 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, acid properties and catalysis by niobium oxide nanostructured materials†
M.
Luisa Marin
ab,
Geniece L.
Hallett-Tapley
a,
Stefania
Impellizzeri
a,
Chiara
Fasciani
a,
Sabrina
Simoncelli
ac,
José Carlos
Netto-Ferreira
ad and
Juan C.
Scaiano
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie, Ottawa, K1N 6N5, Canada. E-mail: scaiano@photo.chem.uottawa.ca
bInstituto Universitario Mixto de Tecnología Química (UPV-CSIC), Universitat Politècnica de València, Avenida de los Naranjos s/n, 46022 Valencia, Spain
cINQUIMAE and Departamento de Química Inorgánica, Analítica, y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, 1428 Buenos Aires, Argentina
dDivisão de Metrologia Química, Instituto Nacional de Metrologia, Qualidade e Tecnologia-INMETRO, Duque de Caxias, 25250-020 Rio de Janeiro, Brazil
First published on 9th April 2014
Abstract
Several forms of niobium oxide were prepared, including nanostructured mesoporous materials, and their acidity properties were comprehensively investigated and compared with commercially available materials. The composites were characterized by a variety of techniques, including XRD, TEM, N2 adsorption and Hammett acid indicator studies. The acidity of the niobium oxide derivatives was also investigated by the ability of the materials to successfully promote the halochromic ring-opening of an oxazine-coumarin probe that was specifically designed for use in fluorescence imaging studies. The ring-opening reaction was easily monitored using UV-visible, fluorescence and NMR spectroscopy. Single molecule microscopy was employed to gain a more in-depth understanding of the niobium oxide acid catalysis pathway. Using this technique, the rate of niobium oxide mediated protonation was estimated to be 1.8 × 10−13 mol m−2 s−1. Single molecule analysis was also used to obtain a detailed map of Brønsted acid sites on the niobium oxide surface. The active sites, located by multiple blinking events, do not seem to be localized on any area of the material, but rather randomly distributed throughout the solid state surface. As the reaction proceeds, the sites with the highest acidity and accessibility are gradually consumed, making the next tier of acid sites available for reaction. The phenomenon was more closely characterized by using time lapsed reactivity maps.
Introduction
There has been a surge in interest in heterogeneous catalysis over the past decades due to the inherent advantages of using solid catalysts. Such materials can be easily removed from the reaction mixture and also promote the minimization of chemical waste. Hydrated niobium oxide (Nb2O5·nH2O) is no exception. This amorphous, water tolerant solid acid is comprised of distorted NbO6 octahedral and NbO4 tetrahedral geometries,1,2 with the former responsible for the Brønsted acidity of these materials (equivalent to >70% H2SO4) as a result of H2O coordination to the highly polarized Nb–O (Scheme 1).1–4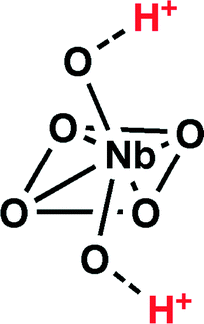 | ||
| Scheme 1 Distorted octahedral geometry of NbO6. | ||
Due to the remarkable acidity of these materials, many opportunities exist for the use of this solid acid catalyst in a variety of systems where harsher chemicals are typically employed (e.g., concentrated H2SO4) and where the removal of excess acid following reaction is desired, but not always feasible. As such, the Brønsted acid character of Nb2O5·nH2O has been exploited in the catalysis of many industrially relevant reactions, including esterification,3 saccharide dehydration3,4 and hydrolysis.3,5 Nb2O5·nH2O holds several advantages over traditional acid catalysts; in addition to ease of removal, the acidity of niobium-based catalysts can be easily tailored using a variety of methodologies. Several studies have examined the effects of temperature on the acidity of Nb2O5·nH2O, where temperatures lower than 473 K are desired to achieve maximum Brønsted acidity; within conditions above this limit, Nb2O5·nH2O Brønsted acidity rapidly decreases due to the increasing crystalline character of the material.2,6,7 As a result, acid doping of hydrated niobium oxides (e.g.; H3PO4, HCl, HNO3)7,8 has been developed to allow for applications outside of such critical temperature ranges, and has also been exploited as a means of enhancing the acid catalytic capabilities of Nb2O5·nH2O.
In the current contribution, we explore the properties of a family of five Nb2O5·nH2O solids in order to compare the Brønsted acidity of these materials to commercially available alternatives. Initial trials were focused on the characterization of the materials. Later, an emissive halochromic oxazine-coumarin system (1) was used to probe the Nb2O5·nH2O acidic character with spectroscopic and single molecule microscopy techniques.
Experimental
Materials
Dichloromethane and acetonitrile were purified with a LC Technology Solutions Inc. SPBT-1 Bench-Top Solvent Purification System. Compound 1 was synthesized according to literature procedures.9 NbCl5 and Nb(OEt)5 used in the preparation of T-I and T-II were purchased from Sigma-Aldrich and used as received. Commercial Nb2O5 (Puratronic®, 99.9985%) was purchased from Alfa Aesar and used as received. Pluronic (P123) and dodecylamine 98% were used as structure-directing agents and were purchased from Sigma-Aldrich. Milli-Q water (resistivity 18.2 MΩ cm−1 at 25 °C; 0.22 μm filter) was used in all preparations and reactions requiring aqueous conditions.Nb2O5·nH2O type I (T-I)10. 5 g of NbCl5 (18.5 mmol) were dissolved in 99% ethanol (10 mL) under constant stirring. To this solution NH4OH (1 M) was slowly added to the aforementioned solution until a white precipitate was observed. The precipitate was then separated from the solution by centrifugation (3000 rpm), washed four times using Milli-Q water and dried at 120 °C for 24 hours.
Nb2O5·nH2O type II (T-II)1. 5 g of NbCl5 (18.5 mmol) were dissolved in 200 mL Milli-Q water. Immediately after addition, the yellow powder became white. After stirring for 3 h at room temperature, the white precipitate was centrifuged (3000 rpm) and washed four times with Milli-Q water until the filtrate was at neutral pH. The obtained solid was dried at 120 °C for 24 hours.
Nb2O5·nH2O type III a and b (T-IIIa and T-IIIb)11. 1.5 g of P-123 were dispersed in 11 mL of Milli-Q water and 45 mL of HCl (2 M) at 40 °C. 5 g of Nb(OEt)5 (15.7 mmol) was added to solution under stirring. The resulting mixture was kept at 40 °C for 24 hours and then precipitated in a Teflon-lined autoclave at 100 °C for 48 hours. The obtained solid was filtered, washed with Milli-Q water and dried in air at room temperature. Excess surfactant was removed by Soxhlet extraction first by washing with 1% HCl in 95% EtOH (300 mL) and then with 95% EtOH only (120 mL). The final, template-free mesoporous Nb2O5·nH2O was dried in the oven at 120 °C for 18 hours.
Nb2O5·nH2O type IV (T-IV)12. A mixture of Nb(OEt)5 (3.25 g, 10.3 mmol) and dodecylamine (0.57 g, 3.1 mmol) was heated until a homogeneous solution was obtained. Subsequent addition of 65.4 mL of Milli-Q water with manual stirring resulted in the formation of a gel-like precipitate. Further addition of 27.6 μL (0.3 mmol) concentrated HCl afforded a white precipitate. The reaction mixture was allowed to stand overnight, sealed and heated at 40 °C for 30 hours, 60–65 °C for 66 hours, 80 °C for 48 hours, and 95–100 °C for 5 days. The solid was then cooled down, collected by filtration and dried at 95–100 °C for 2 hours, 120 °C for 48 hours, and 140 °C for 48 hours. The white solid was then washed four times to remove the presence of excess surfactant. Each washing cycle was conducted for 24 hours under vigorous stirring followed by Büchner filtration. 645 mg (3.4 mmol) of p-toluenesulfonic acid in 13 mL of ether were added to 100 mL of MeOH for the first wash. For the second wash, 65 mg (0.3 mmol) of p-toluenesulfonic acid in 3 mL of ether were added to 100 mL of MeOH. Third and fourth washings were done using MeOH (100 mL) only. The final, template-free mesoporous Nb2O5·nH2O was dried into the oven at 120 °C for 24 hours.
Methods
The synthesis of 1 was monitored by thin-layer chromatography, using aluminum sheets coated with silica (60, F254). NMR spectra were recorded at room temperature with a Bruker Avance 300 instrument. Mass spectral analysis was performed with a 6890N Network GC System equipped with a 5973 Mass Selective Detector from Agilent Technologies. Absorbance spectra were recorded using a Cary 50 UV-visible spectrophotometer. Fluorescence spectra were taken using a Photon Technology International (PTI) spectrofluorimeter using slit widths of 2 nm at the excitation source and 2 nm/5 nm at the detector. Diffuse reflectance spectra were recorded on a Cary 100 UV-visible spectrophotometer fitted with a Labsphere Inc. DRA-30I diffuse reflectance accessory. Nitrogen adsorption/desorption isotherms for the mesoporous Nb oxides were determined from N2 adsorption at 77 K in an AUTOSORB-6 apparatus. The samples were previously degassed for 4 hours at 523 K and 5 × 10−5 bar. The adsorption branch was used to determine the pore size distribution according to the Barret–Joyner–Helender (BJH) methodology. Mesopore volume was measured at the plateau of the adsorption branch of the nitrogen isotherm, P/Po = 0.8.11 Gas adsorption at higher P/Po is mainly due to interparticle condensation. Small-angle powder X-ray diffraction (XRD) analysis was carried out with a Rigaku Ultima IV diffractometer using a CuKα radiation (k = 1.541836 Å), operating at 40 kV and 30 mA, at a scanning velocity of 0.03° min−1 in the 0.7° < 2θ < 10° range. TEM images were acquired using a JEOL JEM-2100F field emission transmission electron microscope equipped with an ultra-high resolution pole piece operating at 200 kV. Thermogravimetric analysis was collected on a TA Instruments Q5000 IR Thermogravimetric Analyzer.Niobium oxides as Brønsted acid catalysts
Acid strength measurements on the Nb2O5 solids discussed in this work were tested using previously published methods.12,13 In short, 3 drops of a 0.1% solution of each indicator in benzene was added to 100 mg of each material in a clean, dry, 2 mL glass vial. Colour changes were visually observed and recorded as positive (observed indicator colour change) or negative (no observable colour change).Acid-catalyzed ring-opening of a halochromic oxazine-coumarin probe
Two mg of each Nb2O5 solid were placed into a 1 cm × 1 cm quartz cuvette containing 3 mL of a 7.5 μM CH3CN solution of compound 1. Each cuvette was sealed to prevent loss of solvent and the absorption and emission spectra were recorded to detect the formation of the ring-opened emissive isomer.Single-molecule fluorescence microscopy
Imaging fluorescence experiments were performed in a borosilicate reaction chamber with an area of 4.2 cm2 per well (Thermo Scientific). Coverslips (18 mm × 18 mm; Fisher Scientific) were cleaned in piranha solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 H2O2:H2SO4) for 1 hour and then rinsed thoroughly with water. The slides were dried with N2 prior to functionalization. The surface was functionalized by spin coating a dispersion of T-I in methanol (2 mg in 2 mL) at 1000 rpm for 40 s. The cover slides were then placed in the chamber and submerged in 1 mL of Milli-Q water, to which 2 μL of a 10 nM solution of compound 1 in CH3CN were added. TIRF images were acquired on an Olympus FV1000 TIRF microscope (Olympus; Japan) using an oil immersion Plan Apo objective 100× NA1.45 (Olympus; Japan) and a highly sensitive Rolera EM-C2 camera (Q-Imaging, Surrey, Canada). TIR illumination was achieved by focusing a 633 nm He–Ne, CW laser onto the back focal plane of the objective. The power density of the excitation laser at the sample was estimated to be 40 W cm−2. A Chroma ET600 excitation filter was used (Chroma Technology Corporation, Bellows Falls, USA). The emission was collected by a camera after direction through a dichroic mirror (Chroma ZT640) and an emission filter (Chroma ET655) to remove light from the excitation source. The pixel size of the image corresponds to 156 nm. Fluorescence spectra were recorded with a Fluorescent Lifetime Imaging System (PicoQuant, Berlin, Germany). The instrument is equipped with a frequency doubled, picosecond pulse diode laser (637 nm, 93 ps, 40 MHz, LDH-P-FA-640L; PicoQuant). The laser beam was collimated and focused through a fiber-coupling unit. A beam splitter (Z638rdc, Chroma) was used to separate excitation and emission light. The emission signal was collected by a Shemrock SR-163 spectrograph (Andor Technology, South Windsor, USA).
:3 H2O2:H2SO4) for 1 hour and then rinsed thoroughly with water. The slides were dried with N2 prior to functionalization. The surface was functionalized by spin coating a dispersion of T-I in methanol (2 mg in 2 mL) at 1000 rpm for 40 s. The cover slides were then placed in the chamber and submerged in 1 mL of Milli-Q water, to which 2 μL of a 10 nM solution of compound 1 in CH3CN were added. TIRF images were acquired on an Olympus FV1000 TIRF microscope (Olympus; Japan) using an oil immersion Plan Apo objective 100× NA1.45 (Olympus; Japan) and a highly sensitive Rolera EM-C2 camera (Q-Imaging, Surrey, Canada). TIR illumination was achieved by focusing a 633 nm He–Ne, CW laser onto the back focal plane of the objective. The power density of the excitation laser at the sample was estimated to be 40 W cm−2. A Chroma ET600 excitation filter was used (Chroma Technology Corporation, Bellows Falls, USA). The emission was collected by a camera after direction through a dichroic mirror (Chroma ZT640) and an emission filter (Chroma ET655) to remove light from the excitation source. The pixel size of the image corresponds to 156 nm. Fluorescence spectra were recorded with a Fluorescent Lifetime Imaging System (PicoQuant, Berlin, Germany). The instrument is equipped with a frequency doubled, picosecond pulse diode laser (637 nm, 93 ps, 40 MHz, LDH-P-FA-640L; PicoQuant). The laser beam was collimated and focused through a fiber-coupling unit. A beam splitter (Z638rdc, Chroma) was used to separate excitation and emission light. The emission signal was collected by a Shemrock SR-163 spectrograph (Andor Technology, South Windsor, USA).
Results and discussion
Nb2O5·nH2O characterization; TEM, XRD and nitrogen adsorption isotherms
Synthetic Nb2O5·nH2O T(I-IV), as well as the commercially available Nb2O5·nH2O were characterized to gain a thorough understanding of the acidic characteristics of these materials. The crystalline state of Nb2O5·nH2O is well known to directly influence the Brønsted acidity of these materials,2,6,7 therefore TEM images were obtained of all six samples (Fig. S1, ESI†). Analysis of the TEM images appears to illustrate an amorphous solid in all cases, typical of hydrated niobium oxides and necessary for achieving optimal Brønsted acidity. The amorphous nature of the Nb2O5·nH2O was more closely examined using XRD spectroscopy. As can be observed in Fig. 1, the XRD patterns obtained following analysis of the commercial Nb2O5·nH2O sample and T-I are considerably different. Specifically, the XRD spectrum of the commercial solid presents sharp defined diffraction peaks, while that of the synthesized derivative presents two broad diffraction peaks at approximately 25° and 53°. Likewise, XRD spectra obtained for T-II to T-IV (Fig. S2, ESI†) also present broadened diffraction patterns, with the mesoporous materials showing one diffraction at approximately 2.5°. Such broadening of the XRD diffraction patterns is characteristic of amorphous solids supporting the conclusions drawn from TEM imaging, and suggesting that the five synthetically prepared niobium oxides may be useful as Brønsted acid catalysts. Conversely, XRD spectroscopy indicates that the commercial Nb2O5·nH2O is more crystalline in nature, illustrated by the well-defined diffraction pattern. Such attributes suggest that this material may be considerably less acidic than the synthesized derivatives.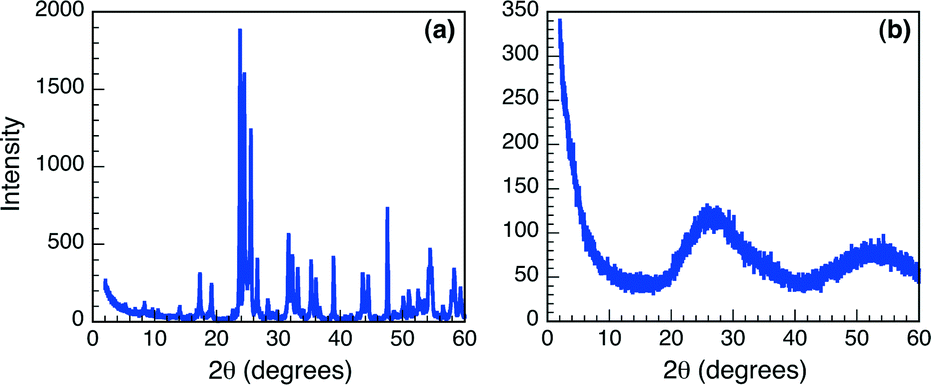 | ||
| Fig. 1 XRD spectra of (a) commercial Nb2O5·nH2O and (b) T-I. | ||
The pore size of the mesoporous materials (T-III and T-IV) was assessed using nitrogen adsorption isotherms due to the distinctive nitrogen uptake as a result of the capillary condensation of nitrogen inside of the mesopores. Table S1 (ESI†) presents the average surface area and pore size as calculated from the isotherm data for the three niobium oxide materials with mesoporous structure and demonstrates the porosity of the solids. Importantly, the overall surface area and pore size variations are not only dependent on the method of preparation, but also tend to vary from batch to batch within the same synthetic methodology, as shown for T-IIIa and T-IIIb. Both of these mesoporous niobium oxides were prepared using P-123 as a templating agent; nonetheless, the surface area (528 m2 g−1 and 295 m2 g−1, respectively) and pore size (24.4 Å and 9.57 Å) are significantly different, trends that are not uncommon in mesopore synthesis.14 As such, both solids were employed for further testing as Brønsted acid catalysts.
Assessment of niobium oxide acidity
In order to assess and quantify the overall acidity of both the commercial and synthesized niobium oxides, both acid indicator studies and thermogravimetric analysis (TGA) of the samples were undertaken. The acidity of solid samples is commonly reported as a function of Hammett acidity (H0).15 Using this method, the solid samples are placed in the presence of indicator dyes with known pKa values, allowing for one to establish a defined pKa range for the sample of interest. Preliminary assessments of the acidity of the niobium oxide materials were carried out using six indicators. The indicators and respective results obtained by this method of qualitative testing are reported in Table 1, which shows that the pKa range of the Nb2O5·nH2O are largely variable, with the commercial Nb2O5 being the least acid with H0 > +5.0. The tabulated Hammett acidity values clearly illustrate that all of the synthesized Nb2O5·nH2O are acidic in nature.| Indicator | Max. pKa | Comm. Nb2O5 | T-I | T-II | T-IIIa | T-IIIb | T-IV |
|---|---|---|---|---|---|---|---|
| a (+) = observable colour change; (−) = no observable colour change. | |||||||
| Methyl red | +5.0 | − | + | + | + | + | + |
| Methyl yellow | +3.3 | − | + | + | + | + | + |
| Crystal violet | +0.8 | − | + | + | + | + | + |
| Dicinnamal-acetone | −3.0 | − | − | + | + | + | + |
| Chalcone | −5.6 | − | − | + | + | + | − |
| Anthraquinone | −8.6 | − | − | − | − | − | − |
| H 0 | > +5.0 | −3.0 to +0.8 | −8.6 to −5.6 | −8.6 to −5.6 | −8.6 to −5.6 | −5.6 to −3.0 | |
Assessment of niobium oxide thermal stability
Knowing that increasing temperatures greatly affects the crystallinity, and, thus, Brønsted acidity of the Nb2O5·nH2O matrix, thermogravimetric analysis was carried out to examine the influence of increasing temperature on the aforementioned solid samples. The TGA traces obtained over a 500 °C temperature range are reported in Fig. 2. The traces illustrate that T-I undergoes the largest thermal degradation, as is represented by the largest loss of sample mass (% weight), followed by T-IIIa and there is no weight loss from the commercial sample. These results seem to suggest that the commercial Nb2O5 is the most robust material to be examined in this study and the least affected by temperature. Moreover, the TGA results suggest that the commercial sample is considerably crystalline in nature and, as such, void of any Brønsted acid characteristics, as corroborated by the indicator studies. All other Nb2O5·nH2O samples showed some thermal degradation upon exposure to extreme temperature conditions, which may be due in part to changes in the physical properties of the solid state matrix (i.e. amorphous to crystalline), loss of mesoporous structure and also partially due to loss of H2O, as H2O coordination is critical in Brønsted acidity activation in Nb2O5.2,7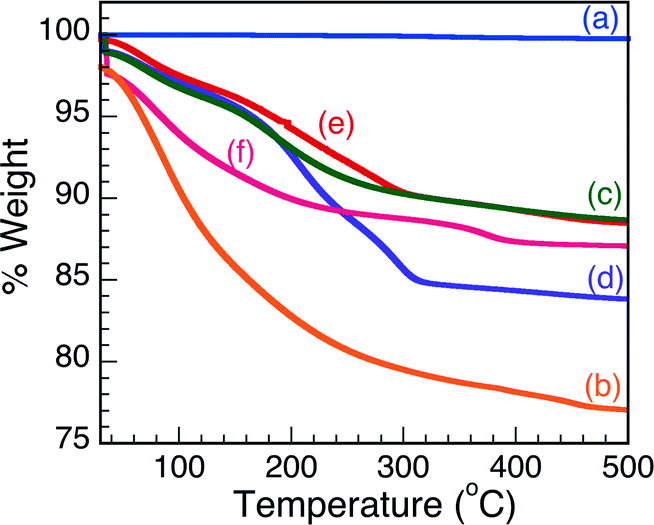 | ||
| Fig. 2 Thermogravimetric analysis of (a) comm. Nb2O5·nH2O, (b) T-I, (c) T-II, (d) T-IIIa, (e) T-IIIb and (f) T-IV. | ||
Catalytic Nb2O5·nH2O fluorescence activation
The design of the halochromic compound 1 is based on the established properties of switchable oxazines. These compounds are known to switch reversibly between isomeric states with distinctive structural properties and spectroscopic signature upon light or chemical stimulation.16 Covalent conjugation of the oxazine moiety with a coumarin fluorophore generates an emissive switchable bimolecular system, in which the fluorescence of the latter can be modulated in response to the pronounced structural and electronic modifications associated with either the photo- or chemically induced transformation of the former.9,17The absorption spectrum of 1 in CH3CN shows a band centered at 410 nm. The addition of acid, such as trifluoroacetic acid (TFA), opens the oxazine ring and permanently generates compound 2 (Fig. 3). Within this transformation, the coumarin functionality is brought in conjugation with the cationic fragment of the generated isomer, shifting its absorption band bathochromically by ca. 180 nm. A fluorescence band centered at 645 nm can then be observed by selectively exciting 2 at λex of 530 nm. Therefore, the transformation of 1 into 2, encouraged by the addition of acid, can be exploited to activate fluorescence and allows the investigation of materials with distinctive acidic properties with relatively simple experimental setups.
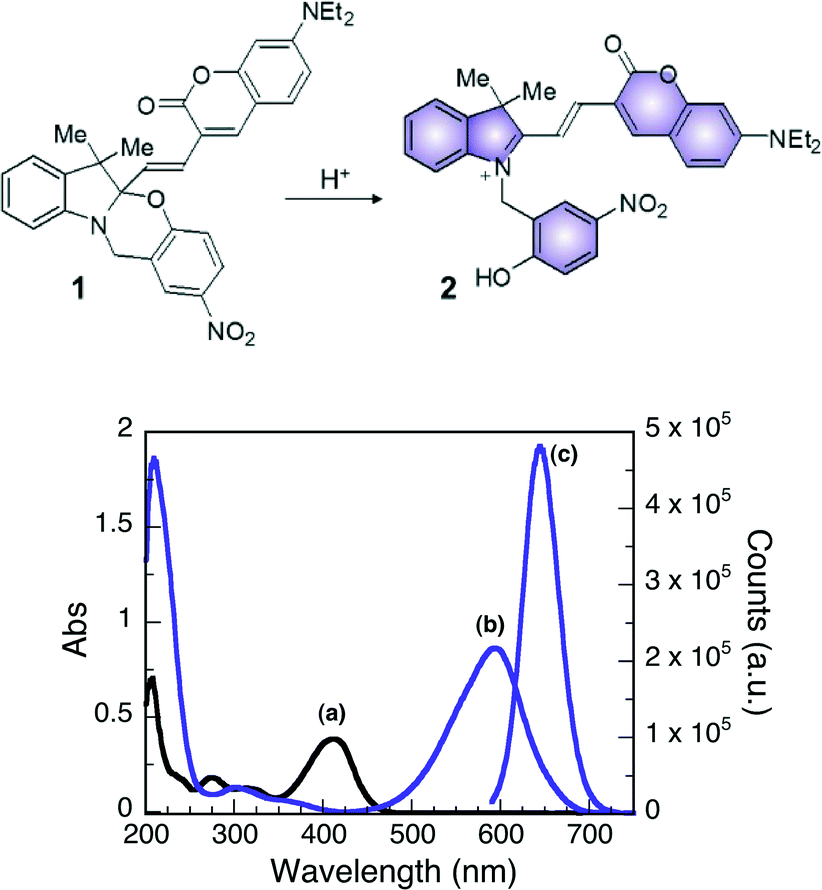 | ||
| Fig. 3 (Top) Structures of 1 and 2. (bottom) Absorption spectra of 1 (10 μM, CH3CN, 25 °C) before (a) and after (b) the addition of 10 equivalents of TFA. Emission spectrum (c, λex = 570 nm, CH3CN, 25 °C, right scale) of 1 after the addition of 10 equivalents of TFA. | ||
The use of an emissive probe with switchable character offers several advantages with respect to other pH-sensitive fluorophores (e.g. coumarin-6) which are strongly emissive either before or after the addition of acid and show only a discrete shift of the emission wavelength in response to protonation.18 The observation of stimuli-generated fluorescence in a dark background (such is observed for 1), rather than the shifting of the emission of a fluorescent substrate (coumarin-6), offers the opportunity to image fluorescent elements with improved signal to noise ratio, allowing the detection and identification of structural features with higher precision and the investigation of Nb2O5·nH2O-mediated acid catalysis at the molecular level.
To further investigate the niobium oxides discussed herein as Brønsted acid catalysts, 2 mg of each solid were placed in 3 mL of a 7.5 μM CH3CN solution of compound 1. In all cases, the reaction mixture rapidly changed colour (from pale yellow to blue/purple) upon the addition of the five synthesized Nb2O5·nH2O materials (Fig. 4), indicating the formation of 2 upon ring-opening reaction, while the addition of commercial Nb2O5 resulted in no observable colour change. These results support the absence of Brønsted acidity in the commercial niobium sample. Trifluoroacetic acid was also used as a control and yielded positive results.
 | ||
| Fig. 4 Photographs illustrating the colour change observed upon ring opening of 1 in the presence of Brønsted acidic Nb2O5·nH2O. | ||
The halochromic opening of 1 was monitored using both absorption and fluorescence spectroscopy. Fig. 5 and S4 (ESI†) present the UV-visible spectra obtained upon addition of TFA or the Nb2O5·nH2O catalysts discussed in Table 1. TFA was first added to a CH3CN solution of oxazine-coumarin 1 as a control experiment. The reaction immediately turned deep blue in colour and was left to equilibrate for 1 h. Subsequent spectroscopic measurements revealed a shift in the absorption spectrum from 410 nm to 595 nm (Fig. S4A†), due to the formation of 2, which slowly continued over the monitoring period of 5 days. Similarly, the niobium oxide catalysts presented in Table 1 were added to a fresh solution of 1. Implementing commercial Nb2O5 as a heterogeneous Brønsted acid catalyst afforded no appreciable change in the absorption maximum (Fig. 5A), indicating no conversion to the ring-opened isomer 2. The results obtained using the commercial sample further suggest that this Nb2O5·nH2O is void of any Brønsted acid properties.
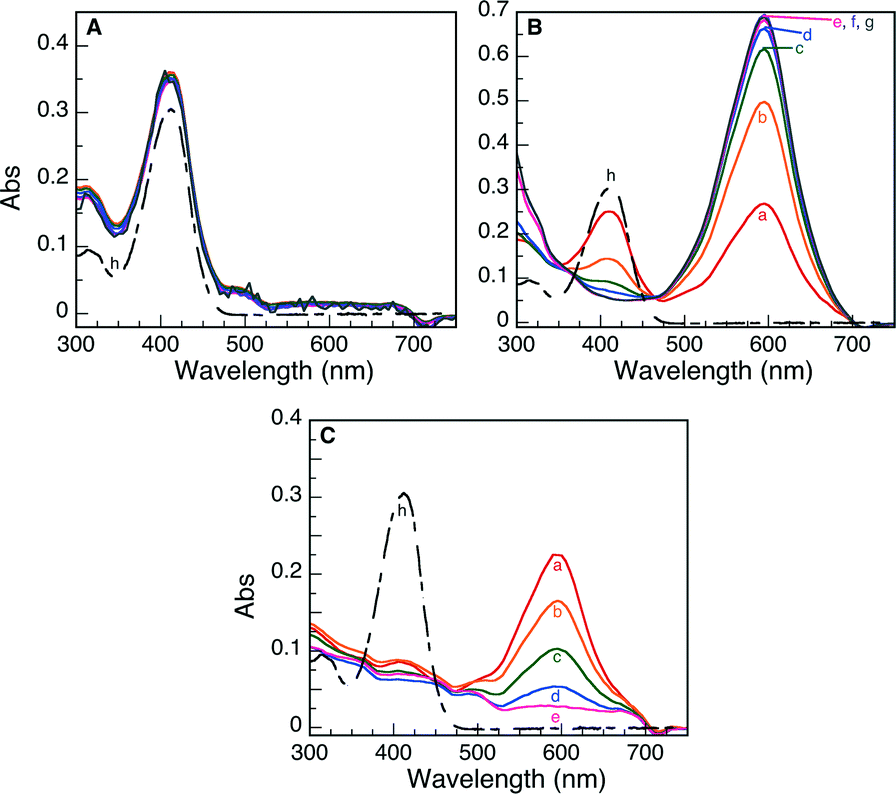 | ||
| Fig. 5 UV-visible spectra monitoring the Brønsted acid-induced ring opening of 1 to 2 using (A) commercial Nb2O5·nH2O, (B) T-I and (C) T-IIIa as heterogeneous catalysts. The reactions were monitored as a function of time: (a) 0 h, (b) 1 h, (c) 3 h, (d) 5 h, (e) 24 h, (f) 3 d, (g) 5 d and (h) initial absorption of 1. | ||
In the case of the five synthetically prepared Nb2O5·nH2O catalysts, a distinct variation in the UV-visible spectrum was detected upon addition of each heterogeneous material to a solution of the oxazine-coumarin probe 1. Following addition of T-I and T-IIIa as Brønsted catalysts (Fig. 5B and C), a subsequent shift in the absorption spectrum was observed, similar to that seen in TFA control studies, indicative of oxazine ring-opening to form compound 2. In the case of catalyst T-I, the formation of 2 slowly progresses over 5 days, whereas when catalyst T-IIIa was employed, rapid formation of 2 was preceded by a gradual decrease of the absorption band ascribed to ring-opened isomer 2 (the decrease in the observed absorbance is only attributed to adsorption on the catalyst surface rather than hydrolysis of 2 as is illustrated by the control experiments presented in Fig. S5†).
Moreover, after 5 days, both catalysts appeared blue in colour, implying that adsorption of the probe molecule onto the acidic sites of the Nb2O5·nH2O catalysts is integral in the catalytic ring-opening process. The more rapid decline in the absorption of 2 in the presence of T-IIIa may be simply due to the mesoporous nature of this material and the more rapid and favourable adsorption of the probe molecule into the support matrix.
Results similar to T-IIIa were obtained for the remaining three Nb2O5·nH2O solids (T-II, T-IIIb and T-IV) and are presented in Fig. S4.† In all cases, the solid catalyst turned blue after 5 days of reaction and the absorption spectra attributed to ring-opened oxazine 2 decreased as a function of monitoring time, again suggesting that adsorption of the probe onto the catalyst surface may constitute a fundamental step in the catalysis pathway.
The formation of the emissive isomer 2 was also monitored via fluorescence spectroscopy. An appearance of fluorescence at 645 nm following the addition of TFA (Fig. S6A†) resulted from protonation of compound 1 to generate 2, which continued to form over the timescale of the experiment (4 days). Fig. 6 presents the emission spectra obtained when commercial Nb2O5·nH2O, T-I and T-IIIa were added as heterogeneous Brønsted acid catalysts. These trends closely mimic those observed for UV-visible spectroscopy, where no changes in the fluorescence spectra were observed when the commercial material was used (over a monitoring period of 4 days). On the other hand, an emission band at 645 nm was detected after 1.5 h following addition of both T-I and T-IIIa. The emission at 645 nm slowly increased as a function of time in the presence of T-I. On the contrary, rapid protonation of 1, followed by an ensuing decrease in intensity of the 645 nm emission over 4 days, was observed for catalyst T-IIIa.
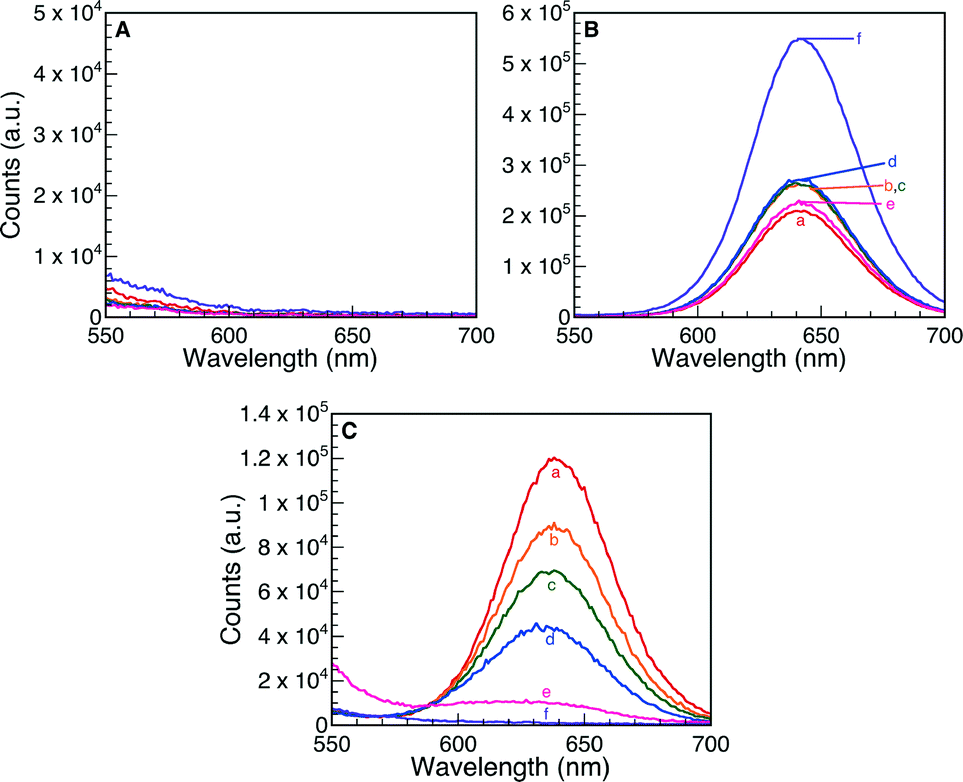 | ||
| Fig. 6 Fluorescence spectra monitoring the Brønsted acid ring opening of 1 to 2 using (A) commercial Nb2O5·nH2O, (B) T-I and (C) T-IIIa as heterogeneous catalysts. The reactions were monitored as a function of time: (a) 1 h, (b) 3 h, (c) 4 h, (d) 6 h, (e) 30 h and (f) 4 d. Note changes in vertical scale in the various panels. | ||
Fig. S6† presents the remaining fluorescence spectra for Nb2O5·nH2O T-II, T-IIIb and T-IV. In all cases, protonation of 1 manifests as the growth of a fluorescence band at 645 nm due to formation of compound 2; however, the emission progressively decreases over 4 days, likely due to adsorption of ring-opened oxazine 2 onto or into the support matrix.
As previously discussed, the change in colour of the synthetically-prepared Nb2O5·nH2O catalysts, in conjunction with the observed variations in both the UV-visible and fluorescence spectra over time, seems to indicate a permanent adsorption of the probe onto the support surface. The rate at which this process occurs was found to be dependent upon the catalyst used, where the mesoporous Nb2O5·nH2O (T-IIIa, T-IIIb and T-IV) demonstrated more rapid adsorption, likely due to incorporation of the molecule into the porous structure. In order to further investigate this process, time-lapsed 1H NMR was performed on a solution of 1 following the addition of T-IIIa in CD3CN. Although the solution coloured instantaneously, the 1H NMR spectrum (Fig. S8a, ESI†) shows broadened peaks that can be only attributed to the starting material 1 (Fig. S7†), and not to its protonated form 2 (chemical shifts from 2 can be observed by adding 10 equivalents of TFA to a solution of 1 in CD3CN, Fig. S7†). The spectra in Fig. S8† represent only the 8.5 to 4 ppm region of the NMR to allow for a more specific comparison of the peaks that are relevant to this discussion. Further confirmation of the exclusive presence of 1 is provided by the analysis of the diastereotopic protons Hy and Hz, which are known to shift from 4.7 ppm in 1 to 5.8 ppm in 2 (Fig. S7 and S8†). In addition, the signals decrease in intensity and lose definition over time (Fig. S8b–d), in agreement with the previous observation of increasing colouration of the solid material. The solid catalyst was separated by centrifugation and analyzed by diffuse reflectance and fluorescence spectroscopy; the corresponding spectra (Fig. S9†) illustrate the presence of a broad band centered at ~630 nm and a broad emission centered at 720 nm, respectively, further confirming the adsorption of compound 2 onto the support surface. Broadening of absorption and emission curves when measured in the solid state is commonly observed due to differing dielectric constants of the solvent and solid state media.19
In order to test the proposal that the halochromic probe was, in part, incorporated into the mesopores of T-IIIa (as suggested by absorption and emission spectroscopy), time-controlled 1H NMR analysis of a mixture of 1 and the non-mesoporous T-I was also carried out for comparison. In the case of T-I, broad signals were also detected that can be attributed to 1, together with the progressive diminution of their intensity (Fig. S10†) and the contemporaneous colouration of the solid catalyst. However, no noteworthy variations in the NMR spectrum could be observed 2 h after catalyst addition (Fig. S10b†), while 1H signals are already significantly different from the initial recordings (Fig. S8b†) 2 h after the addition of T-IIIa. These results confirm that compound 1 adsorbs differently on the catalyst surface depending on the type of Nb2O5·nH2O used, as was also observed during spectroscopy measurements (Fig. 5, 6, S4 and S6†). Furthermore, measurements performed using T-IIIa show that the chemical shifts remained constant for the duration of the experiment (up to 96 hours, with signals that are barely visible), whereas after 4 days from the initial addition of T-I to 1, the 1H NMR peaks shift to resemble those of 2 (Fig. S10c;† as compared with the TFA control). This comparative study suggests that in the case of T-IIIa, product 2 has likely been incorporated into the pores of the material.
Based on these observations, compound 1 is believed to be initially adsorbed onto the surface of the catalyst (due to the total absence of 1H NMR signals from 2) and, once adsorbed, to undergo the proposed Brønsted acid catalyzed ring-opening reaction directly on the surface to generate 2. The instantaneous purple/blue colouration of the solution when the niobic acid catalysts are added to 1 is attributed to an initial stage of the reaction, where the blue colouration is likely the result of a combined effect that includes the reaction of molecules in close proximity to the most exposed acidic sites on the catalyst surface, and the large absorption extinction coefficient of product 2.9
Recyclablity of the Nb2O5·nH2O catalysts was also briefly examined using catalyst T-I as the probe. This particular composite was chosen due to its lack of mesoporosity, thus eliminating consideration of substrate adsorption into the pores of the material. As is shown in Fig. S11,† the amount of ring-opened product 2 obtained following one reuse of the material suggests good catalyst reusability and minimal loss of the Nb2O5·nH2O acidic properties.
Catalysis at the single molecule level
The catalytic protonation of compound 1 (and ring opening) over T-I was investigated by allowing the exposure of the particles to the reagent solution while monitoring the fluorescence signal of the product in a TIRF microscope (Fig. S12†). T-I dispersed in methanol was deposited on a cleaned cover glass by spin coating and then mounted at the bottom of a 1.25 mL reaction chamber. The particles were immersed in 1 mL of Milli-Q water. Upon addition of 2 μL of a 100 nM CH3CN solution of compound 1, bright fluorescent spots appeared over the particles (Fig. 7). Catalytic sites over the particles were reconstructed by accumulating the spot intensity of a movie of 500 frames in length (frame time = 100 ms). The 3D representation of the active sites (Fig. 7, right) shows that events are detected all over the particle without having a preferred position. A fraction of the sites on the catalyst surface seems to be responsible for the activity. | ||
| Fig. 7 Transmission (left) and TIRF (center) images of the same T-I particle immobilized on a cover glass and immersed in a 0.2 nM solution of compound 1 with 633 nm laser excitation (see ESI† Video S1). (Right) 3D representation of the accumulated spot intensity over the same particle reconstructed from a movie of 500 frames in length (frame time = 100 ms). | ||
Typical trajectories at selected bright spots are shown in Fig. 8A. Characterization of bright spots was performed by measuring the emission spectra in situ (Fig. 8C), ascribed to product 2. No fluorescent events were observed in control experiments performed without catalyst or without reagent or in the surrounding solution when compound 1 was added, indicating that the formation of the fluorescent product 2 can only be attributed to the Brønsted acidity of the T-I catalyst. Counting the single “turn ON” events of fluorescent molecules over the catalyst allows for an estimation of the reaction rate constant for the Brønsted acid-catalyzed ring opening of compound 1 and was found to be ~1.8 × 10−13 mol m−2 s−1, as illustrated in Fig. 8B.
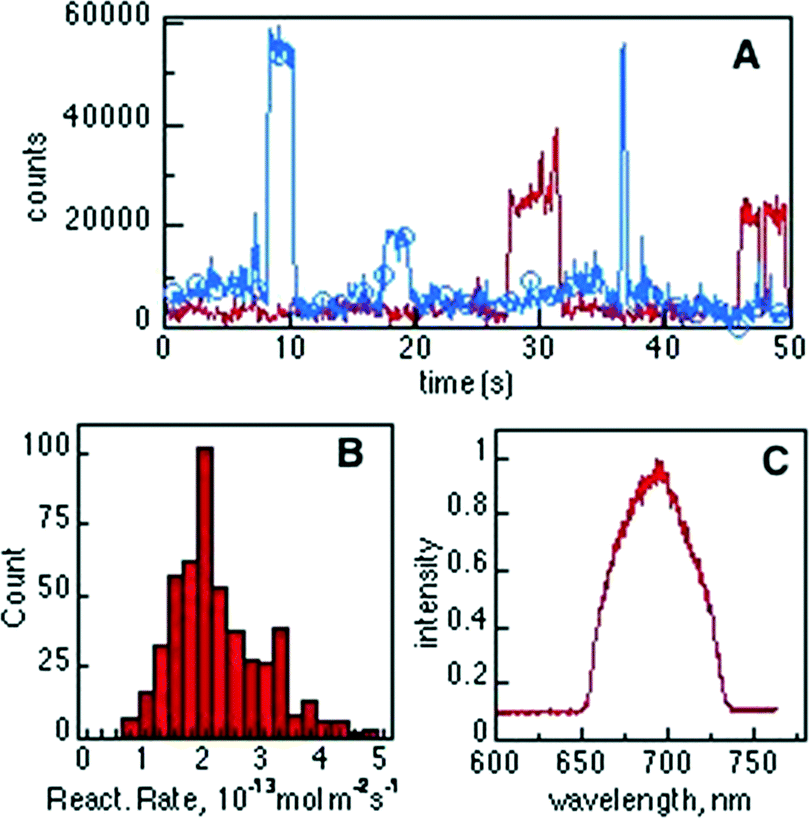 | ||
| Fig. 8 (A) Typical fluorescence intensity trajectories observed for the “turn ON” of molecules of product 2 over a single T-I particle. (B) Distribution of initial reaction rates for the Brønsted acid-catalyzed transformation of compound 1 into 2 (n = 500) and (C) spectral information of the detected bright spots during “turn ON” events measured by passing the epi fluorescent signal through a spectrograph. | ||
The diffusion coefficient of a small molecule in water, for example of fluorescein, is in the order of 5 × 10−6 cm2 s−1,20 thus, within the TIRF excitation region (~200 nm) the diffusion time is on the μs timescale, being much shorter than the 100 ms imaging frame of our experiments. However, the ON time distribution histogram (not shown) shows an average ON time of around 2.5 s. This observation leads to the assumption that molecules of product 2 reside over the Nb2O5 catalyst sites and are observable before photobleaching occurs.
Deactivation of catalyst activity is an important concern for industry applications of any type of catalytic reaction and therefore using microscopic techniques for analyzing adsorption of the product is of wide importance.
In order to establish how robust the catalytic sites are, we carried out a study in which a sequence of 500 images was treated as in Fig. 7 (right), but accumulating separately the first 250 images from the subsequent set of 250 images. This corresponds to two sets of 25 seconds each as shown in Fig. 9, for a data set different from that in Fig. 7 (maps for the spot in Fig. 7 are available in Fig. S13†). Subtraction of the two sets gives the image shown in Fig. 9C and from a side-view in Fig. 9D, where positive signals represent loss of activity and negative signals activity gained. We were initially puzzled by the latter observation, as we did not expect additional acid sites to appear or be released with time. Interestingly, statistics calculated from Fig. 9D show that the average of all peaks is approximately zero within experimental error; this means that active sites that become inactive are replaced by new sites nearby. As we are using diffraction-limited optical methods, this effectively means new sites are ~300 nm or more from sites losing activity. Sites that appear at the same location will not be counted in this method as their bright blinks simply replace the previous ones. Visual analysis of enlarged images suggest that new sites are not very far from old ones. The simplest interpretation is that early acidity maps reveal hot spots on the catalyst, where ‘hot spots’ are characterized by their high acidity and easy accessibility; as these are used up by the chemistry of Fig. 3, the next tier of sites becomes active, effectively appearing in these maps as if acid sites had been created, when in fact they were available all along, they simply become active as their better competitors are used. We find that the time-lapse maps of Fig. 9 are a convenient representation of what otherwise can only be examined from TIRF videos.
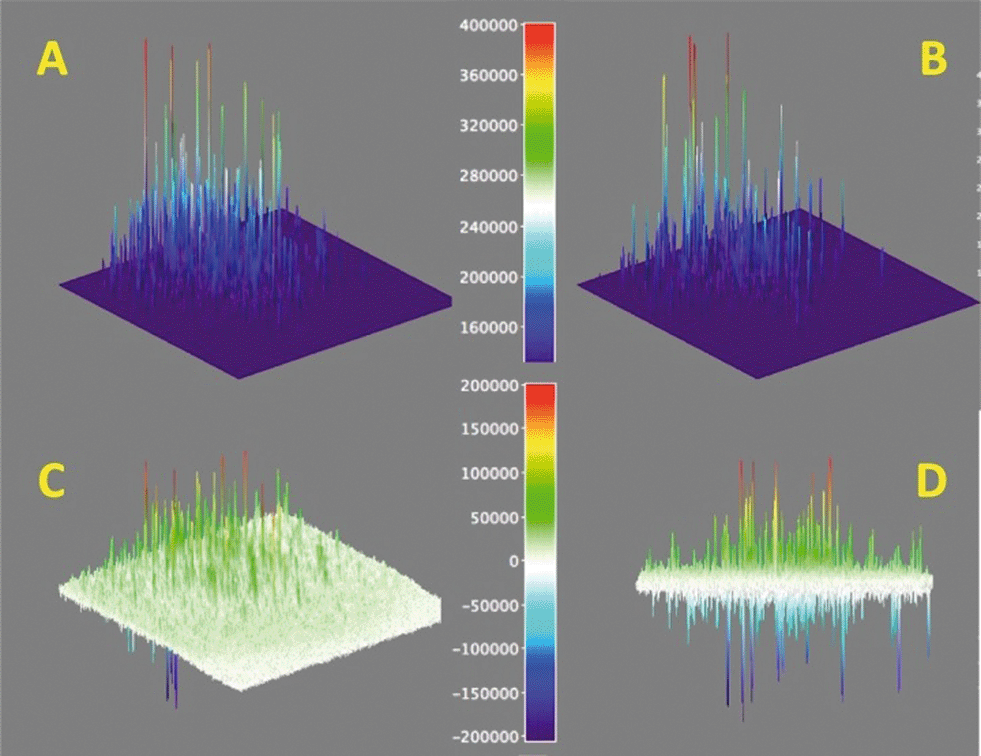 | ||
| Fig. 9 Protonation activity as shown by the conversion of 1 → 2 and detection of the fluorescence from 2. Surface maps A and B show the accumulated signals for 25 seconds and the subsequent 25 seconds, while map C shows the difference of A minus B expanded approximately ×2 (see colour coded scale). Panel D shows a different view of panel C, showing positive and negative peaks. Note that this figure is for a different spot than that in Fig. 7. | ||
Conclusions
Five Nb2O5·nH2O materials have been synthesized using a combination of both literature and newly developed synthetic procedures. These materials were determined to be amorphous in nature by way of XRD and TEM imaging, while N2 adsorption studies confirmed the mesoporous characteristics of samples T-III and T-IV. Acid indicator studies employing the Hammett acidity function of all the samples discussed within this contribution confirmed the acidic nature of the five synthesized Nb2O5·nH2O materials. Surprisingly, the commercial Nb2O5 was found to be void of any acidic characteristics. Commercial Nb2O5 and Nb2O5·nH2O T-I–T-IV were tested in the halochromic ring opening of oxazine-coumarin 1. The prepared Nb2O5·nH2O derivatives rapidly converted oxazine-coumarin 1 to the open-form 2, as was observed in both UV-visible and fluorescence spectroscopy, while commercial Nb2O5 was found to be inactive in this reaction, likely due to its lack of acidic properties. Through NMR spectroscopy, the ring-opening of 1 was suggested to require adsorption of the closed form 1 onto the material surface to facilitate effective substrate protonation and form the Brønsted acid catalyzed ring opened product 2. Total internal reflectance microscopy was used to monitor the acid induced ring opening of 1 with single molecule resolution. The rate constant for the Brønsted acid catalyzed coumarin-spyrooxazine ring opening was estimated to be 1.8 × 10−13 mol m−2 s−1. Moreover, using single molecule techniques, it was determined that the adsorption of substrate 1 and subsequent ring opening is distributed over the Nb2O5·nH2O surface, with the emissive open form 2 exhibiting an average ON time of 2.5 s. The reaction is suggested to proceed with a gradual exchange of the initial ‘hot spots’, characterized by high acidity and easy accessibility, for the next tier of acid sites as the most reactive locations are depleted.Acknowledgements
The authors wish to thank the Natural Sciences and Engineering Research Council (NSERC) and the Canada Research Chairs program. The University of Ottawa International Office provided grants to study niobium. M. L. Marin thanks the financial support of the Generalitat Valenciana (BEST/2012/233). Thanks are due to the Government of Canada and NSERC for a Banting Postdoctoral Fellowship to S. Impellizzeri and a Vanier Sholarship to C. Fasciani. S. Simoncelli acknowledges a DFAIT fellowship from ELAP (Emerging Leaders in the Americas Program) to support her visit to Canada.Notes and references
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS PubMed.
- K. Tanabe and S. Okazaki, Appl. Catal., A, 1995, 133, 191–218 CrossRef CAS.
- T. Armaroli, G. Busca, C. Carlini, M. Giuttari, A. M. R. Galletti and G. Sbrana, J. Mol. Catal. A: Chem., 2000, 151, 233–243 CrossRef CAS; C. Carlini, M. Giuttari, A. M. R. Galletti, G. Sbrana, T. Armaroli and G. Busca, Appl. Catal., A, 1999, 183, 295–302 CrossRef.
- G. S. Nair, E. Adrijanto, A. Alsalme, I. V. Kozhevnikov, D. J. Cooke, D. R. Brown and N. R. Shiju, Catal. Sci. Technol., 2012, 2, 1173–1179 Search PubMed; M. Marzo, A. Gervasini and P. Carniti, Carbohydr. Res., 2012, 347, 23–31 CrossRef CAS PubMed.
- E. I. Ko and J. G. Weissman, Catal. Today, 1990, 8, 27–36 CrossRef CAS.
- S. Okazaki and A. Kurosaki, Catal. Today, 1990, 8, 113–122 CrossRef CAS; S. Okazaki and N. Wada, Catal. Today, 1993, 16, 349–359 CrossRef.
- T. Ushikubo, I. Iizuka, H. Hattori and K. Tanabe, Catal. Today, 1993, 16, 291–295 CrossRef CAS.
- E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 2010, 1, 3506–3509 CrossRef CAS.
- N. Uekawa, T. Kudo, F. Mori, Y. J. Wu and K. Kakegawa, J. Colloid Interface Sci., 2003, 264, 378–384 CrossRef CAS.
- A. I. Carrillo, J. García-Martínez, R. Llusar, E. Serrano, I. Sorribes, C. Vicent and J. Alejandro Vidal-Moya, Microporous Mesoporous Mater., 2012, 151, 380–389 CrossRef CAS PubMed.
- Y. Rao, M. Trudeau and D. Antonelli, J. Am. Chem. Soc., 2006, 128, 13996–13997 CrossRef CAS PubMed.
- M. Yurdakoc, M. Akcay, Y. Tonbul and K. Yurdakoc, Turk. J. Chem., 1999, 23, 319–327 CAS; I. Matsuzaki, M. Nitta and K. Tanabe, J. Res. Inst. Catal., Hokkaido Univ., 1969, 17, 46–53 Search PubMed.
- V. Meynen, P. Cool and E. F. Vansant, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS PubMed.
- M. Yurdakoc, M. Akcay, Y. Tonbul and K. Yurdakoc, Turk. J. Chem., 1999, 23, 319–327 CAS.
- M. Tomasulo, S. Sortino, A. J. P. White and F. M. Raymo, J. Org. Chem., 2005, 70, 8180–8189 CrossRef CAS PubMed; M. Tomasulo, S. Sortino and F. M. Raymo, Org. Lett., 2005, 7, 1109–1112 CrossRef PubMed.
- M. Petriella, E. Deniz, S. Swaminathan, M. J. Roberti, F. M. Raymo and M. L. Bossi, Photochem. Photobiol., 2013, 89, 1391–1398 CrossRef CAS PubMed; S. Swaminathan, M. Petriella, E. Deniz, J. Cusido, J. D. Baker, M. L. Bossi and F. M. Raymo, J. Phys. Chem. A, 2012, 116, 9928–9923 CrossRef PubMed; E. Deniz, M. Tomasulo, J. Cusido, I. Yildiz, M. Petriella, M. L. Bossi, S. Salvatore and F. M. Raymo, J. Phys. Chem. C, 2012, 116, 6058–6068 Search PubMed.
- S. Corrent, P. Hahn, G. Pohlers, T. J. Connolly, J. C. Scaiano, V. Fornés and H. García, J. Phys. Chem. B, 1998, 102, 5852–5858 CrossRef CAS.
- G. L. Hallett-Tapley, C. O. L. Crites, M. Gonzalez-Bejar, K. L. McGilvray, J. C. Netto-Ferreira and J. C. Scaiano, J. Photochem. Photobiol., A, 2011, 224, 8–15 CrossRef CAS PubMed; D. A. Skoog, F. J. Holler and T. A. Nieman, Principles of Instrumental Analysis, Harcourt Brace & Company, Orlando, FL, 5th edn, 1998 Search PubMed.
- B. Fu, F. E. Curry and S. Weinbaum, Am. J. Physiol., 1995, 269, H2124–H2140 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of commercial Nb2O5·nH2O, T-I, T-II, T-IIIa, T-IIIb and T-IV; XRD spectra of T-II, T-IIIa, T-IIIb and T-IV; pore size and surface area data as determined by nitrogen adsorption isotherms; nitrogen adsorption isotherms of commercial Nb2O5·nH2O, T-IIIa, T-IIIb and T-IV; UV-visible spectra monitoring the Brønsted acid ring opening of 1 to 2 using TFA, T-II, T-IIIb and T-IV as heterogeneous catalysts; fluorescence spectra monitoring the Brønsted acid ring opening of 1 to 2 using TFA, T-II, T-IIIb and T-IV as heterogeneous catalysts; 1H NMR spectra and peak attribution of 1 and 2 in CD3CN; 1H NMR spectra of 1 in CD3CN after the addition of T-IIIa; diffuse reflectance and solid state fluorescence spectra of product 2 adsorbed onto T-IIIa; 1H NMR spectra of 1 in CD3CN after the addition of T-I; experimental set up for single-molecule fluorescence observation under TIRF illumination for the Brønsted acid-catalyzed isomerization of 1 to 2. See DOI: 10.1039/c4cy00238e |
| This journal is © The Royal Society of Chemistry 2014 |