 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ageing induced improvement of methane oxidation activity of Pd/YFeO3
Ye
Lu
a,
Sylvain
Keav
a,
Valentina
Marchionni
b,
Gian Luca
Chiarello†
a,
Alfonsina
Pappacena
c,
Marco
Di Michiel
d,
Mark A.
Newton
d,
Anke
Weidenkaff‡
a and
Davide
Ferri
*b
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Solid State Chemistry and Catalysis, Ueberlandstrasse 129, CH-8600 Dübendorf, Switzerland
bPaul Scherrer Institut, CH-5232 Villigen PSI, Switzerland. E-mail: davide.ferri@psi.ch; Tel: +41 56 310 2781
cUniversità di Udine, Dipartimento di Scienze e Tecnologie Chimiche, via Cotonificio 108, I-33100 Udine, Italy
dESRF, 71 Rue des Martyrs, F-38000 Grenoble
First published on 10th April 2014
Abstract
The ageing characteristics of flame-made 2 wt% Pd supported on YFeO3 were analysed in comparison with a Pd/Al2O3–CeO2–ZrO2 three-way catalyst (TWC) with respect to structural changes and catalytic performance for methane oxidation under stoichiometric reaction conditions. Thermal treatment under lean conditions (air, 900 °C) resulted in slight decrease in the methane oxidation activity of the TWC. In marked contrast, YFeO3-supported Pd catalysts exhibit an increase in activity after such treatment. Activity enhancement is even higher when the treatment was performed under stoichiometric conditions (air–fuel equivalence ratio, λ = 1, 900 °C). To explain this observation, in-depth characterization (BET, STEM, OSCC, XAS, and CO chemisorption) of fresh and aged catalysts was performed. Both thermal and stoichiometric ageing cause a severe sintering of the support particles and the phase transformation from hexagonal to orthorhombic YFeO3. Despite the absence of a mixed Pd–YFeO3 phase, the growth of Pd particles appears to be limited under the λ = 1 atmosphere. In contrast to thermally aged catalysts where large PdO particles are formed, well-defined metallic Pd nanoparticles of 10–20 nm are present after stoichiometric ageing along with higher methane oxidation activity. Although it is tempting to conclude that metallic Pd is active for methane oxidation under the given conditions, reversible and periodic partial oxidation of the large metallic particles is observed in modulation excitation high energy X-ray diffraction (HXRD) experiments designed to simulate the oscillating redox conditions experienced during operation. These results indicate that large Pd particles exhibit improved methane oxidation activity but equally confirm that activity under stoichiometric conditions is the result of a delicate equilibrium dictated by the bulk-Pd/surface-PdO pair.
Introduction
The exhaust of modern stoichiometric engines fuelled with natural gas is controlled using Pd-based three-way catalysts (TWC).1 Pd is considered the precious metal of choice for oxidation of methane,2,3 the major component of natural gas. During operation, the atmosphere around the TWC is periodically varied between slightly reducing and slightly oxidizing that allows maintenance of the emission levels within those demanded by the existing stringent regulations.4 Although the TWC for natural gas vehicles is typically derived from the gasoline TWC, their chemistries can display significant differences.5,6 However, similarly to its gasoline counterpart, the TWC for natural gas applications suffers from deleterious ageing effects. Its activity progressively declines with mileage as a result of several intersecting phenomena such as thermal deactivation upon exposure to temperatures as high as 1000 °C and poisoning by fuel additives and impurities.7–14 Therefore, the growing interest in natural gas, bio-gas and synthetic natural gas as interim CO2-poor fuel has driven efforts aiming at designing tailored TWC systems for this specific application.PdO is considered essential for CH4 oxidation under lean conditions.15 Selection of an appropriate oxidic support stabilizes Pd2+ against spontaneous reduction at high temperature.16–18 The decomposition of PdO to Pd is considered to cause catalyst deactivation during reaction.19–21 The role of metallic Pd remains an interesting and much debated issue,22 which is even more fundamental in the case of the TWC because of the oscillating redox reaction conditions. Metallic Pd is observed by X-ray absorption spectroscopy on a model TWC comprising Al2O3 and ceria–zirconia when the catalyst is exposed to high temperature (800–900 °C)5 and a mixed PdO–Pd state needs to be accounted for in light-off experiments under stoichiometric reaction conditions with methane as the hydrocarbon.23 Pd reduction and sintering can be partly prevented by the formation of PdO at increasing O2 concentration, for example during fuel cut-off.24 However, methane oxidation activity under lean conditions was recently shown to increase linearly with Pd particle size ranging from 1 to 20 nm,25 which is in line with previous observations.26,27 Ageing under reaction conditions (O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4 = 4) also produced a more active Pd/Al2O3 with larger Pd particles (7–16 nm).28 Restructuring of PdO crystallites upon reduction resulted in increased catalytic activity.15,29,30
:CH4 = 4) also produced a more active Pd/Al2O3 with larger Pd particles (7–16 nm).28 Restructuring of PdO crystallites upon reduction resulted in increased catalytic activity.15,29,30
Perovskite-type oxides of the general formula ABO3 have long been recognized as oxidation catalysts31–34 and reported as potential NOx storage/reduction catalysts (NSR),35 TWC,36–38 soot combustion catalysts39 and catalysts for natural gas lean burn engines.40 In addition, they are also promising oxygen storage materials.37,41,42 In line with the search for metal oxides able to stabilize Pd2+ against reduction, they can host noble metals in an unusual oxidation state within their lattice. As a direct consequence, in terms of potential TWC components they offer the possibility of stabilizing Pd against sintering under the redox oscillations experienced during operation as has been demonstrated for use with gasoline engines.43 In a very similar way, formation of a Pt-containing perovskite-type oxide (BaPtO3 and Ba2CePtO6) after ageing of the Pt/Ba/CeO2 NSR catalyst was shown to maintain Pt in a well dispersed state, with the reduced Pt state and the activity being able to be restored after mild reduction.44 Despite the growing literature on the topic, which often assumes that the noble metal will adopt the coordination state of the B-site cation, and criticism of the self-regenerative function,45 this property needs to be considered with care as it most likely exists within only a limited choice of phase compositions46–49 and is clearly temperature dependent.43 Nevertheless, even in the absence of noble metal incorporation, LaFeO3 is able to exert a strong stabilization effect that makes Pt more resistant against thermal ageing and consequently more active than when it is deposited on γ-Al2O3.50 Recently, hexagonal YFeO3 has been shown to be a promising host for Pd for CO oxidation51 and for methane oxidation under lean52 and stoichiometric conditions.53 YFeO3 was shown to be superior to YMn0.5Fe0.5O3 with respect to the reversible incorporation of Pd within the YFeO3 lattice.48 YFe1−xPdxO3−δ (x = 0.1) exhibited the self-regenerative property at temperatures below 500 °C where the coordinatively unsaturated Pd reversibly segregates out of and re-incorporates within YFeO3 in consecutive redox treatments.51 Further, the coordination of Pd with YFeO3 changes at high temperature. The fine dispersion of Pd in the catalyst after calcination at 700 °C, where Pd is likely in the coordination state that leads to the self-regenerative CO oxidation catalyst,51 is lost after calcination at 800 °C in favour of segregated PdO, while the hexagonal structure (of the support) is still maintained.52 Well dispersed Pd nanoparticles on YFeO3 grow to well defined particles of ca. 20 nm after repeated methane oxidation cycles under stoichiometric conditions,54 questioning the protective function of the perovskite-related material in case of high temperature treatments of YFeO3. However, activity enhancement for methane oxidation was observed in parallel to the formation of the 20 nm Pd particles. Such an enhancement has been reported also for the La–Mn–Ce–Pd–O perovskite-type oxide where Pd is observed to segregate after ageing under simulated exhaust of a natural gas engine.55
Given the increased methane oxidation activity of Pd/YFeO3 after exposure to 850 °C concurrent to the growth of Pd particles that suggests possible activation after ageing, herein we address the structure and the catalytic performance of flame-made 2 wt% Pd/YFeO3 after treatment under lean and stoichiometric conditions at 900 °C. A sample of Pd/YFeO3 with four-fold lower Pd content was also prepared and aged under identical conditions. The methane oxidation activity under stoichiometric conditions is compared with that of a conventional model TWC.
Experimental
Catalyst synthesis
Pd/YFeO3 (nominal 0.5 and 2 wt% Pd) was synthesized by flame spray synthesis (FSS) using an acetylene flame. Briefly, a mixture of deionized water and N,N-dimethylformamide (DMF, 99%, Sigma-Aldrich, 1:3) was used as solvent of the metal nitrate precursors and sprayed in an acetylene–air flame.53 After synthesis, the samples were calcined in air at 700 °C for 2 h. The powder TWC was composed of Pd, alumina and ceria–zirconia (Pd/ACZ; 1.6 wt% Pd; calcined at 500 °C; SSA = 135 m2 g−1) and was kindly provided by Umicore.
Heat treatments, in the following designated concisely as ageing, were carried out at 900 °C for 2 h. Thermal ageing (AT) was carried out in static air, while stoichiometric ageing (AS) was performed using a gas mixture comprising 7000 ppm CO, 1300 ppm CH4, 1600 ppm NOx and 5300 ppm O2 in He (λ = 1) at 100 mL min−1 in the plug-flow reactor used for catalytic activity measurements. In the following, the samples are labelled 2Pd/YFO-x, 0.5Pd/YFO-x and Pd/ACZ-x, where x is F (fresh), AT or AS.
Characterization
The specific surface area was determined by the single-point BET method in 30 vol% N2/He using a QuantaChrome ChemBET 3000 instrument. Prior to measurements, all samples were degassed in pure N2 at 200 °C for 30 min.The phase composition of fresh and aged catalysts was analysed by X-ray diffraction (X'Pert Pro PANalytical) using monochromatic Cu Kα1 radiation (λ = 1.54 Å). The diffraction patterns were recorded between 20° and 80° (2θ) with an angular step interval of 0.0167°. Phase identification was performed using the ICDD reference database in the X'Pert HighScore Plus software.
The morphology of the catalysts was observed using a Jeol 2200FS transmission electron microscope equipped with a 200 kV field emission gun and a high-angle annular dark field detector in STEM mode providing images with atomic number (Z1.5–1.8) contrast.56 The local composition was determined by energy dispersive X-ray spectroscopy (EDS). The samples were prepared by evaporation of an alcohol suspension on a copper grid coated with a holey carbon film. The palladium particle size distribution was obtained by measuring up to 200 particles using the software ImageJ.57
The oxygen storage capacity complete (OSCC) defined as the total amount of desorbed oxygen was determined under static conditions using the thermogravimetric method.58,59 The samples were treated with 5 vol% H2/Ar at 400 °C for 70 min using a TGA Q500 setup (TA Instruments). The measured weight loss was converted into the corresponding oxygen content assuming that the sample is fully oxidized in its initial state.
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) data were collected at a resolution of 4 cm−1 using a VERTEX 70 FT-IR spectrometer (Bruker) equipped with a high temperature reaction chamber (HVC-DRP2, Harrick) and a liquid-nitrogen cooled MCT detector. Prior to CO adsorption, the samples were exposed to 20 vol% O2/Ar (50 mL min−1) at 500 °C for 30 min, followed by reduction at 300 °C for 30 min in 10 vol% H2/Ar. After changing to Ar and cooling to room temperature, a background spectrum was obtained prior to CO adsorption in 5 vol% CO/Ar.
Extended X-ray absorption fine structure (EXAFS) spectroscopy data were obtained in the transmission mode at the Pd K-edge (24.35 keV) at the SuperXAS beamline of the Swiss Light Source (PSI, Switzerland). The spectrum of the Pd foil was simultaneously collected for energy calibration. The required sample amount was homogeneously mixed with polyethylene and pressed into pellets. The spectra were energy and background corrected and normalized with the software Athena.60
Time-resolved high energy XRD data (86.8 keV, λ = 0.143 Å, ΔE/E ~ 1.4 × 10−3) were collected at beamline ID15B of the ESRF (Grenoble, France) using a digital flat 2D panel detector (Pixium 4700) mounted at 900 mm from the sample position. In a concentration modulation experiment, the sample was mounted in a homemade stainless steel cell and was subjected to a sequence of 50 alternate and equally long pulses (25 s) of 1 vol% O2/He and 1 vol% CO/He at 400 °C at 100 mL min−1.61 A series of 100 time-resolved diffraction patterns were collected during a full modulation period (T = 50 s). The catalyst powder (ca. 50 mg, 50–100 μm) was fixed between two quartz wool plugs in a homemade stainless steel reactor cell equipped with graphite windows. The temperature of the sample was recorded using a thermocouple inserted at the entrance of the catalyst bed and was constant within ±1 °C during the isothermal pulse experiment. The outlet of the cell was connected to a mass spectrometer (OmniStar, Pfeiffer) that was used to follow signals of m/z 28 (CO), 32 (O2), 44 (CO2) and 4 (He) and simultaneously to read the sample temperature. The 5000 time-resolved XRD data collected in each experiment were averaged over the number of modulation periods into a single period prior to phase sensitive detection (PSD):
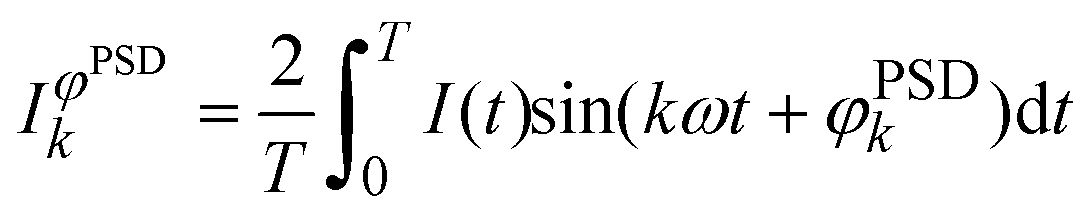 | (1) |
sin(θ/2)/λ, with θ being the scattering angle and λ the wavelength of the incident radiation) and time.62–64 According to the theory of modulation enhanced diffraction,65 the integral of eqn (1) can be more explicitly (and correctly) written to contain two terms, one of which depends only on the component of the system that is stimulated, whereas the second one contains both this component and the static one. PSD with k = 2 should provide only the response of the former component, but we did not find any difference other than intensity by using k = 1, which is shown here. The Simpson series was used to calculate the integral of eqn (1). The resulting new set of phase-resolved data contains only the response of species responding with the same frequency of the external stimulation. A critical point is that the changes must be reversible to the external stimulus, for example a change in concentration of the gas phase. PSD removes static signals – typically overwhelming – thus revealing the information pertaining to what are often weak signals. This method is especially useful to study real catalysts with relevant (low) Pd content, which is either silent or unlikely to be distinguished using conventional spectroscopic/diffraction methods. The resulting new set of phase-resolved data essentially contains the response of the signals (species) that are responding with the same frequency of the external stimulation (CO vs. O2 concentration modulation) and on the delay with which they respond to the external stimulus.
Three-way catalytic activity
The light-off of the catalysts was measured in an exhaust mixture comprising 7000 ppm CO, 1300 ppm CH4, 1600 ppm NOx and 5300 ppm O2 (He balance; 100 mL min−1) between 25 °C and 850 °C. The measurements were carried out in a quartz-tube reactor (d = 6 mm) using catalyst particles (150–200 μm; 100 mg) diluted with the same volume of sea sand and firmly fixed in the middle of the reactor between two quartz wool plugs. Prior to the reaction, the catalysts were treated in 20 vol% O2/He (50 mL min−1) at 700 °C for 2 h. The gas hourly space velocity (GHSV) was approximately 60000 h−1. The outlet of the reactor was analysed using a gas chromatograph (Agilent 3000A Micro GC) equipped with two columns (PoraPLOT-Q for CH4, CO2, H2O and N2O; molecular sieve 5 Å for CO and NO).
Results and discussion
Characterization of fresh and aged catalysts
The structural properties of fresh and aged catalysts are summarized in Table 1. The phase composition of each sample was determined by XRD. Since the same crystal phases were identified in 0.5Pd/YFO and 2Pd/YFO, only the XRD patterns of 2Pd/YFO and Pd/ACZ are presented in Fig. 1. 2Pd/YFO-F exhibits the patterns of hexagonal YFeO3, while no information is obtained on the Pd-containing phase due to the overlap with the YFeO3 phase and its good dispersion (Table 1).53 Irrespective of the ageing atmosphere, hexagonal YFeO3 is converted into the orthorhombic phase, which was reported to occur around 800 °C.66–68 Despite the high temperature treatment, no evidence of the presence of a Pd-containing phase is obtained. On the contrary, the inset of Fig. 1 demonstrates that metallic Pd is formed as a consequence of stoichiometric ageing. The Pd(111) and Pd(200) reflections of fcc-Pd appear at ca. 40 and 47°, respectively. The phase transition of YFeO3 upon exposure to 900 °C is accompanied by a considerable loss of surface area (Table 1). Likely as a consequence of both phase transition and particle growth processes, the oxygen storage capacity complete (OSCC) is also severely diminished compared to the remarkable value measured for the fresh catalyst. The OSCC values of the aged 2Pd/YFO catalysts are ca. ten times higher than that of the thermally aged support suggesting that the OSCC of the aged catalysts is primarily dictated by the presence of Pd. This is also confirmed by the decreased OSCC value obtained with lower Pd loading that is still ca. 4–8 times higher than that of the support depending on the ageing procedure.| Entry | Condition | SSA (m2 g−1) | XRDa | OSCCb (μmol O2 g−1) | Particle sizec (nm) |
|---|---|---|---|---|---|
| a h: hexagonal, o: orthorhombic. b Oxygen storage capacity complete, determined by TG analysis. c Average Pd particle size estimated by STEM. The standard deviation σ is shown in brackets. d Metastable monoclinic Y2O3. e See ref. 53. | |||||
| Pd/ACZ | F | 135 | ACZ | 150 | 8 [3] |
| AT | 70 | ACZ + PdO | 157 | 52 [27] | |
| AS | 97 | ACZ + Pd | 188 | 13 [4.4] | |
| 2Pd/YFO | F | 19 | h + Y2O3d | 504 | 3.5 [1.4] |
| AT | 3 | o + Y2O3 + h | 86 | 91 [35] | |
| AS | 6 | o + Y2O3 + h | 88 | 19 [7.6] | |
| 0.5Pd/YFO | F | 18 | h + Y2O3d | 424 | — |
| AT | 3 | o + Y2O3 + h | 39 | 101 [37.5] | |
| AS | 6 | o + Y2O3 + h | 67 | 18 [5.7] | |
| YFO | Fe | 19 | h + Y2O3d | 416 | — |
| AT | 3 | o + Y2O3 + h | 8 | — | |
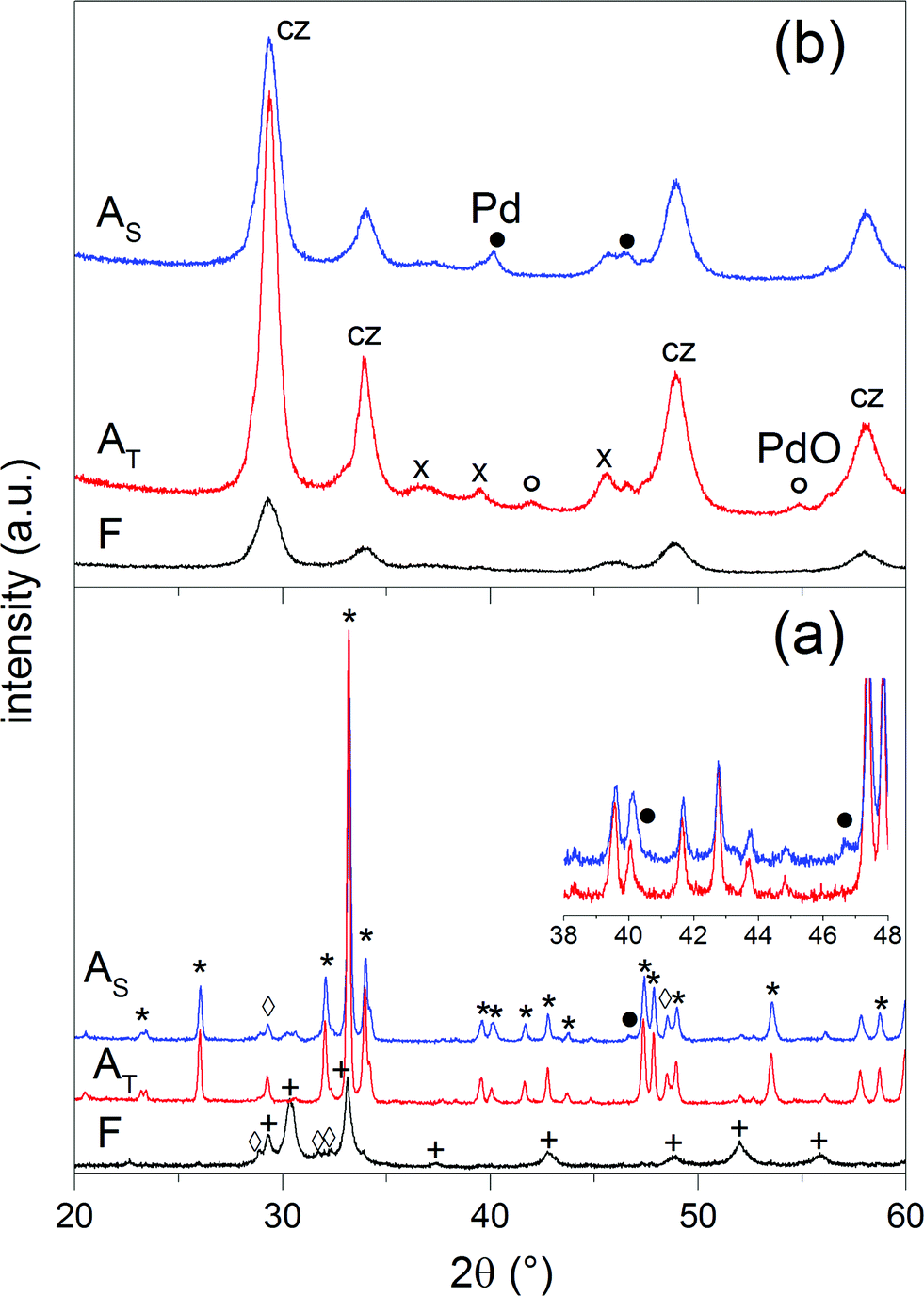 | ||
| Fig. 1 XRD patterns of fresh and aged (a) 2Pd/YFO and (b) Pd/ACZ. (*) Orthorhombic YFeO3, (+) hexagonal YFeO3, (◊) Y2O3, (×) γ-Al2O3, (cz) ceria–zirconia, (●) Pd, and (○) PdO. | ||
For comparison, the XRD patterns of fresh and aged Pd/ACZ (Fig. 1) correspond to those of γ-Al2O3 and cubic CZ and are very similar, confirming the thermal stability of ACZ.69 However, the general sharpening of the diffraction peaks of CZ after both ageing procedures is indicative of some extent of sintering. This is especially evident in Pd/ACZ-AT and is confirmed by the larger extent of surface area loss of this sample compared to Pd/ACZ-AS. A (weak) peak corresponding to PdO becomes visible at ca. 55° in Pd/ACZ-AT as well. Similarly to 2Pd/YFO-AS, the XRD pattern of Pd/ACZ-AS shows reflections of metallic Pd (peaks at 40 and 47°), confirming the reduction of Pd under these conditions.
HAADF-STEM was used to confirm the textural changes occurring upon ageing and to characterize the morphology of the Pd-containing phase. The STEM images of 2Pd/YFO are shown in Fig. 2d–f. Identical conclusions can be drawn for 0.5Pd/YFO (Fig. 2g–i). The sintering of YFeO3 after exposure to 900 °C is rather obvious from these data. Similarly, Pd particle growth is also observed to be accompanied by a change in shape that depends on the ageing procedure. Pd particles appear mostly ‘wetting’ and spreading on YFeO3 after thermal ageing (Fig. 2e; Table 1). Plate-like Pd particles were observed to form in an oxygen containing environment at T > 800 °C on Al2O3, while re-dispersion of Pd occurred on more coordinating metal oxides such as ZrO2.70 However, the particles are not always easily recognizable and preclude any correct determination of particle size distribution. Assuming that the shape of Pd particles present in 2Pd/YFO-AT is similar to that on Al2O3, this would indicate that the anchoring nature of YFeO3 is lost upon ageing, in agreement with recent observations on Pd/YFeO3 calcined at 700 °C and 800 °C.52 The average particle size of the finely dispersed fresh catalyst increases from ca. 3.5 nm to ca. 19 nm in 2Pd/YFO-AS (Fig. 2f) and the particle size distribution broadens (Table 1). Additionally, Pd nanoparticles with a different morphology from those obtained upon thermal ageing are obtained. The large fraction of hexagonal projections detected in the STEM images indicates that Pd particles possess a (truncated) cubo-octahedral shape exposing (111) and (100) facets.71 The change in morphology upon stoichiometric ageing is similar to the one observed after three reaction cycles under λ = 1 conditions.53 Hence, structural changes after repeated reaction cycles and stoichiometric ageing are of similar nature.
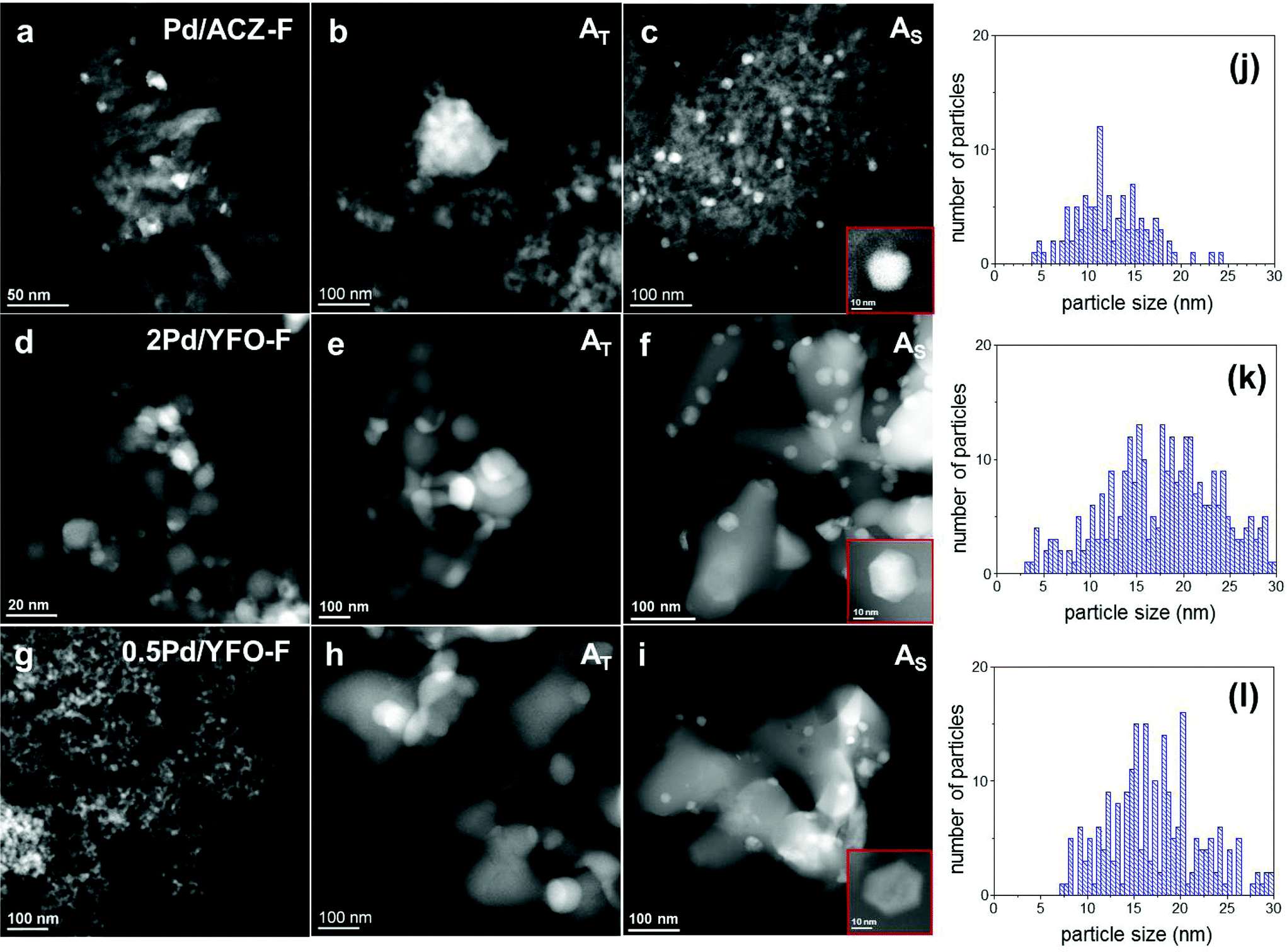 | ||
| Fig. 2 HAADF-STEM images of fresh and aged (a–c) Pd/ACZ, (d–f) 2Pd/YFO, and (g–i) 0.5Pd/YFO and particle size distributions of (j) Pd/ACZ-AS, (k) 2Pd/YFO-AS and (l) 0.5Pd/YFO-AS. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
The different particle shape and the smaller particle size observed for 2Pd/YFO-AS confirm previous results obtained on Pt/Al2O3 catalysts used for NO oxidation that were thermally aged at 800 °C or under a simulated lean diesel environment (propene, CO, NOx and O2) at the same temperature for 5 h.72 Ageing under reaction conditions caused formation of truncated cubo-octahedral Pt particles with reduced size compared to the thermally aged catalyst. Hence, ageing under a reactive atmosphere produces smaller particles than thermal ageing at the same temperature.73
The STEM observations on the evolution of the structure of the Pd phase on YFeO3 are confirmed and compared with the analysis of the Pd/ACZ reference catalyst. No significant change in the size of the metal oxide is observed that could support the variations of XRD intensity. The average Pd particle size increases from 8 nm in Pd/ACZ-F to 52 nm in Pd/ACZ-AT (Fig. 2a and b). The growth of Pd particles is again less severe under stoichiometric conditions, with the average particle diameter being 13 nm in Pd/ACZ-AS. Both round-shaped and truncated particles can be found in this sample (Fig. 2c). The particle size distribution of Pd/ACZ by STEM is, however, greatly influenced by Pd particles deposited on CZ that are difficult to detect.
The oxidation state and coordination environment of Pd were investigated by X-ray absorption spectroscopy. The XANES spectra of fresh and aged 2Pd/YFO and Pd/ACZ recorded at the Pd K-edge are shown in Fig. 3a. The spectra of a Pd foil and bulk PdO are also given for reference. No detectable metallic Pd is present in the fresh catalyst.53 The white line of 2Pd/YFO-F is very close to that of Pd/ACZ-F which is typical of a supported PdO catalyst. Nevertheless, the non-phase shift corrected k3-weighted Fourier transform EXAFS spectrum of 2Pd/YFO-F (Fig. 3b) presents only a clear Pd–O coordination shell at 1.6 Å, while the second Pd–O–Pd shell clearly appears in Pd/ACZ-F at ca. 3.0 Å. The difference is due to the smaller particle size measured for the YFO-based catalyst (Table 1). The XANES and FT-EXAFS data of the thermally aged catalysts are also characteristic of PdO, but the stronger contribution of the Pd–O–Pd shell compared to the fresh catalysts confirms PdO particle growth upon ageing. After stoichiometric ageing, the presence of the Pd–Pd coordination shell at 2.5 Å at the expense of the Pd–O phase characterizes the 2Pd/YFO and Pd/ACZ catalysts. This confirms that Pd is in the metallic state, in agreement with the XRD evidence and the Pd sintering observed by STEM. Based on the presence of the Pd–Pd shell, we can observe that 2Pd/YFO-AT also exhibits a contribution from metallic Pd that is evident from the small peak at ca. 2.5 Å. The presence of an additional scattering feature at 2.1 Å in the FT-EXAFS data of 2Pd/YFO-AS could be assigned to the formation of a Pd–Fe alloy.48
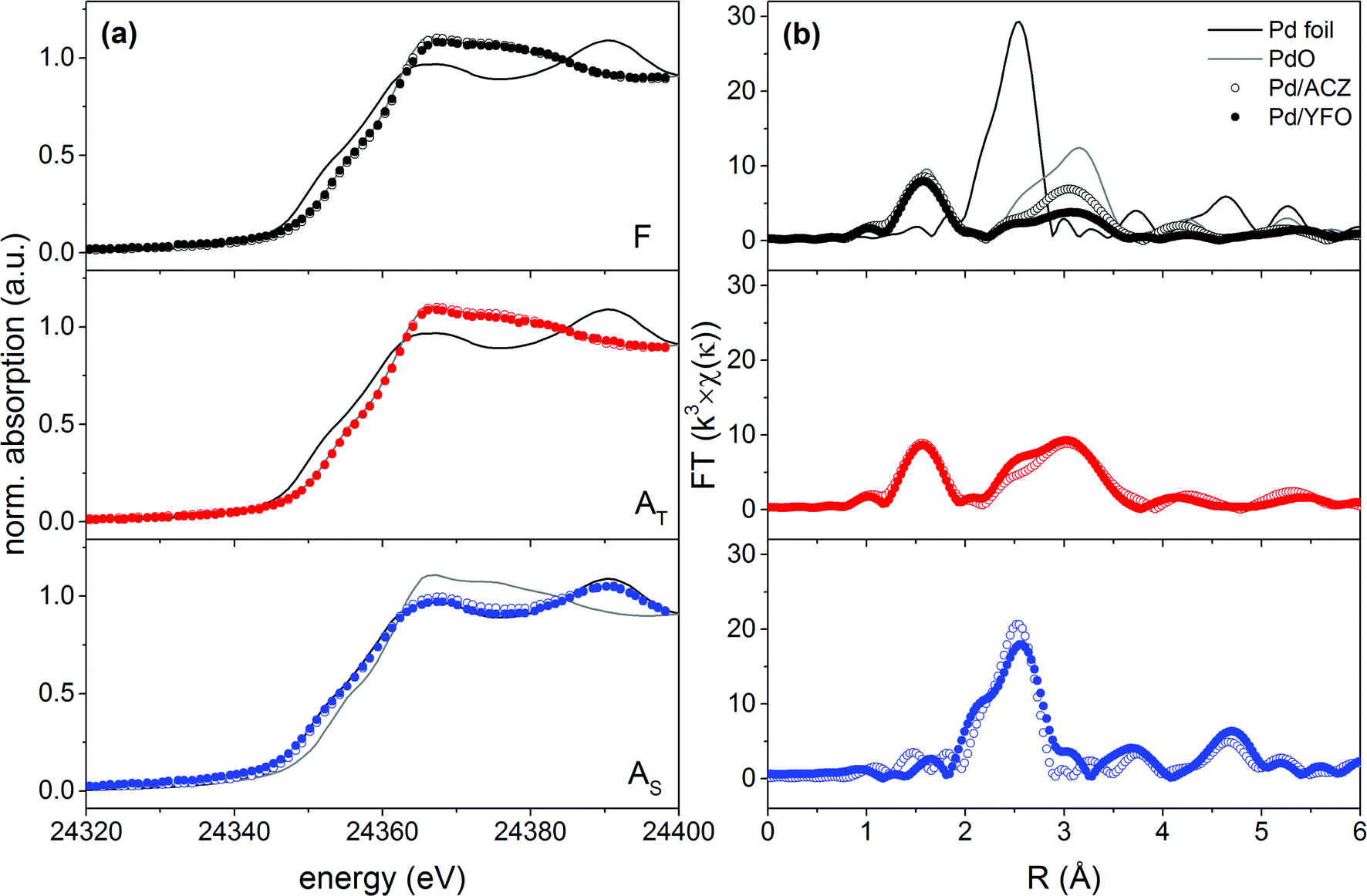 | ||
| Fig. 3 (a) XANES and (b) non-phase shift corrected k3-weighted FT-EXAFS spectra of fresh and aged 2Pd/YFO and Pd/ACZ. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
The dispersion of Pd particles was also qualitatively characterized by CO adsorption using infrared spectroscopy (Fig. 4). After reduction of the fresh catalysts at 300 °C, adsorbed CO on Pd exhibits three main signals at 2090 cm−1 (linearly bonded CO on Pd0 corner atoms, COL), 2000–1950 cm−1 (μ2-bridge bonded species on Pd(100) facets or edge sites) and 1950–1800 cm−1 (a broad band resulting from μ3-hollow/multiple bonded CO on Pd(111) planes) which were assigned according to the available literature.71,74,75 Pd/ACZ-AS is the only sample displaying a signal at 2125 cm−1 that could be attributed either to the presence of ionic Pdδ+ (ref. 76) or to coordination of CO to Ce3+.77 The major difference between the spectra of fresh 2Pd/YFO, 0.5Pd/YFO and Pd/ACZ lies in the overall intensity of the signals (Fig. 4), which varies in the order Pd/ACZ > 2Pd/YFO > 0.5Pd/YFO. The higher intensity of Pd/ACZ-F signals is caused by its larger specific surface area (Table 1), since more Pd can be probed by CO and by the IR radiation, whereas the larger intensity of signals of 2Pd/YFO-F over those of 0.5Pd/YFO-F is attributed to the difference in Pd loading at identical surface area values. The intensity ratio between the signals of COL and that of bridge-bonded CO (A2090/A1990) can be taken as an indicator of the contribution of CO adsorbed on Pd particle edges and corners relative to terrace sites. A larger A2090/A1990 ratio corresponds to a smaller average particle size, which correlates well with the particle size trends measured from STEM images (Table 1). 0.5Pd/YFO-F exhibits the largest A2090/A1990 ratio among all samples, thus explaining the difficulty in estimating its Pd particle size from STEM (Fig. 2g).
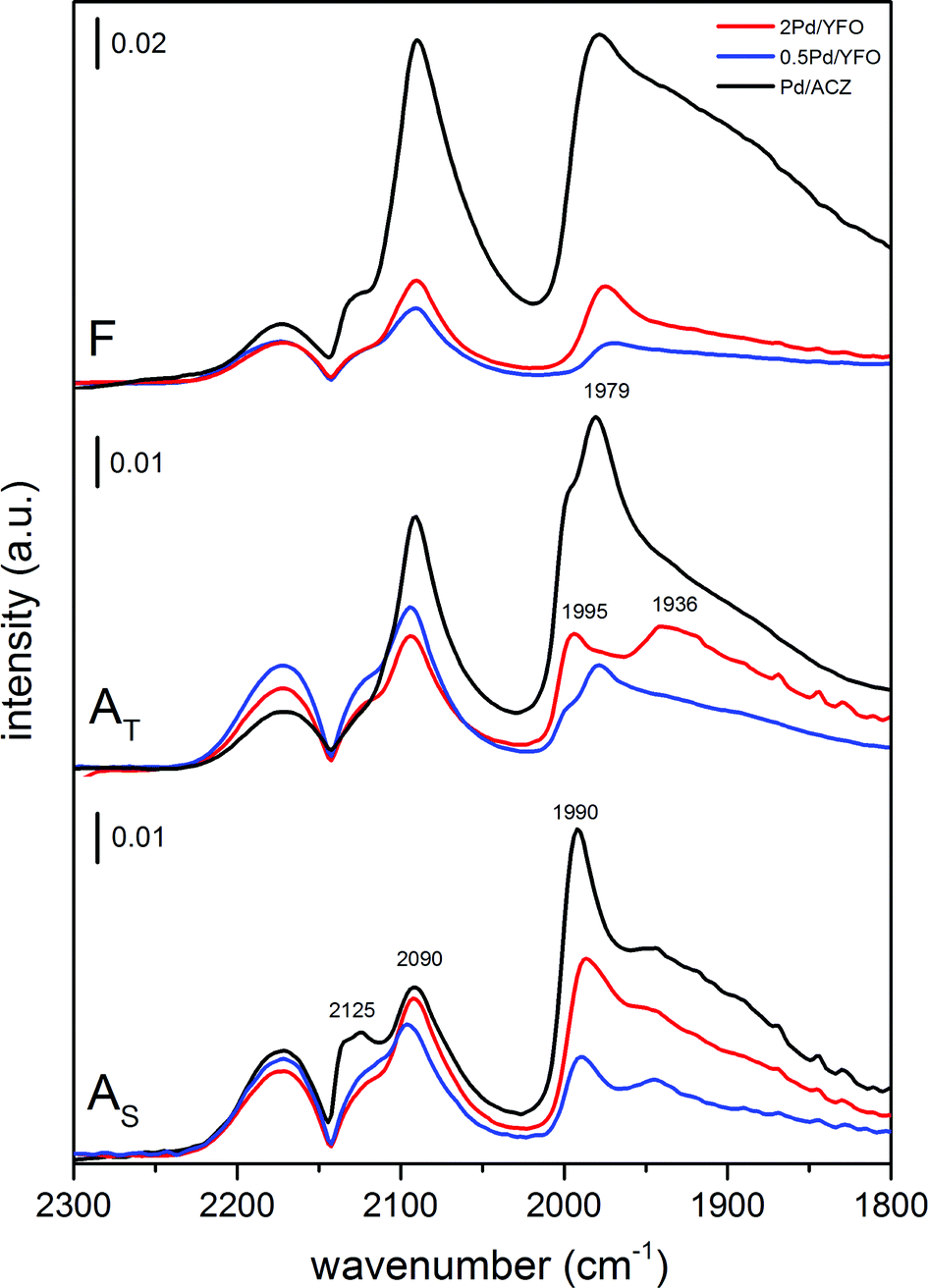 | ||
| Fig. 4 DRIFTS spectra of adsorbed CO on fresh and aged 2Pd/YFO, 0.5Pd/YFO and Pd/ACZ. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
Ageing causes a severe loss of IR signal intensity in Pd/ACZ (note the scale bar in Fig. 4) and of the A2090/A1990 intensity ratio, reflecting the loss of Pd dispersion. The intensity changes associated with the YFO-based catalysts are strikingly less evident. The position of the signals corresponding to COL (2090 cm−1) and hollow bonded CO (1930–1800 cm−1) in the aged samples is similar to those observed in fresh catalysts. The signals of the bridge-bonded CO species vary more significantly from stoichiometric ageing to thermal ageing. In the AS catalysts, the signal is slightly shifted to 1990 cm−1 compared to fresh samples (1980 cm−1), but spectra of 2Pd/YFO-AS, 0.5Pd/YFO-AS and Pd/ACZ-AS are very similar, likely reflecting a comparable morphology but different sizes of the Pd particles. Two to three signals corresponding to μ2-bridge-bonded CO species are visible below 2000 cm−1 in the spectra of AT catalysts. 2Pd/YFO-AT presents a pronounced signal at ca. 1936 cm−1 that could be assigned to isolated bridge species.77,78 Comparison between STEM and DRIFTS data for these catalysts is also more difficult. STEM suggests the presence of large PdO particles (confirmed by EXAFS) that are challenging to clearly detect, whereas pre-reduction of the samples prior to CO adsorption may affect the aspect of the particles. Reduction of the large PdO particles of 2Pd/YFO-AT may cause the formation of islands of metallic Pd that could account for the signal at 1936 cm−1. Therefore, the difference between the spectra of AT and AS catalysts is attributed to the different morphologies obtained after reduction at 300 °C.
Time-resolved high energy XRD
The surface dynamics of the fresh and aged catalysts was addressed using high energy X-ray diffraction combined with the modulation excitation approach.61,79 High energy XRD is a suitable method because it provides information on the whole material in a snapshot and in a time-resolved manner. It has been shown that subtle structural changes occurring during reaction that are not visible in XRD can be enhanced if the experimental data are collected under pulsed conditions.61In this work, the modulation experiment consists of alternate and repeated CO vs. O2 pulses that should simulate the protocol typically used to determine the dynamic OSC.80 We restrict the discussion to fresh and aged 2Pd/YFO catalysts whose time-resolved XRD patterns recorded at 400 °C are dominated by the hexagonal and orthorhombic YFeO3 structures, respectively (Fig. 5a). Similarly to the XRD of Fig. 1, and despite the exposure to high temperature during ageing and the resulting structural changes mentioned above, there remains still no clear indication about the Pd-containing phase because of the systematic overlap of Pd and PdO reflections with those of the support oxide. A completely different picture is obtained from the phase-resolved data of Fig. 5b, where reversible structural variations during the CO–O2 pulses become clearly visible. By comparison with ICSD diffraction data (Fig. 5b, top), the reflections that became visible at 2.78 Å−1 (corresponding to 2θ = 40° for CuKα radiation) and 3.22 Å−1 (46.5°) are assigned to Pd metal, whereas the broader features at 2.36 Å−1 (33.6°) and 3.74 Å−1 (54.6°) are assigned to PdO reflections. The assignment is also corroborated by the opposite sign of the signals of Pd and PdO. The reflections of the Pd-containing phases experience only an amplitude change in response to the modulation. On the contrary, the weaker changes associated with the dominant reflections of YFeO3 involve both amplitude and d spacing variations. The signals exhibit a complex differential profile that is given by the overlap of various close reflections. In agreement with similar experiments conducted on Pd/ACZ,61 the d spacing variation during the modulation experiment is associated with the OSC characteristics of the material, therefore oxygen release in CO (decrease of Q) and oxygen uptake in O2 (increase of Q).
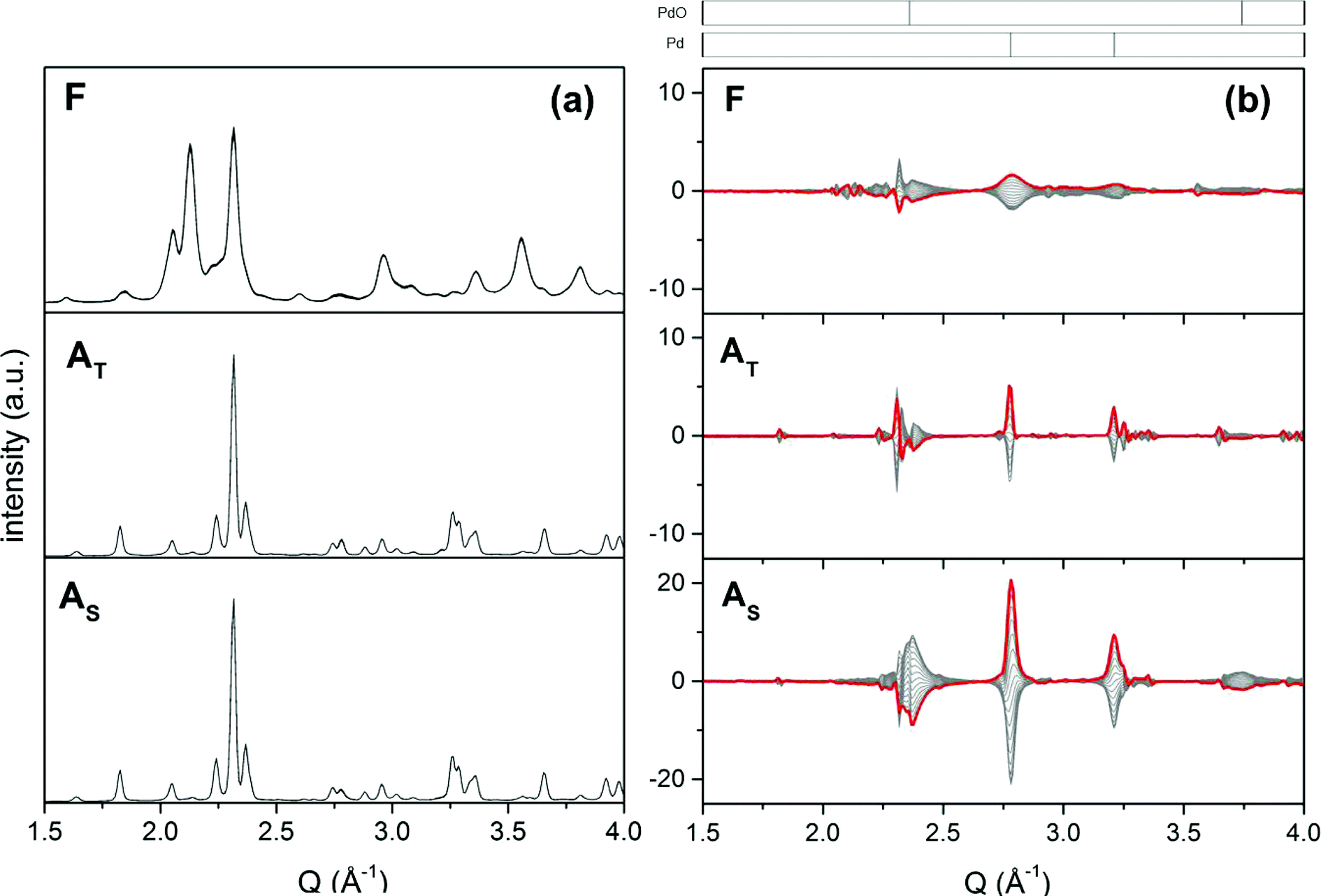 | ||
| Fig. 5 (a) Time-resolved high energy XRD data of fresh and aged 2Pd/YFO obtained in a CO vs. O2 modulation experiment at 400 °C. (b) Corresponding phase-resolved data. The red profile is the in-phase pattern. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
The major difference between the catalysts is the amplitude of the changes emphasized by PSD. The overall intensity of the changes observed in 2Pd/YFO-F is small compared to that of the aged catalysts. Only the oscillations of hexagonal YFeO3(111) at 2.32 Å−1 (2θ = 33°) and Pd(111) at 2.78 Å−1 are obvious. This is due to lack of long range crystal order in the fresh catalyst because of the nano-size of the support and Pd. The reflection of Pd(111) in the fresh catalyst appears very broad for the same reason. The difference in the YFeO3 crystal structure (hexagonal and orthorhombic) may also influence the extent of the observed changes related to the support oxide, because the surface oxygen content of the material is structure dependent.67 The changes around the Pd and PdO reflections are most pronounced in 2Pd/YFO-AS, whereas those associated with orthorhombic YFeO3 are evident only in 2Pd/YFO-AT as a result of the lesser contribution from PdO. Thus, the oxygen exchange properties of the aged catalysts rely mainly on the oxidation and reduction of Pd particles in agreement with the above observations on the low OSCC values.81 We cannot establish quantitatively how relevant is the structural variation observed for YFeO3.
The temporal evolution of the Pd(200) reflection (3.22 Å−1) for the three catalysts is reported in Fig. 6a. No kinetic data for the two YFeO3 phases are shown because of the high level of noise and the very small changes induced by the modulation experiment that allow only for a qualitative evaluation of the data. Pd(200) is selected because the strongest Pd(111) reflection (2.78 Å−1) interferes with those of the YFeO3 phases, especially in 2Pd/YFO-AT. The data also clearly evidence the difference in amplitude discussed above. It is clear from the kinetic data that the Pd and PdO reflections show opposite behavior in the phase-resolved data of Fig. 5b because Pd species reduce and re-oxidize in the CO–O2 pulses. Besides the marked difference in amplitude of the changes, the delay at which changes occur in the CO pulse is also different. Whereas PdO of 2Pd/YFO-AT reduces immediately in the presence of CO (Fig. 6a), the development of the Pd(200) reflection sets in only after ca. 10 s in 2Pd/YFO-F and 2Pd/YFO-AS and is completed within a few seconds.
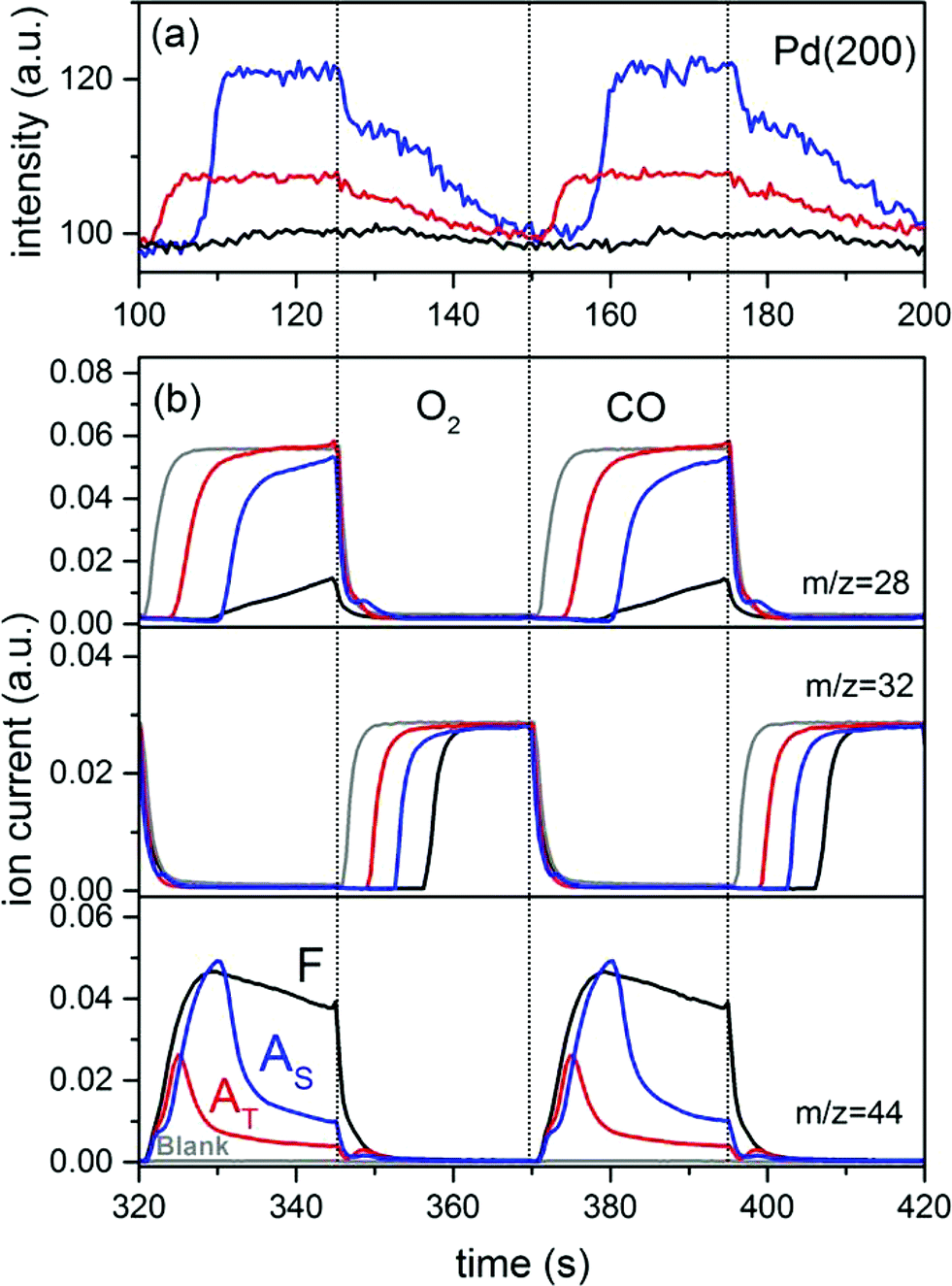 | ||
| Fig. 6 (a) Temporal evolution of the Pd(200) reflection obtained from the time-resolved high energy XRD data of the CO vs. O2 modulation experiment at 400 °C. (b) Corresponding online MS data. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
The structural changes are reversible and Pd re-oxidation occurs in all catalysts. However, it is a slower process than reduction and encompasses the whole O2 pulse. The corresponding MS data (Fig. 6b) show that CO2 evolution takes place over the whole CO pulse in the case of the fresh catalyst. In agreement with the temporal evolution of the Pd(200) reflection, the only little gas phase CO that is observed appears after ca. 10 s from admittance of CO and in correspondence of the decrease of CO2 evolution. Therefore, 2Pd/YFO-F is the most active to oxidize CO, which is facilitated by the rather high SSA and high OSCC value compared to the aged catalysts (Table 1). Ageing causes an earlier CO breakthrough in the gas phase and a corresponding earlier breakthrough of gas phase O2 in the oxidation pulse. However, 2Pd/YFO-AS is able to consume CO up to about half the total duration of the CO pulse, a fact that is reflected in the broader CO2 profile than that of 2Pd/YFO-AT and which agrees well with the retarded Pd reduction observed in the high energy XRD data. Hence, in this type of experiment 2Pd/YFO-AS demonstrates better oxygen storage capacity than 2Pd/YFO-AT. The catalyst aged under stoichiometric conditions possesses highly reactive surface oxygen species giving high CO2 evolution under oscillating conditions, whereas the thermally aged catalyst is characterized by poorer oxygen exchange properties. Given the similar structure of the support, this difference is associated with the redox properties of the metallic Pd particles of 2Pd/YFO-AS. The discrepancy with the OSCC values is explained by the milder reduction conditions and the repeated redox pulses applied during the pulse experiment compared to the thermogravimetric experiment. Therefore, the metallic Pd particles observed on 2Pd/YFO-AS appear crucial for maintaining the oxygen exchange properties of the catalyst.
Based on the Scherrer equation using the Pd(111) reflection of the in-phase XRD pattern of each catalyst (Fig. 5b, red), the estimation of the Pd crystallite size provides 2.6, 13.5 and 7.6 nm for 2Pd/YFO-F, 2Pd/YFO-AT and 2Pd/YFO-AS, respectively. The estimation is strongly affected by the assumption that all catalysts possess spherical (and unstrained) particles, which does not correspond to the STEM images especially in the case of 2Pd/YFO-AT. Finally, the estimation is also affected by some overlap of the selected reflection with those of YFO3. The values obtained considering these issues are lower than the particle size values determined by STEM (Table 1). The kinetic data of Fig. 6a indicate a slow re-oxidation that is possibly not able to involve the full particle volume. Hence, the estimation of the crystallite size based on the net variation of the Pd(200) reflection indicates that a large fraction of Pd but likely not all Pd responds to the CO–O2 modulation, i.e. is repeatedly oxidized and reduced.
To conclude this section, high energy XRD data provide a qualitative measure of the facility of Pd to undergo a redox cycle. The large metallic Pd particles observed in 2Pd/YFO-AS (ca. 20 nm) are subject to re-oxidation under periodic redox conditions at 400 °C suggesting that this could well be the situation also under stoichiometric reaction conditions.
Three-way catalytic activity
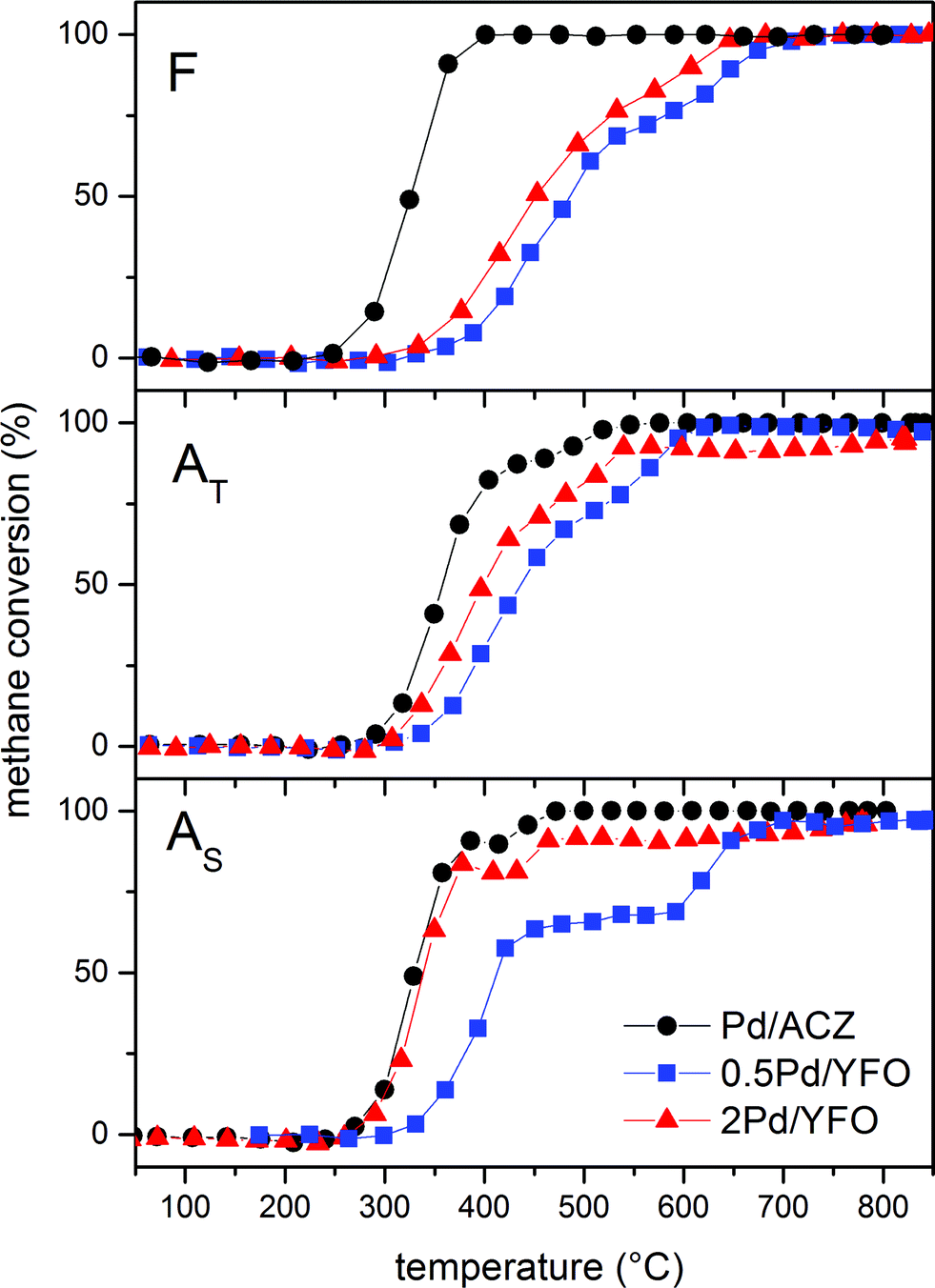
Fig. 7 shows the light-off curves of CH4 obtained for fresh and aged catalysts under stoichiometric conditions. The T50 for CH4 and T50 for CO (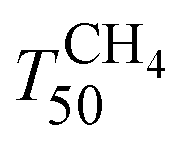 and TCO50) are summarized in Table 2. A systematic improvement in
and TCO50) are summarized in Table 2. A systematic improvement in 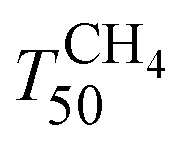 upon ageing is observed in the perovskite-type catalysts. Despite the structural changes observed after ageing involving both Pd and support particle growth,
upon ageing is observed in the perovskite-type catalysts. Despite the structural changes observed after ageing involving both Pd and support particle growth, 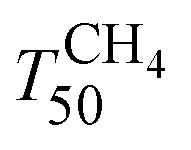 of the 2Pd/YFO-AS catalyst is shifted by more than 100 °C to lower temperature after stoichiometric ageing, attaining those of Pd/ACZ-F and Pd/ACZ-AS. Similar activity improvement by nearly 80 °C is observed also for 0.5Pd/YFO-AS. The effect of thermal ageing is less pronounced, but
of the 2Pd/YFO-AS catalyst is shifted by more than 100 °C to lower temperature after stoichiometric ageing, attaining those of Pd/ACZ-F and Pd/ACZ-AS. Similar activity improvement by nearly 80 °C is observed also for 0.5Pd/YFO-AS. The effect of thermal ageing is less pronounced, but 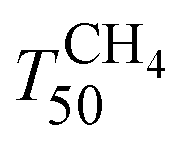 is shifted by 50 °C to lower temperature in both YFO-based catalysts. For comparison, the activity of Pd/ACZ is stable upon ageing. Only thermal ageing increases
is shifted by 50 °C to lower temperature in both YFO-based catalysts. For comparison, the activity of Pd/ACZ is stable upon ageing. Only thermal ageing increases 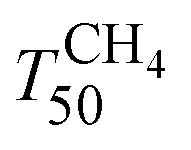 by 40 °C contrary to the behavior of Pd/YFO catalysts. This deactivation is attributed to the more significant sintering of PdO particles (Table 1). In contrast to Pd/ACZ, the aged YFO catalysts do not reach full CH4 conversion at 800 °C, which is attributed to the low surface area of both the Pd and the YFeO3 support and to the limited oxygen storage capacity of the aged catalysts.
by 40 °C contrary to the behavior of Pd/YFO catalysts. This deactivation is attributed to the more significant sintering of PdO particles (Table 1). In contrast to Pd/ACZ, the aged YFO catalysts do not reach full CH4 conversion at 800 °C, which is attributed to the low surface area of both the Pd and the YFeO3 support and to the limited oxygen storage capacity of the aged catalysts.
 | ||
| Fig. 7 CH4 conversion curves of fresh and aged 2Pd/YFO, 0.5Pd/YFO and Pd/ACZ. F, fresh; AT, thermal ageing; AS, stoichiometric ageing. | ||
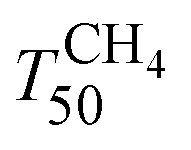
Contrary to CH4, CO oxidation activity of 2Pd/YFO, 0.5Pd/YFO and Pd/ACZ decreases upon ageing (Table 2) and the structural changes produced by thermal ageing appear more severe likely as a result of Pd particle growth.82,83 This is in agreement with observations on Pd-based catalysts aged on a dynamometer.84 Besides, the YFeO3 support alone exhibited deactivation upon consecutive reaction runs.53 Therefore, the activity enhancement is peculiar to CH4 chemistry on Pd. The influence of water on the feedstock still remains to be assessed to ascertain whether a true activity enhancement would also be observed since water has been reported to decrease the catalytic activity of Pd.85,86
It is clear from the characterization and the activity data that activation is coincident with the formation of well-defined metallic Pd particles. Because of the similar structural changes, the activity improvement appears to be of the same nature as that observed for 2Pd/YFO-F during consecutive reaction cycles under stoichiometric conditions.53 The formation of metallic Pd particles is accompanied by the loss of surface area of the catalyst and by the phase transition from hexagonal to orthorhombic YFeO3. However, because the OSCC values after ageing seem to be associated with Pd rather than with the presence of YFeO3, the support does not appear to be responsible for the improved catalytic activity after ageing. This indicates that the degree of coordination of Pd with YFeO3 after prolonged exposure to 900 °C is low and likely not of the same ‘self-regenerative’ nature as LaFeO3. The Pd–YFeO3 interaction was shown to change already from a sample calcined at 700 °C to one calcined at 800 °C, the latter exposing PdO-like particles on hexagonal YFeO3.52 However, no phase transition was observed, indicating that the hexagonal structure may have been stabilized by the interaction with Pd. In contrast, YFe1−xPdxO3−δ (x = 0.1) was shown to be a self-regenerative catalyst for CO oxidation,66 but no high temperature treatment was performed in previous studies. Our interpretation of the low temperature self-regeneration versus the high temperature segregation of Pd is in terms of geometric factors; however, this needs further confirmation. The periodic segregation–incorporation of Pd in YFeO3 is favored at low temperature due to the fact that Pd can adopt a coordination similar to that of tetrahedral Fe ions of hexagonal YFeO3.51 In the flame-made catalyst, Pd is deposited on the YFeO3 oxide already in the fresh state. Some extent of interaction is observed, but it is likely not sufficient to avoid the phase transition to orthorhombic YFeO3.53 In this phase, the segregation of Pd is favored because Y3+ (1.18 Å)87 does not help in stabilizing a Pd3+-like cation in the octahedral Fe sites, contrary to the larger La3+ (1.36 Å)88 in LaFeO3. The strong tendency of Pd3+ to disproportionate to Pd2+ and Pd4+, when it is in an oxide lattice, can be overcome by the relatively small octahedral tilting due to the largest rare earth ion being in the A-site.89
It is tempting to associate the improved catalytic activity with metallic Pd based on the experimental observations. The particle size dependence (structure sensitivity) and the role of metallic Pd vs. PdO are still debated issues in methane oxidation.4,19,90–95 This uncertainty is predominantly associated with the different experimental conditions used for the determination of catalytic activity and the difference in nature of the various catalysts. Moreover, the debate concerns primarily methane oxidation under lean conditions, where it is accepted that PdO is formed during reaction. Few studies have been reported for oxidation under low partial pressures of oxygen as it is the case for a TWC. The characterization data, together with the activity data of Fig. 7, demonstrate that discrete metal particles are favorable for methane oxidation, which is in agreement with various observations by others.25,26,90 This is not only the case for 2Pd/YFO but also for the reference catalyst, a conventional TWC, where particle growth occurs despite the presence of ceria–zirconia, and the activity of Pd/ACZ-AS is unchanged compared to that of Pd/ACZ-F. An important piece of information is derived from the observation that both the large Pd particles and the possibly large PdO particles undergo a reversible reduction–oxidation under redox conditions. Therefore, under the stoichiometric conditions used in this work, oxygen is activated on the large Pd particles of 2Pd/YFO-AS. On the large PdO particles formed in the thermally aged Pd/YFO-AT catalysts where an activity improvement is induced to a lesser extent, the activation phenomenon could be explained with the partial reduction of PdO during reaction, thus forming metallic Pd on the PdO surface.30 EXAFS and DRIFTS data confirm this possibility. Therefore, a mixed PdOx/Pd phase is present during reaction in both aged catalysts.
Finally, the light-off profiles of Pd/ACZ-AS and 2Pd/YFO-AS exhibit a clear loss of CH4 conversion by 10–20% around 350–450 °C (Fig. 7). This is more pronounced in 0.5Pd/YFO-AS (400–650 °C), likely as a consequence of the lower Pd content. This behaviour is characteristic of supported PdO catalysts under lean conditions and is due to spontaneous PdO reduction.19,20,93 The activity loss is slightly more pronounced in the samples aged under stoichiometric conditions because of the presence of large metal particles, thus diminishing the amount of available Pd atoms. These particles can be partially oxidized as demonstrated above, but reduction during reaction occurs at lower temperature because of the low oxygen content in the feed under λ = 1 conditions.23 Further study under reaction conditions should be performed to confirm this behavior and the low temperature reduction of PdO.
Conclusions
The catalytic performance of Pd/YFO with 0.5 and 2 wt% Pd content was determined after high temperature treatment. Two ageing protocols were chosen, thermal ageing at 900 °C in air and stoichiometric ageing at 900 °C under reaction conditions comprising CH4, CO, NO and oxygen. The reference TWC was aged under identical conditions for comparison. A gradual activity improvement was observed from the performance of the fresh Pd/YFO catalyst to the thermally aged one and to the sample aged under stoichiometric conditions. Pd loading affected only the extent of improvement, with the highest loading providing a better activity. After stoichiometric ageing, 2Pd/YFO was as active for methane oxidation as Pd/ACZ. In contrast to Pd/YFO catalysts, Pd/ACZ was less active after thermal ageing. For comparison, CO oxidation activity systematically decreased upon ageing indicating that the observed improved oxidation activity is peculiar to CH4.Structural characterization of the aged catalysts revealed that the activity improvement of Pd/YFO after stoichiometric ageing is accompanied by the formation of well-defined metallic Pd particles of ca. 20 nm. In the case of thermal ageing that produces less activity enhancement, Pd remains predominantly oxidized. The interaction of Pd with YFeO3 is poorer after ageing probably as a result of the phase changes observed in the support that prevent coordination of Pd within the YFeO3 lattice. Therefore, the observed activity improvement is mostly attributed to the changes related to the shape and oxidation state of Pd particles. However, it has been demonstrated that both Pd and YFeO3 remain highly dynamic with respect to exposure to periodic redox conditions. Large metallic and oxidic Pd particles periodically reduce and oxidize. Therefore, this study provides evidence that a mixed PdOx/Pd pair is at work during reaction and that large metal particles are not detrimental to methane oxidation under stoichiometric conditions.
Acknowledgements
Financial support from the Swiss National Science Foundation under project numbers 406240-1261274 (NRP62 ‘Smart materials’) and 200021-138068 and the Centre for Materials Science and Technology (CCMX) is greatly appreciated. ESRF and SLS are acknowledged for beamtime allocation at beamlines ID15B and SuperXAS, respectively.References
- R. M. Heck, R. J. Farrauto and S. T. Gulati, Catalytic air pollution control: Commercial technology, Wiley, 3rd edn, 2009 Search PubMed.
- D. Ciuparu, M. R. Lyubovsky, E. Altman, L. D. Pfefferle and A. Datye, Catal. Rev.: Sci. Eng., 2002, 44, 593 CAS.
- P. Gelin and M. Primet, Appl. Catal., B, 2002, 39, 1 CrossRef CAS.
- D. Bounechada, G. Groppi, P. Forzatti, K. Kallinen and T. Kinnunen, Top. Catal., 2013, 56, 372 CrossRef CAS.
- Y. Lu, S. K. Matam, G. L. Chiarello, P. Dimopoulos Eggenschwiler, C. Bach, M. Weilenmann, A. Spiteri, A. Weidenkaff and D. Ferri, Catal. Commun., 2013, 39, 55 CrossRef CAS PubMed.
- M. Salaün, A. Kouakou, S. Da Costa and P. Da Costa, Appl. Catal., B, 2009, 88, 386 CrossRef PubMed.
- A. Winkler, A. Eyssler, A. Maegli, A. Liati, P. Dimopoulos Eggenschwiler and C. Bach, Fuel, 2013, 111, 855 CrossRef CAS PubMed.
- S. K. Matam, E. H. Otal, M. H. Aguirre, A. Winkler, A. Ulrich, D. Rentsch, A. Weidenkaff and D. Ferri, Catal. Today, 2012, 184, 237 CrossRef CAS PubMed.
- A. Winkler, D. Ferri and R. Hauert, Catal. Today, 2010, 155, 140 CrossRef CAS PubMed.
- M. Honkanen, M. Kärkkäinen, V. Viitanen, H. Jiang, K. Kallinen, M. Huuhtanen, M. Vippola, J. Lahtinen, R. Keiski and T. Lepistö, Top. Catal., 2013, 56, 576 CrossRef CAS PubMed.
- J. Chen, R. M. Heck and R. J. Farrauto, Catal. Today, 1992, 11, 517 CrossRef CAS.
- J. Andersson, M. Antonsson, L. Eurenius, E. Olsson and M. Skoglundh, Appl. Catal., B, 2007, 72, 71 CrossRef CAS PubMed.
- D. L. Mowery, M. S. Graboski, T. R. Ohno and R. L. McCormick, Appl. Catal., B, 1999, 21, 157 CrossRef CAS.
- M. J. Rokosz, A. E. Chen, C. K. Lowe-Ma, A. V. Kucherov, D. Benson, M. C. P. Peck and R. W. McCabe, Appl. Catal., B, 2001, 33, 205 CrossRef CAS.
- R. Burch and F. J. Urbano, Appl. Catal., A, 1995, 124, 121 CrossRef CAS.
- N. van Vegten, M. Macieiewski, F. Krumeich and A. Baiker, Appl. Catal., B, 2009, 93, 38 CrossRef CAS PubMed.
- S. Colussi, A. Trovarelli, G. Groppi and J. Llorca, Catal. Commun., 2007, 8, 1263 CrossRef CAS PubMed.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713 CrossRef CAS PubMed.
- R. J. Farrauto, J. K. Lampert, M. C. Hobson and E. M. Waterman, Appl. Catal., B, 1995, 6, 263 CrossRef CAS.
- J. G. McCarty, Catal. Today, 1995, 26, 283 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227 CrossRef CAS.
- S. K. Matam, G. L. Chiarello, Y. Lu, A. Weidenkaff and D. Ferri, Top. Catal., 2013, 56, 239 CrossRef CAS.
- S. B. Kang, S. J. Han, S. B. Nam, I. S. Nam, B. K. Cho, C. H. Kim and S. H. Oh, Top. Catal., 2013, 56, 298 CrossRef CAS.
- A. Y. Stakheev, A. M. Batkin, N. S. Teleguina, G. O. Bragina, V. I. Zaikovsky, I. P. Prosvirin, A. K. Khudorozhkov and V. I. Bukhtiyarov, Top. Catal., 2013, 56, 306 CrossRef CAS.
- A. Baylet, P. Marecot, D. Duprez, P. Castellazzi, G. Groppi and P. Forzatti, Phys. Chem. Chem. Phys., 2011, 13, 4607 RSC.
- S. K. Matam, M. H. Aguirre, A. Weidenkaff and D. Ferri, J. Phys. Chem. C, 2010, 114, 9439 CAS.
- P. Briot and M. Primet, Appl. Catal., 1991, 68, 301 CrossRef CAS.
- D. Ciuparu and L. Pfefferle, Appl. Catal., A, 2001, 218, 197 CrossRef CAS.
- S. C. Su, J. N. Carstens and A. T. Bell, J. Catal., 1998, 176, 125 CrossRef CAS.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24 CrossRef CAS.
- M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981 CrossRef CAS PubMed.
- W. F. Libby, Science, 1971, 171, 499 CAS.
- R. J. H. Voorhoeve, J. P. Remeika and D. W. J. Johnson, Science, 1973, 180, 62 CAS.
- C. H. Kim, G. Qi, K. Dahlberg and W. Li, Science, 2010, 327, 1624 CrossRef CAS PubMed.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164 CrossRef CAS PubMed.
- N. Guilhaume and M. Primet, J. Catal., 1997, 165, 197 CrossRef CAS.
- K. Zhou, H. Chen, Q. Tian, Z. Hao, D. Shen and X. Xu, J. Mol. Catal. A: Chem., 2002, 189, 225 CrossRef CAS.
- D. Fino, Sci. Technol. Adv. Mater., 2007, 8, 93 CrossRef CAS PubMed.
- D. Fino, N. Russo, G. Saracco and V. Specchia, Prog. Solid State Chem., 2007, 35, 501 CrossRef CAS PubMed.
- Y. Chang and J. G. McCarty, Catal. Today, 1996, 30, 163 CrossRef CAS.
- S. Royer, H. Alamdari, D. Duprez and S. Kaliaguine, Appl. Catal., B, 2005, 58, 273 CrossRef CAS PubMed.
- M. Uenishi, M. Taniguchi, H. Tanaka, M. Kimura, Y. Nishihata, J. Mizuki and T. Kobayashi, Appl. Catal., B, 2005, 57, 267 CrossRef CAS PubMed.
- M. Casapu, J. D. Grunwaldt, M. Maciejewski, A. Baiker, S. Eckhoff, U. Göbel and M. Wittrock, J. Catal., 2007, 251, 28 CrossRef CAS PubMed.
- M. B. Katz, G. W. Graham, Y. Duan, H. Liu, C. Adamo, D. G. Schlom and X. Pan, J. Am. Chem. Soc., 2011, 133, 18090 CrossRef CAS PubMed.
- H. Tanaka, M. Taniguchi, M. Uenishi, N. Kajita, I. Tan, Y. Nishihata, J. Mizuki, K. Narita, M. Kimura and K. Kaneko, Angew. Chem., Int. Ed., 2006, 45, 5998 CrossRef CAS PubMed.
- Y. Lu, A. Eyssler, E. H. Otal, S. K. Matam, O. Brunko, A. Weidenkaff and D. Ferri, Catal. Today, 2013, 208, 42 CrossRef CAS PubMed.
- J. A. Kurzman, J. Li, T. D. Schladt, C. R. Parra, X. Ouyang, R. Davis, J. T. Miller, S. L. Scott and R. Seshadri, Chem. Mater., 2011, 50, 8073 CAS.
- I. Tan, H. Tanaka, M. Uenishi, K. Kaneko and S. Mitachi, J. Ceram. Soc. Jpn., 2005, 113, 71 CrossRef CAS.
- J. P. Dacquin, M. Cabie, C. R. Henry, C. Lancelot, C. Dujardin, S. R. Raouf and P. Granger, J. Catal., 2010, 270, 299 CrossRef CAS PubMed.
- J. Li, U. G. Singh, T. D. Schladt, J. K. Stalick, S. L. Scott and R. Seshadri, Chem. Mater., 2008, 20, 6567 CrossRef CAS.
- A. Eyssler, A. Winkler, O. Safonova, M. Nachtegaal, S. K. Matam, P. Hug, A. Weidenkaff and D. Ferri, Chem. Mater., 2012, 24, 1864 CrossRef CAS.
- Y. Lu, K. A. Michalow, S. K. Matam, A. Winkler, A. E. Maegli, S. Yoon, A. Heel, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2014, 144, 631 CrossRef CAS PubMed.
- S. Yoon, S. Hua, N. Y. Kado, A. Thiruvengadam, J. F. Collins, M. Gautam, J. D. Herner and A. Ayala, Atmos. Environ., 2014, 83, 220 CrossRef CAS PubMed.
- E. Tzimpilis, N. Moschoudis, M. Stoukides and P. Bekiaroglou, Appl. Catal., B, 2009, 87, 9 CrossRef CAS PubMed.
- S. J. Pennycook, Ultramicroscopy, 1989, 30, 58 CrossRef.
- W. S. Rasband, U.S. National Institutes of Health, Bethesda, Maryland, USA, 1997–2012, http://imagej.nih.gov/ij/.
- E. Mamontov, R. Brezny, M. Koranne and T. Egami, J. Phys. Chem. B, 2003, 107, 13007 CrossRef CAS.
- A. Pappacena, K. Schermanz, A. Sagar, E. Aneggi and A. Trovarelli, Stud. Surf. Sci. Catal., 2010, 175, 835 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- D. Ferri, M. A. Newton, M. Di Michiel, S. Yoon, G. L. Chiarello, V. Marchionni, S. K. Matam, M. H. Aguirre, A. Weidenkaff, F. Wen and J. Gieshoff, Phys. Chem. Chem. Phys., 2013, 15, 8629 RSC.
- D. Baurecht and U. P. Fringeli, Rev. Sci. Instrum., 2001, 72, 3782 CrossRef CAS PubMed.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902 CrossRef CAS PubMed.
- D. Ferri, S. K. Matam, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634 RSC.
- D. Chernyshov, W. van Beek, H. Emerich, M. Milanesio, A. Urakawa, D. Viterbo, L. Palin and R. Caliandro, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 327 CrossRef CAS PubMed.
- J. Li, U. G. Singh, T. D. Schladt, J. K. Judith, S. L. Scott and R. Seshadri, Chem. Mater., 2008, 20, 6567 CrossRef CAS.
- L. Wu, J. C. Yu, L. Zhang, X. Wang and S. Li, J. Solid State Chem., 2004, 177, 3666 CrossRef CAS PubMed.
- Y. Zhang, J. Yang, J. Xu, Q. Gao and Z. Hong, Mater. Lett., 2012, 81, 1 CrossRef CAS PubMed.
- T. Suzuki, A. Morikawa, A. Suda, H. Sobukawa, M. Sugiura, T. Kanazawa, J. Suzuki and T. Takada, Toyota Chuo Kenkyusho R&D Rebyu, 2002, 37, 28 CAS.
- N. M. Rodriquez, S. G. Oh, R. A. Dalla-Betta and R. T. K. Baker, J. Catal., 1995, 157, 676 CrossRef.
- T. Lear, R. Marshall, J. A. Lopez-Sanchez, S. D. Jackson, T. M. Klapötke, M. Bäumer, G. Rupprechter, H. J. Freund and D. Lennon, J. Chem. Phys., 2005, 123, 1 CrossRef PubMed.
- S. K. Matam, E. V. Kondratenko, M. H. Aguirre, P. Hug, D. Rentsch, A. Winkler, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2013, 129, 214 CrossRef CAS PubMed.
- R. Rohé and V. Pitchon, Top. Catal., 2001, 16/17, 311 CrossRef.
- P. Gelin, A. R. Siedle and J. T. Yates, J. Phys. Chem., 1984, 88, 2978 CrossRef CAS.
- D. Tessier, A. Rakai and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 741 RSC.
- A. Palazov, C. C. Chang and R. J. Kokes, J. Catal., 1975, 36, 338 CrossRef CAS.
- C. Binet, A. Badri and J. C. Lavalley, J. Phys. Chem., 1994, 98, 6392 CrossRef CAS.
- W. Daniell, H. Landes, N. E. Fouad and H. Knözinger, J. Mol. Catal. A: Chem., 2002, 178, 211 CrossRef CAS.
- R. Caliandro, D. Chernyshov, H. Emerich, M. Milanesio, L. Palin, A. Urakawa, W. van Beek and D. Viterbo, J. Appl. Crystallogr., 2012, 45, 458 CrossRef CAS.
- M. Boaro, F. Giordano, S. Recchia, V. DalSanto, M. Giona and A. Trovarelli, Appl. Catal., B, 2004, 52, 225 CrossRef CAS PubMed.
- R. W. McCabe and R. K. Usmen, Stud. Surf. Sci. Catal., 1996, 101, 355 CrossRef CAS.
- R. Meyer, S. K. Shaikhutdinov and H. J. Freund, Z. Phys. Chem., 2004, 218, 905 CrossRef CAS.
- Z. W. Wang, B. Li, M. S. Chen, W. Z. Weng and H. L. Wan, Sci. China: Chem., 2010, 53, 2047 CrossRef CAS.
- J. H. Baik, H. J. Kwon, Y. T. Kwon, I. S. Nam and S. H. Oh, Top. Catal., 2007, 42–43, 337 CrossRef PubMed.
- D. Ciuparu and L. Pfefferle, Appl. Catal., A, 2001, 209, 415 CrossRef CAS.
- C. F. Cullis, T. G. Nevell and D. L. Trimm, J. Chem. Soc., Faraday Trans., 1972, 68, 1406 RSC.
- X. Cao, C.-S. Kim and H.-I. Yoo, J. Am. Ceram. Soc., 2001, 84, 1265 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- S. J. Kim, S. Lemaux, G. Demazeau, J. Y. Kim and J. H. Choy, J. Mater. Chem., 2002, 12, 995 RSC.
- R. F. Hicks, H. Qi, M. L. Young and R. G. Lee, J. Catal., 1990, 122, 280 CrossRef CAS.
- R. F. Hicks, H. Qi, M. L. Young and R. G. Lee, J. Catal., 1990, 122, 295 CrossRef CAS.
- T. R. Baldwin and R. Burch, Appl. Catal., 1990, 66, 359 CrossRef CAS.
- K. Persson, A. Ersson, S. Colussi, A. Trovarelli and S. G. Järas, Appl. Catal., B, 2006, 66, 175 CrossRef CAS PubMed.
- H. Arai, T. Yamada, K. Eguchi and T. Seiyama, Appl. Catal., 1986, 26, 265 CrossRef CAS.
- J. R. Theis and R. W. McCabe, Catal. Today, 2012, 184, 262 CrossRef CAS PubMed.
Footnotes |
| † Present address: Università degli Studi di Milano, Dipartimento di Chimica, via C. Golgi 19, I-20133 Milano, Italy. |
| ‡ Present address: Institute for Materials Science, University of Stuttgart, Heisenbergstrasse 3, DE-70569 Stuttgart, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |