 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDehydration of xylose and glucose to furan derivatives using bifunctional partially hydroxylated MgF2 catalysts and N2-stripping†
I.
Agirrezabal-Telleria
*a,
Y.
Guo
b,
F.
Hemmann
c,
P. L.
Arias
a and
E.
Kemnitz
b
aDepartment of Chemical and Environmental Engineering, Engineering School of the University of the Basque Country (UPV/EHU), Alameda Urquijo s/n, 48013, Bilbao, Spain. E-mail: iker.aguirrezabal@ehu.es; Fax: +34 946014179; Tel: +34 617912295
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor Straße 2, D-12489, Berlin, Germany
cBAM Federal Institute for Materials Research and Testing, Division 1, Richard Willstaetter Straße 11, D-12489, Berlin, Germany
First published on 11th February 2014
Abstract
The current furfural production yield is low due to the use of non-selective homogeneous catalysts and expensive separation. In this work, partially hydroxylated MgF2 catalysts, synthesized using different water contents, were screened during xylose dehydration in water–toluene at 160 °C. The different Lewis/Brønsted ratios on the MgF2 catalysts showed that under-coordinated Mg can isomerize xylose to xylulose, whilst the surface OH-groups were responsible for the dehydration reactions. The presence of glucose as a co-carbohydrate reduced the furfural selectivity from 86 to 81%, whilst it also led to high 5-hydroxymethylfurfural selectivity. The tests catalyzed by MgF2 in combination with simultaneous N2-stripping showed that a furfural selectivity of 87% could be achieved using low xylose loadings. Moreover, the catalysts regenerated by H2O2 showed high activity during the dehydration tests in water–toluene at 160 °C.
1. Introduction
In the past years, several environmental and economic issues related to petroleum-derived fuels and chemicals have arisen, so the interest for the design of biorefineries and the research related to novel catalytic systems has renewed.1 In this sense, second generation fuels can directly affect the feasibility of integrated strategies to use lignocellulosic wastes as bio-based product sources. Given the high oxygen content of biomass derived carbohydrates, a key process to efficiently reduce it is by dehydration, operating in aqueous phase. Among furan-based chemicals, furfural (FUR) is a widely used chemical for applications as an industrial solvent or precursor for high value-added products. FUR is industrially produced from pentosan-rich biomass (bagasse or corncob) via xylose cyclodehydration, obtaining low FUR yields (50%).2 Three main issues appear in the manufacturing processes of biomass-derived furfural:1. The use of homogeneous H2SO4 type catalysts, increasing the corrosion and separation issues, as well as achieving low furfural selectivity.
2. The separation technology is based on steam, showing high energy requirements for its production and diluting the stripped stream, which increases the subsequent distillation costs.3
3. The furfural production technologies are limited to the upgrading of the hemicellulose in the biomass. In this sense, interesting and feasible routes, such as the glucose dehydration reactions to 5-hydroxymethylfurfural (HMF), are required.
For these reasons, the development of an appropriate catalyst design and sustainable chemical technologies remains of great interest for the growth of furan-based industry.
Among the novel catalysts, most of the recent publications aimed to maximize FUR production by using heterogeneous catalysts and biphasic water–organic solvent systems4 or ionic-liquids.5 The studied materials featured suitable textural and acid-site properties to reduce FUR degradation reactions and thus increase the FUR selectivity.6 Valente and co-workers synthesized and tested several catalysts, ranging from sulfated zirconia,7 acidic cesium salts8 and functionalized micro/mesoporous catalysts.9 Other studies focused on the use of zeolitic structures,10,11 metal oxides12 or sulfonated silica-shells,13 showing high FUR yields.
In most of the studies, the reaction was catalyzed by Brønsted sites. The mechanism of this reaction has been mostly studied under the presence of heterogeneous4 or homogeneous3,14 Brønsted (B) acid-sites. According to the proposed mechanism, Brønsted sites shift the reaction to convert xylose to furfural without any intermediate.15 Recently, the catalytic activity of different heterogeneous/homogeneous Lewis (L) and Brønsted acids,16 SAPO17 and BEA-TUD18 was also reported. Among the catalysts used for carbohydrate conversion, Lewis acid-sites have been mainly applied to upgrade the glucose sugar as feed-source to achieve high HMF yields via its fructose isomer.1,19,20 It is worth noting that some of the different types of Lewis sites might be transformed to Brønsted under the presence of water. In this sense, the acid properties and their nature play an important and complex role when correlating them to the catalytic carbohydrate conversion activity and to the reaction mechanism. In summary, the Lewis/Brønsted surface properties showed high relevance in order to design suitable catalysts to produce furfural.
Among the catalysts featuring bifunctional properties, partially hydroxylated MgF2 catalysts showed interesting properties to study carbohydrate dehydration type reactions, especially due to the tunable Lewis/Brønsted ratios.21 According to the pyridine adsorption results, the under coordinated Mg2+ ions were responsible for the Lewis acidity, whereas the surface hydroxyl groups showed high Brønsted acidity.22,23 During the study of the glycerol acetylation catalytic activity, the authors found an optimum L/B ratio to maximize the production of diacetyl and triacetylglycerol. The Brønsted acidity could be modified using different fluorosulfonic precursors under different grafting conditions.24 In this way, tunable L/B ratios were achieved. These types of bifunctional catalysts show interesting applications to study the reaction mechanism and catalytic activity of xylose dehydration. In order to optimize the FUR yield, the competition between dehydration and degradation reactions could be controlled by varying the L/B ratios. In order to exploit the biggest fraction of the carbohydrate fraction in the biomass, glucose upgrading routes are required. However, it has to be taken into account that the use of glucose in xylose feed could considerably reduce the furfural production yield due to enhanced secondary reactions.
According to published reports, the carbohydrate conversion catalytic field can be properly studied under water–toluene biphasic batch conditions.26 However, the industrial application of such conditions would increase solvent–furfural separation and purification costs. For this reason, novel technologies using advanced separation systems, such as easily recyclable solvents, under mixed carbohydrate contents would be advisable. Recent studies focused on reactions in aqueous phase operating at high temperature25 or using different stripping agents such as N2, showing significant FUR yield improvement.26 These conditions allowed stripping of the FUR from the reaction medium at high selectivity, allowing an easier stripping agent–water separation and reducing further FUR purification stages. In order to apply the new separation techniques at the industrial scale, more realistic reaction mixtures must be studied including not only xylose but also sugars derived from lignocellulosic biomass, such as glucose. In this way, a complete upgrading route of a big fraction of the hemicellulosic–cellulosic fraction of biomass could be achieved. In order to achieve higher isomerization and dehydration reactions of xylose and glucose type molecules, bifunctional partially hydroxylated MgF2 catalysts combined with novel separation processes seem an alternative as previously reported.27 In summary, this work aims to find a solution to the catalytic and technological issues of the furfural manufacturing process, as well as to go beyond the current state-of-the-art studying of the aspects previously mentioned:
1. Finding new alternatives in the catalysis field using bifunctional MgF2 catalysts to produce FUR under batch conditions. The tuning of their properties allows control of the simultaneous dehydration and side-reactions (Scheme 1).
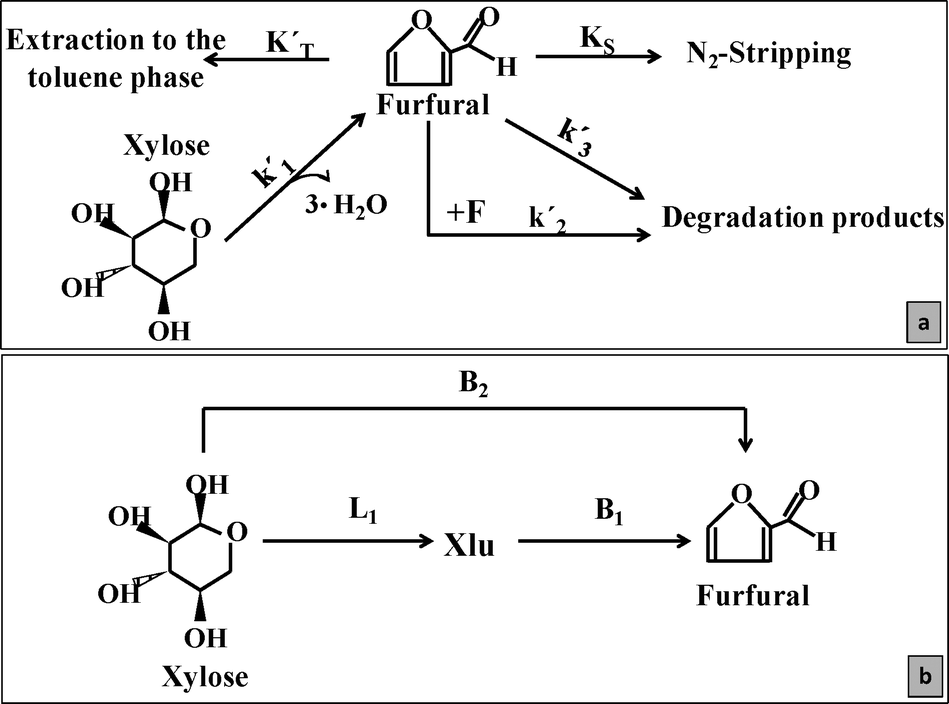 | ||
| Scheme 1 The kinetic and diffusion constants during xylose dehydration to furfural (a). Mechanism and reaction pathways using Lewis and Brønsted catalysts during the dehydration of xylose in water (b). | ||
2. Using a novel FUR separation technology, such as the semi-continuous N2-stripping approach, combined with more selective heterogeneous catalysts than previously reported.27
3. The combination of such concepts using more realistic feed than reported,24 including glucose. Given the Lewis + Brønsted nature of the partially hydroxylated MgF2 catalysts, not only FUR but also HMF could be simultaneously produced. Moreover, the catalyst stability and regeneration tests will prove the potential of such heterogeneous systems to upgrade hemicellulose–cellulose type biomass to interesting molecules. This would provide an alternative route to the current FUR manufacturing catalysis and technology.
2. Materials and methods
2.1 Catalyst preparation
The partially hydroxylated MgF2 catalysts were synthesized using metallic Mg and the fluorolytic sol–gel process: Mg (Aldrich, 99.98%) (7.8 g, 325 mmol) was first dissolved in dried methanol (400 mL) at room temperature overnight. After complete Mg dissolution, the corresponding methanolic HF amount (650 mmol) was added at room temperature. The MgF2 catalyst series were prepared using four different HF concentrations (40, 57, 71 and 87 wt.%, calculated as mHF/(mHF + mwater)). The 40 and 71 wt.% solutions were commercially purchased from Fluka and Sigma-Aldrich, respectively. The 57 and 87% solutions were prepared right before addition. These concentrations correspond to the HF concentrations (wt.%) in the solution to be added to the dissolved Mg. The catalyst, as an example of a HF concentration of 40%, was denoted as M-40. After the addition of HF, the mixtures were vigorously stirred and reacted to form highly viscous transparent sols. These were aged at room temperature overnight, dried under vacuum at increasing temperature until 100 °C for 3 h and maintained at this temperature for 2 h.2.2 Catalyst characterization
19F MAS NMR spectra were recorded on a Bruker AVANCE 400 spectrometer (Larmor frequency: ν19F = 376.4 MHz) using a 2.5 mm MAS probe with 2.5 mm rotors made from ZrO2. The spectra were recorded with a p/2 pulse duration of p1 = 4.0 μs, a spectrum width of 400 kHz, and a recycle delay of 10 s. Up to 60 rotor periods were added before echo detection in rotor synchronized echo experiments. The isotropic chemical shifts δiso of 19F resonances are given with respect to the C6D6 standard. Existent background signals of 19F were suppressed with the application of a phase-cycled depth pulse sequence.28
2.2.2.1 Acid-site qualification. FTIR photoacoustic pyridine adsorption spectroscopy was carried out to qualitatively evaluate the surface acid-sites according to two procedures:
1. Denoted as Pyr-150: the sample (75 mg) was pre-heated at 150 °C under N2 flow for 15 min and then 60 mL of pyridine were injected at 150 °C into the sample tube. The sample was flushed with nitrogen for additional 15 min to remove physisorbed pyridine. Sample spectra were recorded at room temperature using a MTEC cell and a FTIR system 2000 (Perkin-Elmer). Spectra of the samples without pyridine adsorption were also measured as the background.
2. Denoted as RT-Pyr: the FTIR pyridine adsorption spectroscopy measurements at room temperature (RT-Pyr) were carried out in a Thermo Scientific Nicolet iS10 spectrometer using pressed (107 Pa) sample discs. A movable quartz sample holder permits the adjustment of the sample disc in the infrared beam for spectra acquisition. Samples were degassed at 150 °C under high vacuum (2 h). The addition of accurately known pyridine probe doses (at room temperature) was measured by means of a calibrated volume connected to a pressure gauge to control the probe pressure. IR spectra were recorded for each dosing and the pyridine μmol versus peak area was plotted.
2.2.2.2 Acid-site quantification. Temperature programmed desorption of ammonia (NH3-TPD) was carried out as follows: the sample (0.2 g) was first heated under N2 up to 300 °C and then at 120 °C exposed to NH3. After flushing the excess NH3 at 120 °C with N2 for 1 h and cooling to 80 °C, the TPD program was started (10° min−1 up to 500 °C, keeping for 30 min). Desorbed NH3 was monitored continuously via FTIR spectroscopy (Perkin-Elmer).
For the acid quantification measurements using NMR (denoted as Pyr-NMR), 700 mg of the sample were weighted in a Schlenk flask, followed by an activation step at 200 °C under vacuum for 2 h to remove physisorbed water. Then, 30 μl of 15N-pyridine (~367 μmol) were added and the mixture was stirred for 30 min at 150 °C to ensure homogeneous pyridine distribution. Physisorbed pyridine was removed under vacuum at 150 °C for 1 h. Rotors for MAS NMR experiments were carefully filled in the glovebox with a mixture of the sample and NH4Cl (15N enrichment, 9.5%) in a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For quantitative investigations, 15N-NMR single pulse experiments with presaturation and 1H–15N CPMAS (cross-polarization with magic angle sample spinning) experiments are necessary, as described elsewhere.2915N MAS NMR single pulse spectra were recorded at a Larmor frequency of 60.8 MHz. The MAS frequency was 6.5 kHz. The 15N 90° pulse length was 6.2 μs. The repetition time in the single pulse spectra was set to 5 s with previous saturation. 1H high power decoupling (TPPM) was applied and 3072 scans were accumulated. 15N chemical shifts (δ) are reported relative to CH3NO2 with NH4Cl as the secondary standard (δ = −341 ppm).30 The 1H–15N CPMAS experiments are needed for the determination of the T1 corrections of the time optimized 15N MAS NMR spectra using the Torchia method.31 The sample spinning frequency was 6.5 kHz and the spectra were recorded using a 1H 90° pulse length of 6.5 μs, a contact time of 2 ms, and a repetition time of 3 s. The 15N spin lock field was held constant while the 1H spin lock field was ramped down to 50% of its initial value. Mostly 4096 scans were accumulated, and high power decoupling (TPPM) was applied.
:1. For quantitative investigations, 15N-NMR single pulse experiments with presaturation and 1H–15N CPMAS (cross-polarization with magic angle sample spinning) experiments are necessary, as described elsewhere.2915N MAS NMR single pulse spectra were recorded at a Larmor frequency of 60.8 MHz. The MAS frequency was 6.5 kHz. The 15N 90° pulse length was 6.2 μs. The repetition time in the single pulse spectra was set to 5 s with previous saturation. 1H high power decoupling (TPPM) was applied and 3072 scans were accumulated. 15N chemical shifts (δ) are reported relative to CH3NO2 with NH4Cl as the secondary standard (δ = −341 ppm).30 The 1H–15N CPMAS experiments are needed for the determination of the T1 corrections of the time optimized 15N MAS NMR spectra using the Torchia method.31 The sample spinning frequency was 6.5 kHz and the spectra were recorded using a 1H 90° pulse length of 6.5 μs, a contact time of 2 ms, and a repetition time of 3 s. The 15N spin lock field was held constant while the 1H spin lock field was ramped down to 50% of its initial value. Mostly 4096 scans were accumulated, and high power decoupling (TPPM) was applied.
2.3 Catalytic tests and product analyses
The batch tests were carried out in a high-pressure Teflon lined reactor (BR-500 from Berghof) stirred at 500 rpm. The reactor was first loaded with 0.6 g of the catalyst, 150 mL of water and 150 mL of toluene. In order to keep the reaction medium in liquid-phase, the system pressure was maintained at 15 bar. Once the solution was heated to 160 °C, the corresponding xylose solution was added using a Gilson 305 pump to reach a xylose initial concentration of 20 g L−1 of aqueous phase (0.44 M). Reaction samples were taken at different intervals using a needle valve.The N2-stripping tests were carried out in a 2 L reactor (Autoclave Engineers), with controlled electric-heating and stirred at 500 rpm (Scheme 1). In a typical SC experiment operating at 180 °C and 10 bar, the reactor was first loaded with the corresponding amount of MgF2 catalyst and heated up to 190 °C with deionized water (75% of the total initial reactor volume). The remaining 25% of the total reactor volume was fed from a nitrogen-pressurized vessel (to reach the corresponding initial xylose concentration, X0). This set-up allowed the feed solution to be held at room temperature until the desired temperature was reached in the reactor to minimize the initial feed degradation. During the N2-stripping tests, mass-flow controlled nitrogen was bubbled into the liquid bottom at 150 mL min−1 (STP). This gas flow stripped the water–furfural vapour stream. The gaseous flow was later fed to a condenser (cooled by the Peltier effect at 10 °C), where gas and liquid streams were separated. The condensate was continuously weighted. Automatic control valves were used to regulate the reactor-pressure and the liquid level in the condenser.
Xylose conversion (XX) was calculated at different intervals and FUR selectivity (FURS) was calculated as the mol of FUR obtained per mol of converted xylose. Glucose conversion (GX) and HMF selectivity (HMFS) were calculated in the same manner. In all cases, carbohydrate conversion values were calculated using the sum of reactants in the reactor phase: in the water phase during the water–organic batch tests, and in the reactor during the N2-stripping tests. Moreover, the FURS/HMFS values in the batch tests were calculated as the sum in the water and organic phase; whilst the FURS/HMFS for the N2-stripping tests was calculated as the sum of the FUR/HMF present in the reactor and in the condensate. Product yield (FURY or HMFY) was calculated as conversion × selectivity.
The carbohydrate content in the reactor was quantified using a HPLC module ICS-3000 from Dionex coupled to an AS40 autosampler. X and G were quantified using a CarboPac PA20 3 × 150 mm column at 30 °C and 0.5 mL min−1 using 8 mM of NaOH as the mobile phase. Detection was performed using an electrochemical cell, with integrated amperometry and the Standard Carbohydrate Quad method. FUR and HMF were quantified in a 1260 Infinity module from Agilent. The products were separated in a Zorbax SB-C18 column (3.5 μm, 3.0 × 150 mm) at 1 mL min−1 and 35 °C using water as the eluent.
Secondary-products were identified by GC-MS (6890 GC and 5973-Mass Selective Detector from Agilent) using a DB-FFAP column, helium as the carrier gas at 1 mL min−1 and an injection volume of 1 μL. The 1H (νLarmor(1H) = 400.1 MHz) and 13C (νLarmor(13C) = 100.6 MHz) liquid-NMR spectra of the reaction products were recorded on a Bruker AVANCE II 400 under standard conditions for NMR parameters. All spectra were obtained using the specific signals of deuterated benzene as the reference standard in melted off locked-in tubes.
3. Results and discussion
3.1 Catalyst characterization
According to the reported data,21 the partially hydroxylated MgF2 containing different HF concentrations showed L/B ratios from 0.4 to 1.0 (based on the area of the pyridine vibration bands) for M-40 and M-71, respectively. The L/B ratio differences were mainly attributed to the different concentrations of OH groups on the sample surfaces. Troncea et al. reported the highest OH content for the M-40 sample, caused by the higher water content in the aqueous HF used for the synthesis.21| Sample | S BET (m2 g−1) | Pore (Å) | Pore vol. (m3 g−1) | Acidity Pyr-NMRa (μmol g−1) | Ratio L/B | NH3-TPDb (μmol g−1) | (TOF × 106)c mmolX μmolACID−1 h−1 | X X (%) | FURSd (%) | FURYd (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Lewis | Brønsted | ||||||||||
| a Acidity of Lewis and Brønsted sites quantified by pyridine adsorption at 150 °C followed by 15N-NMR analysis. b Total acidity quantified by NH3 adsorption followed by TPD up to 550 °C. c TOF value calculated on converted xylose after 4 h of reaction and based on Pyr-NMR analyses. d Xylose conversion (XX), FUR selectivity (FURS) and yield (FURY) after 8 h. | ||||||||||||
| M-40 | 271 | 27.5 | 0.19 | 303 | 303 ± 45 | — | High | 450 | 10.9 | 82 | 44 | 36 |
| M-57 | 284 | 27.1 | 0.17 | 438 | 302 ± 34 | 136 | 2.2 | 481 | 4.5 | 60 | 69 | 41 |
| M-71 | 460 | 26.9 | 0.30 | 334 | 258 ± 24 | 76 | 3.4 | 567 | 6.3 | 63 | 86 | 54 |
| M-87 | 465 | 25.2 | 0.27 | 308 | 256 ± 19 | 52 | 4.9 | 645 | 3.6 | 35 | 88 | 31 |
| M-100 | 467 | 23.2 | 0.27 | — | — | — | — | — | — | 32 | 43 | 14 |
| Amberlyst | <1 | — | — | — | — | — | Low | 2550 | 0.8 | 90 | 44 | 39 |
The surface-property differences on the MgF2 catalysts were also confirmed by solid-state 19F-MAS-NMR measurements. The MgF2 samples showed a dominant 19F signal centered at δ = −197 ppm, at nearly the same position as that of crystalline MgF2. During the synthesis of partially hydroxylated MgF2 samples, the competition between fluorolysis and hydrolysis of Mg(OCH3)2 was enhanced due to the addition of water on the MgFX(OH)2−X xerogel. The asymmetric shape and the shifting of the −197 ppm signal were previously attributed to the presence of un-substituted oxygen species on its surface.23,32 Among the materials prepared here, the presence of abundant OH groups on M-40 shifted the main peak to −196.7 ppm and created a shoulder in the lower field of the spectrum (δ = −179.3 ppm). On the other hand, the presence of minor OH contents on M-71 reduced the asymmetric shape of the −197.4 ppm signal.
According to the TG-MS data, the MgF2 catalysts showed a very broad exothermal peak in the DTA curves, characteristic of the sol–gel procedure. As depicted in Fig. 1, the M-40 and M-71 catalysts showed an overall weight loss of circa 13%, assigned to the loss of strongly bonded water (100–350 °C) and to the OH surface groups (350–500 °C). As observed in the m18 profiles, a considerable increase of the surface OH incorporation for the sol prepared using 40% HF concentration was achieved compared to M-71. Based on the SBET values for each catalyst and assuming that all strongly bonded OH groups were entrapped in the pores, the M-40 sample showed the highest OH surface concentration (0.71 mmol OH m−2). As observed in Fig. 1, the MS signal also detected some other traces corresponding to fluorine (m19) or methanol (m31).
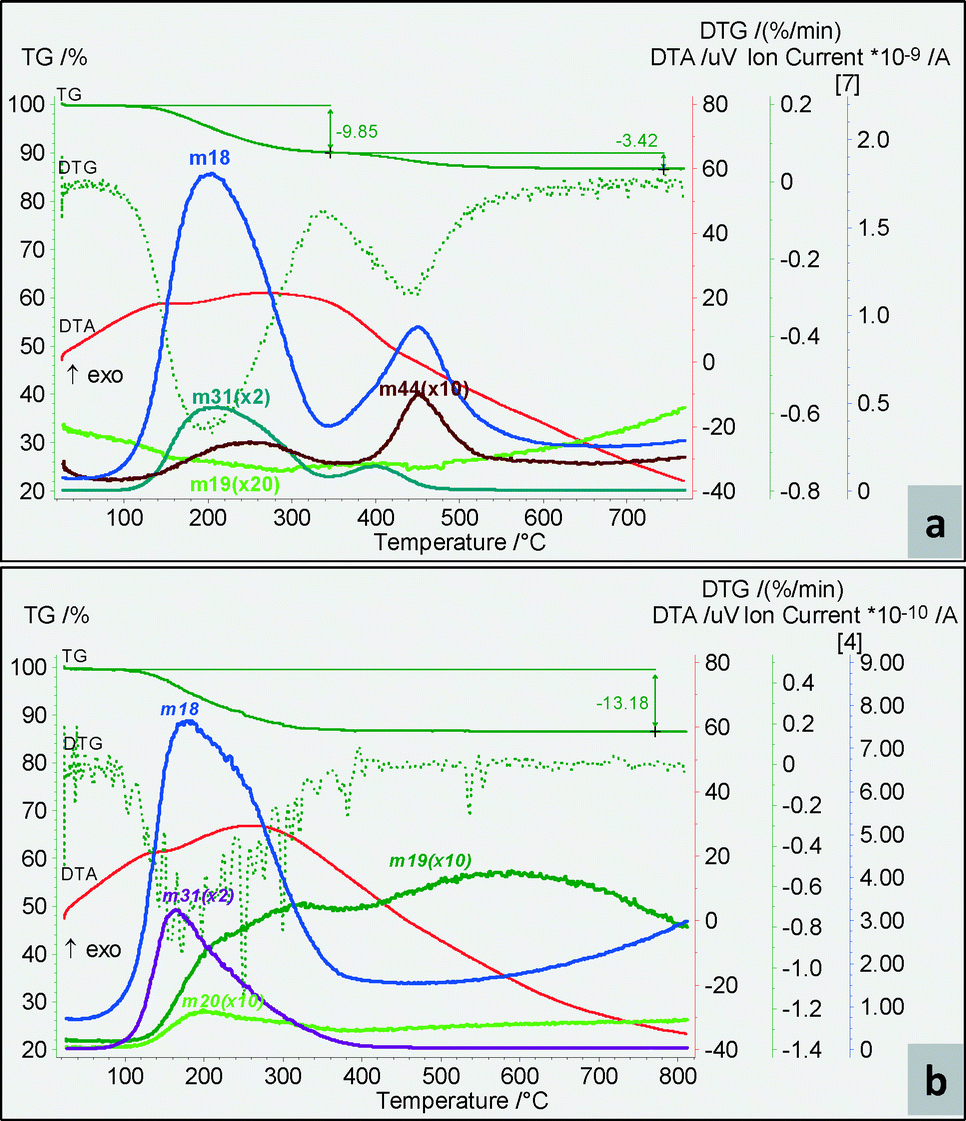 | ||
| Fig. 1 TG, DTG, DTA and the MS profiles of M-40 (a) and M-71 (b). | ||
According to the scanning electron microscope picture in Fig. 2A, the M-71 sample, as well as the rest of the catalysts, presented a particle sizes around 0.5–3 m. However, no further information could be extracted from the transmission electron microscope image (Fig. 2B).
 | ||
| Fig. 2 SEM (A) and TEM (B) images of the partially hydroxylated M-71 fresh sample. | ||
3.1.2.1 Acid-site qualification. The qualitative and quantitative analyses of the acidity of heterogeneous catalysts are determined to correlate these properties to the catalytic activity during xylose dehydration to FUR. The acid properties of the catalysts were first qualitatively evaluated adsorbing pyridine at room temperature (RT-Pyr).
According to the RT-Pyr FTIR measurements (not shown), the Lewis sites on all the samples showed high pyridine protonation capacity, whilst just the M-57 Brønsted sites were detected. During these analyses, the pyridine diffusivity and its protonation capacity were considerably reduced, meaning that mainly strong acid-sites were capable of showing pyridine vibration bands. The capacity to protonate Brønsted sites by RT-Pyr (measured by the adsorption peak areas) was as follows: M-40 < M-87 < M-71 < M-57. It seems that a minimum Brønsted-site content or strength was required to detect Brønsted sites using the RT-Pyr technique.
On the other hand, the addition of pyridine at higher temperature (denoted as 150-Pyr) allowed evaluation of nearly the totality of the Lewis or Brønsted acid-types on the catalyst surface by enhancing its diffusion to the acid-site and its protonation for IR detection. As observed in Fig. 3, despite the high OH concentration on the M-40 surface (based on the TG-MS data), it seems that these Brønsted sites were not strong enough to protonate pyridine and even xylose (as it will be discussed later). This can be explained by the following: the M-40 sample contained the highest OH content but lower fluorine than M-71. As usually observed for Mg(OH)2, which is mainly basic, the lower fluorine content on M-40 could also be reflected as lower Brønsted acidity. On the other hand, as observed in Fig. 3, the catalysts with lower OH contents (M-57, M-71, and M-87) showed both Lewis and Brønsted sites in the IR spectra. In other words, although these catalysts had fewer surface OH groups, their Brønsted acidity was stronger than that of the OH-rich sample, M-40.
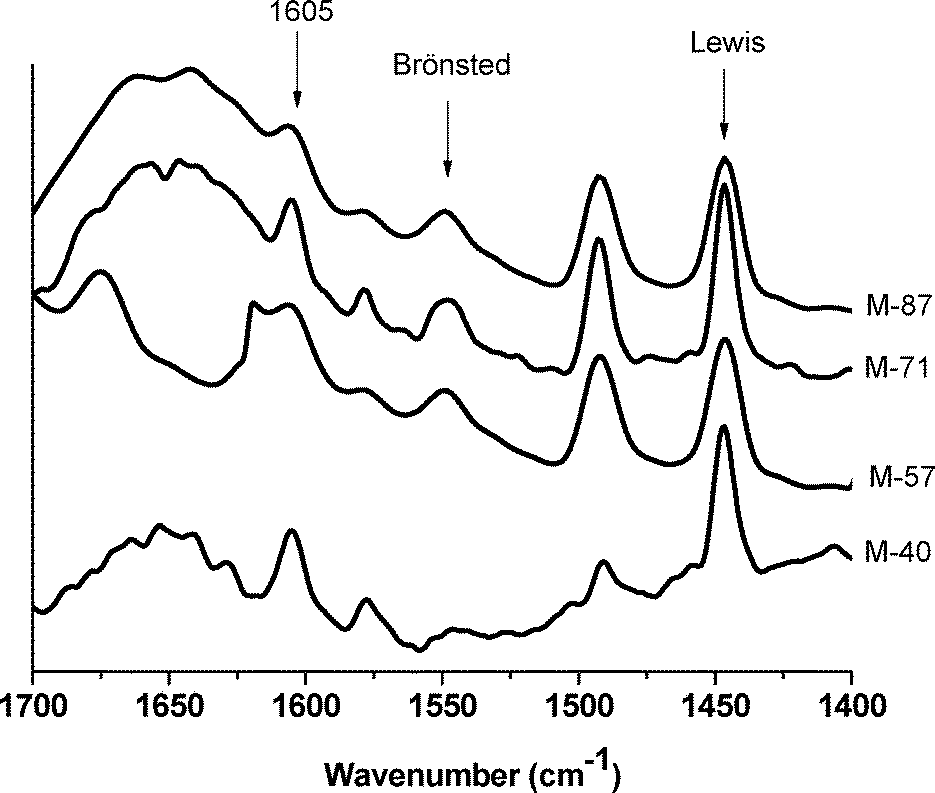 | ||
| Fig. 3 The FTIR spectra of the M-40 and M-71 samples after pyridine adsorption at 150 °C. | ||
In general, the shifting (to a higher wavenumber range) of the vibration band at 1605 cm−1 states the strength of the Lewis sites. However, this band appeared at the same position in the spectra of all the samples.
3.1.2.2 Acid-site quantification. The pyridine adsorption-IR is a powerful method to study the surface acidity. However, it cannot be used as a quantification method by simply calculating the vibration peak area ratio.21 The presence of Lewis sites requires the use of these techniques, in order to avoid the quantification errors associated to inactive sites such as in the aqueous-phase titrations.33 This was already reported on a broad range of Lewis/Brønsted catalysts.16 For this reason, the total acidity was quantified using two different procedures: pyridine adsorption followed by 15N-NMR (Pyr-NMR) and NH3 adsorption followed by TPD (NH3-TPD).
According to the NH3-TPD data, the fresh MgF2 samples showed a FTIR intensity for desorbed NH3 starting at 200 °C and decaying at ~400 °C. As observed in Table 1, the quantification of the total acid sites by NH3-TPD showed higher values than Pyr-NMR. One possible explanation is that the smaller NH3 molecule can diffuse and be adsorbed on the acid-sites which are not accessible to pyridine. Moreover, the TPD carried out up to 550 °C ensures that all adsorbed NH3 is removed, whilst during pyridine adsorption analyses (as observed later) some of the entrapped pyridine may not be quantified. However, taking into account the presence of adsorbed water, the NH3-TPD analyses showed low reproducibility. Therefore, only Pyr-NMR values will be used for further discussion (explained below). Moreover, Pyr-NMR is the only available analysis to differ the Lewis/Brønsted acid-site quantities on these MgF2 catalysts.
The Pyr-NMR spectra allowed selective separation of the physisorbed pyridine (centered at −80 ppm), Lewis sites (−100 ppm), Brønsted sites (−174 ppm) and the standard NH4Cl (−341 ppm). The Pyr-NMR results confirmed the FTIR data previously discussed using the 150-Pyr technique. M-40 showed no Brønsted acidity, proving that the presence of more OH groups on M-40 reduced the pyridine protonation capacity of the Brønsted sites. On the other hand, M-57 showed the highest content of Brønsted acid-sites (136 μmol g−1). In general, the change in the concentration of Brønsted acidity measured by Pyr-NMR showed the same trend as the Lewis acidity (Table 1).
Additionally, the RT-Pyr analyses were attempted to quantify the acid-site content by dosing small quantities of pyridine and recording its IR spectra. The corresponding extinction coefficients were derived for quantification; however, given the low protonation capacity of the Brønsted acid-sites at this temperature, the total values were far below the Pyr-NMR values. This is a powerful method compared to the more sophisticated Pyr-NMR techniques, but not appropriate for these catalysts.
3.2 Catalytic activity during carbohydrate conversion
First of all, and in order to prove the heterogeneous nature of the MgF2 catalysts, the liquid solution after the reactions at 160 °C was analyzed by means of 19F liquid NMR. As observed in Fig. A1, ESI† the presence of fluorinated compounds is negligible, confirming the heterogeneous nature of the tested catalysts. | ||
| Fig. 4 The evolution of (a) the conversion of xylose and (b) the selectivity to FUR versus reaction time during the xylose dehydration tests of the partially hydroxylated sample series at 160 °C in water–toluene. | ||
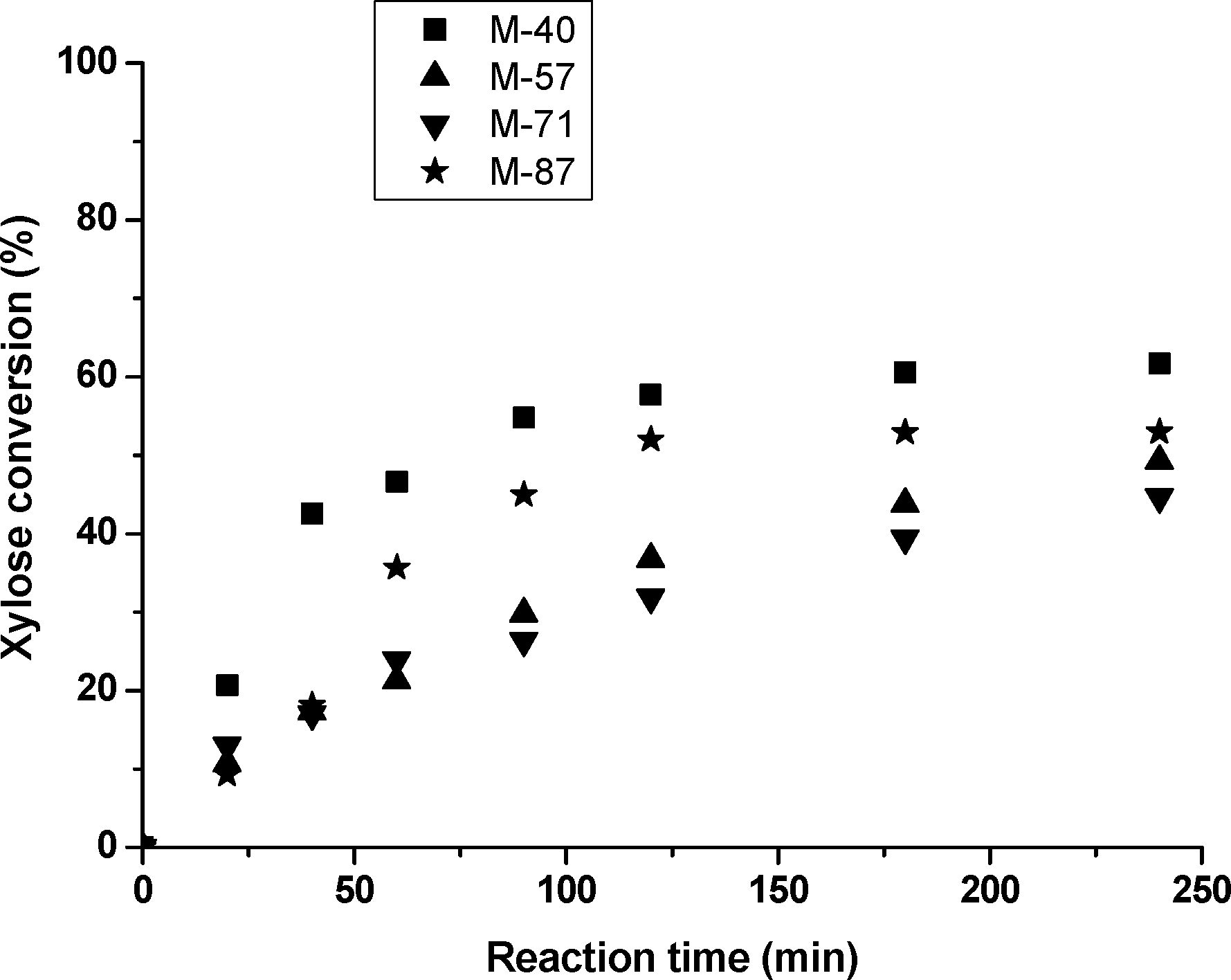
As observed in Fig. 4a, M-40 (dominant Lewis acid-sites) showed the highest xylose conversion, with a TOF value of 10.9 × 10−6 mmolX μmolLEWIS−1 h−1. The TOF values, based only on the Lewis sites, of the rest of the catalysts were as follows: 6.5, 8.1 and 4.3 × 10−6 mmolX μmolLEWIS−1 h−1 for M-57, M-71 and M-87, respectively. However, as observed in Table 1, the presence of OH groups (as Brønsted) reduced the xylose dehydration activity, and thus reduced the TOF values (calculated based on the total acidity of the fresh catalysts) for all the catalysts except for M-40. The double dosing in weight of the catalysts would presumably double the TOF values as well. Among the rest of the catalysts, M-87 showed surprisingly low xylose conversion values. Although this sample exhibited higher total acidity measured by NH3-TPD, its lower Lewis acid-site content may lead to lower xylose conversion rates. On the other hand, it must be pointed out that the xylose conversion values of M-57 and M-71 had very slight differences.
In order to evaluate the effect of MgF2 catalysts on the activity (xylose conversion and furfural selectivity), experiments without any catalyst were also carried out. These tests showed a xylose conversion of 23% and a furfural selectivity of 21% (yield of 5% after 8 h of reaction) at 160 °C in water–toluene, confirming the presence of MgF2 catalysts to improve furfural production.
When bifunctional L + B catalysts such as MgF2 are in contact with xylose, competitive reactions occur and the reaction rates showed clear differences compared to pure Brønsted acids, such as homogeneous H2SO414 or Amberlyst.27 As observed in Fig. 4b, it clearly proved that catalysts containing lower L/B ratios than M-40 could significantly improve the FUR production selectivity (FURS). In these cases, the reactions were catalyzed by a combination of Lewis and Brønsted sites, and thus the FUR production shifted to the route showing the highest FURS (B2 route). In general, it was observed that catalysts presenting high L/B ratios (such as 10.9 for M-40) reduced the furfural production yield. On the contrary, by reducing this ratio to 3.6 (for M-87), FURS values could be considerably increased. However, the optimum L/B ratio was found for the M-71 catalyst, showing intermediate Lewis acidity and higher OH concentration than M-40 or M-57, thus increasing the FUR yield values.
As reported for other pure Brønsted catalysts, the FURS plots present a maximum at the beginning of the reaction, which then drops along the reaction path due to side-reactions.27 In the MgF2 system used in this work, however, the presence of intermediates (such as xylulose, evidenced in Fig. A2, ESI† and quantified as represented in Fig. A3, ESI†) reduced the initial FUR selectivity whilst xylulose can be dehydrated and thus the FURS increased all along the tests (Fig. 4b). Moreover, as observed in Fig. 4b, the FURS decayed after 6 h of reaction for the M-57 sample. Even if xylulose was quantified at this point, FUR side-reactions were enhanced at this point, leading to lower FUR yield values.
Another important reaction parameter was the acid-site density, measured as total acid sites per surface area, showing the following trend: 1.1, 1.5, 0.7 and 0.6 μmol m−2 for M-40, M-57, M-71 and M-87, respectively. The higher FURS observed for M-71 and M-87 could also result from a lower acid-site density, together with an optimized L/B ratio for furfural production (Table 1). Taking into account that the loss of FURS is directly attributed to the side-reactions, such as resinification, higher acid-site densities could enhance these reactions with samples such as M-57. On the other hand, the lower FURS observed for M-40 was directly attributed to the low protonation capacity of the weak Brønsted acid sites on its surface (nearly insignificant) or for the reaction medium H3O+.
As observed in Table 1, even if the M-87 sample showed the highest FURS values, its low xylose conversion values considerably reduced the furfural yield (FURY). In general, the catalyst showing the highest selectivity (M-71) also showed the highest furfural yield values, even after 20 h of reaction. As previously described, furfural yield values strongly depend on the nature and amount of acid-sites. Lewis sites increase xylose conversion values but reduce the FUR selectivity and consequently the yield values. On the contrary, xylose conversion rates are lower for Brønsted sites but yield values are compensated due to higher selectivity values.
Based on the xylose conversion data, the differences were mainly attributed to the change of the reaction mechanism in the presence of Lewis or Brønsted sites. The presence of xylulose (evidenced by the 1H-NMR spectra as shown in Fig. A2, ESI† and quantified as shown in Fig. A3, ESI†) during the reactions showed that Lewis sites catalyzed the xylose conversion to its isomer (L1) by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O rearrangement,34 whilst the presence of Brønsted sites (B1) was required to further dehydrate xylulose to FUR (Scheme 1).15 On the other hand, xylose can be directly dehydrated to FUR in the presence of a strong Brønsted site (B2). A plausible mechanism for the direct xylose-to-furfural reaction consists of transforming the hydroxyl groups of the pentose to H2O+ and the elimination of two 1,2 and one 1,4 water molecules.3,14 In this case, samples such as M-57, M-71 or M-87 could produce FUR via L1 + B1 or via B2 routes (due to their relatively low L/B ratio). However, dehydration reactions using M-40 could only be catalyzed by weaker Brønsted acid species, i.e. the H3O+ ions in the reaction medium. As reported, the B2 route increases the FURS, but the overall conversion rate is slower than the L1 + B1 route.15 These reactions were embraced as a general k′1 in Scheme 1. Moreover, the FUR can also be degraded by condensation reactions with fragments (k′2) or produce FUR resins under the presence of strong acids (k′3), mainly Brønsted.
O rearrangement,34 whilst the presence of Brønsted sites (B1) was required to further dehydrate xylulose to FUR (Scheme 1).15 On the other hand, xylose can be directly dehydrated to FUR in the presence of a strong Brønsted site (B2). A plausible mechanism for the direct xylose-to-furfural reaction consists of transforming the hydroxyl groups of the pentose to H2O+ and the elimination of two 1,2 and one 1,4 water molecules.3,14 In this case, samples such as M-57, M-71 or M-87 could produce FUR via L1 + B1 or via B2 routes (due to their relatively low L/B ratio). However, dehydration reactions using M-40 could only be catalyzed by weaker Brønsted acid species, i.e. the H3O+ ions in the reaction medium. As reported, the B2 route increases the FURS, but the overall conversion rate is slower than the L1 + B1 route.15 These reactions were embraced as a general k′1 in Scheme 1. Moreover, the FUR can also be degraded by condensation reactions with fragments (k′2) or produce FUR resins under the presence of strong acids (k′3), mainly Brønsted.
According to the 1H-NMR spectra xylulose was present as the only intermediate. As reported in the literature, when Lewis catalysts are present xylulose appears as the only intermediate for furfural production. On the other hand, catalysts showing more Bronsted sites show also the direct xylose conversion route. In this case, as the M-71 catalyst also presented Lewis sites, it also showed the presence of xylulose in the reaction solution.
In order to identify the possible reaction by-products, the soluble phase was analyzed by means of 1H-NMR. Taking into account the low xylose concentration detected and its slow dehydration to furfural,35 it seemed that xylose was favorably isomerized to xylulose (see Fig. A2, ESI†) under the presence of partially hydroxylated MgF2 catalysts. The combination of Lewis sites (M-40) and weak Brønsted (H3O+) can also convert the pentose carbohydrates to fragments such as formic acid (8.4 ppm, derived from the O3 protonation36), glycolaldehyde (2.2/3.2 ppm) or dihydroxyacetone (4.4 ppm) by retro-aldol reactions (RA). Moreover, other side-products might be derived from the dehydration reactions of xylose/xylulose or from dihydroxyacetone to pyruvaldehyde (2.33 ppm), as well as several other minor fragments at 2.1–2.5 ppm. A detailed characterization and quantification of all the sub-products during reaction is very difficult. Moreover, the presence of humins and coke-deposits makes a detailed carbon balance analysis difficult.
Concerning the effect of the solvent on the activity data, efficient FUR isolation is required to increase the FURS. In this sense, the xylose conversion rate of the MgF2 catalysts only changed slightly under monophasic conditions in water, but showed lower FURS values (M-40: 26%, M-57: 33%, M-71: 42% and M-87: 44%). However, the use of an aprotic solvent such as DMSO (a Lewis base with a pKa of 35) induced significant changes. As observed in Fig. 5, XX values stopped below 60% after 4 h. Even if the Lewis site might be partially suppressed, M-40 showed again the highest activity. The behavior of M-87 was the only different one compared to water–toluene data. This could be attributed to its very low L/B ratio. However, the FURS obtained in DMSO (10%) were below the ones obtained in water.
 | ||
| Fig. 5 The reaction time vs. xylose conversion of the partially hydroxylated sample series at 160 °C in DMSO. | ||
:1 w/w X:G feed at 160 °C in water–toluene. As observed in Fig. 6a, the presence of glucose slightly increased the XX compared to the one measured with just the X feed for the tests using M-71. Moreover, the conversion of glucose took place at slower catalytic rates. During the X + G tests, the most significant changes were observed for the FURS at 160 °C in water–toluene (Fig. 6b). The FUR condensation reactions with glucose and HMF intermediates were enhanced26 and FURS reduced from 44 to 38% for M-40 and from 86 to 81% for M-71 after 20 h of reaction in water–toluene. The tests at 180 °C using X + G feed showed a slightly lower FURS and a considerable HMFS decrease (Table 2). When the FUR is not quickly isolated from the reaction medium under batch conditions, the resinification effects could considerably reduce the FUR and HMF selectivities. Given the “high” L/B ratio of these MgF2 catalysts, this effect was less important than in processes operated with higher Brønsted content. On the other hand, the “entropy effects” related to higher-temperatures27 showed no effects on the FURS.
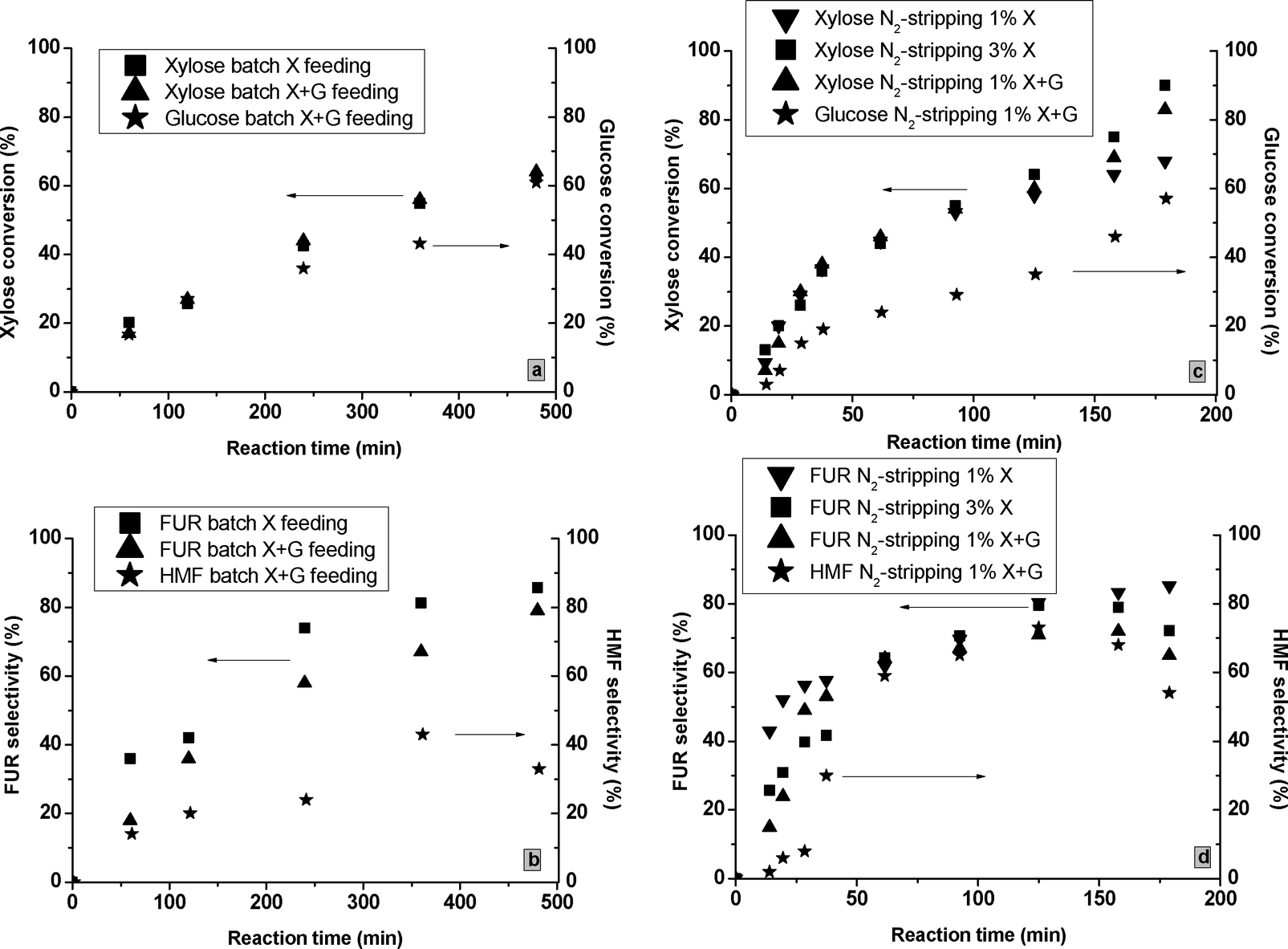 | ||
| Fig. 6 The xylose and glucose conversions and FUR and HMF selectivities for the tests catalyzed by M-71 under batch conditions at 160 °C in water–toluene (Fig. 6a and b) and N2-stripping at 180 °C in water (Fig. 6c and d). | ||
| Catalyst | Tests | T (°C) | Feed | X X (%) | FURSc (%) | G X (%) | HMFSc (%) | TOF 106e (mmolX−G μmolACID−1 h−1) |
|---|---|---|---|---|---|---|---|---|
| a Batch tests performed with a 1/1 v/v water/solvent ratio. b N2-stripping performed using 150 mL min−1 of N2 and pressure calculated according to the corresponding vapor pressure. c Xylose (XX) or glucose (GX) conversion, FUR selectivity (FURS) and HMF selectivity (HMFS) after 20 h of reaction. d Xylose conversion for the N2-stripping tests after 3 h of reaction. e Turnover frequency for xylose or glucose after 4 h of reaction and for xylose using the X + G feed. SD: overall standard deviation: ~6%. | ||||||||
| M-40 | Water–toluenea | 160 | X | 82 | 44 | — | — | 10.9 |
| M-71 | 94 | 86 | — | — | 6.3 | |||
| M-71-R1 | 98 | 59 | 8.1 | |||||
| M-71-R2 | 82 | 52 | 6.2 | |||||
| M-40 | Water–toluenea | 180 | X | 90 | 47 | — | — | 13.4 |
| M-71 | 99 | 75 | — | — | 8.5 | |||
| M-40 | Water–toluenea | 160 | G | — | — | 99 | 23 | 11.6 |
| M-71 | — | — | 85 | 39 | 6.1 | |||
| M-71 | 180 | G | — | — | 92 | 45 | 9.4 | |
| M-71 | Water–1-butanola | 160 | G | — | — | 78 | 58 | 3.5 |
| M-71 | 180 | — | — | 99 | 66 | 9.1 | ||
| M-40 | Water–toluenea | 160 | X + G | 99 | 38 | 99 | 28 | 11.2 |
| M-71 | 97 | 81 | 88 | 40 | 6.9 | |||
| M-71 | Water–toluenea | 180 | X + G | 99 | 73 | 99 | 12 | 9.3 |
| M-71 | N2-strippingbd | 160 | X (1%) | 64 | 59 | — | — | — |
| 180 | X (1%) | 68 | 87 | — | — | — | ||
| X (3%) | 90 | 73 | — | — | — | |||
| X + G (1%) | 83 | 65 | 58 | 53 | — | |||
| X + G (3%) | 91 | 59 | 82 | 35 | — | |||
This study aims to provide a real solution to the carbohydrate fractions present in the biomass. In this sense, catalysts such as MgF2, containing Lewis and Brønsted sites, show interesting properties to isomerize the glucose to fructose and dehydrate fructose to HMF via Brønsted acid-sites.
The tests at 160 °C performed in water–toluene confirmed the low extraction efficiency of toluene, showing HMFS of 40% for M-71 (Table 2). The tests carried out at 180 °C slightly increased the HMFS to 45% for M-71. On the other hand, the use of 1-butanol as a co-solvent, which can also be obtained through the fermentation of biomass derived carbohydrates, considerably increased the HMFS (58% at 160 °C and 66% at 180 °C). The HMF partition coefficient of solvent–water increased from 0.1 to 2.6 for toluene and 1-butanol, respectively. This higher extraction efficiency decelerated the HMF yield-loss reactions, such as rehydration to levulinic acid and formic acid and the formation of condensation products.39 Thus, the combination of suitable reaction solvents and partially hydroxylated MgF2 seemed promising to optimize the production of HMF from glucose.
3.3 Stability and regenerability of the MgF2 catalysts under batch conditions
The stability of the catalysts was evaluated in terms of structure and acidity. According to the textural properties of the used catalysts, due to morphological changes all the catalysts suffered a strong decrease of SBET (from 460 to 150 m2 g−1 for M-71), whilst the pore size increased from 26.9 to 54.8 Å for M-71. The explanation for the decrease of the surface area and the reduction of the pore size is a similar phenomenon as explained in section 3.1.1 for the fresh catalysts. The catalysts prepared using higher water/HF ratios created bigger particle aggregates, and thus these materials showed a decrease of the BET surface area and an increase of the pore size. In the same manner, the experimental tests were carried out in water-phase. Moreover, the conditions were harsher (160 °C) than the fresh sample synthesis conditions (80 °C). This way, M-71 also suffered a similar decrease of the surface area and an increase of the pore size due to the formation of bigger aggregates. This was strictly related to the physical properties of the MgF2 partially hydroxylated materials. In order to produce structurally stable MgF2 catalysts under hydrothermal conditions, further work is required in this field. Moreover, according to the liquid 19F-NMR analyses (Fig. A1, ESI†), no fluorinated species were found, proving the stability of the MgF2 catalyst.On the other hand, the stability of the acid-sites was determined to evaluate the long-term feasibility of the heterogeneous catalysts, especially concerning their industrial application. The catalysts used during the catalytic tests (using X feed) showed a brown color derived from deposited coke or humins. According to previous studies, the catalysts could be efficiently regenerated by calcination at 280 °C.40 This allowed avoidance of any interference of the coke on the acidity analyses. However, the coke on MgF2 catalysts could not be removed at this temperature. Moreover, given the limited thermal stability of the acid-sites, milder oxidation procedures using H2O2 on the MgF2 catalysts were further researched.41 In a typical regeneration step, 2 g of as-recovered catalysts were treated with 20 g of 30% H2O2 at 100 °C in Teflon-lined flasks under severe stirring for 4 h. The materials were filtered, washed with ethanol and dried at 100 °C. The CHN elemental analyses of the treated M-71 proved that 98% of the coke layer could be efficiently removed. According to the Pyr-NMR data, the M-71 sample preserved 80% of its total acidity. This acidity was mainly attributed to the Lewis sites, which corresponded to 78% of the acid-sites on fresh M-71. The type of acidity was also checked by 150-Pyr, showing a strong decay of mostly Brønsted acidity. This fact proved that Lewis sites were more stable than Brønsted under hydrothermal conditions. Apparently, the leaching of the surface hydroxyl groups was enhanced in water at 160 °C. According to the acidity data on used catalysts calcined at 300 °C, only 57% of the total acid-sites were recovered after the calcination treatment.
The regenerated catalysts were further tested at 160 °C in water–toluene (see Table 2). Given the change of the acid-properties of a sample containing more Lewis sites and the decrease of total acidity, the TOF value of the catalyst regenerated in the first run (M-71-R1) increased from 6.3 to 8.1 × 10−6 mmolX μmolACID−1 h−1. A second run showed even lower acid-content, which decreased the xylose conversion activity. As expected, an increase of the L/B ratio had an opposite effect on the FUR production, reducing the FURS from 86% to 59 and 52% for fresh M-71, M-71-R1 and M-71-R2, respectively. Given the low catalyst amount used in each test (0.4 g) and the loss on reactor walls and filtration in each regeneration step, additional regeneration tests could not be carried out.
3.4 N2-stripping of furfural
The current FUR manufacturing process uses between 16:1 (Quaker Oats) and 30:1 (Rosenlew) steam to product weight ratio.3 The high energy requirements, the separation stages and the use of homogeneous catalysts make this process environmentally unsustainable. The use of N2 as the stripping agent could improve the manufacturing process by reducing the heating costs and it can be easily separated and recycled from the stripped stream due to its high volatility.
The developed system in this study allowed selective production of FUR and HMF from xylose and glucose, respectively. Future environmentally sustainable processes could upgrade these furan derivatives to higher value-added compounds, such as methyltetrahydrofuran42 or dimethylfuran.39
4. Conclusions
Partially hydroxylated MgF2 catalysts showed high activity during the dehydration of pentoses and hexoses to furanic aldehydes. The bifunctional properties of the MgF2 catalysts allowed tuning of the Lewis/Brønsted acid ratio and thus control of the parallel reactions. FUR selectivity could be maximized up to 86% at 160 °C in water using toluene as a co-solvent. When glucose was used as a feed source, high HMF selectivity (66%) could be achieved using 1-butanol as an extracting agent. In general, Brønsted sites were required to achieve high FUR and HMF selectivities. The industrial application and sustainability of the batch process in water–solvent is quite limited. The tests catalyzed by MgF2 in combination with simultaneous N2-stripping showed that a FUR selectivity of 87% could be achieved using low xylose concentrations. The combination of xylose + glucose feed showed that high FUR and HMF selectivities could still be achieved. Moreover, the catalysts regenerated by H2O2 showed high activity during the dehydration tests in water–toluene at 160 °C. In general, this work proved that partially hydroxylated catalysts show high activity to convert xylose to furfural. Moreover, the N2-stripping tests showed several advantages with respect to biphasic systems, such as higher product purity and selectivity and thus reduced separation stages. As described throughout this work, the design of an appropriate MgF2 catalyst and the use of N2-stripping proved that more sustainable chemical technologies could be developed to achieve environmentally friendly chemical industrial technologies and processes.Acknowledgements
This work was supported by funds from the Spanish Ministerio de Economía y Competitividad (CTQ-2012-38204-C03-03) and from the Gobierno Vasco (Programa de Formación de Personal Investigador del Departamento de Educación, Universidades e Investigación). The graduate school “fluorine as key element” GRK 1582 founded by the Deutsche Forschungsgemeinschaft DFG is gratefully acknowledged. The authors thank Dr. M. Feist for performing thermal analysis measurements and S. Bäßler for NH3-TPD and Pyr-adsorption measurements.Notes and references
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002 CAS.
- A. S. Mamman, J. M. Lee, Y. C. Kim, I. T. Hwang, N. J. Park, Y. K. Hwang, J. S. Chang and J. S. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2, 438 CrossRef CAS PubMed.
- K. J. Zeitsch, in Sugar Series, Elsevier, The Netherlands, 1st edn, 2000, vol. 13, p. 1 Search PubMed.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414 CrossRef CAS PubMed.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93–99 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, J. Requies, M. B. Güemez and P. L. Arias, Appl. Catal., B, 2012, 115–116, 178 Search PubMed.
- A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, Catal. Lett., 2007, 114, 151 CrossRef CAS PubMed.
- A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, Carbohydr. Res., 2006, 341, 2946 CrossRef CAS PubMed.
- A. S. Dias, S. Lima, P. Brandao, M. Pillinger, J. Rocha and A. A. Valente, Catal. Lett., 2006, 108, 179 CrossRef CAS PubMed.
- S. B. Kim, S. J. You, Y. T. Kim, S. Lee, H. Lee, K. Park and E. D. Park, Korean J. Chem. Eng., 2011, 28, 710 CAS.
- C. Moreau, R. Durand, D. Peyron, J. Duhamet and P. Rivalier, Ind. Crops Prod., 1998, 7, 95 CrossRef CAS.
- A. Chareonlimkun, V. Champreda, A. Shotipruk and N. Laosiripojana, Bioresour. Technol., 2010, 101, 4179 CAS.
- G. H. Jeong, E. G. Kim, S. B. Kim, E. D. Park and S. W. Kim, Microporous Mesoporous Mater., 2011, 144, 134 CAS.
- M. J. Antal, T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724 CrossRef CAS.
- R. Weingarten, G. A. Tompsett, W. C. Conner and G. W. Huber, J. Catal., 2011, 279, 174 CrossRef CAS PubMed.
- S. Lima, A. Fernandes, M. M. Antunes, M. Pillinger, F. Ribeiro and A. A. Valente, Catal. Lett., 2010, 135, 41 CrossRef CAS.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141 CrossRef CAS PubMed.
- Y. J. Pagán-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930 CrossRef.
- Y. R. Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef PubMed.
- S. B. Troncea, S. Wuttke, E. Kemnitz, S. M. Coman and V. I. Parvulescu, Appl. Catal., B, 2011, 107, 260 CrossRef CAS PubMed.
- E. Kemnitz, S. Wuttke and S. M. Coman, Eur. J. Inorg. Chem., 2011, 4773 CrossRef CAS PubMed.
- S. Wuttke, S. M. Coman, G. Scholz, H. Kirmse, A. Vimont, M. Daturi, S. L. M. Schroeder and E. Kemnitz, Chem.–Eur. J., 2008, 14, 11488 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, F. Hemmann, C. Jäger, P. L. Arias and E. Kemnitz, J. Catal., 2013, 305, 81 CrossRef CAS PubMed.
- Q. Jing and X. Y. Lu, Chin. J. Chem. Eng., 2007, 15, 666 CrossRef CAS.
- I. Agirrezabal-Telleria, J. Requies, M. B. Güemez and P. L. Arias, Green Chem., 2012, 14, 3132 RSC.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Güemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478 CrossRef CAS PubMed.
- D. G. Cory and W. M. Ritchey, J. Magn. Reson., 1988, 80, 128 Search PubMed.
- F. Hemmann, G. Scholz, K. Scheurell, E. Kemnitz and C. Jaeger, J. Phys. Chem. C, 2012, 116, 10580 CAS.
- D. M. Grant and R. K. Harris, Encyclopedia of Nuclear Magnetic Resonance, John Wiley & Sons Ltd., Chichester, U. K., 1996, vol. 5, p. 3247 Search PubMed.
- D. A. Torchia, J. Magn. Reson., 1978, 30, 613 CAS.
- G. Scholz, C. Stosiek and E. Kemnitz, J. Fluorine Chem., 2011, 132, 1079 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033 RSC.
- J. Mikkola, R. Sjöholm, T. Salmi and P. Mäki-Arvela, Catal. Today, 1999, 48, 73 CrossRef CAS.
- R. O'Neill, M. N. Ahmad, L. Vanoye and F. Aiouache, Ind. Eng. Chem. Res., 2009, 48, 4300 CAS.
- M. R. Nimlos, X. H. Qian, M. Davis, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 11824 CrossRef CAS PubMed.
- J. H. Zhang, L. Lin and S. J. Liu, Energy Fuels, 2012, 26, 4560 CrossRef CAS.
- H. Jadhav, E. Taarning, C. M. Pedersen and M. Bols, Tetrahedron Lett., 2012, 53, 983 CrossRef CAS PubMed.
- Y. Román-Leshkov, J. N. Cheda and J. A. Dumesic, Science, 2006, 312, 1933 Search PubMed.
- I. Agirrezabal-Telleria, J. Requies, M. B. Güemez and P. L. Arias, Appl. Catal., B, 2014, 145, 34 CrossRef CAS PubMed.
- X. Shi, Y. Wu, H. Yi, G. Rui, P. Li, M. Yang and G. Wang, Energies, 2011, 4, 669 CrossRef CAS.
- J. P. Lange, E. van der Heide, B. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00129j |
| This journal is © The Royal Society of Chemistry 2014 |