NaF regulated aqueous phase synthesis of aromatic amides and imines catalyzed by Au/HT†
Qianqian
Wang
ab,
Youquan
Deng
a and
Feng
Shi
*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Center for Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: fshi@licp.cas.cn; Fax: +86 931 8277088; Tel: +86 931 4968142
bGraduate School of the Chinese Academy of Sciences, Beijing, 100049, China
First published on 17th March 2014
Abstract
An Au/HT catalyst was found to be an efficient heterogeneous catalyst for the coupling reaction of aromatic alcohols and amines. The amides and imines can be selectively synthesized with up to 99% yield with or without the addition of NaF as the cocatalyst.
Nitrogen-containing compounds such as amides and imines play a key role in organic and biological chemistry.1 Generally, the alkyl amides are produced through the reaction of amides and alkyl halides with the addition of stoichiometric amounts of inorganic bases or by the reaction of a carboxylic acid or ester and an amine.2–5 These methods often suffer from drawbacks such as the generation of inorganic waste.6 For the production of imines, the condensation of amines with aldehydes or ketones is an effective way.7 However, this method is usually limited by the use of dehydrating agents or apparatus. In the last years, the direct synthesis of amides and imines via oxidative coupling of alcohols with amines has developed rapidly due to its high atom efficiency.5,8–12 This procedure involves the following steps. First, the alcohol is converted into the corresponding aldehyde by catalytic oxidation. Afterwards, the hemiaminal is generated via the coupling of amine and aldehyde. Finally, the hemiaminal is converted to amides or imines.
In 2007, Milstein reported the transformation of alcohols and amines into amides and hydrogen catalyzed by a ruthenium pincer complex.13 Subsequently, a series of Ru catalyst systems were developed.14,15 On the other hand, homogeneous catalysts show high activity for imine formation from alcohols and amines, too.16,17 Although homogeneous catalysts are very efficient for generating amides or imines selectively, there are disadvantages such as ligand dependence, complicated operation and non-reusability.16 A more efficient and feasible heterogeneous catalyst for the coupling reaction of alcohols and amines is highly desirable.18 In 2009, Satsuma and co-workers reported a heterogeneous Ag/Al2O3 catalyst, which can catalyze the amide synthesis from secondary amines and alcohols with a good yield, and the catalyst can be recycled.17 Since then, Au/DNA,19 PICB-Au,20 OMS-2,21 Au/PVP,22 Au/HT,23 Au–Pd/resin24 and Au/HAP25 have been reported. Although there are gratifying achievements in the direct coupling of amines and alcohols catalyzed by heterogeneous catalysts, one limitation is the low yield when aromatic amines and aromatic alcohols are used as starting materials. In the existing literature reports, normally a high alcohol/amine ratio is needed for the coupling reactions of alcohols and amines to form substituted amides in heterogeneous catalyst systems.
Hydrotalcite, especially Mg–Al hydrotalcite, is widely used as a catalyst in different reactions.26,27 In addition hydrotalcite supported nanometal catalysts have gradually played an important role in the field of catalysis, such as oxidation of alcohols in the presence of oxygen.28,29 Here, we report an Au/HT catalyst system, which was prepared by a precipitation–deposition–reduction method, for the synthesis of alkyl amides and imines. Importantly, alkyl amides and imines can be selectively generated by tuning the catalyst systems with or without the addition of NaF as the cocatalyst.
In this work, water was chosen as the reaction medium because it is considered as a “green solvent” in organic chemistry, and it has attracted extensive attention in the last years.30,31 Additionally, since the first report on the insoluble Diels–Alder reactions using water as a solvent,31 “on-water chemistry” has also been investigated.32–34 In the current work, the synthesis of amides or imines might be an in-water or on-water organic reaction.
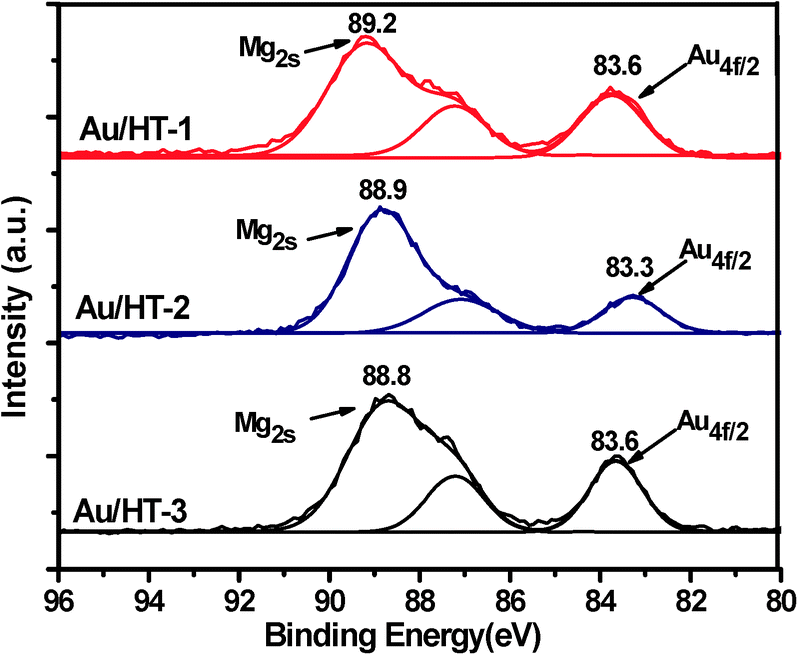
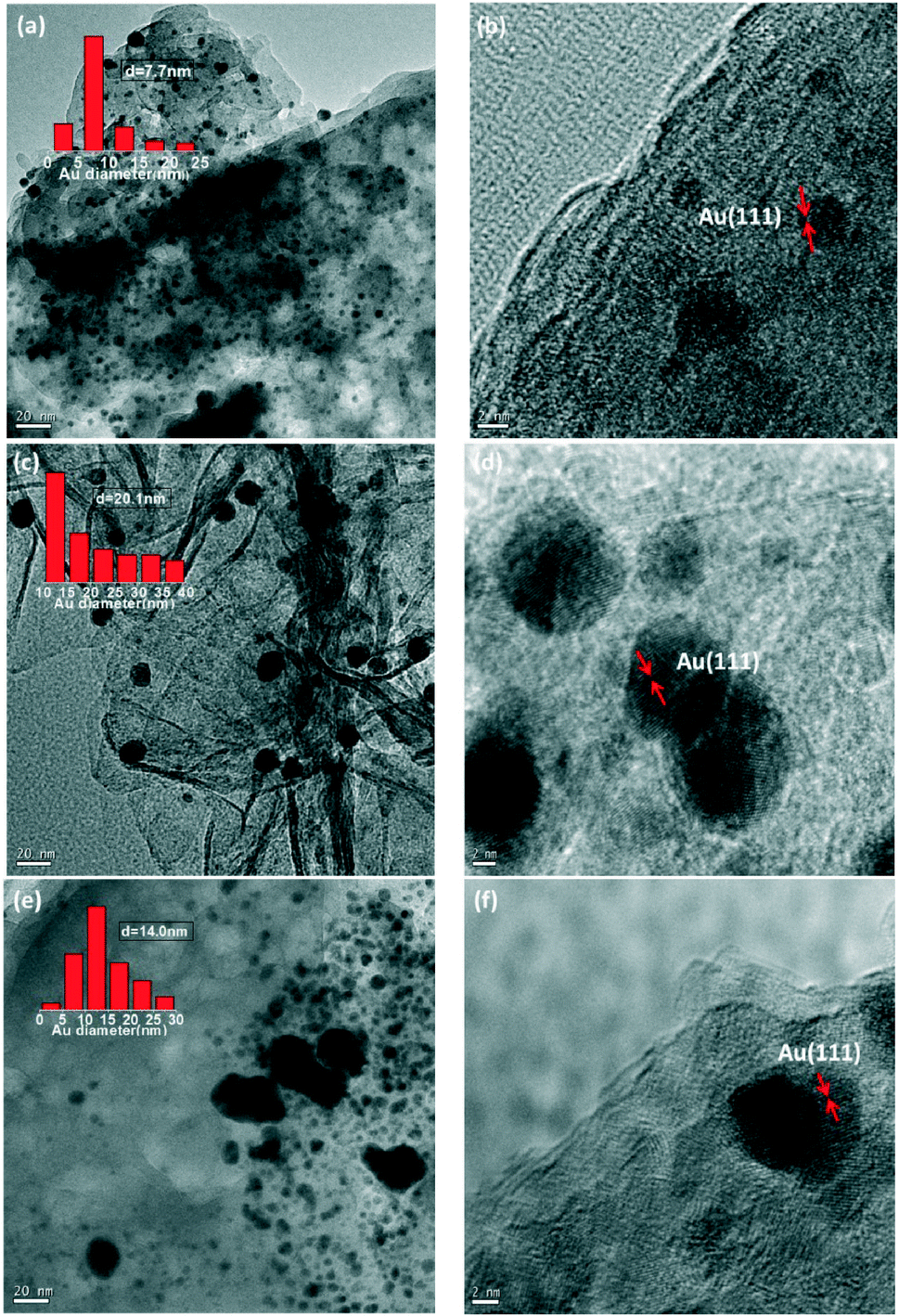
The structure and physicochemical properties of Au/HT-1 (Mg/Al = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), Au/HT-2 (Mg/Al = 3:1) and Au/HT-3 (Mg:Al = 5:1) catalysts were characterized by XRD, XPS, TEM, BET and ICP-AES. From the BET analysis (Table S1†), Au/HT-2 exhibited the largest BET area accompanied by the smallest pore size and pore volume compared with Au/HT-1 and Au/HT-3. In addition, an ordered mesoporous structure was formed in Au/HT-2, which may have contributed to the high surface area and is beneficial for the preparation of the active catalyst. According to the XRD diffraction patterns (Fig. 1), the typical diffraction peaks at 11.7, 23.5, 34.7, and 46.4, which are related to the (003), (006), (009) and (018) reflections of hydrotalcite, were observable. Another featured peak at 38.4 is related to the crystal lattice of Au (111). Noteworthy, the Au (111) diffraction peak in sample Au/HT-2 was much weaker, which suggests that the gold species in Au/HT-2 are less crystallized. X-ray photoelectron spectroscopy (XPS) was used to clarify the chemical state of the gold species formed on the catalyst surface (Fig. 2). It can be seen that the binding energies of Au4f7/2 for Au/HT-1, Au/HT-2 and Au/HT-3 are 83.6, 83.3 and 83.6 eV, respectively. Another peak is attributed to Mg2s and the binding energies are 89.2, 88.9 and 88.8 eV. Therefore, metallic gold formed on the catalyst surface. TEM images showed that the structure of the catalysts comprises nano-Au particles anchored on the surface of hydrotalcite (Fig. 3). It can be seen that all of the supported nano-Au particles are evenly distributed, and the average particle sizes are 7.7, 20.1 and 14.0 nm. So the properties of the supports influence the structure of the final catalyst samples remarkably. ICP-AES analysis showed that the loadings of gold in these catalysts are 3.4 wt%, 3.7 wt% and 3.4 wt%.
:1), Au/HT-2 (Mg/Al = 3:1) and Au/HT-3 (Mg:Al = 5:1) catalysts were characterized by XRD, XPS, TEM, BET and ICP-AES. From the BET analysis (Table S1†), Au/HT-2 exhibited the largest BET area accompanied by the smallest pore size and pore volume compared with Au/HT-1 and Au/HT-3. In addition, an ordered mesoporous structure was formed in Au/HT-2, which may have contributed to the high surface area and is beneficial for the preparation of the active catalyst. According to the XRD diffraction patterns (Fig. 1), the typical diffraction peaks at 11.7, 23.5, 34.7, and 46.4, which are related to the (003), (006), (009) and (018) reflections of hydrotalcite, were observable. Another featured peak at 38.4 is related to the crystal lattice of Au (111). Noteworthy, the Au (111) diffraction peak in sample Au/HT-2 was much weaker, which suggests that the gold species in Au/HT-2 are less crystallized. X-ray photoelectron spectroscopy (XPS) was used to clarify the chemical state of the gold species formed on the catalyst surface (Fig. 2). It can be seen that the binding energies of Au4f7/2 for Au/HT-1, Au/HT-2 and Au/HT-3 are 83.6, 83.3 and 83.6 eV, respectively. Another peak is attributed to Mg2s and the binding energies are 89.2, 88.9 and 88.8 eV. Therefore, metallic gold formed on the catalyst surface. TEM images showed that the structure of the catalysts comprises nano-Au particles anchored on the surface of hydrotalcite (Fig. 3). It can be seen that all of the supported nano-Au particles are evenly distributed, and the average particle sizes are 7.7, 20.1 and 14.0 nm. So the properties of the supports influence the structure of the final catalyst samples remarkably. ICP-AES analysis showed that the loadings of gold in these catalysts are 3.4 wt%, 3.7 wt% and 3.4 wt%.
 | ||
| Fig. 1 XRD diffraction patterns of the prepared catalysts. | ||
 | ||
| Fig. 2 XPS spectra of the prepared catalysts. | ||
 | ||
| Fig. 3 TEM pictures of the prepared catalysts Au/HT-1 (a, b), Au/HT-2 (c, d) and Au/HT-3 (e, f). | ||
Next, we chose the amidation of benzylamine and benzyl alcohol as the model reaction to optimize the reaction conditions (alcohol:amine = 1.5, mol/mol). As given in (Table 1), N-phenylbenzamide is not obtained when using Au/HT-2 as the catalyst and the major product is N-benzylideneaniline (entry 1). This result is similar to a previous report showing that alcohol imination can be realized with Au/Mg2Al–HT as the catalyst and with molecular oxygen as the oxidant.35 It has been shown that amides can be synthesized efficiently with the addition of Cs2CO3.17 However, our reaction was not significantly improved and the selectivity to amide product was only 37% (entry 2). Other bases such as NaOH and Na2CO3 were also tested and the selectivity increased to 96% and 84% but the conversions of aniline were not high enough (entries 3, 4). Interestingly, the conversion and selectivity increased remarkably when NaF was used as the additive and 95% aniline conversion was obtained (entry 5). With the addition of KF and NaBF4 as cocatalysts, the amidation of aniline with benzyl alcohol can be carried out as well (entries 6, 7). The coupling reaction did not occur when NaCl was used as an additive which indicates that the reaction is promoted by fluoride (entry 8). When the ratios of Mg to Al of the catalyst support were changed to 1:1 and 5:1, the conversions and selectivities of the amidation reaction decreased remarkably (entries 9, 10). Therefore, 3:1 is a suitable Mg/Al ratio. In order to explore the catalytic behavior of leached catalyst species, the catalyst was removed by filtration after it reacted for 12 h and the aniline conversion was maintained at 77% after reaction for another 12 h. Therefore, the real catalyst is the supported nano-Au. Additionally, only 50% amide was formed when the catalyst was reused for the second run. ICP-AES analysis showed that the Au loading was 3.1 wt% after use. Possibly the catalyst structure variation but not Au leaching is responsible for the deactivation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Additive | Con. % of 1b | Sel. % of 3b | Sel.% of 4b |
| a Aniline (0.5 mmol), benzyl alcohol (0.75 mmol), Au/HT (50 mg), additive (2 mmol), 2 mL H2O, O2, 40 °C, 12 h. b The conversion and selectivity were detected by GC-FID according to the peak area. c Cs2CO3: 20 mol%. d The catalyst was reused in the second run. | |||||
| 1 | Au/HT-2 | — | 80 | 16 | 84 |
| 2c | Au/HT-2 | Cs2CO3 | 79 | 36 | 74 |
| 3 | Au/HT-2 | NaOH | 45 | 96 | 4 |
| 4 | Au/HT-2 | Na2CO3 | 87 | 84 | 6 |
| 5 | Au/HT-2 | NaF | 95 | 98 | 2 |
| 6 | Au/HT-2 | KF·2H2O | 90 | 95 | 5 |
| 7 | Au/HT-2 | NaBF4 | 83 | 86 | 14 |
| 8 | Au/HT-2 | NaCl | 9 | 3 | 97 |
| 9 | Au/HT-1 | NaF | 69 | 82 | 18 |
| 10 | Au/HT-3 | NaF | 76 | 66 | 34 |
| 11d | Au/HT-2 | NaF | 95 | 50 | 50 |

Then, the generality of the amidation reaction of amines and alcohols was investigated under the optimized reaction conditions and the results are summarized in Table 2. First, N-phenylbenzamide was synthesized with 86% yield (entry 1). Then, we investigated the scope of the amidation of benzyl alcohol and anilines containing different functional groups. Clearly, the catalyst can catalyze the amidation reactions of various aromatic amines, which contain electron-donating as well as electron-withdrawing substituents, to synthesize the corresponding amides with moderate to good isolated yields (46%–81%, entries 2–7). However, a lower yield was obtained when o-toluidine was used as the starting material, which might be caused by steric hindrance. Upon using benzyl alcohol derivatives as starting materials, the catalyst showed good catalytic activity with 47–78% isolated yields of the desired products (entries 8–16). Further, alcohols with electron-donating substituents such as methyl, methoxyl and isopropyl were more favorable for the amidation reactions than those with electron-withdrawing substituents such as chlorine, bromine, fluorine and trifluoromethyl. Similarly, the catalysts exhibited lower activity in the amidation of o-methylbenzyl alcohol compared with m-methylbenzyl alcohol and p-methylbenzyl alcohol (entries 8–10). 4-(Trifluoromethyl)benzyl alcohol and 3,4-(methylenedioxy)aniline were converted into the corresponding amides with 50% and 47% isolated yields (entries 16 and 17).


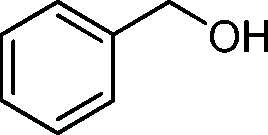
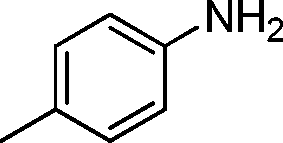






















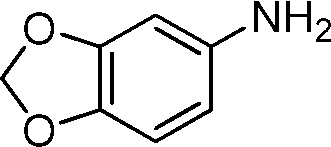
During the optimization of the reaction conditions for the amidation reaction, we found that benzyl alcohol and aniline can be converted into N-benzylideneaniline in the absence of NaF (Table 1, entry 1). It inspired us that our catalyst might be suitable for the imination of amines and alcohols without the addition of NaF. As expected, the anilines with various substituents such as methyl, methoxyl, chlorine, and bromine were converted into the corresponding imines with benzyl alcohol (Table 3, entries 1–9). Moreover, the electron-donating substituents on the aromatic ring are slightly favorable for the imination reaction compared with the electron-withdrawing substituents while steric effects have a negative influence. 80% yield was obtained when 2,5-dimethylaniline was used as the starting material (entry 9). In addition, the reactions of anilines with different aromatic alcohols progressed well with 82–97% yields (entries 10–12).















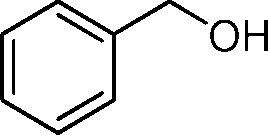

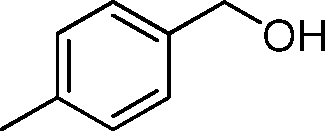
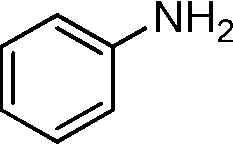

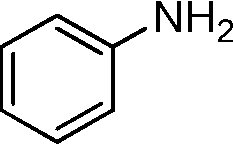

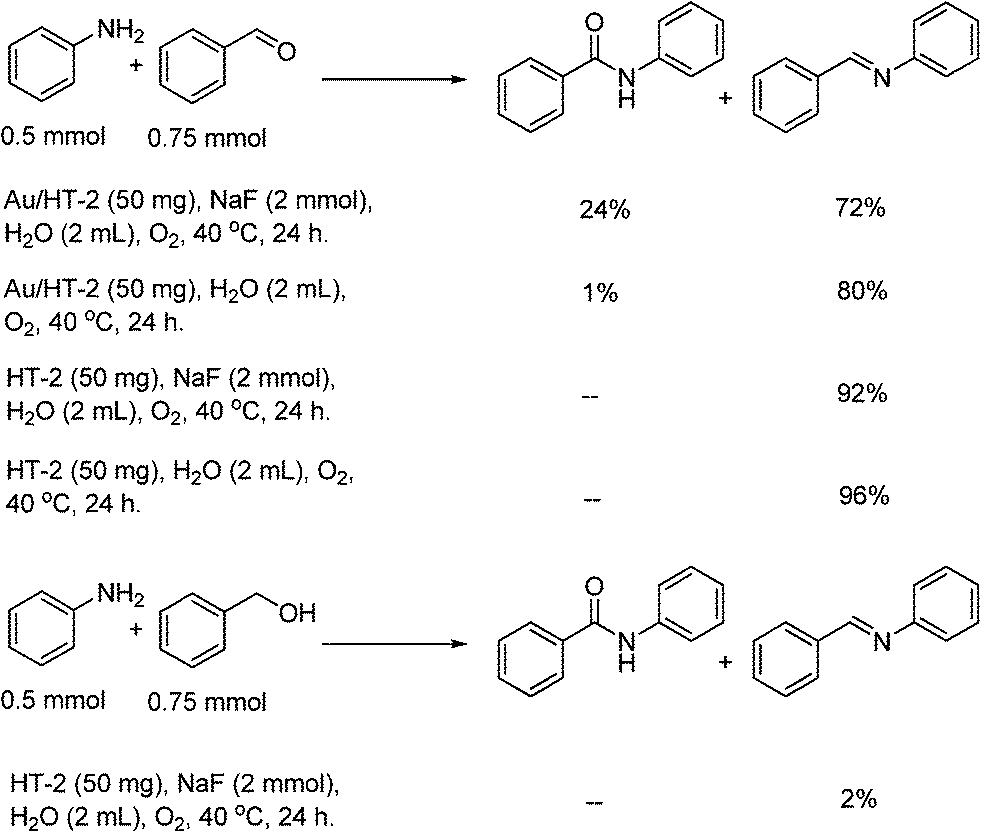
In order to explore the reaction mechanism, several control reactions were used (Scheme 1). Full conversion of aniline was obtained with 24% amide and 72% imine under the given reaction conditions using benzaldehyde as the starting material catalyzed by Au/HT-2 and NaF, and no amidation product was observed when NaF or gold was excluded. Similarly, almost no reaction occurred when benzyl alcohol and aniline were used as starting materials with HT and NaF as catalysts. These results suggest that the co-presence of nano-Au and fluoride is crucial for amide formation. Moreover, the amidation reaction might happen promptly after the formation of hemiaminal on the surface of Au/HT in the presence of fluoride. For the imination reaction, it can be carried out in the absence of fluoride (Scheme 2).
 | ||
| Scheme 1 The mechanism investigation of alcohol amidation and imination with alcohol. | ||
 | ||
| Scheme 2 The proposed reaction mechanism of amidation and imination reactions catalyzed by Au/HT-2 with or without the addition of NaF. | ||
Conclusions
In summary, an active Au/HT catalyst was prepared by a precipitation–deposition–reduction method for the coupling reaction of amines and alcohols. The amidation and imination reactions can be realized efficiently with moderate to good yields with or without the addition of NaF as the co-catalyst. In addition, the mechanism of the reaction was investigated, too.Experimental section
General
All of the solvents and chemicals were obtained commercially and were used as received. Mass spectra were, in general, recorded using an HP 6890/5973 GC-MS. High-resolution TEM analysis was carried out using a JEM 2010 operating at 200 KeV. The catalyst samples after pretreatment were dispersed in methanol, and the solution was mixed ultrasonically at r.t. A part of solution was dropped on the grid for the measurement of TEM images. XRD measurements were conducted by a STADI P automated transmission diffractometer (STOE) equipped with an incident beam curved germanium monochromator selecting Cu Kα1 radiation and a 6° position sensitive detector (PSD). The XRD patterns were scanned in the 2θ range of 10–80°. For the data interpretation the software WinXpow (STOE) and the database of Powder Diffraction File (PDF) of the International Centre for Diffraction Data (ICDD) were used. The XPS measurements were performed using a VG ESCALAB 210 instrument provided with a dual Mg/Al anode X-ray source, a hemispherical capacitor analyser and a 5 keV Ar+ ion gun. All spectra were recorded using non-monochromatic Mg Ka (1253.6 eV) radiation. Nitrogen adsorption–desorption isotherms were measured at 77 K using a Micromeritics 2010 instrument. The pore–size distribution was calculated using the Barrett, Joyner and Halenda (BJH) method from desorption isotherms. The Cu and Al contents of the catalyst were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES) using an Iris advantage Thermo Jarrel Ash device.Typical procedure for HT-2 preparation36
7.69 g (30 mmol) of Mg(NO3)2·6H2O and 3.75 g of (10 mmol) Al(NO3)3·9H2O were dissolved into 100 mL of deionized water. Then the solution was dropwise added into 100 mL of Na2CO3 solution (0.3 mol L−1) after 1 h under magnetic stirring and the pH value of the solution was maintained at 10 by adding aqueous solution of NaOH (1 M). After aging for 1 h at 65 °C, the white solid was filtered and washed with deionized water (2 L). The obtained solid was dried at 100 °C in air overnight and ~3.3 g of HT-2 was obtained. HT-1 and HT-3 were prepared using a similar procedure.General procedure for Au/HT preparation
500 mg of HT was added to 4 mL of HAuCl4 solution (24 mM) under magnetic stirring for 2 min. The pH value of the solution was adjusted to 10 with the addition of aqueous NH3 (~25 wt%), and the mixture was stirred at room temperature for 12 h. Then, it was centrifuged and washed with 100 mL of deionized water. The obtained solid sample was dispersed in 10 mL of deionized water and 20 mL of NaBH4 solution (26 mmol L−1) was added after 1 h under magnetic stirring. The Au/HT catalyst was obtained after centrifugation, washing with deionized water and drying at 80 °C for 1 h.General procedure for the amidation of amines and alcohols
All of the reactions were carried out in a Schlenk tube. Typically, 0.5 mmol of aniline, 0.75 mmol of benzyl alcohol, 50 mg of Au/HT (3.7 wt% Au, 1.9 mol% to aniline) and 2 mmol of NaF were added successively. Then, the reaction mixture was stirred (300 rpm) at 40 °C under 1 atm oxygen (O2 balloon) for 24 h. Then it was cooled down to room temperature. The reaction mixture was analyzed by GC-MS (Agilent 6890–5973)/GC-FID (Agilent 7890A) and 86% yield was obtained by column chromatography (ethyl acetate/petroleum ether = 9/1, v/v; silica gel: 200–300 mesh; white solid; m.p. 162–163 °C). For the reusability test, the catalyst was separated by centrifugation, washed with ethanol and water, and was used for the next run using the same procedure as mentioned above.General procedure for the imination of amines and alcohols
All of the reactions were carried out in a Schlenk tube. Typically, 0.5 mmol of aniline, 0.75 mmol of benzyl alcohol, 50 mg of Au/HT (3.7 wt% Au, 1.9 mol% to aniline) and 2 mmol of NaF were added successively. Then, the reaction mixture was stirred (300 rpm) at 40 °C under 1 atm oxygen (O2 balloon) for 24 h. Then it was cooled down to room temperature and quantitatively analyzed by GC-FID (Agilent 7890A) using biphenyl as the standard material.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21303228).Notes and references
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2008, 109, 259 CrossRef PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20 CAS.
- J. E. Anderson, R. Davis, R. N. Fitzgerald and J. M. Haberman, Synth. Commun., 2006, 36, 2129 CrossRef.
- C. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766 CrossRef CAS PubMed.
- S. Kegns, J. Mielby, U. V. Mentzel, T. Jensen, P. Fristrup and A. Riisager, Chem. Commun., 2012, 48, 2427 RSC.
- T. Ishida, H. Watanabe, T. Takei, A. Hamasaki, M. Tokunaga and M. Haruta, Appl. Catal., A, 2012, 425, 85 CrossRef.
- J. Mielby, A. Riisager, P. Fristrup and S. Kegnaes, Catal. Today, 2013, 203, 211 CrossRef CAS.
- S. K. Klitgaard, K. Egeblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419 RSC.
- X. Y. Liu and K. F. Jensen, Green Chem., 2013, 15, 1538 RSC.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed.
- B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682 CrossRef CAS PubMed.
- J. Zhang, M. Senthilkumar, S. C. Ghosh and S. H. Hong, Angew. Chem., Int. Ed., 2010, 49, 6391 CrossRef CAS PubMed.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem. – Eur. J., 2009, 15, 9977 CrossRef CAS PubMed.
- B. J. Xu, R. J. Madix and C. M. Friend, Chem. – Eur. J., 2012, 18, 2313–2318 CrossRef CAS PubMed.
- Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917 CrossRef CAS PubMed.
- J.-F. Soule, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550 CrossRef CAS PubMed.
- K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544 CrossRef CAS PubMed.
- P. Preedasuriyachai, H. Kitahara, W. Chavasiri and H. Sakurai, Chem. Lett., 2010, 39, 1174 CrossRef CAS.
- J. Zhu, Y. Zhang, F. Shi and Y. Deng, Tetrahedron Lett., 2012, 53, 3178 CrossRef CAS.
- L. L. Zhang, W. T. Wang, A. Q. Wang, Y. T. Cui, X. F. Yang, Y. Q. Huang, X. Y. Liu, W. G. Liu, J. Y. Son, H. S. Oji and T. Zhang, Green Chem., 2013, 15, 2680 RSC.
- W. Wang, Y. Cong, L. Zhang, Y. Huang, X. Wang and T. Zhang, Tetrahedron Lett., 2014, 55, 124 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276 RSC.
- A. Takagaki, K. Iwatani, S. Nishimura and K. Ebitani, Green Chem., 2010, 12, 578 RSC.
- V. V. Costa, M. Estrada, Y. Demidova, I. Prosvirin, V. Kriventsov, R. F. Cotta, S. Fuentes, A. Simakov and E. V. Gusevskaya, J. Catal., 2012, 292, 148 CrossRef CAS.
- K. Ebitani, K. Motokura, T. Mizugaki and K. Kaneda, Angew. Chem., Int. Ed., 2005, 44, 3423 CrossRef CAS PubMed.
- C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC.
- M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24 CrossRef CAS PubMed.
- P. Liu, C. Li and E. J. M. Hensen, Chem. – Eur. J., 2012, 18, 12122 CrossRef CAS PubMed.
- A. Tsuji, K. T. V. Rao, S. Nishimura, A. Takagaki and K. Ebitani, ChemSusChem, 2011, 4, 542 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst preparation procedures and characterization details. See DOI: 10.1039/c4cy00210e |
| This journal is © The Royal Society of Chemistry 2014 |