 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroisomerization of long-chain paraffins over nano-sized bimetallic Pt–Pd/H-beta catalysts†
Frank
Bauer
a,
Karsten
Ficht
ab,
Marko
Bertmer
c,
Wolf-Dietrich
Einicke
a,
Thomas
Kuchling
b and
Roger
Gläser
*a
aInstitute of Chemical Technology, Universität Leipzig, 04103 Leipzig, Germany. E-mail: roger.glaeser@uni-leipzig.de; Fax: +49 341 9736349; Tel: +49 341 9736300
bInstitute of Energy Process Engineering and Chemical Engineering, TU Bergakademie Freiberg, Germany
cInstitute of Experimental Physics II, Universität Leipzig, 04103 Leipzig, Germany
First published on 10th July 2014
Abstract
The hydroisomerization of long-chain n-paraffins was studied in the temperature range of 205–230 °C at pH2 = 50 bar using a pilot-scale trickle-bed continuous-flow reactor over bimetallic catalysts consisting of mixtures of platinum and palladium supported on commercially available nano-sized zeolite beta (nSi/nAl = 12.5 and 25, respectively) extruded with an alumina binder. For n-hexadecane conversion, high yields of isomers (25 and 45 wt.% of mono- and multibranched isomers, respectively) without extensive cracking (>10 wt.%) were obtained at a conversion of 80%. Long-term tests with n-hexadecane and blends of solid n-paraffins for 30–60 days on-stream clearly indicate that a minor loss in catalyst activity can easily be compensated for by increasing the reaction temperature from 220 °C to 225 °C. The zeolite with a “mild acidity” exhibits a low hydrocracking activity with isomerization yields of up to 70 wt.% and high stability over more than 60 days on-stream. Carbonaceous deposits formed during n-paraffin hydroisomerization were investigated by elemental analysis, TGA, ATR-FTIR and 13C MAS NMR spectroscopy, showing the formation of hydrogen-rich coke which leads to pore blocking.
1. Introduction
To reduce the content of linear paraffins in hydrocarbon mixtures, hydroisomerization to their branched isomers over bifunctional catalysts is a more sustainable process in the modern petroleum refining industry than selective hydrocracking (dewaxing) of long-chain n-paraffins.1 Thus, hydroisomerization has been applied for producing high-octane gasoline and low-pour-point diesel as well as for improving the viscosity properties of waxy feedstocks such as slack waxes (“isodewaxing”). Obviously, dewaxing by hydroisomerization is the most adequate process for adjusting the cold flow properties of the oil by conversion, rather than the removal of the n-paraffins.2 Bifunctional catalysts containing a noble metal, typically Pt or Pd, on medium- or wide-pore silicoaluminophosphates or zeolites such as SAPO-11 (ref. 3) and beta4 have shown a high isomerization/cracking ratio at reaction temperatures of 250–300 °C compared to Pt catalysts supported on amorphous SiO2–Al2O3 demanding 345–380 °C.5 In recent years, the catalytic performance of several amorphous and zeolitic catalysts was disclosed for the hydroisomerization of long-chain n-paraffins, including SiO2–Al2O3,6–8 beta,9–11 ZSM-12,12 and ZSM-22.9,13 Generally, catalysts with a high hydrogenation activity and a low degree of acidity are favorable for maximizing hydroisomerization versus hydrocracking, since a strong hydrogenation activity limits the degree of branching by hydrogenating primary isomerization products. Nevertheless, the influence of catalyst properties such as metal dispersion, acid site strength and density, macro/mesopore distribution, or binder properties on the rate and the selectivity of hydroisomerization is manifold and requires a careful design of investigations.5,14In addition to a well-balanced interplay between metal and acidic sites within the bifunctional catalytic mechanism,1 shape selectivity and spatial constraints within zeolitic micropores can control the degree of isomerization. The use of appropriate zeolites can thus result in relatively low-branched paraffin isomers. For instance, the small channels of SAPO-11 hardly allow the formation of multibranched isomers15 which are urgently required for converting long-chain paraffins into lubricants and microcrystalline waxes. For n-heptane isomerization, zeolite Pt/H-beta exhibits a more promising catalytic performance than Pt/HY, possibly due to the unique structure of three-dimensional interconnected channels of the beta-type zeolite.16 On the other hand, the accelerated formation of multibranched isoparaffins on large-pore zeolites such as USY and beta can be accompanied by high hydrocracking yields due to the high acid site density and the strength of these catalysts. With respect to pore size, ordered mesoporous MCM-41-type catalysts have also shown high activity in the isodewaxing process.1 This indicates that pore diffusion effects within the catalysts used for long-chain hydroisomerization reactions have to be reduced, e.g., by the application of mesoporous materials or nano-sized zeolites.
The application of nanocrystals is commonly assumed to reduce the residence time of olefinic intermediates inside the beta zeolite crystal, thereby enhancing the selectivity towards isomerization. Furthermore, bi-metallic Pt–Pd impregnated catalysts are described to exhibit a higher metal dispersion, thus achieving a high catalytic activity at a lower amount of noble metals than regularly required.17 In this study, we report on the catalytic behavior of two nano-sized H-beta catalysts (with different nSi/nAl ratios) loaded with 0.8 wt.% platinum and/or palladium in the hydroisomerization of long-chain paraffins, i.e., n-hexadecane and a C18+ mixture. The effect of noble metal loading and composition, the nSi/nAl ratio of the zeolite component as well as the reaction conditions on catalytic activity, selectivity and stability were studied. Eventually, catalyst deactivation by carbonaceous deposits was investigated by temperature-programmed oxidation, elemental analysis, argon and nitrogen sorption, ATR-FTIR spectroscopy, and 13C MAS NMR spectroscopy.
2. Experimental
2.1 Catalyst preparation
The nano-sized crystal morphology of the commercially available zeolite beta (Zeolyst International Inc., nSi/nAl = 12.5 and 25, respectively) was visualized with a JEOL JSM-6600 scanning electron microscope as cauliflower-like agglomerates in the micrometer range consisting of primary particles <50 nm in size (not shown). The zeolite delivered in the ammonium form was mixed with 15 wt.% binder (γ-alumina), kneaded with the addition of water and nitric acid as peptization agents, extruded in the form of cylindrical pellets (5 mm length, 1.5 mm diameter), dried at 120 °C (6 h), and, in a further step, calcined at 500 °C (3 h) to obtain the zeolite catalyst in its acidic form. The calcined moldings were spray-impregnated (as is typical for industrial preparations and accepting some of the metal content deposited on the binder) with aqueous solutions of H2[Pt(OH)6] and/or Pd(NH3)4(NO3)2 containing the amount of salts required to achieve the desired loading, typically 0.8 wt.%. The impregnated materials were dried at 120 °C, calcined at 500 °C, and finally reduced in flowing hydrogen (299 cm3 cm−3 h−1) at 250 °C (3 h). The desired metal loadings were confirmed by elemental analysis via ICP-OES (PerkinElmer Optima 8000) and the metal dispersions were determined by CO chemisorption (Porotec TPD/R/O 1100) assuming an nCO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nPt or Pd adsorption stoichiometry of 1:1.
:nPt or Pd adsorption stoichiometry of 1:1.
2.2 Catalytic experiments
Kinetic experiments and tests for catalyst activity and stability were performed in a pilot-scale continuous trickle-bed reactor (Fig. 1). The reactor allows a maximum liquid flow of approximately 250 mL h−1. The total volume of the 1.2 m long tubular reactor is 325 mL. The catalyst bed, i.e., a mixture of silicon carbide (d = 100–200 μm) and catalyst (volume ratio of 1:1 referred to the bulk density), was placed between a flow-in and a flow-off layer. The dilution was applied to prevent wall effects, provide plug flow and ensure isothermal conditions within the catalyst bed. The catalyst bulk volume was about 90 mL. Unless otherwise stated, the experiments were carried out at pH2 = 50 bar, nH2/nhydrocarbons = 750:1 and a liquid hourly space velocity (LHSV) of 0.5 h−1. In addition, kinetic experiments on n-hexadecane conversion in hydrogen (at atmospheric pressure, molar gas-to-feed ratio of 7:1) were carried out in a 10-mm-diameter quartz microreactor loaded with 0.2 g of crushed catalyst pellets (0.3–0.6 mm in size).
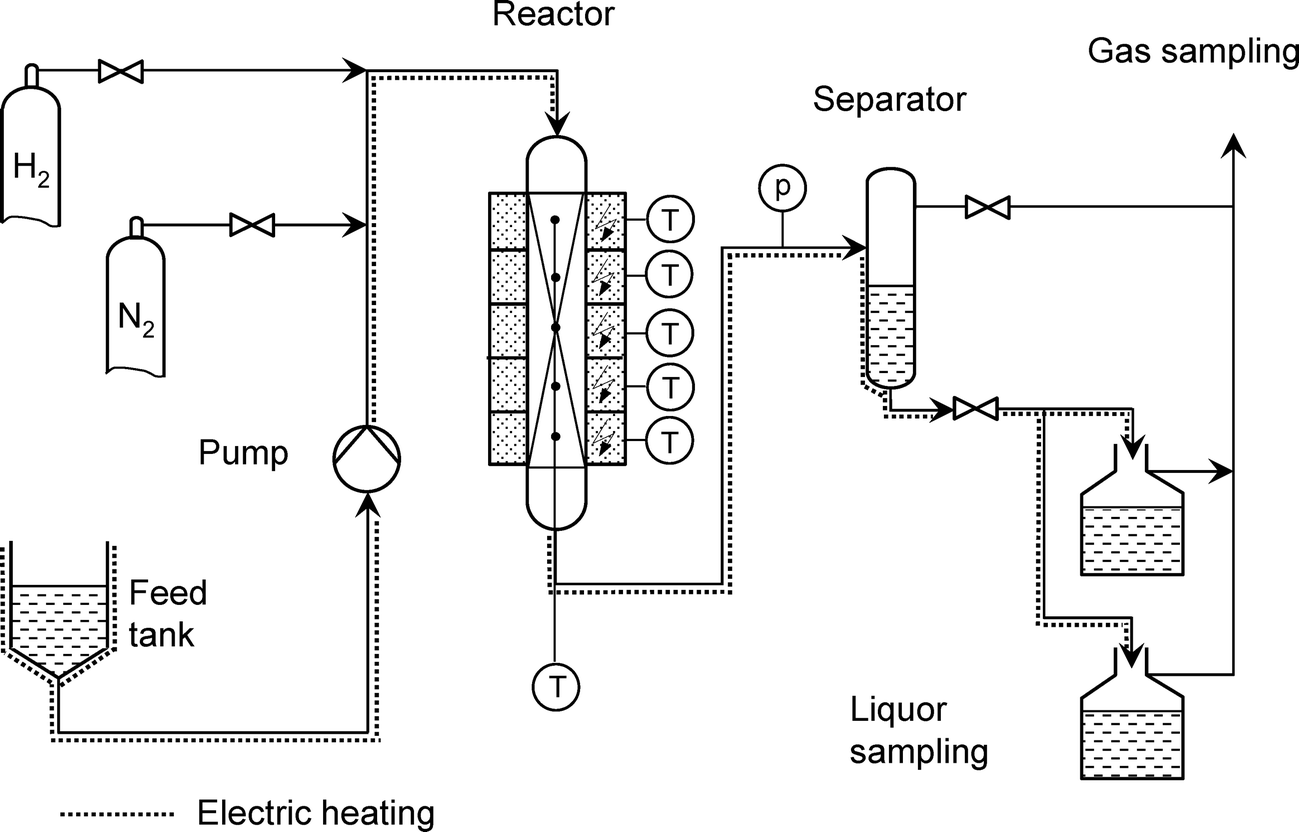 | ||
| Fig. 1 Flow sheet of the continuous-flow apparatus for catalytic experiments using a trickle-bed reactor. | ||
The feedstocks used in this study were n-hexadecane or a solid C18+n-paraffin mixture. The liquid reaction products were collected at room temperature and periodically analyzed by a PerkinElmer GC-FID Autosystem supplied with an on-column injection system using a 30 m long FactorFour VF-5ht UltiMetal capillary column. As shown in Fig. 2, the liquid products of n-hexadecane conversion are divided into monobranched isomers, multibranched isomers and cracking products (carbon number <C13).
 | ||
| Fig. 2 Gas chromatogram of the liquid reaction products from n-hexadecane conversion over Pd–Pt/H-beta (nSi/nAl = 25) at 220 °C. | ||
2.3 Characterization of spent catalysts
Before cooling down the reactor, any retained reaction products have to be thoroughly removed from the catalyst by stripping with low-molecular-weight paraffins (recommended for high-molecular-weight solid paraffin feeds) followed by severe hydrogen flushing at the reaction temperature. Otherwise, misinterpretations of the analysis of the coke deposits on the catalyst cannot be excluded.Nitrogen/argon sorption measurements of fresh and spent catalysts were performed at −196 °C with an ASAP 2010 apparatus by Micromeritics. All samples were degassed at 150 °C before measurement for at least 24 hours at 10−5 mbar. Adsorption and desorption isotherms were measured over a range of relative pressures (p/p0) of 0–1.0. The specific micropore volume and the specific micropore surface area were determined according to the t-plot method. Pore width distribution was determined by nonlocal density functional theory (NLDFT) using the argon adsorption isotherms.
The carbon content of spent catalysts was obtained from elemental analysis (vario Max CHN, Elementar Analysensysteme GmbH). Characterization by thermogravimetric analysis (TGA) was performed on a TGA 7 thermobalance (PerkinElmer). The samples were heated in a stream of synthetic air (10 ml min−1) at a rate of 10 °C min−1 up to 850 °C. Furthermore, the nature of carbonaceous deposits was studied by ATR-FTIR on a Digilab FTS 6000 spectrometer. Solid-state 13C MAS NMR spectra were recorded on a Bruker MSL 500 spectrometer. Magic-angle spinning was performed at a spinning rate of 5 kHz. Solution 13C NMR spectroscopy of hydroisomerized paraffins was performed in CDCl3 on a Bruker Avance DPX 400 spectrometer.
3. Results and discussion
3.1 Effect of Pt–Pd loading and nSi/nAl ratio on catalyst performance
Using a mixture of light paraffins, Roldan et al.18 tested a series of mono- and bimetallic (Pt and/or Pd) catalysts supported on zeolite beta samples for their hydroisomerization performance and found that the bimetallic catalyst is significantly more active than either of the monometallic ones. Based on these findings, we studied the effect of hydrogenation components (Pt, Pd or mixtures thereof) as well as the zeolite's nSi/nAl ratio on the hydroconversion of long-chain paraffins on zeolite H-beta. The catalytic properties of samples loaded with different fractions of the two noble metals, but maintaining an overall metal content of 0.8 wt.%, are given in Table 1. In accordance with the results of Roldán et al.,18 Pd/H-beta was the least active catalyst with an overall conversion of about 10% lower than that of the catalyst containing Pt as the hydrogenation component only. Moreover, the Pd/H-beta catalyst is considerably less selective for isomerization than the Pt-containing analogues. These significant differences in conversion and selectivity could be due to the lower hydro-/dehydrogenation activity of Pd with respect to Pt. Thus, less olefinic intermediates are formed and a lower paraffin conversion is reached. Moreover, the residence time of these olefinic intermediates at the catalyst increases along with the probability of cracking reactions. For all of the other catalysts, the selectivity for iso-C16 isomers was higher than 90% at conversions of >75%, with the Pd-free Pt/H-beta catalyst being the most selective but somewhat less active (64.8% conversion). The gradual increase of conversion and hydroisomerization activity with increasing Pd content in bimetallic Pt–Pd/H-beta catalysts is typically explained by a higher noble metal dispersion (see Table 1) caused by a promoting effect of the more easily reducible palladium oxides present after calcination.17 Therefore, the Pt/Pd ratio of catalysts for further experiments was set to 1:3, i.e., 0.2 wt.% Pt-0.6 wt.% Pd, considering that a partial substitution of platinum by palladium with similar catalytic properties may offer a significant cost reduction for an industrial application.
| Catalyst H-beta with Pt–Pd content (wt.%/wt.%) | Pt–Pd (0.8/0.0) | Pt–Pd (0.6/0.2) | Pt–Pd (0.4/0.4) | Pt–Pd (0.2/0.6) | Pt–Pd (0.0/0.8) |
|---|---|---|---|---|---|
| Metal dispersion (%) | 54 | 77 | 91 | 92 | 53 |
| Conversion (%) | 64.8 | 74.2 | 77.6 | 77.0 | 54.9 |
| Iso-C16 selectivity (%) | 93.8 | 92.3 | 92.9 | 91.2 | 80.4 |
| Cracking selectivity (%) | 6.2 | 7.7 | 7.1 | 8.8 | 19.6 |
As shown in Table 2 for zeolite H-beta with an nSi/nAl ratio of 25, the Pt–Pd amount required to balance with the acidic sites is already achieved at a loading of 0.4 wt.% because the selectivity for iso-C16 is similar to that for the other catalysts with higher overall noble metal loadings. For further experiments, the metal content of Pd–Pt/H-beta catalysts was kept constant at 0.8 wt.% to comply with the higher acid site density of the H-beta sample with an nSi/nAl ratio of 12.5. In case of a too low metal content, the rate of dehydrogenation and hydrogenation can limit the bifunctional hydroisomerization rate. Typically, the density of metal sites is in balance with that of the acid sites and the hydro-/dehydrogenation reactions proceed faster than the skeletal isomerization of olefins on the acid sites.1
:3, nSi/nAl = 25) loaded with different total amounts of noble metals (200 °C, 1 bar, LHSV = 3 h−1)
| Catalyst H-beta with Pt–Pd content (wt.%) | 0.4 | 0.7 | 0.8 | 0.9 | 1.1 |
| Conversion (%) | 71.6 | 76.3 | 77.0 | 77.9 | 78.4 |
| Iso-C16 selectivity (%) | 89.2 | 87.6 | 91.2 | 90.3 | 89.8 |
| Cracking selectivity (%) | 10.8 | 12.4 | 8.8 | 9.7 | 10.2 |
Catalytic experiments applying two H-beta zeolites with different nSi/nAl ratios were performed to study the effect of acid site density on the reaction rate and the selectivity for n-paraffin hydroisomerization. The yields of isomerization products over the two catalysts are plotted against n-hexadecane conversion in Fig. 3. The catalysts behave similarly up to about 70% conversion, and cracking is of minor extent (<10 wt.% yield of cracking products). Significant differences are observed, however, at higher conversions. While for H-beta (nSi/nAl = 12.5), the maximum isomerization yield is around 65%, the “mild acidity” of H-beta with a lower Al content (nSi/nAl = 25) reaches a higher maximum of about 70% and avoids extensive cracking at conversions of up to 90%. With higher nSi/nAl ratios, selectivity and yield are higher, because less olefinic intermediates can interact with acid sites, and consequently, the cracking of intermediates is also slowed down.
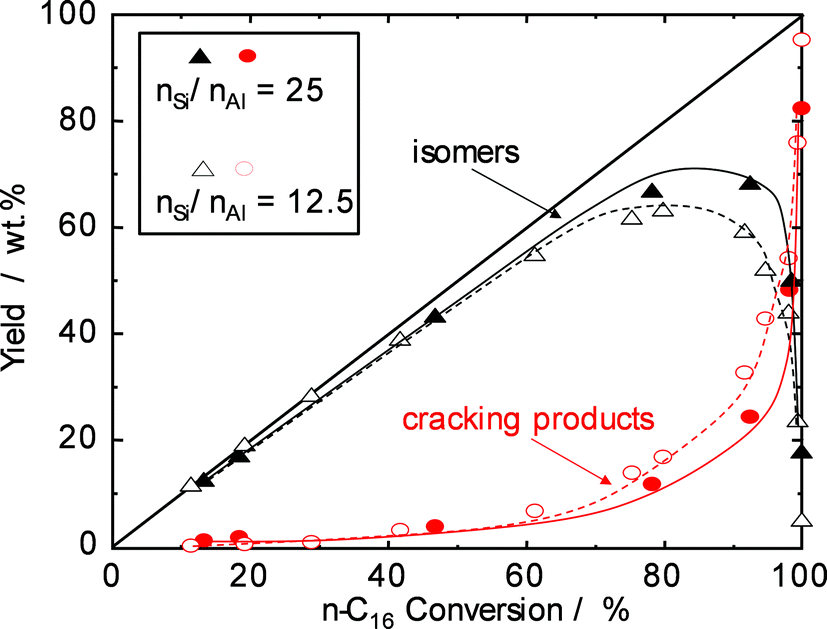 | ||
| Fig. 3 Yield of reaction products as a function of n-hexadecane conversion over Pd–Pt/H-beta samples (nSi/nAl = 25 (solid symbols) and nSi/nAl = 12.5 (open symbols)). | ||
The high selectivity to multibranched isomers even at high conversion makes the Pd–Pt/H-beta (nSi/nAl = 25) catalyst attractive for the selective hydroisomerization of long-chain linear paraffins to improve their cold flow properties. Obviously, the isomerization selectivity is greatly influenced by the acidity of the catalyst, i.e. strength and density of acid sites, and probably also the pore size with associated diffusion effects. While Pt/H-MCM-22 and Pt/H-ZSM-5 catalysts showed lower isomerization selectivity during the hydroisomerization of n-hexadecane due to their strong acidity, Pt/SAPO-11 and Pt/H-beta catalysts with weak and mild acid sites yielded higher selectivities.19 When two Pt/H-beta samples with different nSi/nAl ratios are compared,20,21 the zeolite with the lower nSi/nAl ratio, and consequently with a higher density of acid sites, is more active but less selective for isomerization reactions. It is also conceivable that the density of acid sites has an impact on the residence time of olefinic intermediates within the zeolite pores. In summary, a density of acid sites as low as possible without compromising the overall activity is strongly recommended for hydroisomerization catalysts.
3.3 Effect of reaction pressure
Industrial hydroisomerization reactions are typically carried out at 20–80 bar depending on the catalyst lifetime versus activity. Therefore, the effect of pressure was also studied here. For n-hexadecane hydroconversion on Pt/SiO2–Al2O3, Calemma et al.5 reported a decrease in conversion from about 60% at 20 bar (290 °C) to about 30% at 80 bar (330 °C). Our experiments on Pd–Pt/H-beta revealed a decline of the n-hexadecane conversion from 89% at 30 bar to 53% at 75 bar obtained, however, at a considerably lower reaction temperature of 220 °C. In addition, screening experiments carried out in a stirred microautoclave (100 mL; Andreas Hofer Hochdrucktechnik GmbH, Germany) revealed that at H2 pressures of >80 bar unsatisfactorily low paraffin conversions <10% were obtained. This can clearly be explained by the very low concentrations of olefinic intermediates at high pressures and a higher degree of hydrogen occupation on the catalytically active sites with respect to the hydrocarbon molecules. Although higher hydrogen pressures have a negative effect on n-paraffin conversions and negligible improvements in selectivity and in catalyst stability, industrial specifications may initiate catalyst characterization in the pressure range of 30–50 bar.4,5,8At a H2 pressure of 1 bar and even at the lowest Pt–Pd loading of 0.4 wt.%, no olefins were detected in the reaction product pointing to the high hydrogenation activity of bimetallic Pt–Pd/H-beta catalysts. As Fig. 4 shows, an increase in H2 pressure from 1 to 50 bar requires an increase of about 20 °C to obtain a similar degree of conversion. Moreover, the profiles of conversion over reaction temperature indicate that a temperature increase of about 30 °C is enough to shift from low conversion of about 10% to a nearly complete conversion of n-hexadecane.
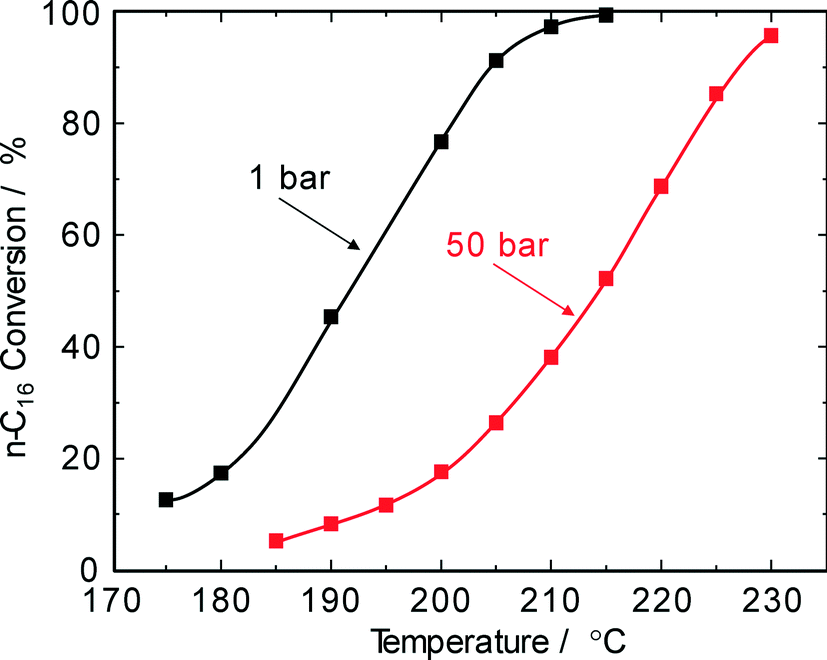 | ||
| Fig. 4 n-Hexadecane conversion on Pd–Pt/H-beta (nSi/nAl = 25) as a function of reaction temperature and pressure (LHSV = 3 h−1). | ||
3.4 Hydroisomerization selectivity
The evolution of the three main different product groups, i.e., monobranched hexadecanes, multibranched hexadecanes, and cracking products, with the reciprocal of space velocity and the yield of n-hexadecane (1-conversion) are shown in Fig. 5 and 6, respectively. Monobranched and multibranched isomers are formed as primary and secondary reaction products, respectively. At a maximum isomerization yield of about 70 wt.% as shown in Fig. 6, the formation of multibranched isomers (achieving a yield of 52 wt.%) is preferred in comparison to those of monobranched hexadecanes with about 18 wt.% and cracking products with 13 wt.%. The high selectivity for isohexadecanes (especially for multibranched hexadecanes) on Pt–Pd/H-beta (nSi/nAl = 25) can be explained by the abovementioned “mild acidity”, i.e., the monobranched olefinic intermediates have a relatively long average residence time within the zeolite pores before they are hydrogenated and desorbed. Thus, these intermediates are subject to further transformations on the acid sites leading to more multibranched isomers, but not to extensive cracking. Similar findings of the effect of acid properties of Pt/H-beta on the maximum isomerization yield are reported by Batalha et al.22 when comparing the zeolite loadings on α-Al2O3 used as a binder. For example, the isomer yield improved from 35 to 80 wt.%, when the zeolite content was reduced from 100 wt.% to 13 wt.%. On the other hand, a higher zeolite content increased the residence time of monobranched isomers on the acid sites, which, in turn, favors the formation of multibranched isomers and cracking.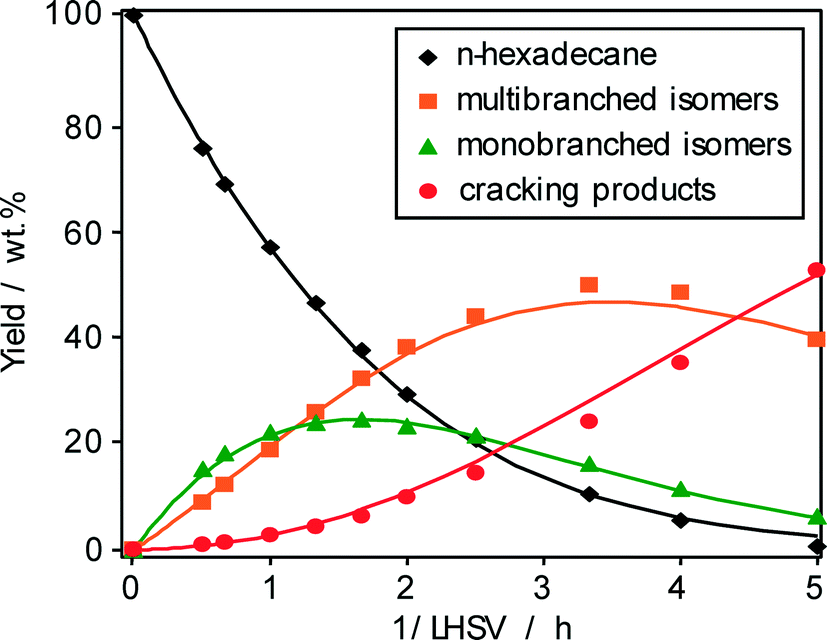 | ||
| Fig. 5 Yield of n-hexadecane and reaction products as a function of 1/LHSV over Pd–Pt/H-beta (nSi/nAl = 25) at 220 °C and 1 bar. | ||
 | ||
| Fig. 6 Yield of reaction products as a function of n-hexadecane conversion over Pd–Pt/H-beta (nSi/nAl = 25). | ||
Fig. 7 summarizes the selectivities for iso-hexadecanes and cracking products including the data for the long-term test for n-hexadecane hydroisomerization (see sectionr 3.5). When these selectivities are plotted versus total n-hexadecane conversion, one identical curve is obtained regardless of the way in which conversion is varied, i.e., by increasing the reaction temperature, space velocity, hydrogen pressure, hydrogen/feed ratio, or time on-stream. Obviously, the selectivity to iso-hexadecanes and cracked products is only a function of the level of conversion. This indicates that differences in activation energy for isomerization and cracking are insignificant, and hydroisomerization/hydrocracking on the Pt–Pd/H-beta samples apparently follows a single consecutive reaction pathway.
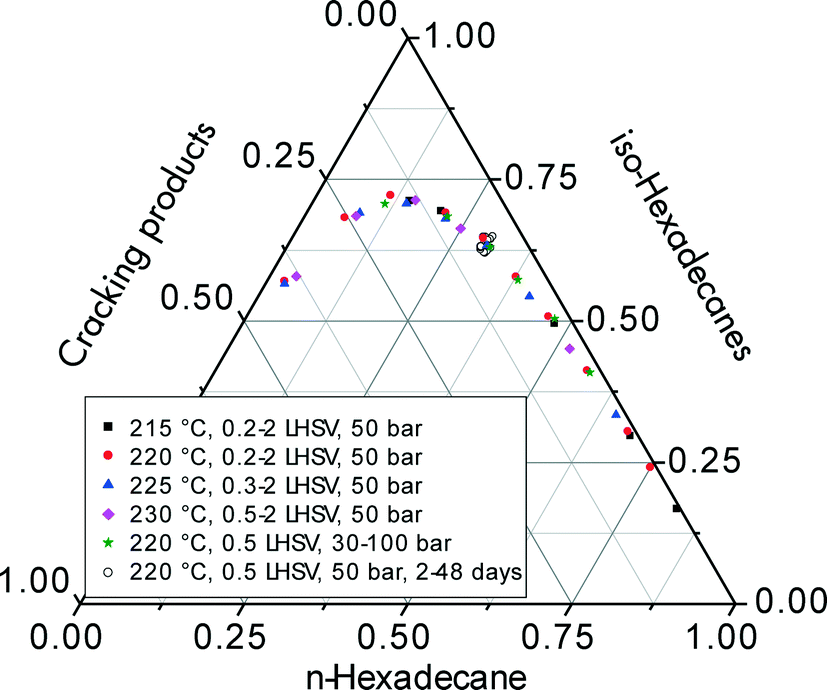 | ||
| Fig. 7 Ternary plot of n-hexadecane, isohexadecanes and cracking product fractions for hydroisomerization over Pt–Pd/H-beta (nSi/nAl = 25) by varying reaction conditions including temperature, space velocity, pressure, and time on-stream. | ||
Using such ternary plots for comparing hydrocracking results in two different Pt/SiO2–Al2O3 catalysts. Kang et al.6 convincingly showed that the data of Calemma et al.5 revealed less cracking of C16 and C28 paraffins at higher conversion levels. In addition to differentiating the performance of several hydroisomerization catalysts, the generation of these characteristic selectivity curves does not necessarily require a large amount of cumbersome kinetic experiments at different temperatures and/or pressures. Provided that the conversion levels cover a wide range, the experimental data for the ternary plots can be obtained even at ambient pressure by varying the space velocity at constant temperature or changing the temperature at constant space velocity.
Apparently, the plots in Fig. 5–7 follow the assumed pattern of consecutive reactions in which the paraffin feed exclusively undergoes successive isomerization steps before cracking. Hence, the kinetic constants of the classical consecutive reaction mechanism (Fig. 8, without dashed arrows) were obtained by fitting such a simplified model to the experimental data for n-paraffin hydroisomerization (Fig. 5, solid lines). The normalized values of the kinetic constants (Fig. 8, given in brackets) indicate that cracking on Pt–Pd/H-beta (nSi/nAl = 25) occurs at a rate at least five times lower than that of isomerization. The rate constants obtained are in accordance with the classical consecutive reaction mechanism, i.e., the isomerization rate constant of monobranched isomers into multibranched isomers should be higher than the rate constant of n-paraffins into monobranched isomers and than the rate constant for cracking.23
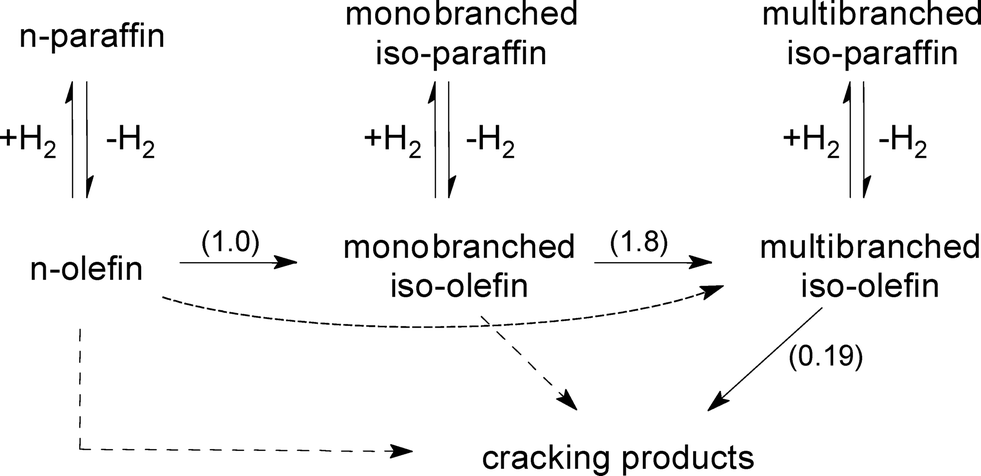 | ||
| Fig. 8 Simplified schematic reaction pathway for the hydroisomerization of n-paraffins. Normalized values of the kinetic constants are given in brackets. | ||
Nevertheless, the estimation of reaction rate constants using a complex network of hydroisomerization and hydrocracking reactions (Fig. 8, including dashed arrows), i.e., with the assumption that n-paraffins and/or monobranched isomers are exposed to severe cracking,4,7,9,24–26 can turn out to be beneficial for explaining, for example, the ratio of mono- to multibranched isomers. Merabti et al.9 hold that a pore size effect is responsible for the less selective production of monobranched isomers on H-beta compared to Pt/H-ZSM-22: the small size of the pore apertures of H-ZSM-22 allows the n-paraffin to react only at the pore openings, while the entrance and diffusion of paraffins inside the larger channels of the H-beta zeolite are expected to be very rapid. The same considerations can be applied to the formation of multibranched isomers during C16 hydroconversion over Pt/SAPO-11, Pt/IZM-2, and Pt/USY catalysts, which is initially suppressed on SAPO-11 and IZM-2 compared to USY.27 While for Pt/SAPO-11 and Pt/IZM-2 a maximum isomerization yield of >80 wt.% (40–50 wt.% monobranched and 30–40 wt.% multibranched isomers) can be obtained, and the isomerization yield for Pt/USY is limited to about 60 wt.% (20 wt.% monobranched isomers and 40 wt.% multibranched isomers) due to its larger pore size and its particular sensitivity to hydrocracking.
3.5 Studies on catalyst stability
For the hydroisomerization of light and long-chain n-paraffins, the content of olefinic intermediates and their residence time within the zeolite crystallites obviously control not only the conversion and the selectivity to mono-/multibranched isomers and cracked products, but also the deposition of carbonaceous materials, i.e., the stability of the catalyst.Pilot plant tests (220 °C, 50 bar, LHSV = 0.5 h−1, nH2/nhydrocarbons = 750:1) up to 62 days on-stream revealed an outstanding isomerization performance of the Pt–Pd/H-beta (nSi/nAl = 25) catalyst for the hydroisomerization of n-hexadecane (Fig. 9). At a conversion of ~70% which remained nearly stable over the whole test period, the selectivity for isomers slightly decreases from 89% to 86%. Negligible catalyst deactivation during n-hexadecane hydroconversion is in accordance with a low coke content of about 0.2 wt.% on the catalyst (see the following chapter). A similarly high isomer selectivity of ~90% is observed for the hydroconversion of a solid n-C18+ paraffin blend. However, the conversion of about 60% under the same reaction conditions (220 °C, 50 bar, LHSV = 0.5 h−1) is significantly lower than that of n-hexadecane. Consequently, the selectivity for isomers is found to be a little higher than n-hexadecane conversion. The lower reactivity of the n-C18+ paraffin blend is more noteworthy, as the relative conversion of n-paraffins within the mixed feeds greatly favors the conversion of hydrocarbons with a higher carbon number, i.e., the kinetic rate constants should increase with the chain length of the paraffins.6,28 Therefore, the lower diffusivity and/or sorption rate rather than the vapor–liquid equilibrium6 are assumed to explain the lower conversion of the n-C18+ paraffin blend.
 | ||
| Fig. 9 Conversions of n-hexadecane (solid symbols) and n-C18+ paraffin feed (open symbols) as well as product selectivities as a function of time on-stream over Pd–Pt/H-beta (nSi/nAl = 25) at 220 °C and 50 bar (LHSV = 0.5 h−1). | ||
To extend the lifetime of the catalyst as much as possible, the chemical industry prefers the regeneration of spent catalysts without burning off the detrimental carbonaceous deposits. Such rejuvenation treatments29,30 typically involve an interruption of the feed stream followed by switching to hydrogen at a temperature slightly higher than the reaction temperature during a short period of time in order to hydrocrack the carbonaceous deposits assumed to deactivate the catalyst. To obtain an accelerated deactivation during pilot-scale studies, the hydroisomerization of the solid n-C18+ paraffin feed was performed at a reduced hydrogen/hydrocarbon ratio (Fig. 10). After stopping the paraffin feed stream, flushing the reactor with hydrogen at a reaction temperature of 220 °C for 12 h was applied for restoring the catalyst activity. For a pilot-scale conversion of methanol on zeolite H-ZSM-5, such a straightforward procedure using a hydrogen-containing recycle gas at 420 °C resulted in a partial reactivation of the spent catalyst.29 Our results on the hydroisomerization of long-chain n-paraffins over Pt–Pd/H-beta (nSi/nAl = 25) indicate, however, that no improvement of catalyst activity can be achieved by hydrogen treatment at the reaction temperature of 220 °C used so far. The original conversion level of about 40% was not restored, i.e., the reactivity of the carbonaceous species towards a regeneration treatment at 220 °C is insufficient. Evidently, the temperature and/or the acidity of H-beta (nSi/nAl = 25) are too low for the required hydrocracking of coke species formed. Nevertheless, the catalytic experiments discussed above imply that the observed activity decrease of about 10% due to carbonaceous deposits can easily be compensated for by increasing the reaction temperature to above 225 °C.
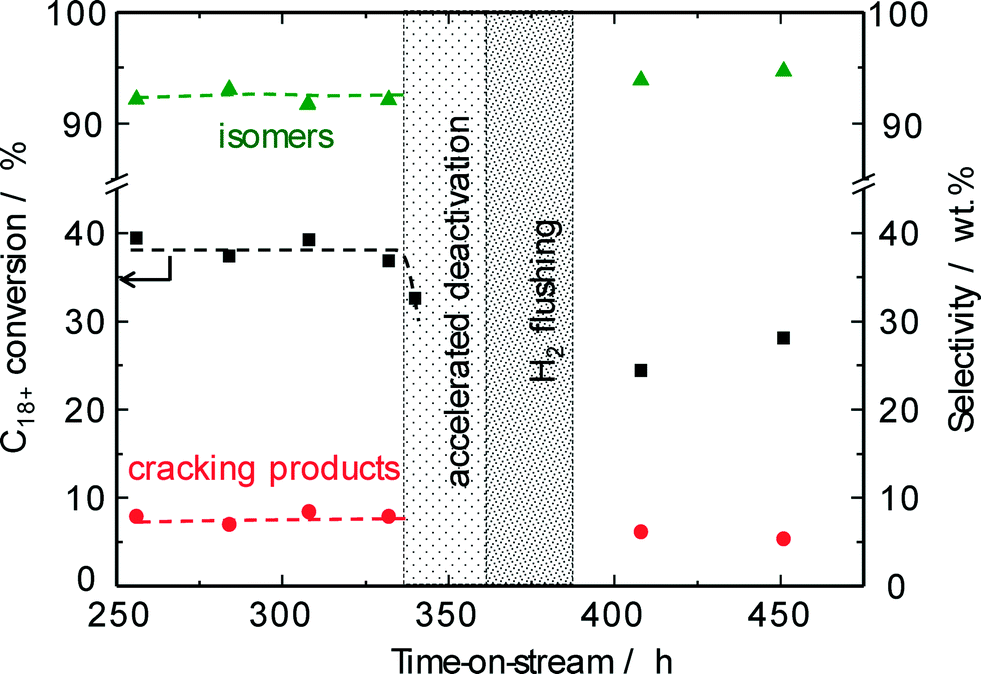 | ||
| Fig. 10 Conversions of n-C18+ paraffin feed and product selectivities as a function of time on-stream over Pd–Pt/H-beta (nSi/nAl = 25) at 220 °C and 50 bar (LHSV = 0.5 h−1). The effect of hydrogen flushing after accelerated carbon deposition shows a marked reduction of the nH2/nhydrocarbons ratio. | ||
3.6 Characterization of spent catalysts
In order to find the appropriate process conditions for a regeneration treatment (see previous section), the nature of carbonaceous deposits formed on the Pt–Pd/H-beta (nSi/nAl = 25) catalyst during the hydroisomerization of paraffins was characterized using various analytical techniques.The porosity of Pt–Pd/H-beta catalysts before and after use in hydroisomerization conversion was probed using argon sorption analysis. The argon adsorption/desorption isotherms of the catalyst used for the C18+ paraffin hydroconversion are shown in the inset of Fig. 11. With a significant argon uptake at a relative pressure (p/p0) below 0.1 which indicates its mainly microporous nature, the isotherm of the zeolite beta-based catalyst is of type IV according to the IUPAC classification and exhibits typical hysteresis between the adsorption and the desorption branches at a relative pressure of 0.4. The textural properties of the fresh and spent catalysts were calculated using the t-plot method and are summarised in Table 3. After 62 days on-stream, both the volume and the surface area of micropores were reduced by about 8%. Although the argon sorption data used in conjunction with the Horvath–Kawazoe model provide realistic pore-size values,31 non-local density functional theory (NLDFT) methods are preferred for obtaining information on the micropores of zeolite catalyst.32 NLDFT argon pore size distributions indicate the presence of micropores with sizes between 0.8 and 1.0 nm (Fig. 10) and clearly identify that coke deposition decreases the micropore surface area and the micropore volume of the Pt–Pd/H-beta catalyst by partial pore blocking.
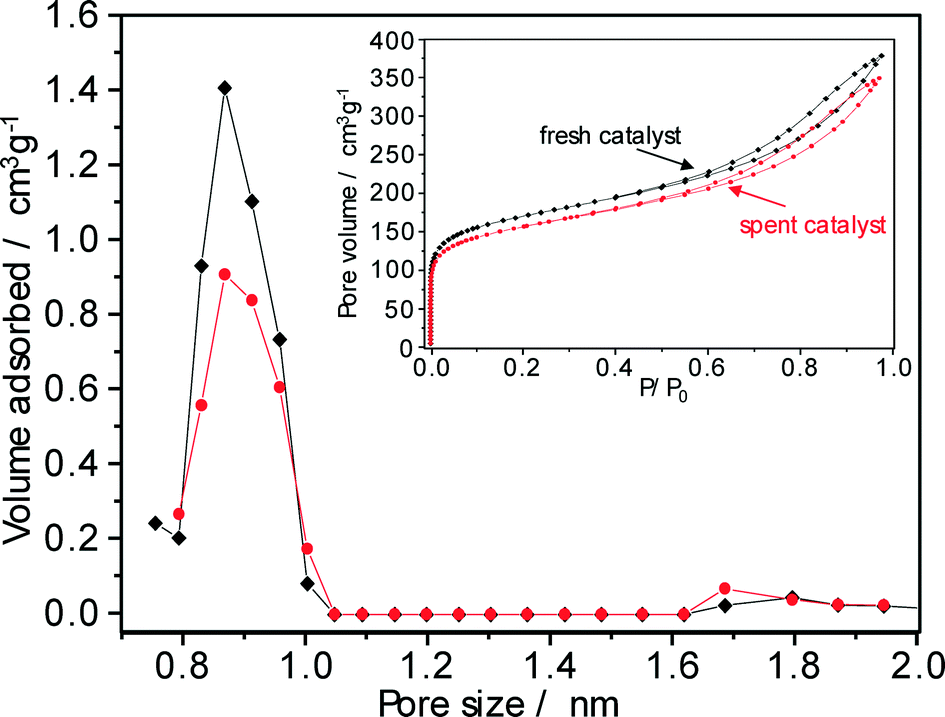 | ||
| Fig. 11 NLDFT pore size distributions of the fresh (squares) and the spent (circles) Pt–Pd/H-beta catalyst derived from Ar sorption isotherms presented in the inset (n-C18+ paraffin feed, 220 °C, 50 bar, LHSV = 0.5 h−1, 62 days on-stream). | ||
| Pt–Pd/H-beta (nSi/nAl = 25) | Fresh sample | After 62 days on-streama |
|---|---|---|
| a Coke content: 4.5 wt.%. b Calculated according to the t-plot method. | ||
| Total specific pore volume (cm3 g−1) | 0.484 | 0.447 |
| Specific micropore volumeb (cm3 g−1) | 0.128 | 0.118 |
| Specific micropore surface areab (m2 g−1) | 281 | 258 |
| External specific surface area (m2 g−1) | 212 | 197 |
In order to assess the formation of carbonaceous deposits during the hydroisomerization of long-chain paraffins, the spent Pt–Pd/H-beta catalysts were also characterized by means of TGA in air. For n-hexadecane conversion at 220 °C, 50 bar and 48 days on-stream, the TGA profile of the “spent” catalyst (Fig. 12) reveals an extremely low coke content (about 0.2 wt.% as determined by elemental analysis). Obviously, for such low amounts of carbonaceous deposits, the determination of the temperature of maximal weight loss as well as the nH/nC ratio is difficult and the spent catalysts are rather characterized by a lower uptake of water than by distinct TGA profiles. For comparison, hydroisomerization of the solid C18+ paraffin feed yielded after 62 days on-stream (220 °C and 50 bar) a spent Pt–Pd/H-beta sample with a coke content of 4.5 wt.%. As can be seen in Fig. 12, coke combustion took place between 250 °C and 400 °C and carbonaceous species were entirely removed around 500 °C. Such low-temperature coke combustion centred at ~330 °C usually facilitates the determination of its chemical nature33 and points to the predominant formation of hydrogen-rich deposits (so called “white” or “soft” coke irrespective of the dark gray color of the spent catalysts). As also shown in Fig. 12, both the carbonaceous species and the hydroisomerized paraffins reveal similar combustion behavior. For hexadecane conversion at 310 °C on Pd/SiO2–Al2O3 catalysts, however, Regali et al.8 observed two TGA maxima: low-temperature and high-temperature combustion at 160–270 °C and at 450–480 °C, respectively. Increasing the metal loading from 0.18 wt.% Pd to 1.20 wt.% Pd resulted in a lower temperature of both maxima. The authors concluded that more reactive coke deposits, presumably with a higher hydrogen content, were formed on the samples with higher Pd loadings, while less reactive coke species are produced at lower Pd loadings. However, the nH/nC ratio of those more reactive coke species on Pd/SiO2–Al2O3 having a CO2 evolution maximum at ~160 °C should be significantly higher than that of the hydroisomerized reaction products having a TGA maximum at ~340 °C (Fig. 12). It should be noted, however, that the formation of “harder” coke species (combustion at T > 400 °C) during the hydroisomerization of n-paraffins is unfavorable, because it reduces the feasibility of any in situ reactivation treatment of spent catalysts.29,30
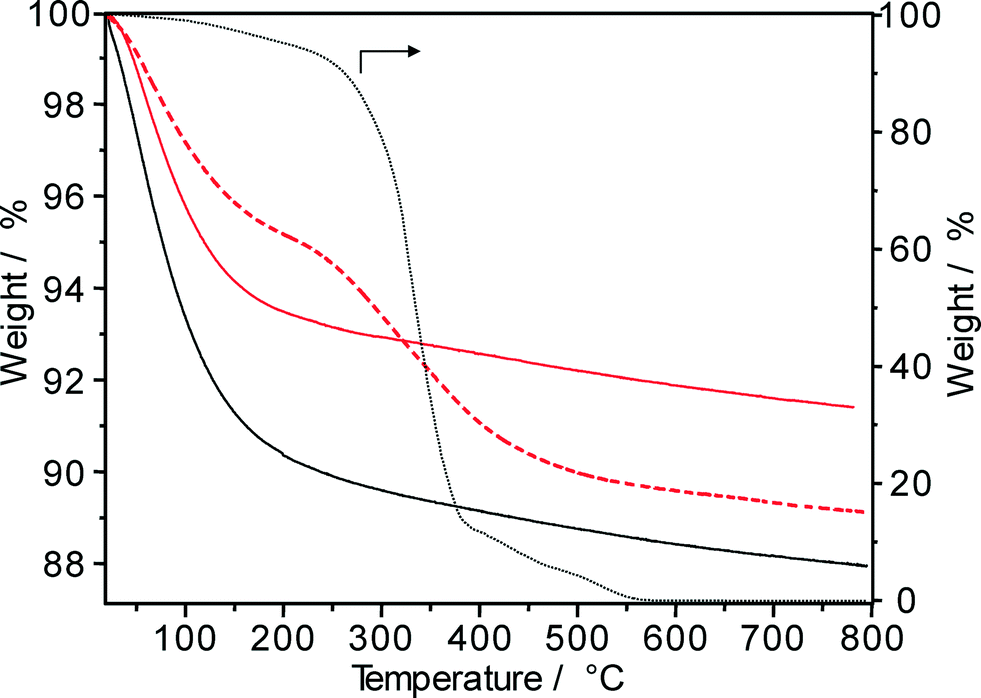 | ||
Fig. 12 TGA profiles of fresh Pd–Pt/H-beta (nSi/nAl = 25) catalyst (—) and samples spent during the hydroisomerization of hexadecane ( ) and n-C18+ paraffin feed ( ) and n-C18+ paraffin feed ( ) at 220 °C, 50 bar, LHSV = 0.5 h−1, and 62 days on-stream. For comparison: TGA profile of hydroisomerized products (⋯). ) at 220 °C, 50 bar, LHSV = 0.5 h−1, and 62 days on-stream. For comparison: TGA profile of hydroisomerized products (⋯). | ||
The 13C MAS NMR spectrum of a Pt–Pd/H-beta sample spent during the hydroisomerization of the C18+ paraffin feed at 220 °C reveals numerous NMR lines in the aliphatic range between 10 and 50 ppm (Fig. 13), as expected. The carbon atoms of paraffinic species may contribute to the observed signals at 10–15, 20–25, and 30–40 ppm. A large fraction of branched hydrocarbons is indicated by the signal at around 30 ppm. Furthermore, the NMR results do not provide indications for the presence of polyaromatic coke (characterized by chemical shifts of about 130 ppm) as well as of oxygen-containing deposits (having chemical shifts between 50 and 90 ppm). Unfortunately, NMR spectroscopy does not precisely disclose the chain length of the hydrogen-rich carbonaceous species.
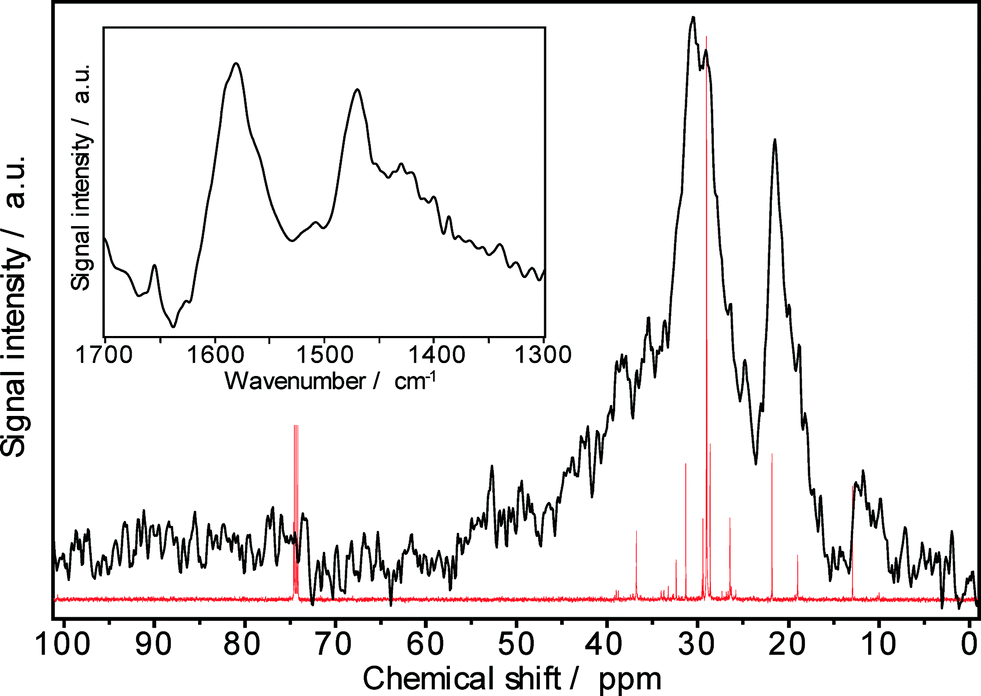 | ||
Fig. 13
13C MAS NMR spectrum of carbonaceous deposits (—) on Pd–Pt/H-beta and 13C NMR spectrum of hydroisomerized products ( ) (inset: FTIR spectrum of coke formed during n-C18+ paraffin hydroisomerization at 220 °C, 50 bar, LHSV = 0.5 h−1, 62 days on-stream). ) (inset: FTIR spectrum of coke formed during n-C18+ paraffin hydroisomerization at 220 °C, 50 bar, LHSV = 0.5 h−1, 62 days on-stream). | ||
The paraffinic nature of the carbonaceous deposits formed during hydroisomerization is also revealed by the FTIR results (inset of Fig. 13). In the characteristic IR region between 1300 and 1700 cm−1, three typical coke bands at 1580 cm−1 (assigned to C–C stretching vibrations of coke species), at 1440–1480 cm−1 (assigned to C–H deformation vibrations of paraffinic coke species), and at 1360–1390 cm−1 (preferentially assigned to CH3 groups of aromatic coke species) can be expected.33 Especially, the band intensity at about 1460 cm−1 can be attributed to the chemical nature of coke, e.g., the nH/nC ratio. As Fig. 13 shows, the intensities at 1580 cm−1 and 1470 cm−1 are very much alike, while the band at 1390 cm−1 is nearly lacking. Therefore, the predominant presence of hydrogen-rich carbonaceous deposits on the Pt–Pd/H-beta catalyst is independently indicated by FTIR spectroscopy and these findings are in accordance with the 13C MAS NMR and TGA results.
4. Conclusions
The hydroisomerization of long-chain n-paraffins over nano-sized zeolite Pt–Pd/H-beta (nSi/nAl = 25) yields high contents of multibranched isomers without extensive hydrocracking at conversions of up to 90%. The activity and the selectivity for hydroisomerization over bimetallic Pt–Pd/H-beta catalysts are superior to those of catalysts containing only one of the two noble metals. The remarkable yield of 52 wt.% for multibranched hexadecanes in comparison to about 18 wt.% for monobranched isomers can be related to a suitable residence time of olefinic intermediates and isoparaffins inside the nanocrystallites of zeolite beta and to its low density of acid sites avoiding severe cracking. A multi-technique characterization approach confirms the formation of hydrogen-rich coke during n-paraffin hydroisomerization on the Pt–Pd/H-beta catalysts and pore blocking by carbonaceous deposits can be held responsible for catalyst deactivation. Consequently, a balance between the metallic de/hydrogenation component and the acid and textural properties of the zeolite component provides the basis for further improvement of highly efficient and stable catalysts for the hydroisomerization of long-chain paraffins.Acknowledgements
The authors thank Dr. U. Decker and I. Reinhard (both from Leibniz-Institut für Oberflächenmodifizierung, Leipzig) for the experimental assistance given to this work.Notes and references
- H. Deldari, Appl. Catal., A, 2005, 293, 1–10 CrossRef CAS PubMed
.
- E. F. Sousa-Aguiar, F. B. Noronhac and A. Faro, Catal. Sci. Technol., 2011, 1, 698–713 Search PubMed
- L. Guo, X. J. Bao, Y. Fan, G. Shi, H. Y. Liu and D. J. Bai, J. Catal., 2012, 294, 161–170 CrossRef CAS PubMed
- A. Soualah, J. L. Lemberton, L. Pinard, M. Chater, P. Magnoux and K. Mojord, Appl. Catal., A, 2008, 336, 23–28 CrossRef CAS PubMed
- V. Calemma, S. Peratello and C. Perego, Appl. Catal., A, 2000, 190, 207–218 CrossRef CAS
- J. Kang, W. P. Ma, R. A. Keogh, W. D. Shafer, G. Jacobs and B. H. Davis, Catal. Lett., 2012, 142, 1295–1305 CrossRef CAS PubMed
- F. Regali, R. S. Paris, A. Aho, M. Boutonnet and S. Jaras, Top. Catal., 2013, 56, 594–601 CrossRef CAS PubMed
- F. Regali, L. F. Liotta, A. M. Venezia, V. Montes, M. Boutonnet and S. Jaras, Catal. Today, 2014, 223, 87–96 CrossRef CAS PubMed
- R. Merabti, L. Pinard, J. L. Lemberton, P. Magnoux, A. Barama and K. Moljord, React. Kinet., Mech. Catal., 2010, 100, 1–9 CAS
- N. Batalha, L. Pinard, S. Morisset, J. L. Lemberton, Y. Pouilloux, M. Guisnet, F. Lemos and F. R. Ribeiro, React. Kinet., Mech. Catal., 2012, 107, 285–294 CrossRef CAS PubMed
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, J. Catal., 2013, 307, 122–131 CrossRef CAS PubMed
- S. Mehla, K. R. Krishnamurthy, B. Viswanathan, M. John, Y. Niwate, K. Kumar, S. M. Pai and B. L. Newalkar, J. Porous Mater., 2013, 20, 1023–1029 CrossRef CAS
- K. B. Chi, Z. Zhao, Z. J. Tian, S. Hu, L. J. Yan, T. S. Li, B. C. Wang, X. B. Meng, S. B. Gao, M. W. Tan and Y. F. Liu, Pet. Sci., 2013, 10, 242–250 CrossRef CAS PubMed
- A. Rufer, A. Werner and W. Reschetilowski, Chem. Eng. Sci., 2012, 87, 160–172 CrossRef PubMed
- C. H. Geng, F. Zhang, Z. X. Gao, L. F. Zhao and J. L. Zhou, Catal. Today, 2004, 93–5, 485–491 CrossRef PubMed
- T. Matsuda, K. Watanabe, H. Sakagami and N. Takahashi, Appl. Catal., A, 2003, 242, 267–274 CrossRef CAS
- E. Blomsma, J. A. Martens and P. A. Jacobs, J. Catal., 1997, 165, 241–248 CrossRef CAS
- R. Roldan, F. J. Romero, C. Jimenez-Sanchidrian, J. M. Marinas and J. P. Gomez, Appl. Catal., A, 2005, 288, 104–115 CrossRef CAS PubMed
-
W. Huang, D. Li, X. Kang, Y. Shi and H. Nie, Hydroisomerization of n-hexadecane on zeolite catalysts in Recent Advances in the Science and Technology of Zeolites and Related Materials, Pts A - C, ed. E. VanSteen, M. Claeys and L. H. Callanan, 2004, vol. 154, pp. 2353–2358 Search PubMed
- A. Chica and A. Corma, Chem. Ing. Tech., 2007, 79, 857–870 CrossRef CAS
- G. Talebi, M. Sohrabi, R. L. Keiski, M. Huuhtanen, S. J. Royaee, S. Maghsoudi and H. Iammverdizadeh, J. Chil. Chem. Soc., 2008, 53, 1413–1419 Search PubMed
- N. Batalha, S. Morisset, L. Pinard, I. Maupin, J. L. Lemberton, F. Lemos and Y. Pouilloux, Microporous Mesoporous Mater., 2013, 166, 161–166 CrossRef CAS PubMed
- M. J. Girgis and Y. P. Tsao, Ind. Eng. Chem. Res., 1996, 35, 386–396 CrossRef CAS
- J. C. Chavarria, J. Ramirez, H. Gonzalez and M. A. Baltanas, Catal. Today, 2004, 98, 235–242 CrossRef CAS PubMed
- V. Calemma, S. Peratello, F. Stroppa, R. Giardino and C. Perego, Ind. Eng. Chem. Res., 2004, 43, 934–940 CrossRef CAS
- F. Regali, L. F. Liotta, A. M. Venezia, M. Boutonnet and S. Jaras, Appl. Catal., A, 2014, 469, 328–339 CrossRef CAS PubMed
- F. M. Mota, C. Bouchy, E. Guillon, A. Fecant, N. Bats and J. A. Martens, J. Catal., 2013, 301, 20–29 CrossRef PubMed
- C. Gambaro, V. Calemma, D. Molinari and J. Denayer, AIChE J., 2011, 57, 711–723 CrossRef CAS
- F. Bauer, H. Ernst, E. Geidel and R. Schodel, J. Catal., 1996, 164, 146–151 CrossRef CAS
- L. Pinard, P. Bichon, A. Popov, J. L. Lemberton, C. Canaff, F. Mauge, P. Bazin, E. F. S. Aguiar and P. Magnoux, Appl. Catal., A, 2011, 406, 73–80 CrossRef CAS PubMed
- S. Storck, H. Bretinger and W. F. Maier, Appl. Catal., A, 1998, 174, 137–146 CrossRef CAS
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS PubMed
-
F. Bauer and H. G. Karge, Characterization of Coke on Zeolites in Molecular Sieves - Characterization II, ed. H. G. Karge and J. Weitkamp, Springer-Verlag, Berlin, 2007, vol. 5, pp. 251–364 Search PubMed
Footnote |
| † In memoriam: Hellmut G. Karge (1931–2013) |
| This journal is © The Royal Society of Chemistry 2014 |