involved in iron-catalysed ethylene polymerization. Thus, in a series of spectroscopic studies on the activation of [FeCl2(BIP)] complexes with MAO or trialkylaluminum reagents, Bryliakov and Talsi identified the formation of two types of bimetallic Fe/Al products.7 While MAO gives rise to relatively stable cationic complexes [Fe(μ-X)(μ-Me)AlMe2(BIP)]+ (X = Cl, Me), simple aluminum alkyls (trimethyl or triisobutylaluminum) lead to isostructural but neutral species [Fe(μ-X)(μ-Alkyl)2Al(Alkyl)2 (BIP)] (X = Cl, Alkyl) that are unstable at room temperature. Based on these results, the authors suggested that, depending on the activator used, either cationic (A) or electroneutral (B) propagating species could be formed upon dissociation of the AlR3 unit (Fig. 1) containing innocent or singly reduced BIP ligands, respectively, but sharing in common a high-spin Fe(II) ion. Intriguingly, it was suggested that, given the similar electronic configuration of the metal centre in both intermediates, they should exhibit similar catalytic behaviours.6b,7b
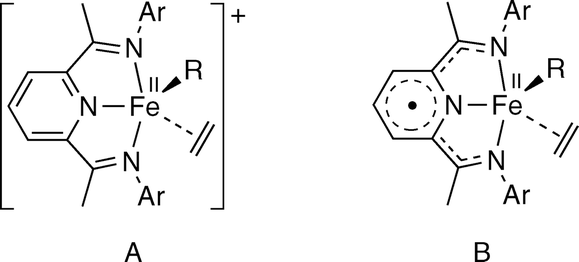 | ||
| Fig. 1 Cationic and neutral propagating species proposed for iron-catalysed ethylene polymerization. | ||
We have reported recently that the iron dialkyl complex [Fe(CH2SiMe3)2(iPrBIP)] (1) reacts with trimethylaluminum (TMA) in a stepwise manner (Scheme 1).8 NMR studies showed that the first step, reduction to the monoalkyl complex [Fe(CH2SiMe3)(iPrBIP)], 2, is accomplished with one equivalent amount of TMA per Fe atom. With a second equivalent of TMA the CH2SiMe3 group is exchanged to afford the monomethyl complex [Fe(Me)(iPrBIP)], 3. Very likely, this sequence of reactions is closely related to the activation of Fe–BIP complexes by TMA; therefore the reactivity of complexes 2 and 3 towards ethylene would be highly relevant in order to understand the mechanism of the catalytic polymerization reaction.
 | ||
| Scheme 1 | ||
Although 2 and 3 are paramagnetic, this type of complex gives rise to informative 1H NMR spectra, and this provides a direct means of investigating their interaction with ethylene. When the latter is bubbled through solutions of complexes 2 or 3 in C6D6 at room temperature, no polyethylene is formed but GC analyses revealed the formation of mixtures of oligomers (>95% α-olefins), characterized by a Flory–Schulz molecular weight distribution coefficient of 0.57. Once ethylene is consumed, slow isomerization of the α-olefins occurs as indicated by the gradual growth of new peaks in the GC of the mixtures. Precise quantification of the catalytic activities in the absence of aluminum alkyls was hampered by the sensitivity of these catalysts to impurity traces in ethylene. However, 1H NMR showed that samples containing 8 μmol of 2 or 3 consumed 5 mL of ethylene (ca. 25 equiv.) within 60 and 45 min, respectively, indicating turnover frequencies in the 20–40 h−1 range. Although these activities are quite low, they demonstrate that neutral monoalkyl complexes have a distinct reactivity towards ethylene, different from the ethylene polymerization observed in dual Fe/Al catalytic systems. In addition, the ability of 2 and 3 to oligomerize ethylene contrasts with the lack of catalytic activity of their cobalt analogues.9
At room temperature, ethylene has no effect on the 1H NMR spectra of 2 and 3, i.e., the oligomerization reaction proceeds while the shape and position of the signals of the complexes remain unaltered. When the oligomerization of 2 is monitored, any active intermediates remain below the detection threshold, presumably because the initial ethylene insertion into the Fe–CH2SiMe3 bond is considerably slower than the catalytic process. However, in the case of 3, at least two sets of low-intensity signals corresponding to as many different intermediates were observed during the catalytic reaction (Fig. 2). The relative intensity of these signals evolves during the experiment. After 20 min, they represent up to 30% of the original intensity of 3, but as ethylene is depleted they decay and disappear within several hours.
 | ||
| Fig. 2 1H NMR monitoring of the reaction of 3 with ethylene (25 equiv.). Shadowed regions contain signals of complexes arising from ethylene insertions into the Fe–C bonds. Dotted lines mark the position of the signals of the ethyl (n = 0) and butyl complexes (n = 2), taken from ref. 10. | ||
As shown in Fig. 2, most of the new signals appear in regions that are close to the analogous signals of 3. This is the kind of spectral analogy that would be expected if the new signals correspond to alkyl complexes arising from ethylene insertion reactions. Furthermore, the positions very closely match the chemical shifts reported by Chirik for the ethyl and butyl complexes [Fe(R)(iPrBIP)] (R = Et, n-Bu), marked with dotted lines in the figure.10 The slight deviations with regard to the literature values are systematic and can be attributed to small differences in experimental variables such as temperature or concentration. The identification of the main intermediate signals corresponding to these two alkyl complexes is confirmed by the observation of two signals at δ 149.6 and 140.6 ppm that have no correlation with the spectrum of 3 but match the chemical shifts reported for the β-hydrogen atoms of the ethyl and butyl groups. Actually, the ethyl and butyl complexes are expected to be the most abundant intermediates in an ethylene oligomerization cycle involving chain propagation by consecutive ethylene insertions and termination by chain transfer to the monomer, which suggests that this mechanism also operates in this system. These compounds are known to be thermally unstable in solution, and this is also in agreement with the disappearance of the signals once ethylene has been consumed. Note that several low-intensity paramagnetic signals are observed after 5 h. These are different from those observed in the initial stages and we tentatively assign them to branched alkyl species participating in the olefin isomerization process.
Although the accumulation of detectable amounts of intermediates indicates that 3 is more reactive than 2, only a fraction of the former actually reacts with ethylene (in spite of this being initially in a large excess), indicating that the ethylene insertion into the Fe–C bond is still somewhat slower for the methyl complex than for the higher alkyl iron intermediates. The origin of this effect is unclear but could be related to the somewhat higher thermodynamic stability of transition metal–carbon bonds in methyl complexes than in ethyl or higher alkyls.11
Prior to insertion into the Fe–C bond of 3, ethylene has to coordinate to the iron centre. Although, as mentioned before, the 1H NMR spectrum of 3 is not altered by the presence of ethylene at 25 °C, variable temperature NMR experiments provided evidence for the reversible Fe⋯C2H4 interaction (Fig. 3). Injection of ethylene (0.6 mL) in a toluene-d8 solution of 3 (10 μmol; ethylene/3 = 2.4) at −70 °C causes the disappearance of all paramagnetic signals. The signals of 3 are observed again when the sample is warmed up to −50 °C, although they appear broader and their intensity is ca. 25% lower than the original. They recover their initial intensity and width as the temperature is increased. Simultaneously, the ethylene signal broadens and shifts to higher frequency. At 5 °C, all signals of 3 display normal intensities, positions and widths although that of ethylene remains broad. These observations are consistent with the formation of an NMR-silent ethylene adduct, 3·C2H4, which is thermodynamically favoured at low temperature but begins to dissociate above −50 °C, allowing rapid exchange between free and coordinated ethylene. The ethylene binding constant, estimated from the decrease of the intensities of the signals of 3 between −50 and −35 °C, is ca. 8.5 L mol−1, corresponding to ΔG0 ≈ −1 kcal mol−1, in good agreement with preliminary DFT calculations, see the ESI.† The room temperature spectrum is consistent with essentially full dissociation of 3·C2H4, but ethylene oligomerization indicates that the complex remains thermodynamically accessible as a reaction intermediate. Although evidence for the reversible binding of ethylene to a Fe(II) alkyl complex has been reported before,12 this is the first time that such an interaction is observed with a catalytically active Fe–BIP complex.
 | ||
| Fig. 3 Selected regions of the 1H NMR spectrum of 3 recorded at different temperatures, in the absence (bottom) and in the presence of ethylene (2.4 equiv.). Signals in the diamagnetic region marked as * correspond to the solvent (hexane) and the residual peak of the solvent (toluene-d8). Signals D, D′, F and G correspond to 3 (see Fig. 1). Vertical scales have been optimized to improve clarity. | ||
Compared with their cationic analogues,6a2 and 3 are orders of magnitude less active and produce lower molecular weight products. However, since the electronic configuration and stereochemical environment of the iron centre is very similar in both types of compounds, it is very likely that the mechanisms of chain propagation and transfer are also qualitatively similar in both cases. It is also noteworthy that, although the cobalt analogues of 2 and 3 are catalytically inactive, higher alkyls [Co(CH2CH2R)(iPrBIP)] readily undergo chain transfer to ethylene.13 This suggests an analogy between the Fe and Co systems: in both cases, going from the cationic to the neutral systems gives rise to an ethylene insertion barrier, while chain transfer remains a competitive process.
In summary, we have shown that iron monoalkyl complexes 2 and 3 catalyse ethylene oligomerization to linear α-olefins. For the first time, we detected the reversible interaction of iron alkyls with the monomer and observed the formation of alkyl iron complexes arising from consecutive ethylene insertions. Although these observations support the idea that the similar electronic configuration of the iron centre in isostructural neutral and cationic monoalkyl derivatives should lead to comparable catalytic activity,7,8 the low catalytic activity of 2 and 3 and the low molecular weight of the products rule out the direct involvement of neutral iron monoalkyls in two-component ethylene polymerization catalysis.
Acknowledgements
This work was supported by the Spanish Government, Junta de Andalucía and the European Union (projects CTQ2012-30962, FQM5074 and FEDER funds). MAC thanks CSIC for an I3P predoctoral fellowship. We thank CESGA for granting access to computational resources (ICTS proposal no. 262).Notes and references
- L. Li and P. T. Gomes, in Olefin Upgrading Catalysis with Nitrogen-based Metal Complexes, ed. G. Giambastiani and J. Cámpora, Springer, Dordrecht, 2011, vol. II, pp. 77–198 Search PubMed.
- See for example: (a) J. V. Obligacion and P. J. Chirik, Org. Lett., 2013, 15, 2680 CrossRef CAS PubMed; (b) K. Sylvester and P. J. Chirik, J. Am. Chem. Soc., 2009, 131, 8772 CrossRef CAS PubMed; (c) A. B. Biernesser and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553 CrossRef CAS PubMed.
- (a) S. Blanchard, E. Derat, M. Desage-El Murr, L. Fensterbank, M. Malacria and V. Mouriés-Mansuy, Eur. J. Inorg. Chem., 2012, 376 CrossRef CAS; (b) P. J. Chirik and K. Weighardt, Science, 2010, 327, 794 CrossRef CAS PubMed.
- (a) W. Zhang, W. H. Sun and C. Redshaw, Chem. Soc. Rev., 2012, 42, 8988 Search PubMed; (b) A. Boudier, P. A. R. Breuil, L. Magna, H. Olivier-Borbigou and P. Braunstein, Chem. Commun., 2014, 50, 1398 RSC.
- Q. Knjinenburg, S. Gambarotta and P. H. M. Budzelaar, Dalton Trans., 2006, 5442 RSC.
- (a) M. W. Bowkamp, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 127, 9660 CrossRef PubMed; (b) A. M. Tondreau, C. Milsman, A. D. Patrick, H. M. Hoyt, E. Lobkovsky, K. Weighardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 15046 CrossRef CAS PubMed.
- (a) K. P. Bryliakov, N. V. Semikolenova, V. A. Zakharov and E. P. Talsi, Organometallics, 2004, 23, 5375 CrossRef CAS; (b) K. P. Bryliakov, E. P. Talsi, N. V. Semikolenova and V. A. Zakharov, Organometallics, 2009, 28, 3225 CrossRef CAS.
- M. A. Cartes, A. Rodríguez-Delgado, P. Palma, E. Álvarez and J. Cámpora, Organometallics, 2014, 33, 1834 CrossRef CAS.
- (a) T. M. Kooistra, Q. Knijinenburg, J. M. M. Smits, A. D. Horton, P. H. M. Budzelaar and A. W. Gal, J. Am. Chem. Soc., 2001, 40, 4719 CAS; (b) N. Kielgrewe, W. Steffen, T. Blömker, G. Kehr, R. Fröhlich, B. Wibbling, G. Erker, J.-C. Wasilike, G. Wu and G. Bazan, J. Am. Chem. Soc., 2005, 127, 13955 CrossRef PubMed; (c) M. J. Humphries, K. P. Tellmann, V. C. Gibson, A. J. P. White and D. J. Williams, Organometallics, 2005, 24, 2309 CrossRef.
- R. J. Trovitch, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2009, 130, 11631 CrossRef PubMed.
- J. A. M. Simoes and J. L. Beauchamp, Chem. Rev., 1990, 90, 629 CrossRef CAS.
- J. Volbeda and M. W. Bouwkamp, Organometallics, 2009, 28, 209 CrossRef CAS.
- V. C. Gibson, K. P. Tellman, M. J. Humphreys and D. F. Wass, Chem. Commun., 2002, 2136 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00612g |
| This journal is © The Royal Society of Chemistry 2014 |