 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sortase A catalyzed reaction pathways: a comparative study with six SrtA variants†
Tobias
Heck
,
Phu-Huy
Pham
,
Alpaslan
Yerlikaya
,
Linda
Thöny-Meyer
and
Michael
Richter
*
Empa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Bioactive Materials, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: michael.richter@empa.ch; Tel: +41 58 765 7868
First published on 2nd June 2014
Abstract
Sortase A from Staphylococcus aureus (SrtA) has emerged as a useful enzymatic tool to covalently link proteins in a site-directed manner. Despite the proposed specificity of the SrtA-catalyzed reaction for target proteins carrying the amino acid motif LPXTG, enzymatic side reactions are frequently encountered impairing the yield and purity of the transpeptidation product. In this comparative study we investigated reactions of six different variants of SrtA with soluble protein substrates. Besides the transpeptidation reaction generating the desired heterodimeric product, all tested SrtA variants also catalyzed the formation of byproducts (i.e. homooligomeric and hydrolyzed species of the target protein) to different extents. We found that not only the choice of the enzyme, but also the composition and the context of the sorting motif within the particular amino acid sequence of the target protein had a critical impact on the apparent initial rates of the coupling reactions. In addition to the natural sorting signal LPETG, the motif LAETG was also recognized by the investigated SrtA variants. Even though the overall coupling rates with the LAETG-containing target proteins were rather slow, the formation of unwanted byproducts was largely suppressed under these conditions.
Introduction
Sortase A enzymes constitute a family of membrane-anchored enzymes that catalyze the covalent attachment of specific target proteins to the growing cell wall of Gram-positive bacteria by peptide bond formation.1 Despite their transpeptidase function in vivo sortases are systematically classified within the cysteine endopeptidases (EC 3.4.22), which are commonly associated with hydrolysis rather than formation of peptide bonds. Sortase A from Staphylococcus aureus (SrtA)2 is the best studied sortase to date and has gained particular attention because it can be used as an in vitro tool to facilitate site-directed, covalent modifications of proteins under mild reaction conditions.3,4 Thus, the so-called “sortagging” approach5 represents an enzymatic alternative to commonly applied chemical coupling techniques such as native chemical ligation and click chemistry.6,7 SrtA is described to recognize proteins carrying a specific LPXTG amino acid sorting motif, where X can be any of the naturally occurring amino acids.2,8,9 The enzymatic reaction comprises the initial formation of a thioester intermediate, which is formed between the Cys184 residue in the SrtA active site and the carbonyl carbon atom of the peptide bond between threonine and glycine of the sorting motif.10,11 Nucleophilic attack by the terminal amino group of an oligoglycine-functionalized compound – a pentaglycine crossbridge of lipid II in nature – on the thioester of the acyl-enzyme intermediate leads to generation of a new amide bond between the SrtA-bound target protein moiety and the coupling partner. The formed transpeptidation product is eventually released and the active site of the enzyme becomes available for another reaction cycle.The transpeptidation reaction catalyzed by SrtA enables access to a great diversity of site-specific coupling reaction with respect to target proteins and acceptor molecules, provided that they can be engineered with the LPXTG sorting motif and an N-terminally exposed oligoglycine tail, respectively.3 Due to this versatility on the one hand and the proposed specificity for the sorting motif on the other hand the SrtA-catalyzed reaction has been widely used to create covalent protein conjugates with new functionalities, such as fusions of two distinct protein species,12,13 site-specifically labeled proteins,14,15 cyclized proteins,16,17 or proteins immobilized on solid supports.18–20 A series of protocols has recently been published describing the production of SrtA and the design of target proteins as well as the experimental setup for various SrtA-based transpeptidation scenarios.21–24
Two soluble versions of SrtA from S. aureus are commonly used as catalysts in synthetic reactions.9,25 They differ by the extent of truncation of the N-terminal transmembrane domain, which in vivo anchors the sortase in the bacterial membrane. In H6-SrtAΔ59 and H6-SrtAΔ25 the N-terminal amino acid residues 1–59 and 1–25, respectively, are deleted from the enzymes and replaced with a hexahistidine tag allowing for straight-forward affinity purification. Both truncated variants of SrtA have been described to show virtually identical coupling activities.22,25 Furthermore, a C-terminally His-tagged SrtA variant (SrtAΔ59-H6) and various mutants thereof with supposedly improved catalytic parameters for the coupling of two fluorophore-labeled oligopeptides have been recently identified using a yeast display approach.26
The possibility of employing proteolytic enzymes as catalysts for the formation of peptide bonds and their impact in organic synthesis has been widely investigated.27,28 Under so-called kinetically controlled reaction conditions, serine and cysteine proteases that react via a covalent acyl-enzyme intermediate not only function as hydrolases, but can also catalyze the formation of amide bonds from activated amide or ester substrates (“acyl donors”) and amine nucleophiles (“acyl acceptors”).28 In the case of the SrtA-catalyzed transpeptidation reaction no initial activation of the rather non-activated peptide structure of the acyl donor (represented by the LPXTG-containing target protein) is needed to facilitate peptide bond formation with the oligoglycine-modified acyl acceptor component. Besides transpeptidation, side reactions are an important issue frequently encountered in protease-catalyzed peptide synthesis under kinetically controlled conditions because additional nucleophiles may compete with the desired nucleophilic acceptor for the thioester carbonyl group of the acyl-enzyme intermediate. On the one hand the presence of competing amine nucleophiles may lead to unwanted intermolecular coupling reactions among the reactants, on the other hand hydrolysis of the activated acyl donor is likely to occur in aqueous reaction systems.28,29 The formation of such byproducts has also been reported in SrtA-catalyzed reaction systems, but in most cases no further characterization of the generated protein species has been performed.30–32 Möhlmann and coworkers investigated different approaches to overcome byproduct formation during the SrtA-catalyzed site-specific attachment of a fluorophore-labeled oligoglycine probe to an antibody Fab fragment.32 In their study, the authors observed hydrolysis of the sorting motif as well as unspecific covalent crosslinking of the light and heavy antibody chains leading to drastically reduced yields of the desired transpeptidation product. In this case, the formation of crosslinked byproducts could be attributed to the side chain amino group of one lysine residue on the heavy antibody chain, which was ideally positioned to attack the acyl-enzyme intermediate formed between SrtA and the sorting motif at the C-terminus of the light antibody chain. Two strategies proved successful to suppress the formation of byproducts to a large extent: (i) reaction engineering employing increased concentrations of the oligoglycine-modified acceptor probe at a slightly basic pH of 7.5, and (ii) substrate engineering exchanging the potential lysine nucleophile on the heavy antibody chain for a non-nucleophilic arginine residue.
In the present study we compared the catalytic properties of six variants of SrtA in coupling reactions involving two soluble target proteins. Particular attention was paid on the progress of the desired transpeptidation reaction along with competing side reactions catalyzed by the different enzymes. All tested SrtA variants originate from S. aureus and are readily described in the scientific literature;9,25,26,33 for a summary of the SrtA constructs we refer to Table 1 and the ESI† (Fig. S1). Furthermore, we investigated the impact of engineering the sorting motif on the interplay of site-specific transpeptidation and unwanted crosslinking reactions catalyzed by the six SrtA variants. The green fluorescent protein GFPuv,34,35 a mutant of wild-type GFP from Aequorea victoria, was considered an appropriate model target protein because (i) GFP has been widely used as fusion partner in SrtA-catalyzed reactions due to its stability and unique fluorescent properties, and (ii) the formation of GFP-derived byproducts by SrtA has been observed, but not further investigated in several independent studies.30,31,36–38 Between the employed SrtA variants we not only observed drastic differences in the apparent rates of coupling, but also in the propensity of the enzymes to catalyze site-directed transpeptidation rather than formation of intermolecularly crosslinked byproducts. Furthermore, we found that the occurrence of side reactions could be minimized by modifications introduced within and upstream of the sortase-recognition sequence.
Results and discussion
SrtA catalyzes intermolecular crosslinking of GFPuv
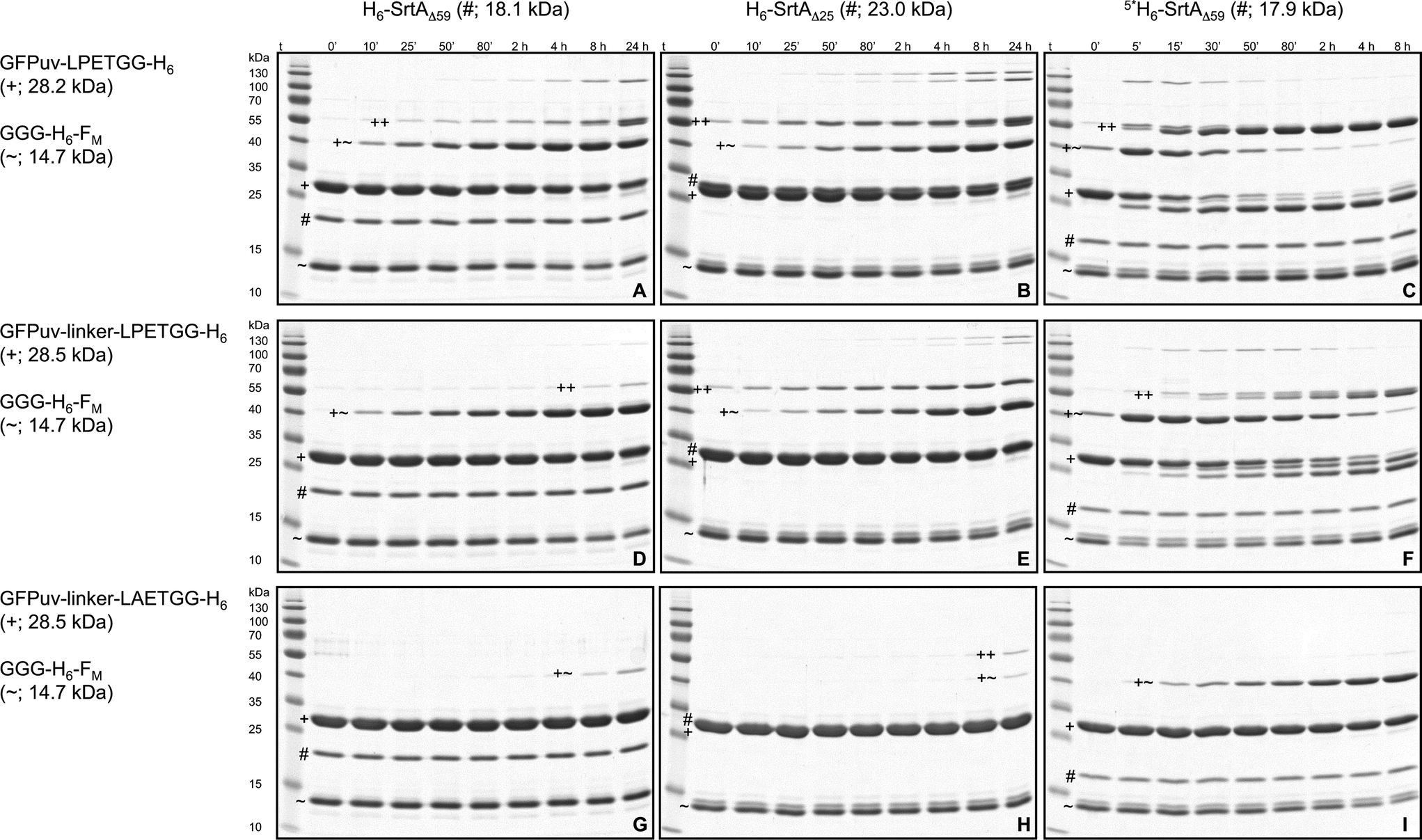
In experiments using a sortase variant from S. aureus ATCC6538 (H6-SrtAΔ59) and hexahistidine-tagged GFPuv-LPETGG-H6 as the sole substrate in the absence of an oligoglycine-modified acceptor molecule, we observed the time-dependent formation of new protein species (Fig. 1, gel A). After 8 hours of incubation, two weakly separated bands corresponding to proteins with molecular weights of approximately 55 kDa could be distinguished by SDS-PAGE analysis of the reaction mixtures. The appearance of these bands suggested the covalent linkage of GFPuv monomers (28 kDa) by the action of SrtA, yielding two distinct dimeric forms. Similar protein species generated in SrtA-catalyzed coupling reactions with green-fluorescent protein variants have previously been observed,30,31,36–38 but further efforts to investigate their origin have not been undertaken. In order to study the formed protein species in more detail, we analyzed the entire reaction mixture by electrospray ionization mass spectrometry (ESI-MS). We found masses of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 066.5 and 28022.0 Da corresponding with good accuracy to H6-SrtAΔ59 (18067.3 Da) and the mature form of GFPuv-LPETGG-H6 after removal of the N-terminal methionine residue (28022.3 Da),39,40 respectively. Furthermore, masses of 55088.5 Da and 54133.5 Da were detected. The first mass is in close accordance with the molecular weight of dimeric GFPuv-LPET-GFPuv-LPETGG-H6 (55089.8 Da) substantiating the SrtA-catalyzed crosslinking of two GFPuv-LPETGG-H6 monomers with the concomitant loss of one C-terminal GG-H6 fragment (Fig. 2, species 1). The latter mass indicates the subsequent loss of another GG-H6 fragment and a water molecule from the C-terminus of the previously formed GFPuv dimer (Fig. 2, species 1). This strongly suggests that a cyclized dimeric form of GFPuv (expected mass: 54134.8 Da) had been generated (Fig. 2, species 2). Neither by SDS-PAGE nor by ESI-MS analysis of the reaction mixtures we found an indication of a long-lived GFPuv-SrtA reaction intermediate of 45.2 kDa as has been shown by mass-spectrometric analysis of SrtA-catalyzed reactions with short oligopeptide probes.41
066.5 and 28022.0 Da corresponding with good accuracy to H6-SrtAΔ59 (18067.3 Da) and the mature form of GFPuv-LPETGG-H6 after removal of the N-terminal methionine residue (28022.3 Da),39,40 respectively. Furthermore, masses of 55088.5 Da and 54133.5 Da were detected. The first mass is in close accordance with the molecular weight of dimeric GFPuv-LPET-GFPuv-LPETGG-H6 (55089.8 Da) substantiating the SrtA-catalyzed crosslinking of two GFPuv-LPETGG-H6 monomers with the concomitant loss of one C-terminal GG-H6 fragment (Fig. 2, species 1). The latter mass indicates the subsequent loss of another GG-H6 fragment and a water molecule from the C-terminus of the previously formed GFPuv dimer (Fig. 2, species 1). This strongly suggests that a cyclized dimeric form of GFPuv (expected mass: 54134.8 Da) had been generated (Fig. 2, species 2). Neither by SDS-PAGE nor by ESI-MS analysis of the reaction mixtures we found an indication of a long-lived GFPuv-SrtA reaction intermediate of 45.2 kDa as has been shown by mass-spectrometric analysis of SrtA-catalyzed reactions with short oligopeptide probes.41
 | ||
| Fig. 1 SDS-PAGE analysis of SrtA-catalyzed reactions with GFPuv target proteins. The six gels show reactions of the target proteins GFPuv-LPETGG-H6 (A–C) and GFPuv-linker-LPETGG-H6 (D–F) with the SrtA variants H6-SrtAΔ59 (A, D), H6-SrtAΔ25 (B, E) and 5*SrtAΔ59-H6 (C, F). The symbols #, + and ++ designate the bands corresponding to the particular SrtA variant, the GFPuv target protein and the crosslinked homodimeric species of GFPuv, respectively. Due to the unusual migration behavior of SrtA on SDS-PAGE gels the bands corresponding to H6-SrtAΔ25 and the monomeric GFPuv target proteins could not be separated. A summary of SDS-PAGE gels for the reaction analysis of all six SrtA variants with the different GFPuv target proteins is shown in the ESI† (Fig. S3a–f, left columns). | ||
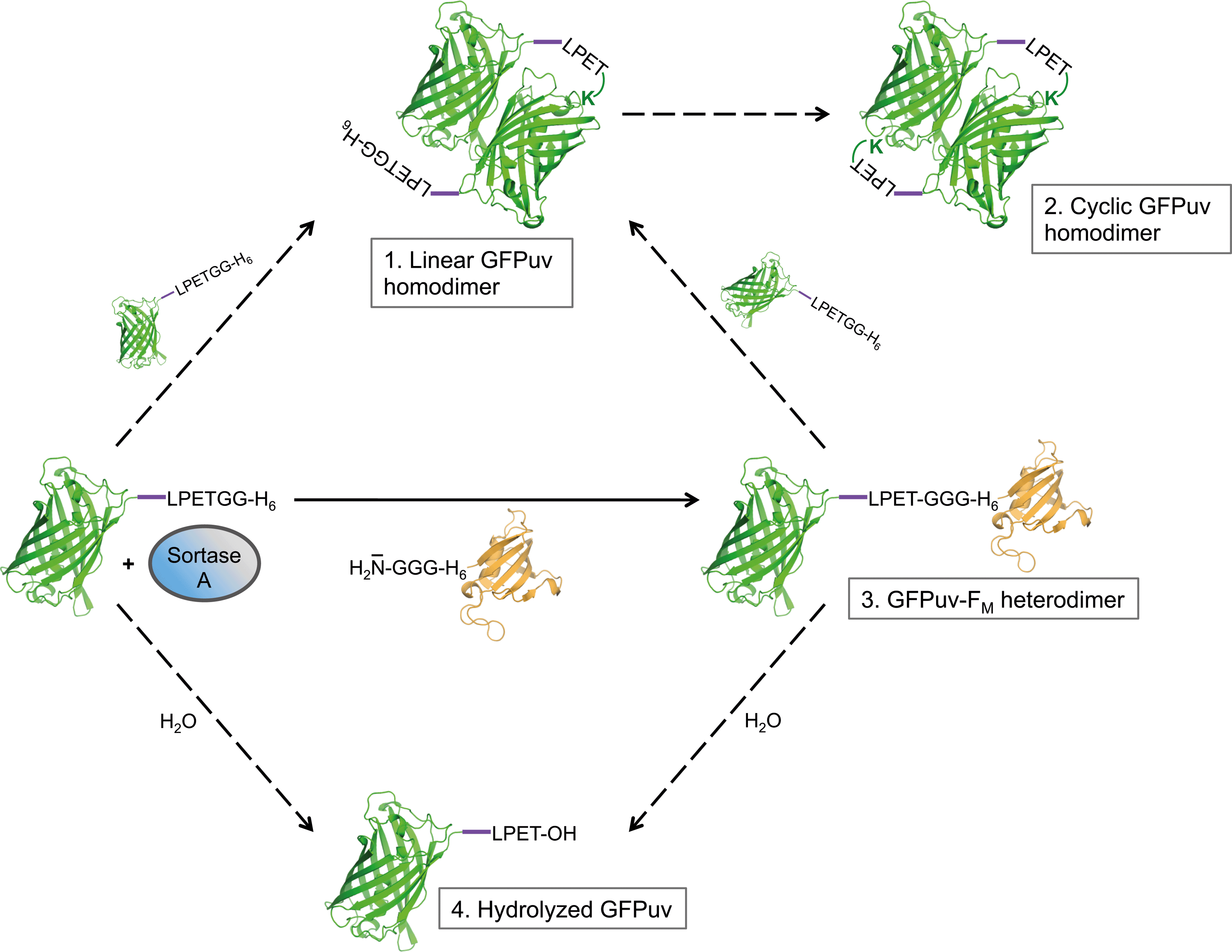 | ||
| Fig. 2 Schematic representation of SrtA-catalyzed reactions with the target protein GFPuv engineered with a C-terminal LPETG sorting motif; the model was deduced from results obtained in the present investigation. The site-specific transpeptidation reaction in the presence of the acceptor protein GGG-H6-FM is indicated with a solid arrow, side reactions (intermolecular GFPuv crosslinking and hydrolysis) are marked with dashed arrows. The 5-amino acid linker added upstream of the sorting motif in some GFPuv target proteins is depicted in purple. | ||
Based on the results obtained from SDS-PAGE analysis and ESI-MS measurements we propose that GFPuv-LPETGG-H6 serves a dual role as acyl donor and acyl acceptor in the SrtA-catalyzed reaction. On the one hand GFPuv-LPETGG-H6 represents a target protein for SrtA-catalyzed reactions due to the presence of an easily accessible LPETG sorting motif close to the C-terminus. On the other hand the protein, despite the absence of an N-terminal oligoglycine stretch, evidently acts as a nucleophile acceptor causing the formation of covalently crosslinked GFPuv dimers. It is reported that GFP from A. victoria associates into a dimeric assembly in the crystalline state (PDB ID: 1GFL) and also in solution.42 On this basis we suggest that once the GFPuv-SrtA reaction intermediate has formed the activated carbonyl group in the thioester bond of this complex is prone to nucleophilic attack by an appropriately positioned lysine ε-amino group located on the neighboring GFPuv subunit within the GFPuv dimer. The fact that the ε-amino group of an exposed lysine residue can replace the natural oligoglycine nucleophile of common SrtA-catalyzed reactions has already been confirmed in recent investigations.32,41 This reaction gives rise to formation of a stable intermolecular isopeptide bond at the threonine residue of the sorting motif and prevents further recognition of the target protein by SrtA.32 In the case of GFPuv, the symmetric arrangement of subunits provides the basis for the successive formation of two isopeptide bonds by SrtA at opposite sides of the crosslinked GFPuv dimer. This sequence of intermolecular coupling reactions likely explains the emergence of a linear and a cyclized homodimeric species of GFPuv that we propose based on the results obtained by SDS-PAGE and ESI-MS measurements (Fig. 2, species 1 and 2).
Intermolecular GFPuv crosslinking can be reduced by engineering the sorting motif
Due to the proposed specificity of the SrtA-catalyzed reaction for the LPXTG sorting motif on the target protein and the N-terminal oligoglycine stretch on the nucleophilic acceptor molecule this enzymatic approach has been widely applied for the site-specific modification of proteins. However, if intermolecular crosslinking of target proteins by isopeptide-bond formation occurs – as shown in the present study for GFPuv or by Möhlmann and coworkers for an antibody Fab fragment32 – the portion of target protein available for site-specific transpeptidation with the oligoglycine-modified acceptor can be drastically diminished as the enzymatic reaction proceeds. Hence, the generation of isopeptide bonds between target protein molecules not only reduces the yield, but also compromises the purity of the desired transpeptidation product.32 Intermolecular crosslinking by isopeptide-bond formation can largely be overcome by using the oligoglycine-functionalized acceptor moiety in high molar excess over the LPETG-containing donor protein. However, this approach is unfavorable as it raises costs for the coupling reaction and complicates the subsequent purification of the transpeptidation product. With the aim to reduce intermolecular crosslinking of GFPuv-LPETGG-H6 by a protein engineering approach we initially exchanged the structurally most flexible lysine residues located close to the termini of the protein (K3 and K238) for arginine. The resulting mutants GFPuv-LPETGG-H6 K3R and GFPuv-LPETGG-H6 K238R were incubated with H6-SrtAΔ59, and samples taken from the reaction mixture at different time points were analyzed by SDS-PAGE. Since no significant reduction of the SrtA-catalyzed crosslinking reaction was observed with the two mutated GFPuv target proteins (data not shown) we inserted a 5-amino acid linker (GGGGS)22 between the GFPuv core and the sorting motif (GFPuv-linker-LPETGG-H6) in order to increase the distance and conformational flexibility between the potential crosslinking sites on the GFPuv dimer. Using GFPuv-linker-LPETGG-H6 we observed a significant decline in the formation of crosslinked GFPuv homodimers by SrtA as compared to reactions carried out with GFPuv-LPETGG-H6 (Fig. 1, cf. gels A and D). To further test the specificity of SrtA for recognition of the sorting motif we exchanged the LPETG sequence on GFPuv-LPETGG-H6 and GFPuv-linker-LPETGG-H6 by LAETG (Fig. S2†). With both target proteins the change of a single amino acid (P to A) in the sorting motif drastically reduced, but not completely abolished the crosslinking activity of SrtA (Fig. S3a,† left column). This observation is in agreement with a screen of small peptide substrates done by Kruger and coworkers, who showed that besides the favored amino acid proline in the second position of the sorting motif, the sequence LAXTG may also serve as a weak recognition site for SrtA.43Comparison of intermolecular GFPuv crosslinking by SrtA variants
Two versions of SrtA derived from S. aureus are most frequently used as catalysts for site-directed transpeptidation reactions in vitro. H6-SrtAΔ59 and H6-SrtAΔ25 essentially differ by the composition of their N-terminal regions connecting the catalytic core of the enzyme with the hexahistidine tag (Fig. S1†). Despite this structural difference both SrtA variants have been described as functionally identical and are therefore used interchangeably.22,25 In comparative crosslinking experiments using the GFPuv target proteins described above (see also Fig. S2†) we observed considerable differences in the crosslinking reaction catalyzed by H6-SrtAΔ59 and H6-SrtAΔ25 (Fig. 1, cf. gels A, D and B, E). Besides the aforementioned covalently crosslinked homodimeric species of GFPuv that were equally formed with both SrtA variants, H6-SrtAΔ25 generated a substantial quantity of GFPuv oligomers with molecular masses >100 kDa that were clearly visible on coomassie stained SDS-PAGE gels (Fig. 1). Furthermore, H6-SrtAΔ25, unlike H6-SrtAΔ59, showed only a minor reduction in crosslinking activity with GFPuv-linker-LPETGG-H6 as compared to reactions with GFPuv-LPETGG-H6 (Fig. 3). Apart from the obvious structural difference in the N-terminal regions of H6-SrtAΔ59 and H6-SrtAΔ25, the used sortase constructs (derived from S. aureus strains ATCC 6538 and Newman, respectively) showed an additional minor discrepancy at amino acid position 167 within the SrtA core domain (Fig. S1†). In order to cross-check that this single mutation does not affect the crosslinking activities of H6-SrtAΔ59 and H6-SrtAΔ25, we created two point mutants containing interchanged amino acids at position 167 (H6-SrtAΔ59 E167G and H6-SrtAΔ25 G167E). We found that both SrtA mutants generated the same patterns of intermolecularly crosslinked GFPuv species on SDS-PAGE gels as the corresponding SrtA wild-type variants (data not shown). From these results we conclude that the variations of the N-terminal regions in H6-SrtAΔ59 and H6-SrtAΔ25 are solely responsible for the differences observed in the crosslinking activities of the enzymes with the GFPuv target proteins. Moreover, we tested whether the localization of the hexahistidine tag at either the N- or C-terminus of the employed SrtAΔ59 variant affects the outcome of the crosslinking reaction. As shown in Fig. 3 we found no significant change in intermolecular GFPuv crosslinking when either H6-SrtAΔ59 or SrtAΔ59-H6 was used as the catalyst (cf. also Fig. S3a and c,† left columns).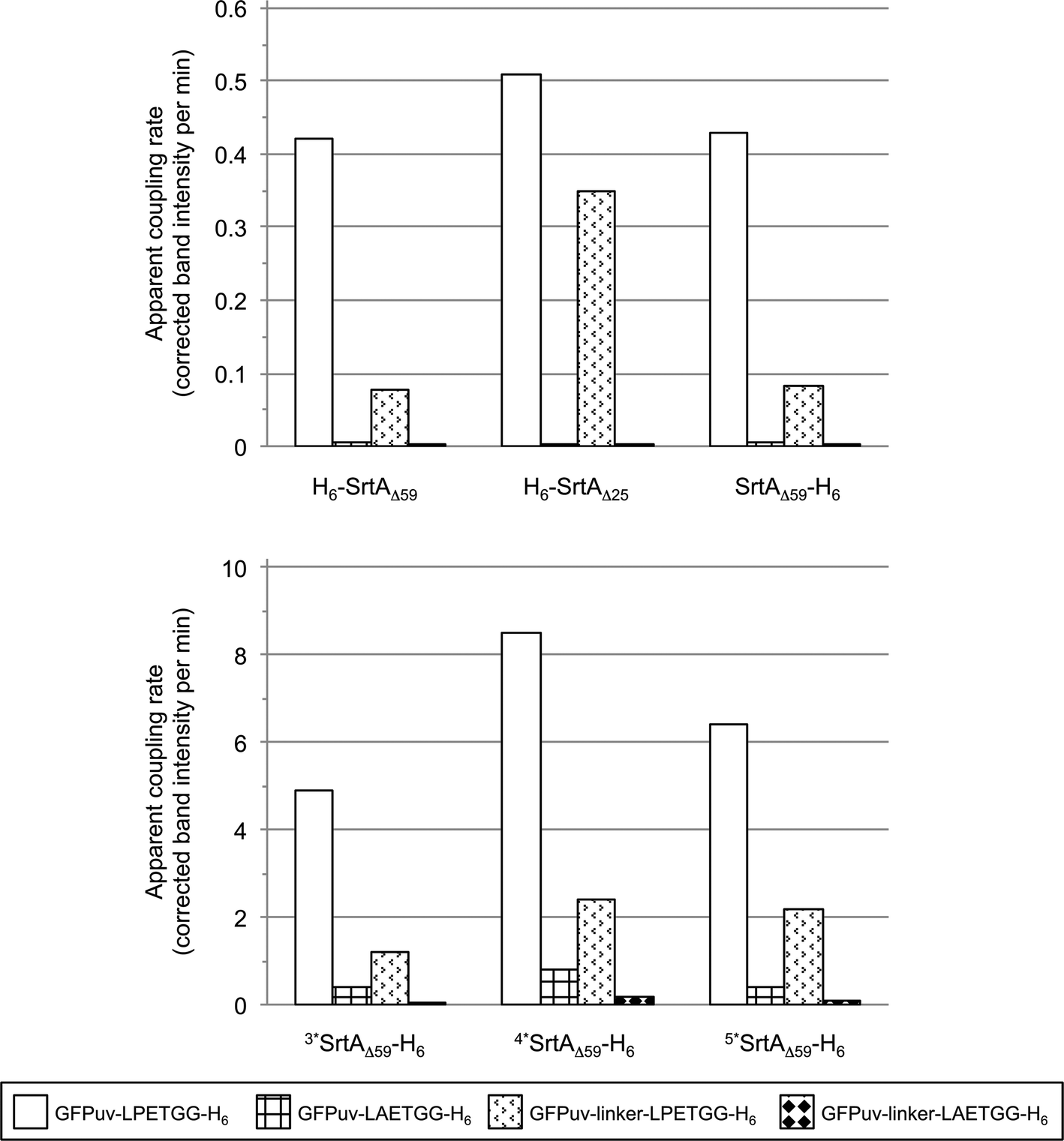 | ||
| Fig. 3 Comparison of SrtA-catalyzed reactions with the four tested GFPuv target proteins (depicted in different patterns). The bars display the apparent initial rates of covalent GFPuv homodimer and -oligomer formation as determined by digital image analysis of SDS-PAGE gels (Fig. S3, left columns and S4a†). | ||
In the following experiments, we investigated intermolecular crosslinking of the GFPuv target proteins using three point mutants of the SrtAΔ59-H6 variant as catalysts (3*: P94S,D160N,D165A; 4*: P94S,D160N,D165A,K196T; 5*: P94R,D160N,D165A,K190E,K196T). These SrtA mutants have been described recently to catalyze the site-directed fusion of short-length oligopeptides with drastically improved catalytic rates as compared to the respective wild-type enzyme SrtAΔ59-H6.26 As calculated from the SDS-PAGE gels depicted in Fig. 1 and S3d–f† (left columns) all three mutants catalyzed the formation of intermolecular crosslinks between the GFPuv target proteins at considerably higher apparent rates than SrtAΔ59-H6 (Fig. 3). In contrast to reactions performed with the wild-type SrtA variants, where the crosslinked GFPuv species represented the only reaction products visible on coomassie stained SDS-PAGE gels, particularly 4*SrtAΔ59-H6 and 5*SrtAΔ59-H6 catalyzed the degradation of the GFPuv target proteins into at least two different byproducts of lower molecular weight (Fig. 1, gels C and F). By ESI-MS analysis of a reaction mixture containing 5*SrtAΔ59-H6 (expected mass: 17853.1 Da, found mass: 17855.0 Da) and GFPuv-linker-LPETGG-H6 (expected mass: 28337.6 Da, found mass: 28341.0 Da) dominant masses of 27404.5 and 27385.5 Da were identified. The first mass can likely be assigned to the GFPuv target protein after hydrolysis of the sorting motif between amino acids T and G (expected mass: 27400.7 Da), whereas the latter mass indicates loss of an additional water molecule and may hence indicate formation of a cyclic monomeric form of GFPuv (expected mass: 27382.7 Da). Apart from intermolecular crosslinking among substrate molecules, substrate hydrolysis represents another commonly observed side reaction when sortases,32 but also other proteolytic enzymes27–29 are employed for synthetic reactions in buffered aqueous solution. In order to minimize the formation of hydrolytic byproducts, protease-catalyzed peptide synthesis has in some cases been successfully performed in non-aqueous reaction media.27 However, it has to be considered that in many cases the structural and functional integrity of the catalyst is compromised by the addition of organic solvents. Particularly in sortase-catalyzed reactions, where it is generally desired to retain the functional state of all protein components involved, the use of organic solvents may likely affect either the sortase or one of the protein substrates and has thus, to the best of our knowledge, not been investigated.
Competition of site-specific transpeptidation and intermolecular crosslinking reactions by SrtA variants
In order to investigate the interplay of SrtA-catalyzed transpeptidation and intermolecular crosslinking by isopeptide-bond formation we incubated each of the six enzyme variants (Fig. S1†) in parallel reaction batches with each of the four previously used GFPuv target proteins (GFPuv-LPETGG-H6, GFPuv-linker-LPETGG-H6, GFPuv-LAETGG-H6 and GFPuv-linker-LAETGG-H6; Fig. S2†) in the presence of an equimolar amount of the triglycine-modified acceptor protein GGG-H6-FM (see Fig. 2). Samples were taken at different time points throughout the enzymatic reactions and analyzed by SDS-PAGE (Fig. 4 and S3a–f,† right columns). With all SrtA variants the simultaneous formation of site-specifically coupled GFPuv-FM heterodimers and intermolecularly crosslinked GFPuv oligomers was observed. The formation of the heterodimeric GFPuv-FM product (expected mass: 41945.2 Da, found mass: 41949.0 Da) was confirmed by ESI-MS analysis of a reaction mixture containing the catalyst H6-SrtAΔ59 as well as the protein substrates GFPuv-linker-LPETGG-H6 and GGG-H6-FM (data not shown). For the sake of completeness it should be mentioned that SrtA did not catalyze the formation of covalently linked GFPuv-FM heterodimers from LPETG-tagged GFPuv and an FM protein variant lacking the N-terminal triglycine acceptor nucleophile.
 | ||
| Fig. 4 SDS-PAGE analysis of SrtA-catalyzed reactions between GFPuv target proteins and the acceptor protein GGG-H6-FM. The nine gels show reactions of the target proteins GFPuv-LPETGG-H6 (A–C), GFPuv-linker-LPETGG-H6 (D–F) and GFPuv-linker-LAETGG-H6 (G–I) with GGG-H6-FM catalyzed by the SrtA variants H6-SrtAΔ59 (A, D, G), H6-SrtAΔ25 (B, E, H) and 5*SrtAΔ59-H6 (C, F, I). The symbols ~, # and + designate the bands corresponding to the acceptor protein GGG-H6-FM, the particular SrtA variant and the GFPuv target protein, respectively. The heterodimeric GFPuv-FM transpeptidation product and the crosslinked homodimeric species of GFPuv are marked with the symbols +~ and ++, respectively. Due to the unusual migration behavior of SrtA on SDS-PAGE gels the bands corresponding to H6-SrtAΔ25 and the monomeric GFPuv target proteins could not be separated. A summary of SDS-PAGE gels for the reaction analysis of all six SrtA variants with the different GFPuv target proteins and GGG-H6-FM is shown in the ESI† (Fig. S3a–f, right columns). | ||
Fig. 5 gives a summary of the apparent initial rates for the SrtA-catalyzed formation of GFPuv-FM heterodimers and competing intermolecular GFPuv crosslinking reactions as calculated from the respective SDS-PAGE gels (Fig. S3a–f, right columns and S4b†). Furthermore, the ratio between the two rates is given for each reaction to express the relative propensity of the sortase to react with the oligoglycine-modified acceptor protein instead of generating isopeptide-crosslinked GFPuv byproducts. Like in the absence of oligoglycine-modified acceptor (Fig. 3) almost identical rates for intermolecular GFPuv crosslinking and transpeptidation reactions were observed with the SrtA variants H6-SrtAΔ59 and SrtAΔ59-H6. This corroborates our previous assumption that the position of the hexahistidine tag at either the N- or C-terminus does not affect the catalytic properties of the sortase (Fig. 5). In reaction mixtures containing GFPuv-LPETGG-H6, GGG-H6-FM and the SrtAΔ59 variants the apparent initial rates of intermolecular GFPuv crosslinking were reduced by a factor of five as compared to the same reactions lacking the triglycine modified acceptor protein (cf.Fig. 3 and 5). The replacement of GFPuv-LPETGG-H6 by GFPuv-linker-LPETGG-H6 led to a further reduction in the initial rate of GFPuv crosslinking whereas the GFPuv-FM transpeptidation product was formed at almost unchanged rates. This tendency is reflected by the increase in the values calculated for the ratio of product divided by byproduct formation in the particular reactions (Fig. 5). In contrast to H6-SrtAΔ59 and SrtAΔ59-H6, H6-SrtAΔ25 catalyzed the formation of isopeptide-crosslinked byproducts from GFPuv-LPETGG-H6 and GFPuv-linker-LPETGG-H6 at constant rates regardless of the absence or presence of GGG-H6-FM (cf.Fig. 3 and 5). These results suggest that in the presence of two competing amine nucleophiles the N-terminally fully truncated SrtAΔ59 variants have a higher propensity to react with the oligoglycine-modified acceptor whereas H6-SrtAΔ25 remains more prone to generate intermolecular crosslinks between the GFPuv target proteins by isopeptide bond formation. With all three wild-type SrtA variants the exchange of the LPETG sorting motif on the GFPuv target protein for LAETG led to approximately 100-fold reduced initial rates of both transpeptidation and intermolecular GFPuv crosslinking.
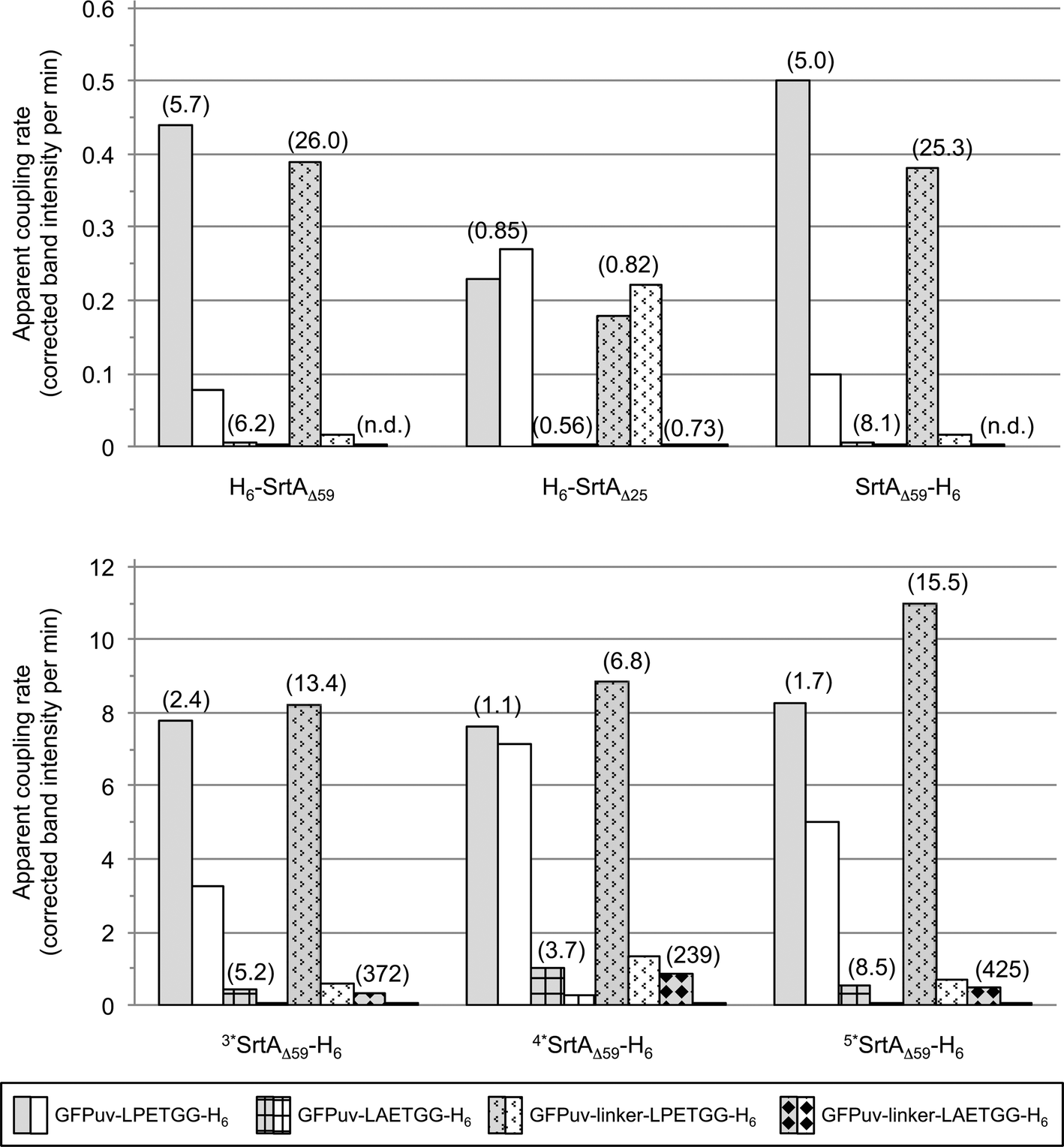 | ||
| Fig. 5 Comparison of SrtA-catalyzed reactions between the four tested GFPuv target proteins (depicted in different patterns) and the acceptor protein GGG-H6-FM. The bars display the apparent initial rates of transpeptidation (grey shaded bars) and covalent GFPuv homodimer and -oligomer formation (white bars) as determined by digital image analysis of SDS-PAGE gels (Fig. S3, right columns and S4b†). Ratios of the initial rate of transpeptidation over the rate of intermolecular GFPuv crosslinking are given in brackets for each combination of reaction. Apparent initial rates and associated ratios for reactions catalyzed by the SrtA mutants 3*SrtAΔ59-H6, 4*SrtAΔ59-H6 and 5*SrtAΔ59-H6 could only be roughly estimated due to the rapid initial progress of the enzymatic reactions. N.d.: ratio not specified because the apparent rate for intermolecular GFPuv crosslinking was below the detection limit. | ||
In agreement with the report by Chen and coworkers26 the three sortase mutants 3*SrtAΔ59-H6, 4*SrtAΔ59-H6 and 5*SrtAΔ59-H6 catalyzed the formation of the desired GFPuv-FM transpeptidation product from the GFPuv target proteins and GGG-H6-FM at considerably higher initial rates than the corresponding wild-type sortase variant SrtAΔ59-H6 (Fig. 4 and 5). At the same time, the formation of isopeptide-crosslinked byproducts in reactions containing the LPETG-tagged GFPuv variants and GGG-H6-FM was much more prominent with the mutant sortases than with SrtAΔ59-H6. As commonly observed in protease-catalyzed peptide synthesis under kinetic control,28Fig. 4 clearly illustrates that the GFPuv-FM transpeptidation product generated by the SrtA mutants accumulated to a maximum before it underwent subsequent enzyme-catalyzed hydrolysis and intermolecular crosslinking reactions. This can be explained by the fact that heterodimeric GFPuv-FM still bears the LPETG sorting motif and is therefore prone to subsequent SrtA-catalyzed reactions (Fig. 2, species 3), whereas the hydrolyzed and isopeptide-crosslinked species of GFPuv represent dead-end byproducts that cannot undergo further conversion by SrtA (Fig. 2, species 1, 2 and 4). In contrast to the three SrtA mutants that transformed most of the initially formed GFPuv-FM transpeptidation product into those undesired byproducts within eight hours, no significant product loss was observed in reactions catalyzed by the wild-type SrtA variants within the monitored 24 hour reaction period (Fig. 4 and S3†). However, considering the basic assumptions for reaction systems under kinetic control28 it is reasonable to propose that independently of the employed SrtA variant the real equilibrium of the presented reaction lies on the side of hydrolysis and isopeptide-crosslinked byproducts. In order to obtain high yields of the desired transpeptidation product it is therefore advisable to stop the catalytic reaction (e.g. by addition of EDTA to inactivate SrtA by calcium complexation) before unwanted side reactions start to dominate the reaction system.
Interestingly, when we used the unusual SrtA target protein GFPuv-linker-LAETGG-H6 in combination with GGG-H6-FM, the apparent initial rates of GFPuv-FM formation by the SrtA mutants were in the same range as the one observed for the reaction of wild-type SrtAΔ59-H6 with the actual target protein GFPuv-linker-LPETGG-H6 and GGG-H6-FM. At the same time, crosslinked oligomers of GFPuv were formed at 4- to 16-fold decreased initial rates from GFPuv-linker-LAETGG-H6. The characteristics of the particular reactions are reflected by the values calculated for the ratio of product divided by byproduct formation shown in Fig. 5. This suggests that the use of sortase target proteins with a non-typical LAXTG sorting motif in combination with one of the SrtA mutants26 may be beneficial to obtain covalently linked protein heterodimers of high purity in reaction systems where undesired isopeptide crosslinking impedes the site-directed transpeptidation reaction.
Experimental section
Construction of plasmids
A summary of plasmids used in this study is given in Table 1. Plasmids were either obtained from the sources indicated in the table or created by standard cloning techniques and site-directed mutagenesis (QuikChange Lightning Multi Site-Directed Mutagenesis Kit, Agilent, Basel, Switzerland). Primer synthesis and sequencing services of the relevant plasmid regions were done by Microsynth (Balgach, Switzerland).| Plasmid name | Plasmid backbone | Protein of interest | E. coli expression strain | Source |
|---|---|---|---|---|
| a The employed FM model protein represents a variant of the human FK 12-binding protein, originally developed for use in gene therapy.44,45 | ||||
| pTH2 | pQE30 | H6-SrtAΔ59 | JM109 | This study |
| pTH28 | pQE30 | H6-SrtAΔ59 E167G | JM109 | This study |
| pMC141 | pET15b | H6-SrtAΔ25 | BL21 Rosetta 2 (DE3) | ref. 33 |
| pTH29 | pET15b | H6-SrtAΔ25 G167E | BL21 (DE3) | This study |
| pTH14 | pET29a | SrtAΔ59-H6 | BL21 (DE3) | ref. 26 |
| pTH15 | pET29a | 3*SrtAΔ59-H6 P94S,D160N,D165A | BL21 (DE3) | ref. 26 |
| pTH16 | pET29a | 4*SrtAΔ59-H6 P94S,D160N,D165A,K196T | BL21 (DE3) | ref. 26 |
| pTH17 | pET29a | 5*SrtAΔ59-H6 P94R,D160N,D165A,K190T,K196T | BL21 (DE3) | ref. 26 |
| pPP2 | pET22b | GFPuv-LPETGG-H6 | BL21 (DE3) | This study |
| pPP4 | pET22b | GFPuv-LAETGG-H6 | BL21 (DE3) | This study |
| pTH24 | pET22b | GFPuv-linker-LPETGG-H6 | BL21 (DE3) | This study |
| pTH27 | pET22b | GFPuv-linker-LAETGG-H6 | BL21 (DE3) | This study |
| pTH25 | pRSET | GGG-H6-FMa | BL21 (DE3) | This study |
| pTH22 | pRSET | H6-FM-LAETGa | BL21 (DE3) | This study |
Expression and purification of sortase variants and target proteins
Chemically competent E. coli cells were transformed with the expression plasmids encoding sortases and target proteins (Table 1). Bacterial cultures were inoculated from 10 mL overnight pre-cultures in 2 L shake flasks containing 500 mL LB Lennox medium (Carl Roth GmbH, Karlsruhe, Germany) with the relevant antibiotics and cultured at 37 °C and 180 rpm. At an optical density (measured at 600 nm) of approximately 0.5, IPTG was added to a final concentration of 0.5 mM to induce protein expression at a reduced temperature of 30 °C. After a cultivation time of four hours cells were harvested by centrifugation and cell pellets were frozen at −20 °C until further use. Thawed cells were resuspended at approximately 10% cell wet weight in lysis buffer (50 mM Tris/HCl pH 7.8, 150 mM NaCl, 20 mM imidazole, 1 mg ml−1 lysozyme, 1/2 tablet of complete protease inhibitor cocktail EDTA-free (Roche, Basel, Switzerland), 8 U mL−1 Benzonase nuclease (Sigma Aldrich, Buchs, Switzerland)) and kept for one hour at 37 °C and 180 rpm. Cells were disrupted on ice by ultrasonication using a Branson Digital Sonifier (Branson Ultrasonic, Urdorf, Switzerland) at 80% output and twelve 10 second pulses interrupted by 10 second breaks. Crude cell extracts were centrifuged for one hour at 20000 g and 4 °C. Histidine-tagged proteins were purified from the supernatants by immobilized metal chromatography (IMAC) using an ÄKTApurifier FPLC system with a 1 mL HisTrap FF column (GE Healthcare, Glattbrugg, Switzerland) at a constant flow rate of 1 mL min−1 and UV detection at 254 nm. After loading the supernatant onto the column in buffer A (50 mM Tris/HCl pH 7.8, 150 mM NaCl, 20 mM imidazole) the bound protein was eluted in a linear gradient from 0 to 100% buffer B (50 mM Tris/HCl pH 7.8, 150 mM NaCl, 1 M imidazole) within 10 min. Protein containing fractions were pooled and the imidazole was removed by exchanging the buffer to 50 mM Tris/HCl pH 7.8, 150 mM NaCl using PD-10 desalting columns (GE Healthcare). The protein solutions were concentrated with Vivaspin 6 centrifugal concentrators (MWCO 10000 Da; Sartorius AG, Göttingen, Germany) until a final protein concentration of >400 μM was reached. Protein concentrations were determined with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA) at a wavelength of 280 nm based on the proteins' specific molar extinction coefficients given in Fig. S1 and S2† (calculated with the ProtParam tool, Expasy, Swiss Institute of Bioinformatics). The protein solutions were stored frozen at −80 °C and thawed on demand.
SrtA-catalyzed reactions
SrtA-catalyzed coupling reactions were carried out in 2 mL Eppendorf tubes in a total reaction volume of 110 μL containing 2 μM of the SrtA variant and 10 μM of the GFPuv target protein in reaction buffer (50 mM Tris/HCl pH 7.8, 150 mM NaCl, 10 mM CaCl2); optionally, 10 μM GGG-H6-FM was added to the mixtures. Depending on the SrtA variant used the reaction mixtures were incubated for 8 or 24 hours at 25 °C under vigorous shaking (650 rpm) in an Eppendorf shaker. Samples of 10 μL were taken at defined time intervals, and the enzymatic reactions were instantly stopped by addition of 4 μL 3× SDS buffer and heating at 95 °C for 5 minutes.SDS-PAGE and ESI-MS analysis of reaction mixtures
Time series of samples were analyzed by SDS-PAGE on 12% polyacrylamide gels applying a constant voltage of 160 V for 70 minutes. For maximum reproducibility of gel analysis a standardized staining and evaluation procedure was followed: after three washes with distilled water (15 minutes each) gels were stained for 90 minutes with GelCode Blue Stain Reagent (Thermo Fisher Scientific Inc.) and left in distilled water over night. Pictures of the gels were taken with a GelDoc-It TS Imaging System (UVP, Upland, CA, USA) at constant shutter rate and exposure time. For digital image analysis the software ImageJ 1.45s (http://imagej.nih.gov/ij) was used as follows. After sharpening of the pictures and subtraction of the background, grey values were integrated and the band intensity values obtained for each lane were corrected with the band intensity and the molecular weight of the respective sortase. Apparent initial rates for intermolecular crosslinking of GFPuv target proteins and transpeptidation reactions were calculated from time plots of the corrected band intensities of the formed protein species (Fig. S4a and b†). In the case of reactions involving H6-SrtAΔ25 and the target proteins GFPuv-linker-LPETGG-H6 and GFPuv-linker-LAETGG-H6, the sortase could not be separated from the GFPuv variants on the SDS-PAGE gels. Thus, in order to correct the band intensities for the formed protein species as described above we estimated the band intensity of H6-SrtAΔ25 from values obtained for reactions carried out with the target proteins GFPuv-LPETGG-H6 and GFPuv-LAETGG-H6.For ESI-MS analysis enzymatic reactions were stopped by addition of 10 mM EDTA when sufficient substrate conversion was reached (as judged from the previously analyzed SDS-PAGE gels). Samples were desalted on C4 ZipTips and measured by the Functional Genomics Center Zürich (FGCZ, Zürich, Switzerland) in a mass range between 500 and 3000 Da. The m/z data were converted into MS data using the MaxEnt1 software. The expected masses for each protein species was calculated from the respective amino acid sequence with the ProtParam tool (Expasy, Swiss Institute of Bioinformatics).
Conclusions
The covalent fusion of functionally intact proteins in a defined orientation is a challenging task that can be elegantly achieved using the enzyme SrtA. Since the establishment of SrtA as a tool for the in vitro modification of proteins several SrtA variants originating from S. aureus have been described and treated as equally performing catalysts.22,25 These wild-type SrtA variants H6-SrtAΔ59, SrtAΔ59-H6 and H6-SrtAΔ25 share identical amino acid sequences of the catalytic cores, but differ by the length of their truncated N-termini derived from the membrane anchor of the full-length enzyme and by the position of the His-tag located at either the N- or the C-terminus. In the present report, we demonstrate that despite the general applicability of all tested SrtA variants for site-directed transpeptidation reactions with appropriately engineered target proteins the enzymes vary in their propensities to form a variety of crosslinked byproducts throughout the coupling reaction. In the investigated reaction system using GFP-derived target proteins, H6-SrtAΔ25 catalyzed intermolecular crosslinking by isopeptide bond formation between target proteins at significantly higher rates than the N-terminally shortened variants H6-SrtAΔ59 and SrtAΔ59-H6. Taking both transpeptidation and competing crosslinking reactions into account, our results suggest that the most commonly employed SrtA variants H6-SrtAΔ59 and H6-SrtAΔ25 should not per se be regarded as functionally equivalent enzymes as proposed in the literature.22,25 Furthermore, we included in our study three SrtA mutants that had been described to have considerably improved catalytic properties with regard to transpeptidation reactions using small peptide substrates.26 On the one hand, we could reproduce similar results monitoring transpeptidation reactions between two distinct protein substrates. On the other hand, besides accelerated transpeptidation also competing crosslinking and hydrolysis reactions among the LPETG-tagged target proteins were catalyzed by the SrtA mutants at considerably higher rates than by the wild-type enzymes. This issue obviously leaves room for further improvement of the synthetic properties of the SrtA mutants by protein engineering approaches.Apart from the particular sortase variant used for the transpeptidation reaction, the amino acid composition of the sorting motif and its context within the sequence of the target protein can significantly affect the outcome of the transpeptidation reaction. In the case of the GFPuv target proteins investigated in this study the introduction of a 5-amino acid linker between the core of the protein and the sorting motif presumably conferred additional flexibility to the C-terminal stretch of the protein. Using this GFPuv-linker-LPETGG-H6 target protein for the SrtA-catalyzed reaction, intermolecular crosslinking by isopeptide-bond formation was largely suppressed in favor of transpeptidation product formation. Interestingly, an additional reduction of competing side reactions was achieved when GFPuv target proteins carrying an LAETG sorting sequence instead of the pristine LPETG motif were transformed by the SrtA mutants.
Summing up, we conclude that apart from the reaction conditions (e.g. choice of pH and reactant concentrations) the interplay of SrtA-catalyzed transpeptidation and crosslinking reactions is also influenced by the chosen enzyme variant itself and the composition of the target protein carrying the sorting motif. In the investigated reaction system the formation of intermolecularly crosslinked byproducts between GFPuv target proteins could be reduced to a minimum by (i) engineering the amino acid sequence next to and within the sorting motif, and by (ii) comparing the properties of various SrtA variants within the particular transpeptidation scenario. A similar comparative approach might be useful to optimize other SrtA-catalyzed transpeptidation reactions, in particular when byproduct formation by isopeptide-crosslinking among target proteins is observed.
Acknowledgements
Plasmid pMC141 and plasmids named pTH14, pTH15, pTH16 and pTH17 herein were kindly provided by Marie Elliot (McMaster University, Hamilton, ON, Canada) and Brent Dorr (Harvard University, Cambridge, MA, USA), respectively. We thank Serge Chesnov (FGCZ) for ESI-MS analysis of protein samples, Frederik Hammes (Eawag) for fruitful discussions and reading of the manuscript, and Urs Bünter (Empa) for the preparation of high-resolution illustrations.References
- T. Spirig, E. M. Weiner and R. T. Clubb, Mol. Microbiol., 2011, 82, 1044–1059 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS.
- M. W.-L. Popp and H. L. Ploegh, Angew. Chem., Int. Ed., 2011, 50, 5024–5032 CrossRef CAS PubMed.
- S. Tsukiji and T. Nagamune, ChemBioChem, 2009, 10, 787–798 CrossRef CAS PubMed.
- M. W.-L. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707–708 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier Ltd, Oxford, 2nd Revised edn, 2008 Search PubMed.
- T. Heck, G. Faccio, M. Richter and L. Thöny-Meyer, Appl. Microbiol. Biotechnol., 2013, 97, 461–475 CrossRef CAS PubMed.
- O. Schneewind, D. Mihaylova-Petkov and P. Model, EMBO J., 1993, 12, 4803–4811 CAS.
- H. Ton-That, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424–12429 CrossRef CAS.
- H. Ton-That, S. K. Mazmanian, K. F. Faull and O. Schneewind, J. Biol. Chem., 2000, 275, 9876–9881 CrossRef CAS PubMed.
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. L. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 CrossRef CAS PubMed.
- D. A. Levary, R. Parthasarathy, E. T. Boder and M. E. Ackerman, PLoS One, 2011, 6, e18342 CAS.
- H. T. Ta, S. Prabhu, E. Leitner, F. Jia, D. von Elverfeldt, K. E. Jackson, T. Heidt, A. K. N. Nair, H. Pearce, C. von zur Muhlen, X. Wang, K. Peter and C. E. Hagemeyer, Circ. Res., 2011, 109, 365–373 CrossRef CAS PubMed.
- J. M. Antos, G.-L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W.-L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Angew. Chem., Int. Ed., 2012, 51, 9377–9380 CrossRef CAS PubMed.
- J. M. Antos, M. W.-L. Popp, R. Ernst, G.-L. Chew, E. Spooner and H. L. Ploegh, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed.
- J. G. M. Bolscher, M. J. Oudhoff, K. Nazmi, J. M. Antos, C. P. Guimaraes, E. Spooner, E. F. Haney, J. J. Garcia Vallejo, H. J. Vogel, W. Van't Hof, H. L. Ploegh and E. C. I. Veerman, FASEB J., 2011, 25, 2650–2658 CrossRef CAS PubMed.
- L. Chan, H. F. Cross, J. K. She, G. Cavalli, H. F. P. Martins and C. Neylon, PLoS One, 2007, 2, e1164 Search PubMed.
- T. Ito, R. Sadamoto, K. Naruchi, H. Togame, H. Takemoto, H. Kondo and S.-I. Nishimura, Biochemistry, 2010, 49, 2604–2614 CrossRef CAS PubMed.
- A. Sinisi, M. W.-L. Popp, J. M. Antos, W. Pansegrau, S. Savino, M. Nissum, R. Rappuoli, H. L. Ploegh and L. Buti, Bioconjugate Chem., 2012, 23, 1119–1126 CrossRef CAS PubMed.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 CrossRef CAS PubMed.
- M. W.-L. Popp, J. M. Antos and H. L. Ploegh, Curr. Protoc. Protein Sci., 2009, ch. 15, Unit 15.13 Search PubMed.
- C. S. Theile, M. D. Witte, A. E. M. Blom, L. Kundrat, H. L. Ploegh and C. P. Guimaraes, Nat. Protoc., 2013, 8, 1800–1807 CrossRef CAS PubMed.
- M. D. Witte, C. S. Theile, T. Wu, C. P. Guimaraes, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1808–1819 CrossRef CAS PubMed.
- U. Ilangovan, H. Ton-That, J. Iwahara, O. Schneewind and R. T. Clubb, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6056–6061 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- F. Bordusa, Chem. Rev., 2002, 102, 4817–4867 CrossRef CAS PubMed.
- V. Schellenberger and H.-D. Jakubke, Angew. Chem., Int. Ed. Engl., 1991, 30, 1437–1449 CrossRef.
- T. Heck, V. S. Makam, J. Lutz, L. M. Blank, A. Schmid, D. Seebach, H.-P. E. Kohler and B. Geueke, Adv. Synth. Catal., 2010, 352, 407–415 CrossRef CAS.
- J. M. Antos, G. M. Miller, G. M. Grotenbreg and H. L. Ploegh, J. Am. Chem. Soc., 2008, 130, 16338–16343 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- S. Möhlmann, C. Mahlert, S. Greven, P. Scholz and A. Harrenga, ChemBioChem, 2011, 12, 1774–1780 CrossRef PubMed.
- A. Duong, D. S. Capstick, C. Di Berardo, K. C. Findlay, A. Hesketh, H.-J. Hong and M. A. Elliot, Mol. Microbiol., 2012, 83, 992–1005 CrossRef CAS PubMed.
- H. Fukuda, M. Arai and K. Kuwajima, Biochemistry, 2000, 39, 12025–12032 CrossRef CAS PubMed.
- A. Crameri, E. A. Whitehorn, E. Tate and W. P. C. Stemmer, Nat. Biotechnol., 1996, 14, 315–319 CrossRef CAS PubMed.
- X. Guo, Z. Wu and Z. Guo, Bioconjugate Chem., 2012, 23, 650–655 CrossRef CAS PubMed.
- K. Park, J. Jung, J. Son, S. H. Kim and B. H. Chung, Chem. Commun., 2013, 49, 9585–9587 RSC.
- Z. Wu, X. Guo, J. Gao and Z. Guo, Chem. Commun., 2013, 49, 11689–11691 RSC.
- P. H. Hirel, M. J. Schmitter, P. Dessen, G. Fayat and S. Blanquet, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 8247–8251 CrossRef CAS.
- L. Zhang, H. N. Patel, J. W. Lappe and R. M. Wachter, J. Am. Chem. Soc., 2006, 128, 4766–4772 CrossRef CAS PubMed.
- S. Dasgupta, S. Samantaray, D. Sahal and R. P. Roy, J. Biol. Chem., 2011, 286, 23996–24006 CrossRef CAS PubMed.
- F. Yang, L. G. Moss and G. N. Phillips Jr., Nat. Biotechnol., 1996, 14, 1246–1251 CrossRef CAS PubMed.
- R. G. Kruger, B. Otvos, B. A. Frankel, M. Bentley, P. Dostal and D. G. McCafferty, Biochemistry, 2004, 43, 1541–1551 CrossRef CAS PubMed.
- M. M. Kämpf, E. H. Christen, M. Ehrbar, M. Daoud-El Baba, G. Charpin-El Hamri, M. Fussenegger and W. Weber, Adv. Funct. Mater., 2010, 20, 2534–2538 CrossRef.
- C. T. Rollins, V. M. Rivera, D. N. Woolfson, T. Keenan, M. Hatada, S. E. Adams, L. J. Andrade, D. Yaeger, M. R. van Schravendijk, D. A. Holt, M. Gilman and T. Clackson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7096–7101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00347k |
| This journal is © The Royal Society of Chemistry 2014 |