 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals
Peter J.
Deuss
a,
Katalin
Barta
*a and
Johannes G.
de Vries
*ab
aStratingh Institute for Chemistry, Rijksuniversiteit Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.barta@rug.nl; johannes.devries@catalysis.de; j.g.de.vries@rug.nl
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
First published on 23rd January 2014
Abstract
The transition from a petroleum-based infrastructure to an industry which utilises renewable resources is one of the key research challenges of the coming years. Biomass, consisting of inedible plant material that does not compete with our food production, is a suitable renewable feedstock. In recent years, much research has been focused on developing new chemical strategies for the valorisation of different biomass components. In addition to the many heterogeneous and enzymatic approaches, homogenous catalysis has emerged as an important tool for the highly selective transformation of biomass, or biomass derived platform chemicals. This Perspective provides an overview of the most important recent developments in homogeneous catalysis towards the production and transformation of biomass and biomass related model compounds. The chemical valorisation of the main components of lignocellulosic biomass – lignin and (hemi)cellulose is reviewed. In addition, important new catalyst systems for the conversion of triglycerides and fatty acids are presented.
 Peter J. Deuss | Peter J. Deuss completed his Bachelor and Masters courses at the University of Amsterdam. He then moved to the University of St. Andrews where he completed his PhD research in the group of Paul Kamer on the application of artificial metalloenzymes in transition metal catalysed reactions. Following a career development position at the LMB in Cambridge (UK), he moved to the Rijksuniversiteit Groningen as EC fellow as part of the SuBiCat Initial Training Network (ITN). His current research focuses on the development of sustainable homogeneous catalysts for the valorisation of lignin. |
 Katalin Barta | Katalin Barta is Assistant Professor of Green and Sustainable Chemistry at the Stratingh Institute, University of Groningen, where her research focuses on the conversion of renewable resources by homogeneous and heterogeneous catalysis. After finishing her master's thesis at ELTE Budapest, Hungary (with István T. Horváth), she gained her PhD at RWTH-Aachen, Germany with Walter Leitner. She then carried out post-doctoral research in the group of Peter Ford at University of California, Santa Barbara and was Associate Research scientist at the Center for Green Chemistry and Engineering at Yale University. |
 Hans de Vries | Hans de Vries received a PhD from the University of Groningen in 1979. After a postdoc at Brandeis University, Waltham, USA, his first job was as a medicinal chemist with Sandoz in Vienna and in London. From 1988–2013 he worked for DSM in Geleen, The Netherlands as a Principal Scientist in the area of homogeneous catalysis. In 1999 he was appointed part-time professor at the University of Groningen. In 2014, he commenced a new career as professor at the Leibniz Institute for Catalysis in Rostock, Germany. His research interests are in the area of homogeneous hydrogenation, metal-catalysed C–C and C–X bond formation, combinatorial catalysis, nanocatalysis and catalytic conversion of renewable resources and platform chemicals. |
1. Introduction
In the last century, fossil fuels have been the resource of choice for most of our chemical and energy needs. However, due to the predicted decline in the long-term availability of these supplies and their increasing demand, interest in renewables has been re-discovered in the last decade.1 Biomass is inexpensive and globally accessible. The available feedstocks should be sufficient to replace a significant portion of the non-renewable raw materials now required by the chemical industry.1d,2 Moreover, currently, a large portion of biomass originating from inedible plant material is considered to be waste and is burned to provide heat. There is an increasing need for a better utilisation of these waste streams in the future.3 Selective biomass conversion to biofuels and useful chemicals requires significant improvements in the current chemical approaches and technologies. These novel strategies will play a key role in the implementation of the bio-based economy relying on renewable resources and have a significant environmental and societal impact.1,4Two main approaches have been envisioned for the valorisation of biomass resources.1d,5 The “drop-in strategy” relies on converting biomass into bulk chemicals used in the current production processes. This approach is highly attractive and economically viable, as it allows for the application of previously developed technologies in an already existing infrastructure. The second approach involves complete substitution of conventional bulk chemicals by new, biomass-based platform chemicals. This, “emerging strategy”, which is probably more preferred in the long term, takes advantage of the higher degree of functionality inherently present in bio-polymers and could create new materials, with advantageous properties. These new reaction pathways should ultimately lead to processes requiring fewer reaction steps, in agreement with the principles of green chemistry.4a,6
Catalysis will play a key role in enabling both the short term “drop-in strategies” and longer term “emerging strategies”.1d However, the development of new approaches for the conversion of renewables still represents a great challenge. Conventionally, value added chemicals are obtained stepwise, starting from more simple structures, obtaining more complex ones. Thus, existing catalytic systems are typically focused on the addition of functionality to simple molecules, comprising mainly reduced carbon. The valorisation of biomass or biomass-derived feedstocks firstly requires the selective depolymerisation of complex, highly oxygenated biopolymers or oligomers followed by selective defunctionalisation (Fig. 1).7 In order to enable these novel pathways, fundamentally new catalysts, chemical reactions, and mechanistic insights are necessary.
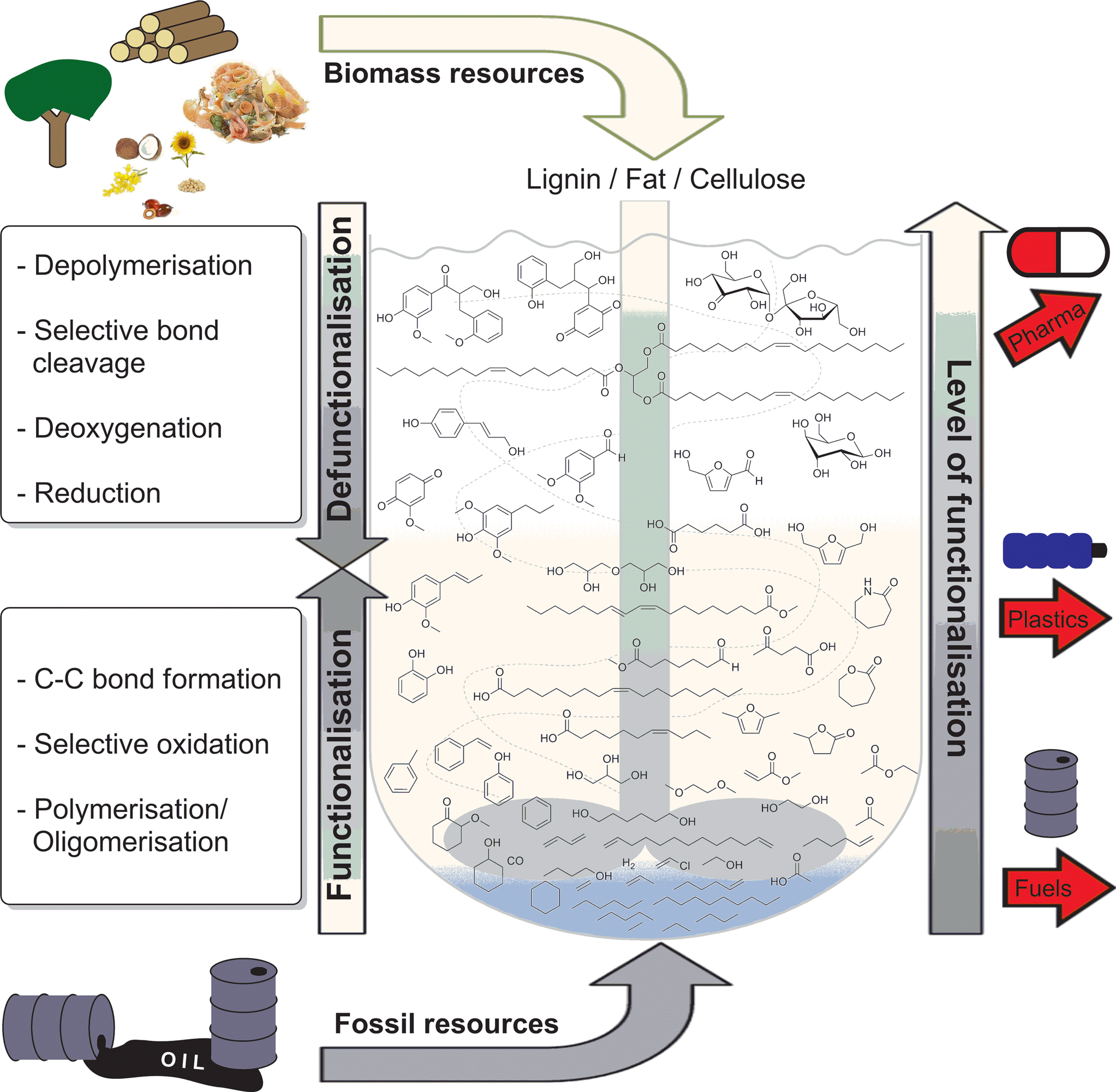 | ||
| Fig. 1 The concept of catalysed defunctionalisation of biomass as opposed to catalysed functionalisation of fossil resources towards platform chemicals for commercial products. | ||
Many significant improvements have already been made in the field of heterogeneous catalysis, especially in the production of biofuels. These have been extensively reviewed for the sugar platform1b,c,4c,e,8a as well as for lignin.1b,8b,9 The current research efforts in homogeneous catalysis have also led to the development of promising new methods and insights. In this Perspective, we attempt to summarize these efforts, focusing on recent developments in the field of homogeneous catalysts for important transformations such as selective depolymerisation, defunctionalisation, or functionalisation. For industrial applications, easily removable/recyclable heterogeneous catalysts are typically preferred.10 However, in the end, it is the catalyst cost per kg of product that really counts and indeed many bulk chemical processes are known in which soluble transition metal complexes are used as the catalyst, in particular those related to CO chemistry. Homogeneous catalysis provides advantages that cannot be equalled by the heterogeneous counterparts.11 Well-defined and carefully designed catalyst structures allow for high levels of activity and selectivity. In addition, the solubility of the catalysts simplifies detailed mechanistic studies, which can be used as a starting point for the improvement of their activity, selectivity and stability. Some spectacular examples are known, where the catalyst activity was improved by many orders of magnitude, which is unparalleled in the world of heterogeneous catalysis.12 Soluble catalysts have the additional advantage of being able to penetrate better to the linkages targeted for depolymerisation in the polymeric materials that are the constituents of lignocellulosic biomass. One particular challenge in the transformation of biomass derived feedstocks is the co-existence of competing pathways that lead to decreased product yields and selectivities at the temperatures usually applied for defunctionalisation and/or depolymerisation. Homogeneous catalysts operating under milder reaction conditions hold the potential to ameliorate these hurdles in process development. Thus, homogeneous transition metal based catalysts are expected to serve as an important tool in the valorisation of biomass.
In this Perspective, we consider the three main types of biomass derived starting materials for the production of value added chemicals, based on the availability and suitability as sustainable resources (Fig. 2). First, the chemistry related to the valorisation of the main components of lignocellulose: lignin, (hemi)cellulose and in particular, the constituent sugars, will be discussed. The first section encompasses the production of mostly aliphatic platform chemicals, from glucose and fructose, and their further conversion into “drop-in” chemicals. The second section describes efforts for valorisation of the amorphous biopolymer lignin, an important and unique potential source of aromatic platform chemicals. Here, the main challenge is a selective depolymerisation through highly specific bond cleavage reactions. The third section includes catalyst development for the conversion of triglycerides. In this case, the focus lies in the valorisation of this feedstock through selective synthetic pathways for the modification of fatty acids. Fatty acid derived products find increasing applications in polymer and materials science. Other biomass resources, for example suberin, tannins, terpenes and chitin, the application of which has been detailed elsewhere,1c,13 are not further discussed in this perspective.
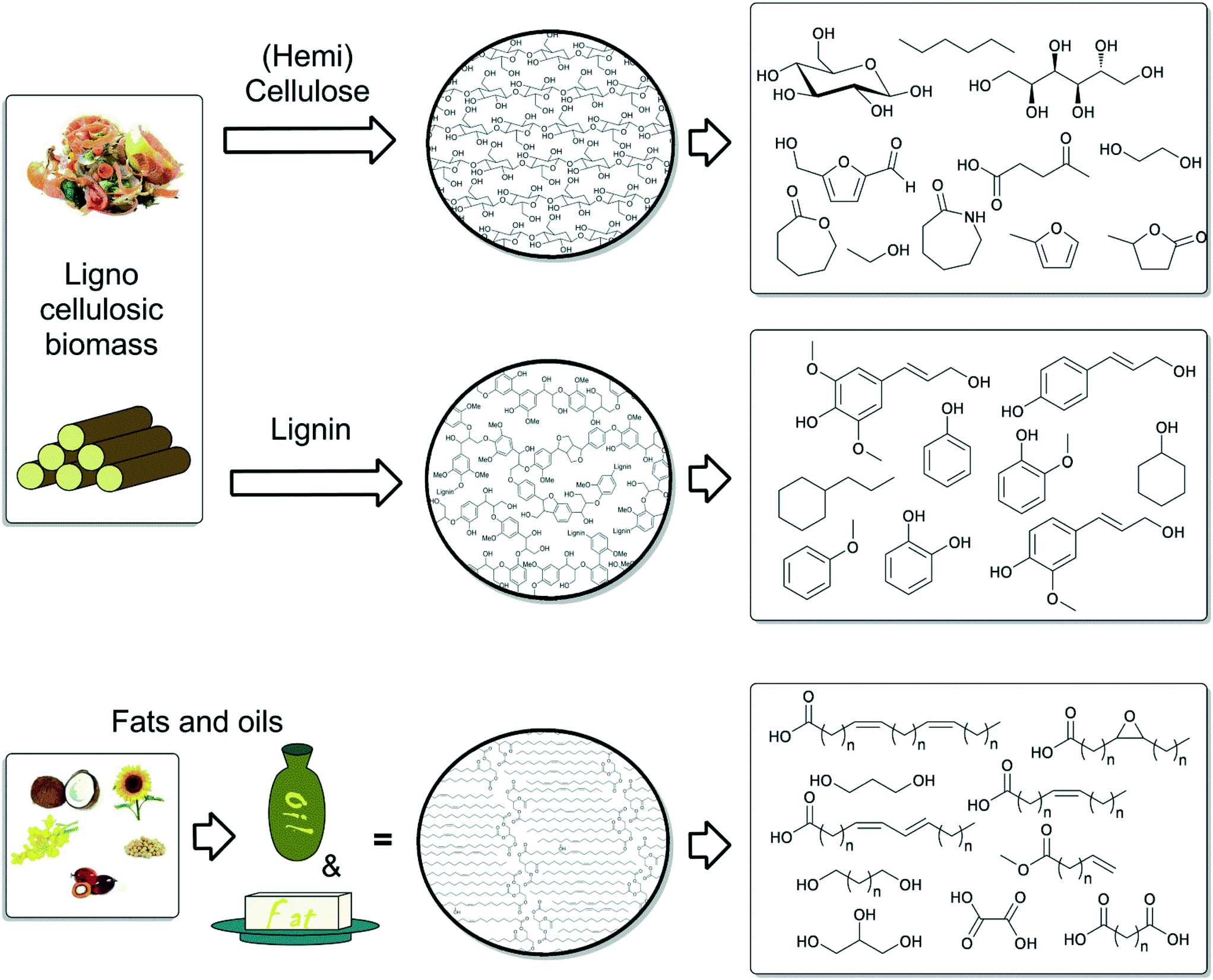 | ||
| Fig. 2 Different sources of biomass and derived chemical products. | ||
2. Valorisation of (hemi)cellulose
The first lignocellulosic bioethanol plants are being built as we write this Perspective. This also means that technology to enzymatically liberate glucose form cellulose has now reached a level where it can compete with starch, or sucrose-derived glucose. The acid-catalysed hydrolysis of cellulose to glucose is also still under investigation. This section will describe the use of homogeneous catalysis in the conversion of cellulose, monomeric sugars and sugar or cellulose-derived platform chemicals, such as levulinic acid (LA) and 5-hydroxymethylfurfural (HMF).2.1 Reactions of sugars
In view of the poor solubility of sugars in anything but water, limited transformations by transition metal complexes have been reported. Most reports describe the use of water-soluble catalytic transition metal complexes in order to overcome this limitation.The dehydration of fructose or glucose to form HMF using Brønsted acids in water suffers from the instability of HMF under these conditions. It is rehydrated and forms levulinic acid. In order to suppress this side reaction, the use of soluble Lewis acids in dipolar aprotic solvents has been developed.14 One of the best results was obtained by Ishida and co-workers who used LaCl3 as a catalyst in DMSO and other similar solvents (Scheme 1). They were able to obtain HMF in a 93% yield.15 Unfortunately, the recovery of HMF from DMSO is not economically feasible and DMSO is not stable under the conditions used for this reaction, and for this reason two-phase systems are preferred.
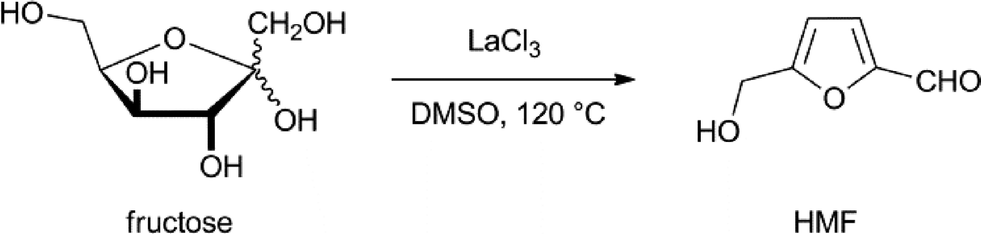 | ||
| Scheme 1 Dehydration of fructose to HMF.15 | ||
Soluble Lewis acids have also been used in the conversion of glucose to lactic acid.16 Recently, it was shown that even cellulose can be converted to lactic acid, in up to a 68% yield, using Pd(NO3)2 as a catalyst at 190 °C.17 When using Er(OTf)3 as the catalyst, lactic acid was obtained from cellulose in close to a 90% yield, at 240 °C.18
Šunjić reported the homogeneous hydrogenation of glucose to sorbitol and of mannose to mannitol using RuCl2(TPPTS)3 as the catalyst.19 In addition, he was able to use the same catalyst for the transfer hydrogenation of these sugars using HCO2H–Et3N mixtures as the reductant. Rajagopal and co-workers reported the transfer hydrogenation of glucose and fructose. They used RuCl2(PPh3)3 as the catalyst and isopropanol as the reductant, in mixtures of water and a polar aprotic solvent, such as DMF or DMA. With glucose, the main product was sorbitol, although glucono-1,5-lactone was formed as a side product in most of the reactions, through an internal redox reaction of glucose.20 The transfer hydrogenation of fructose gave glucitol and mannitol as the only products.
Sinou and co-workers were able to oxidise protected sugars to their respective lactones using benzalacetone as hydrogen acceptor and RuH2(PPh3)4 as the catalyst.21 The group of Bäckvall was able to racemise the remaining secondary alcohol group in doubly protected sugars using (η5-C5Ph5)Ru(CO)2Cl as the catalyst. However, in all these cases, mixtures of the two epimers were formed.22 Grützmacher et al. reported a rhodium-catalysed dehydrogenative coupling between alcohols and water, to produce the corresponding carboxylic acids.23 Cyclohexanone or acetone was used as the hydrogen acceptor. In this way, they were able to convert glucose methyl acetal into the corresponding glucuronic acid in a 66% yield (Scheme 2a). Similarly, the reaction with benzylamine, instead of water, gave the N-benzylamide in a 85% yield. Xiao et al. reported the reductive amination of glucose with phenylalanine and tryptophane via a transfer hydrogenation with HCOOH–Et3N, using an iridacycle catalyst (Scheme 2b).24
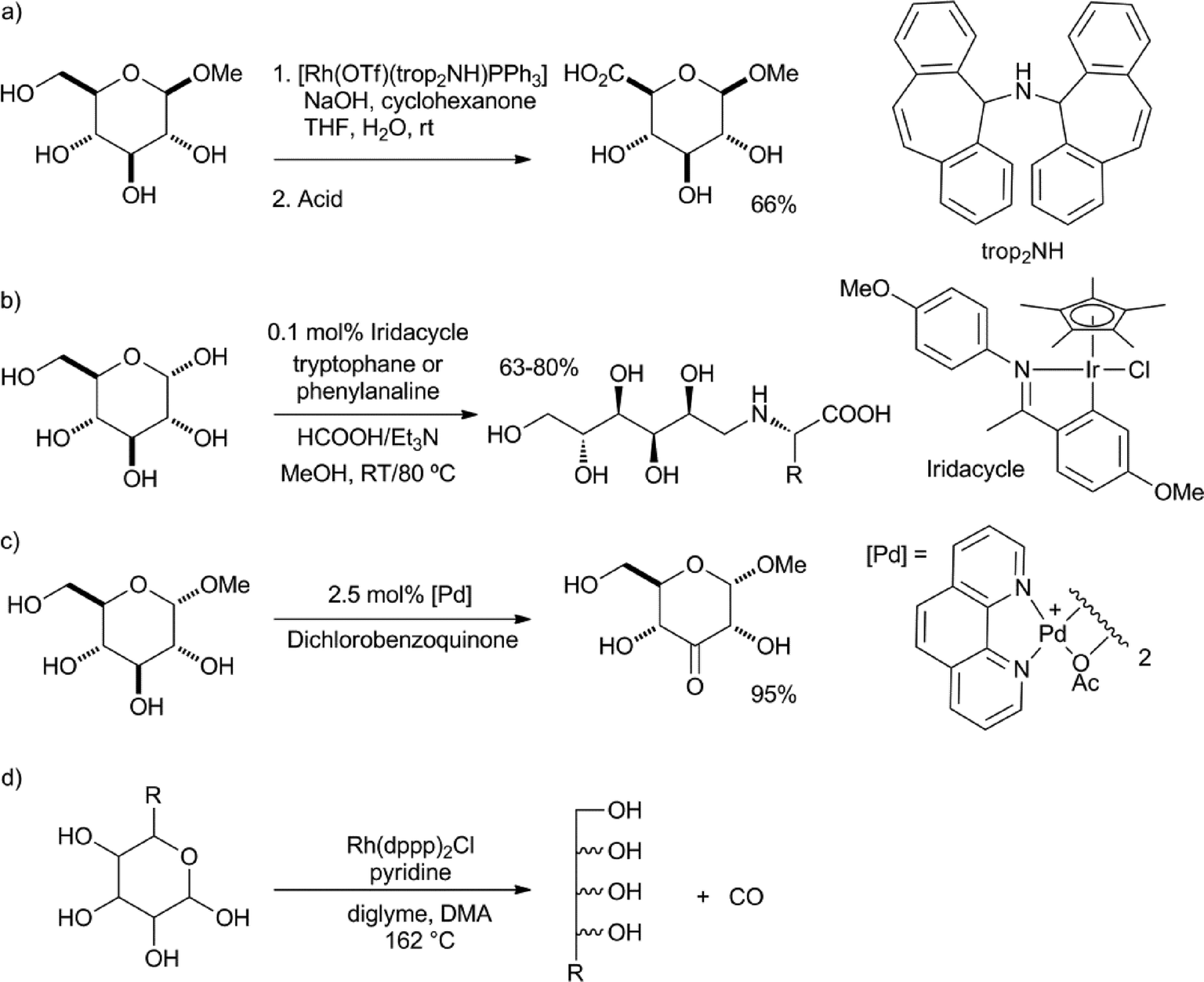 | ||
| Scheme 2 a) Dehydrogenative coupling between glucose methyl acetal and water.23 b) Reductive amination of D-glucose.24 c) Highly selective oxidation of glucose methyl ether.26 d) Rhodium-catalysed decarbonylation of sugars.27 | ||
Minnaard, de Vries and co-workers reported, using the palladium catalyst developed by Waymouth,25 a remarkably selective oxidation of pyranosyl glycosides.26 In spite of the fact that only the hydroxyl group at the anomeric centre was protected, the reaction showed an overwhelming preference for oxidation of the hydroxyl group in the 4-position (Scheme 2c). Madsen and co-workers reported the decarbonylation of hexoses and pentoses, using Rh(dppp)2Cl as the catalyst (Scheme 2d).27 The pentaols and tetraols were isolated in yields of up to 74%, respectively.
2.2 Levulinic acid
The hexoses in lignocellulosic biomass can be converted into the platform chemical LA and formic acid, by treatment with dilute sulfuric acid, at around 200 °C (Scheme 3).28 The yields are surprisingly high (55–75%). The C-5 sugars are simultaneously converted into furfuraldehyde. A process was developed by the company Biofine, which was proven on a pilot scale. A large scale plant was built in Italy, but unfortunately this plant never produced levulinic acid.29 Other companies are working on improved levulinic acid processes.30 | ||
| Scheme 3 Conversion of lignocellulosic biomass into levulinic acid.28 | ||
Levulinic acid can be hydrogenated to γ-valerolactone (GVL) in good yields, using heterogeneous ruthenium catalysts.31 A number of groups have reported the use of homogeneous catalysts for this transformation (Table 1). Several homogeneous ruthenium catalysts were reported, such as RuCl2(PPh3)3, Ru(acac)3/PBu3, and Ru(acac)3/P(n-Oct)3 (entries 1–3).32 The use of an iridium pincer complex led to very high turnover numbers (entry 4).33 Unfortunately, in this process, a stoichiometric amount of base is necessary, making it impractical on a large scale. Fu et al. reported the use of iridium half-sandwich complexes and obtained good yields and turnover numbers in the hydrogenation of LA in water (entry 5).34
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | S/C | T (°C) | H2 (bar) | t (h) | GVL yield (%) | Ref |
| a TPPTS = P(C6H4-m-SO3Na)3. b R = Me, n-Pr, i-Pr, n-Bu, cyclopentyl. | ||||||||
| 1 | RuCl2(PPh3)3 | — | 200 | 180 | 12 | 24 | 99 | 32a |
| 2 | Ru(acac)3/PBu3/NH4PF6 | — | 1559 | 135 | 100 | 8 | 100 | 32b |
| 3 | Ru(acac)3/P(n-Oct)3 | 1000 | 160 | 100 | 18 | 99 | 32c | |
| 4 |
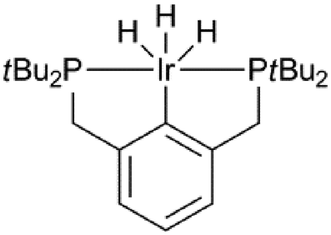
|
KOH (120 mol%) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
100 | 50 | 24 | 98 71 | 33 |
| 100000 |
100 | 100 | 48 | |||||
| 5 |
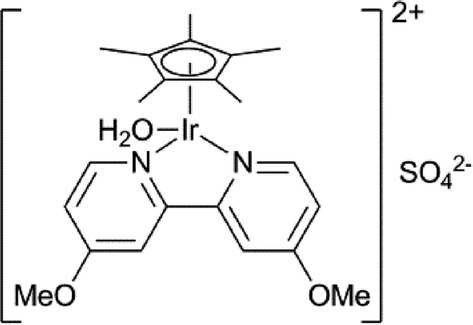
|
— | 10000 |
120 | 10 | 4 | 98 87 | 34 |
| 100000 |
120 | 10 | 36 | |||||
| 6 | HRu(Ph2PC6H4-m-SO3Na)3X (X = Cl, OAc) | — | 100 | 60 | 0.81 | 1.4 | 100 | 35 |
| 7 | Ru(acac)3/TPPTSa | — | 600 | 140 | 70 | 12 | 95 | 32b, 36 |
| 8 | Ru(acac)3/RP(C6H4-m-SO3Na)2b | — | 6370 | 140 | 10 | 4.5 | 39–100 | 37 |
| 100 | 1.8 | 96–100 | ||||||
| 9 | Ru(acac)3/R2P(C6H4-m-SO3Na)b | — | 6370 | 140 | 10 | 4.5 | 42–91 76– | 37 |
| 100 | 1.8 | 92 | ||||||

Water soluble ruthenium catalysts were pioneered by Joo and co-workers; for the hydrogenation of LA they used a ruthenium complex based on mono-sulfonated triphenylphosphine (entry 6).35 Later work was based on catalysts made in situ from Ru(acac)3 and TPPTS (entry 7).32b,36 This methodology allows the easy separation of the catalyst and reuse of the catalyst containing an aqueous phase in consecutive runs. Kühn and co-workers tested a range of different water-soluble ligands in the ruthenium-catalysed hydrogenation of LA; the best results were obtained with TPPTS.38 However, it was noticed that higher turnover frequencies were obtained using ligand-free Ru(acac)3, which presumably is reduced to ruthenium nanoparticles, and with ruthenium on alumina. As the hydrogenation rate is usually enhanced by the use of electron-donating ligands, Mika and co-workers developed the use of mixed phosphines, carrying one (entry 8) or two alkyl groups (entry 9) and one or two sulfonated phenyl groups.32a,37 The best results were obtained with n-BuP(C6H4-m-SO3Na)2 (full conversion after 1.8 h, amounting to a turnover frequency of 3185).
Heeres and co-workers performed a dehydration of glucose to LA and its simultaneous hydrogenation using RuCl3/TPPTS as the hydrogenation catalyst, 94 bar of hydrogen and dilute TFA as the dehydration catalyst, to obtain 19% of LA and 23% of GVL.39 Interestingly, Leitner and co-workers, upon hydrogenation of LA with Ru(acac)3/TRIPHOS at 160 °C, obtained a 95% yield of 1,4-pentanediol (Scheme 4).32c If the hydrogenation of LA with this catalyst was carried out in the presence of NH4PF6 and a small amount of an ionic liquid, 2-methyl-tetrahydrofuran was obtained in a good yield.
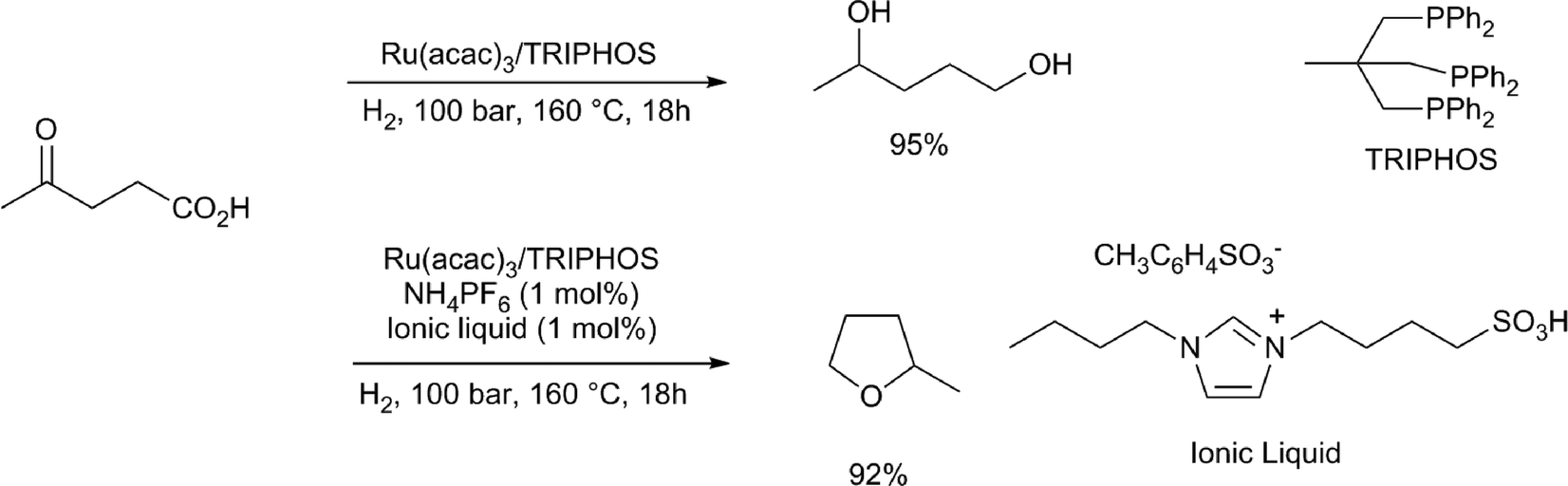 | ||
| Scheme 4 Conversion of levulinic acid into 1,4-pentanediol and 2-methyl-tetrahydrofuran.32c | ||
Several groups have reported the use of transfer hydrogenation for the conversion of LA to GVL (Table 2). This is an attractive option, as formic acid is produced along with LA in similar amounts, in the conversion of lignocellulosic biomass to LA (Scheme 3).

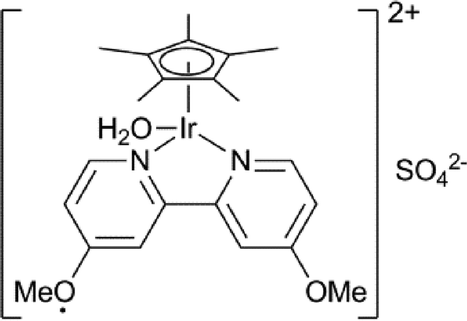
Horvath reported that the addition of the Shvo catalyst to a crude fructose dehydration mixture, containing LA and formic acid, led to the formation of GVL in about a 55% yield.41 Williams used the fact that the primary reduction product of LA, 4-hydroxypentanoic acid, spontaneously ring-closes to GVL, as a method to drive an Oppenauer oxidation of aromatic alcohols with LA as the oxidant. He used combinations of Ru(PPh3)3(CO)H2 with bisphosphine ligands as catalysts and obtained aromatic ketones with excellent yields.42
The group of Xiao performed reductive amination reactions of LA, with anilines and alkylamines, using an iridacycle catalyst with formate as the reductant, to obtain the N-aryl- and N-alkyl-pyrrolidinones in yields between 73–97% (Scheme 5).43 The reaction worked best in a narrow pH range with pH 3.5 as the optimum, which he established by using a mixture of HCO2H and HCO2Na.
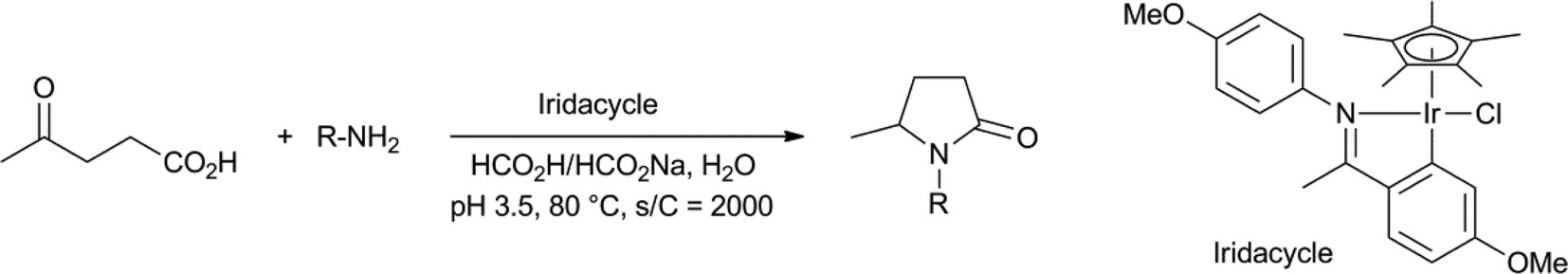 | ||
| Scheme 5 Reductive amination of levulinic acid to N-substituted pyrrolidinones.43 | ||
Two groups have reported a process for the production of adipic acid from levulinic acid. Both processes start with the hydrogenation of LA to GVL. This is usually achieved with heterogeneous ruthenium catalysts in good yields. In the DSM process, GVL is subjected to a gas-phase ring opening reaction, yielding a mixture of isomeric methyl pentenoates (Scheme 6). This mixture was subjected to an isomerising methoxycarbonylation reaction, using the palladium catalyst developed by Lucite for their methyl methacrylate process, giving dimethyl adipate with a selectivity in excess of 99%.44 The acid-catalysed hydrolysis of dimethyl adipate gives adipic acid. Chemists from ICES-Astar, in Singapore, developed a very similar process, in which the ring opening of GVL was performed by adding an acidic catalyst and distilling out a mixture of pentenoic acids.45 Using the same catalyst as in the DSM process, they perform the isomerising hydroxycarbonylation of this mixture of pentenoic acids, to give adipic acid directly.
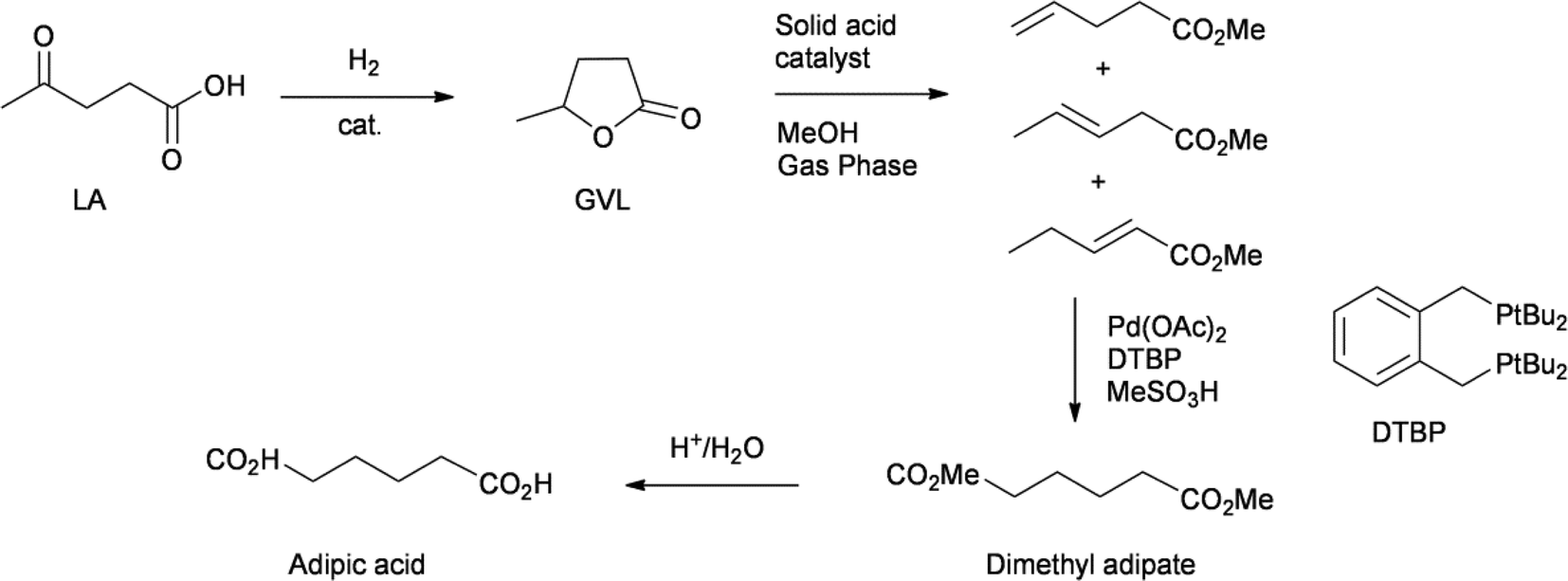 | ||
| Scheme 6 Production of adipic acid from LA.43,44 | ||
2.3 Hydroxymethylfurfural
The treatment of fructose with acidic catalysts at high temperatures leads to the formation of the platform chemical 4-hydroxymethyl-2-furfural (HMF).14 To prevent the consecutive formation of LA from HMF via rehydration, this reaction should be performed in dipolar aprotic solvents like DMF or DMSO, or better, in two-phase aqueous–organic solvent systems. It is also possible to prepare HMF from glucose, or even cellulose and lignocellulose, however, this requires the use of catalytic CrCl2 for the isomerisation of glucose to fructose.46 The oxidation of HMF gives 2,5-furandicarboxylic acid, which is under investigation as a replacement for phthalic acid in polyesters, like PET.46 A number of C–C bond forming reactions have been reported to further add functionality to HMF.47 Sheldon and co-workers reported the carbonylation of HMF, using PdCl2/TPPTS as the catalyst (Scheme 7a).47a In addition to 4-formyl-2-furancarboxylic acid, they also found substantial amounts of 4-methyl-furfuraldehyde, which presumably is formed by decarboxylation of the acid. Feringa, de Vries and co-workers reported the coupling between HMF and two equivalents of isoprene, using the reductive coupling method developed by Krische (Scheme 7b).47b Similar chemistry was performed on other substituted furfurals.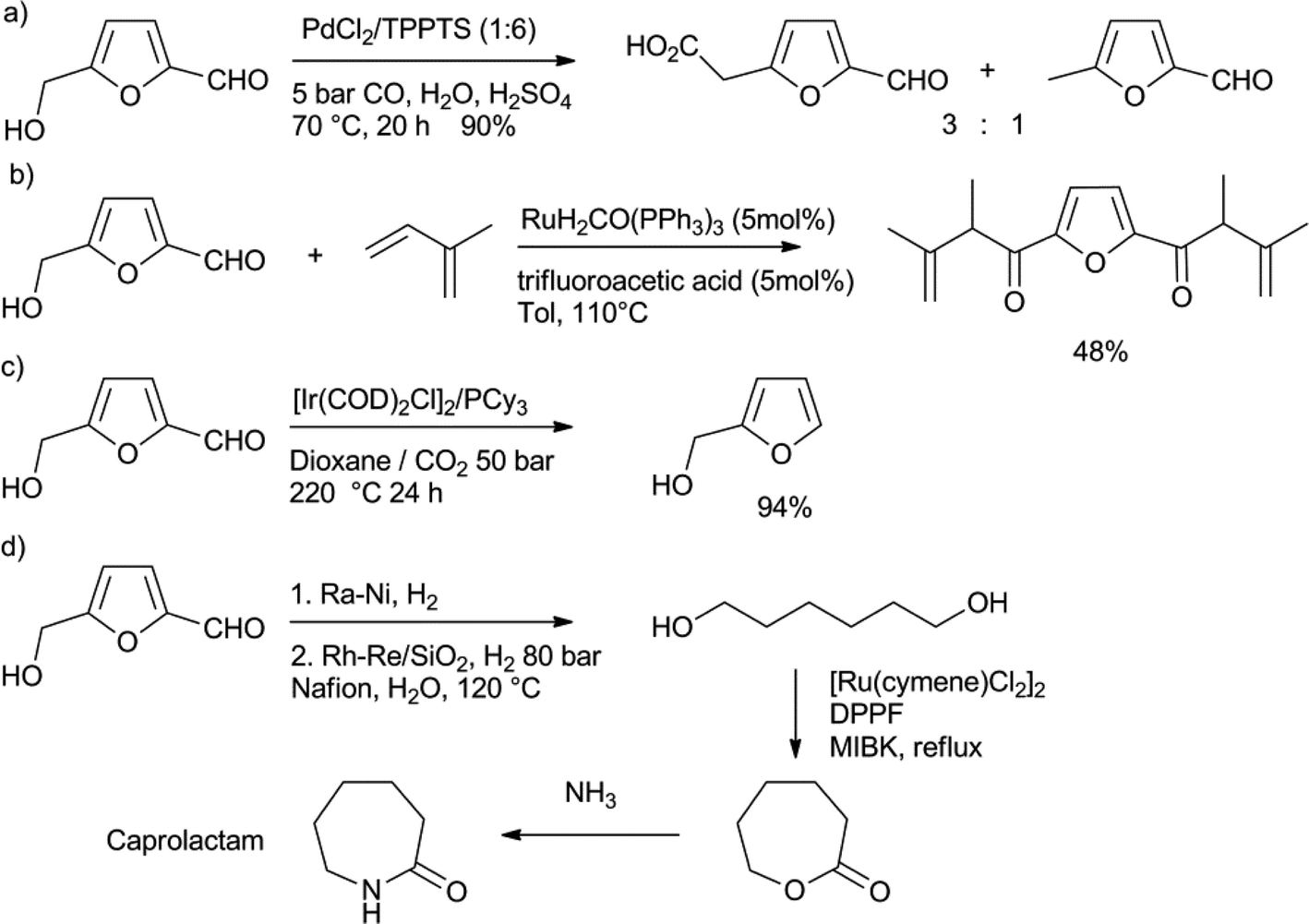 | ||
| Scheme 7 a) Palladium-catalysed carbonylation of HMF.47a b) Reductive coupling of isoprene to HMF.47b c) Decarbonylation of HMF to furfurylalcohol.48 d) Lactonisation of 1,6-hexanediol as the key step in the conversion of HMF into caprolactam.49 | ||
Klankermayer, Leitner and co-workers reported the decarbonylation of HMF, using an iridium catalyst (Scheme 7c).48 In order to achieve a full conversion, very high temperatures were needed, which led to poor selectivities. However, the use of CO2 expanded dioxane as the solvent for this decarbonylation solved this problem and led to the formation of furfurylalcohol in a very good yield. Heeres, de Vries and co-workers described a route to transform HMF into caprolactam, the monomer for nylon-6 (Scheme 7d).49 They were able to hydrogenate HMF to 1,6-hexandiol in two steps. The diol was then subjected to a ruthenium catalysed lactonisation reaction, to form caprolactone in virtually a quantitative yield.50 As caprolactone had been previously converted into caprolactam on an industrial scale, this constitutes an interesting pathway to bio-based nylon-6.
2.4 Isosorbide
The hydrogenation of glucose to sorbitol is practised on an industrial scale. Under the influence of acids or Lewis acids, sorbitol can be dehydrated to isosorbide.51 Recently, the direct conversion of cellulose to isosorbide was also developed.52 Isosorbide is currently being investigated as a diol in bio-based polyesters. Both Beller et al., as well as Vogt et al., developed the direct catalytic amination of isosorbide to a mixture of isomers of the diamine, using a homogeneous ruthenium catalyst (Scheme 8). A number of different bidentate phosphines were tested in this reaction and the Beller group found that xantphos gave the best results, in combination with HRu(PPh3)3(CO)Cl.53 Vogt et al. explored a similar chemistry and tested combinations of Ru3(CO)12 and bidentate phosphines.54 They obtained their best results with the acridine based ligand (1). The diamines can be used as monomers for polyamides and polyurethanes.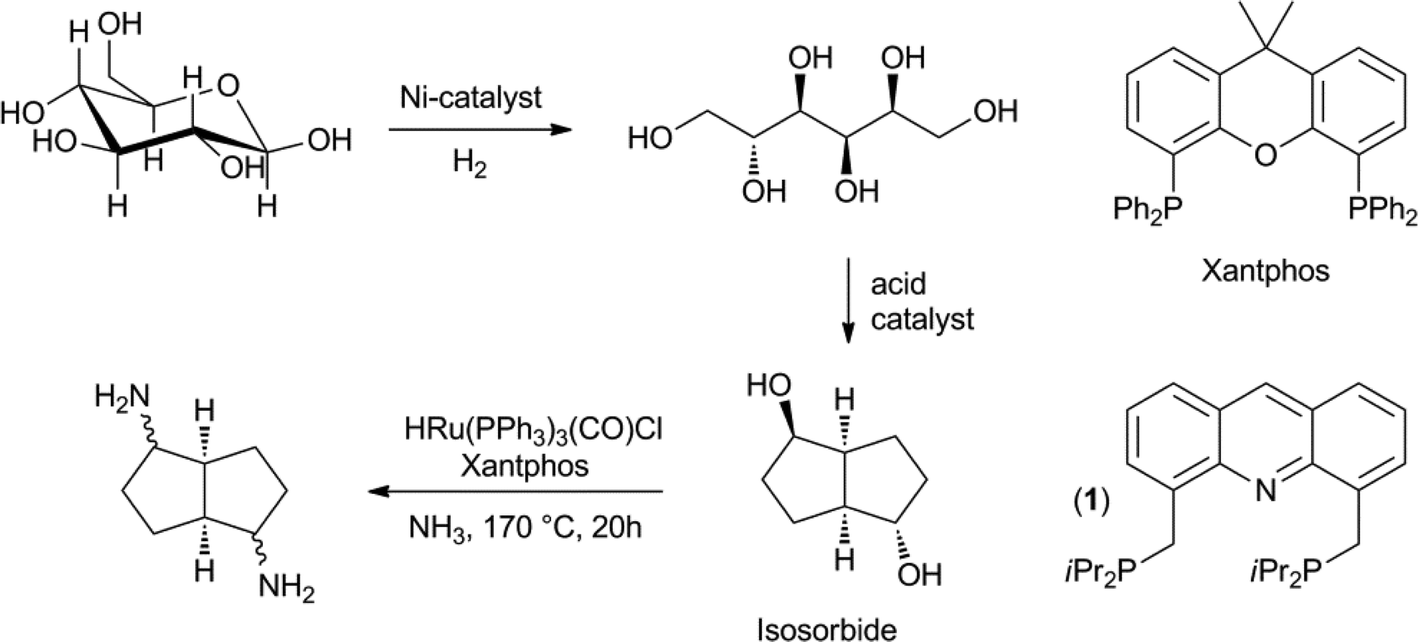 | ||
| Scheme 8 Ruthenium-catalysed direct amination of isosorbide. | ||
3. Selective depolymerisation of lignin
Due to its high aromatic content, lignin has great potential to function as an alternative to non-renewable fossil resources, for the production of aromatic fine chemicals.2a,4b,9,55 However, because of its amorphous structure, lignin is highly refractory towards chemical modification, which represents a major challenge in its efficient utilisation. Lignin consists of mainly propyl-phenolic units that are randomly cross-linked via various types of C–O and C–C linkages. The most common sub-units in lignin are derived from the constituent monolignans: coniferyl alcohol, sinapyl alcohol and coumaryl alcohol (Fig. 3). Lignins are structurally very diverse, and contain different ratios of these building blocks, depending on the plant source. The guaiacyl/syringyl ratio is an important characteristic when considering different plant materials as feedstocks.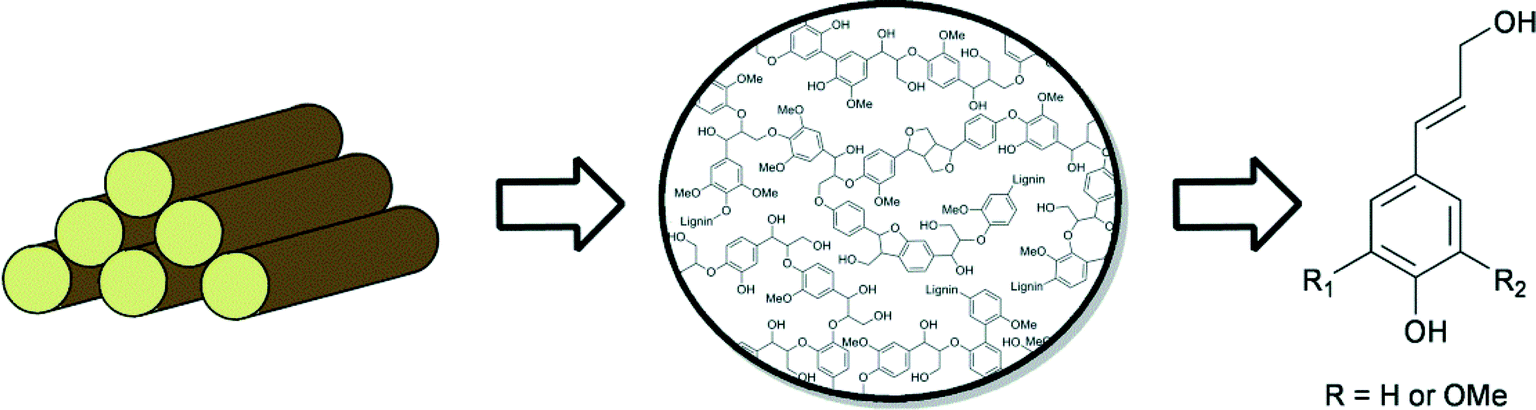 | ||
| Fig. 3 Lignin structure and monomeric units. | ||
Another characteristic that influences the properties of various plant materials is the ratio of the different types of linkages encountered. The most commonly found C–C and C–O inter unit bond motifs are depicted in Fig. 4.9,56 Of these, the β–O–4 motif is by far the most abundant (up to 50%) and is thus an important target for selective depolymerisation efforts. Most other bond motifs also contain aromatic or aliphatic ether sub-units. In a recent paper, Beckham et. al. compared the bond dissociation energies (BDE) of the most prevalent types of bonds in lignin.56d This study found that cleaving the most recurring β–O–4 bond and the rarer α–O–4 bond (3–5%) would be most viable (~70 and ~55 kcal mol−1, respectively). As a comparison, the BDE of the biphenyl-ether type linkage, that makes up 3.5–25% of the bonds in lignin and the β–5 linkage, that makes up 4–10%, are around 115 and 105 kcal mol−1, respectively (precise values determined for differently substituted models). Considering a strategy that allows for the formation of a ketone in the β–O–4 structural unit, either by dehydrogenation or oxidation, further decreases the BDE to below 60 kcal mol−1, making this a desirable approach for depolymerisation via selective bond cleavage.
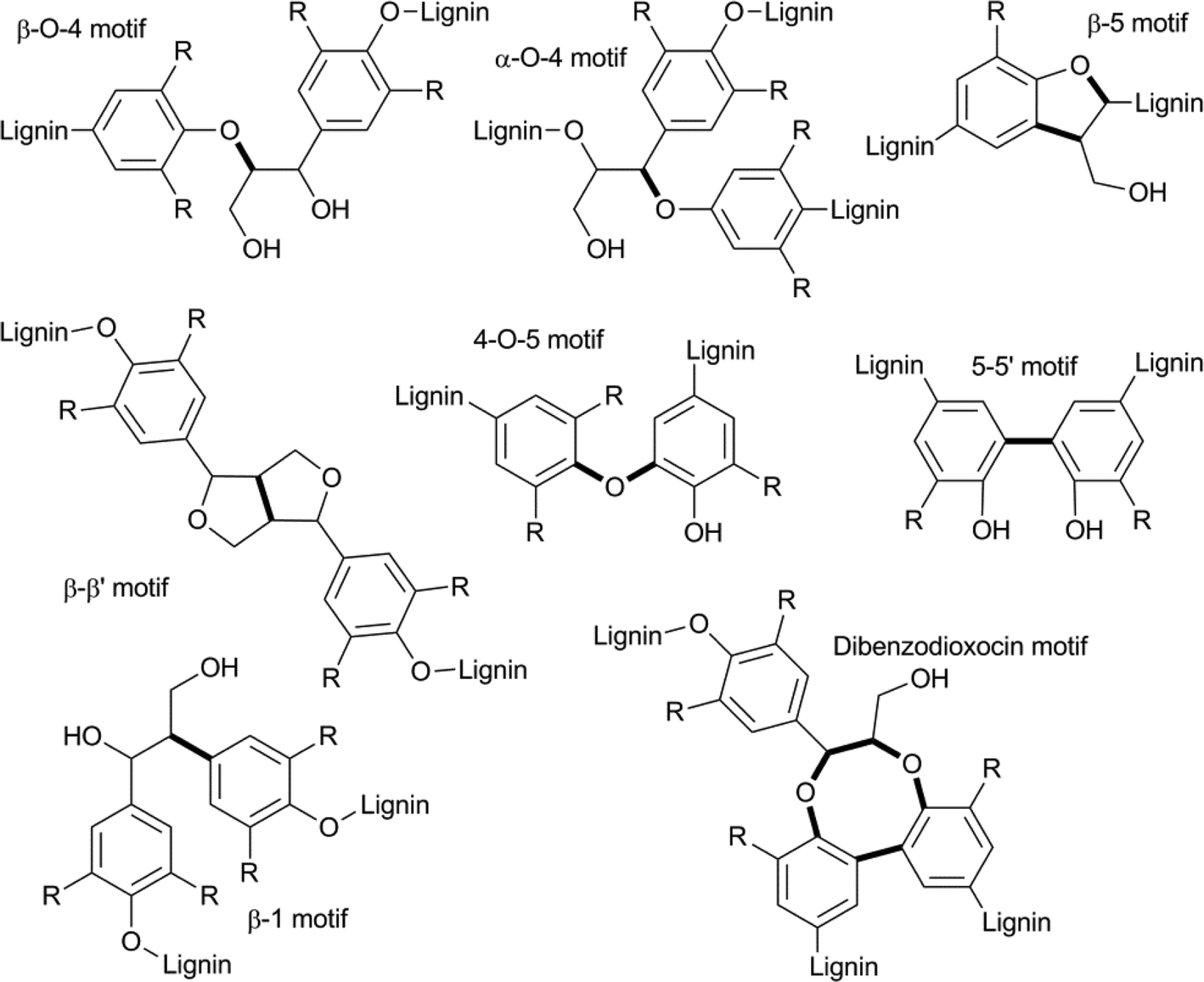 | ||
| Fig. 4 Typical bond-motifs found in lignin and linkages (bold) that serve as potential targets for selective cleavage. | ||
The key to the successful utilisation of lignin is the development of efficient methodologies for its depolymerisation to smaller aromatic units. Additionally, subsequent selective chemical transformations are required, mainly aimed at defunctionalisation of these aromatic intermediates, in order to obtain desired aromatic platform chemicals.2a Historically, compounds like phenols, substituted phenols and several aromatic and aliphatic acids have been produced from lignin.55 Also, more valuable products, like vanillin, can be obtained.57 Further targets may be products derived from coumaryl, sinapyl and coniferyl alcohols, but substituted catechols77 or functionalised styrenes might also become available on a larger scale, which could lead to market applications.2a
One of the challenges in the effective depolymerisation of lignin is that its structure is bulky and highly functionalised. This steric hindrance limits the access of the catalyst to target linkages, especially if the catalyst itself is heterogeneous in nature.7a Although various heterogeneous catalysts for lignin9,59 or lignocellulose8b,60 degradation are known, and allow for direct use of raw biomass in a one-pot approach, these generally require higher reaction temperatures. Soluble transition metal complexes should also become a viable alternative, especially for the efficient conversion of already solubilised bio-polymers using milder reaction conditions. Catalysts capable of penetrating the macromolecular structure of lignin would potentially allow for the use of milder reaction conditions. Described below is a selection of the most promising catalyst systems towards lignin valorisation.
3.1 Reductive approaches for bond cleavage in lignin and model compounds
An elegant method for the redox neutral cleavage of the β–O–4 motif containing lignin model compounds was reported by Bergman, Ellman and coworkers, using Ru(CO)H2(PPh3)3 in combination with xantphos (Scheme 9).61 The hydrogenolysis of the β–O–4 bond was achieved by a tandem α-alcohol dehydrogenation and reductive ether cleavage. Studies showed that the formation of the ketone is essential for the subsequent β–O–4 bond cleavage, as it makes this bond more susceptible to hydrogenolysis by the ruthenium–phosphine complex. Of the phosphine ligands applied; PPh3, PCy3, dppm, dppp, dppbz, dppf and xantphos, only the last imparted a significant activity. Computational studies confirmed that the sequential dehydrogenation was followed by hydrogenolysis.62 A later study showed that, using hydrogen gas, the ruthenium–xantphos complex is capable of the direct hydrogenolysis of the β–O–4 motif containing a ketone at the benzylic carbon instead of an alcohol.63 Different catalysts, consisting of alternative combinations of ruthenium precursors and phosphine ligands, have been reported for the conversion of similar model compounds. Triphos ligands, in combination with [Ru(cod)(methylallyl)2], catalysed the redox neutral cleavage of the same β–O–4 motif.64 With this ruthenium precursor, PPh3, dppe and interestingly also xantphos, did not give a significant cleavage. Of the two triphos ligands that were tested, (Ph2PCH2CH2)2PPh proved most effective. Interestingly, a wider variety of ruthenium precursors and phosphine ligands (e.g. triphenylphosphine) have been successfully applied in the same reaction, when a mixture of potassium t-amylate and ethylacetate was additionally applied.65 In this case, the cleavage could also be coupled to a second hydrogen autotransfer process, by applying a subsequent α-alkylation step with primary alcohols. Of these studies showing a ruthenium catalysed redox neutral β–O–4 cleavage, only James et al. reported on the use of more complex lignin model compounds. These substrates, which more closely resemble the actual lignin motif, additionally contained a γ-OH primary alcohol moiety.63 In this case however, cleavage of the phenyl–ether bond was not successful, due to the formation of stable substrate–ruthenium chelates.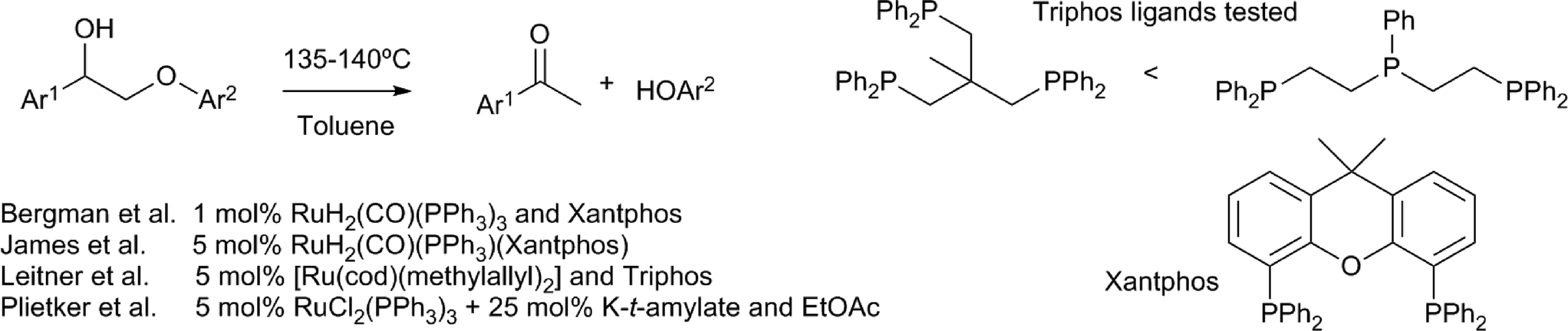 | ||
| Scheme 9 Lignin models representing the β–O–4 moiety and their cleavage by ruthenium–phosphine complexes.61,63–65 | ||
Hartwig and co-workers reported a highly efficient method for the selective cleavage of lignin model compounds containing aryl–ether linkages, using a catalyst prepared in situ from nickel(COD)2 and an N-heterocyclic carbene ligand (Scheme 10).66 Aromatic ring hydrogenation could be prevented by the addition of a base, and this method could be extended to a whole range of substituted diaryl ethers. This methodology was most effective with substrates bearing an electron withdrawing groups. The effective cleavage of electron rich aryl ethers could also be achieved using nickel precursors without stabilising ligands, yielding nickel nanoparticles (Scheme 10).67 Aryl alkyl ether bonds were also cleaved, however, at a lower rate, compared to the diaryl substrates. A study using deuterium labelled aryl alkyl ethers showed that the mechanism for these substrates involves nickel coordination to the aromatic system, followed by C–O insertion and β–H elimination, to release formaldehyde, resulting in an aryl–nickel hydride species. Finally, reductive elimination led to the deoxygenated aromatic compound.68 Homogeneous nickel hydrogenolysis catalysts under micellar conditions were also utilised for aryl ether bond cleavage, but did lead to full reduction of the aromatic systems.69
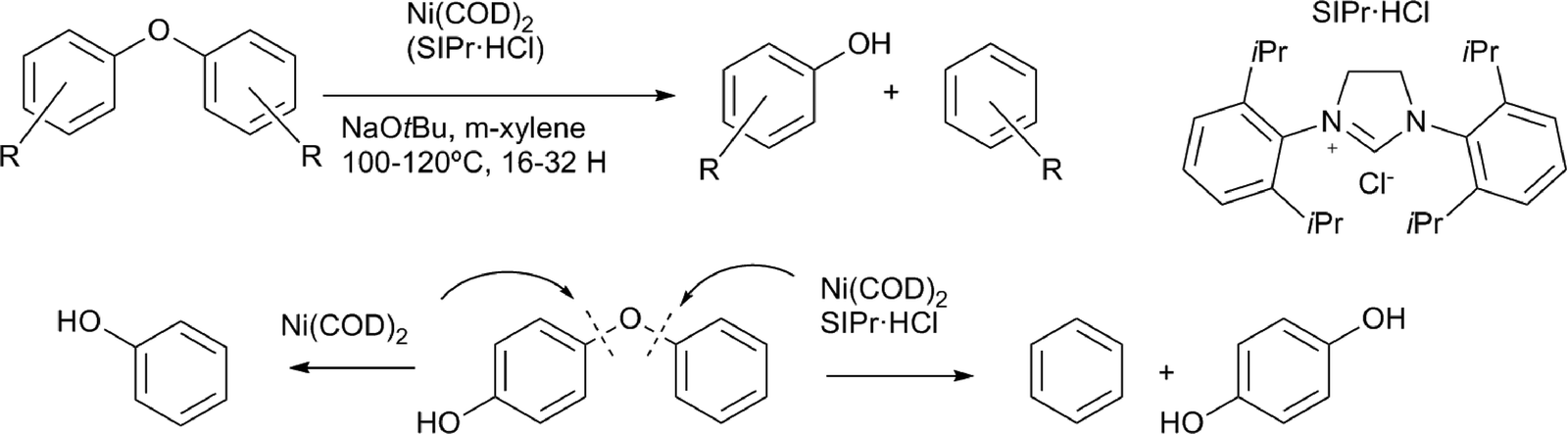 | ||
| Scheme 10 Diaryl ether lignin model compounds cleaved by nickel catalyst systems.66,67 | ||
Recently, the reductive cleavage of aromatic ether bonds using Et3SiH and a base was reported by Grubbs and co-workers.70 This method is stoichiometric in triethylsilane, but does not require any transition metal catalyst. It does require the careful selection of the base and reaction temperature, to avoid ortho-silylation, but the method is regioselective and does not cause reduction of the aromatics.
The cleavage of aryl alkyl and dialkylether compounds was demonstrated by Marks et al. (Scheme 11).71,72 A lanthanide mediated microscopic reverse alkene hydroxylation was coupled to a Pd-nanoparticle catalysed hydrogenation. In this cooperative catalytic system, the ytterbium(III) triflate complex breaks the O–alkyl bond via C–H to O–H hydrogen transfer, to form an alkene intermediate, which is subsequently hydrogenated by palladium.71,72 Interestingly, the used ionic liquid medium stabilises the palladium nanoparticles and prevents sintering. Many different C–O bonds are readily cleaved using this cooperative catalytic system. Compared to the above nickel and ruthenium based systems, the lanthanide system allows for efficient catalyst and solvent recycling by ether extraction or vacuum transfer.
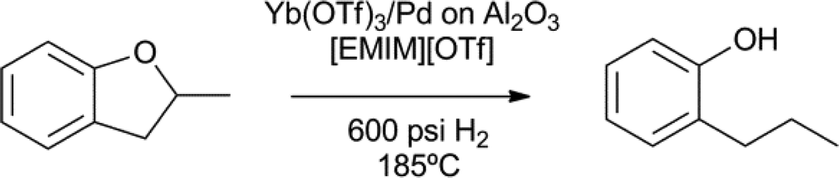 | ||
| Scheme 11 Aryl alkyl and dialkyl ether cleavage by ytterbium combined with a palladium nanoparticle hydrogenation catalyst in an ionic liquid ([EMIM][OTf] = ethylmethyl-imidazolium triflate).71 | ||
Few examples exist in which these homogeneous catalysts were directly applied for the conversion of real lignin samples. James and co-workers attempted lignin depolymerisation using the ruthenium/xanthphos catalyst system, but found no significant activity.63,73 Several ruthenium precursors, containing phosphine and nitrogen ligands, were successfully applied alongside rhodium for the degradation/solubilisation of ethanol organsolv lignin (EOL), with hydrogen.74 The degradation/solubilisation was linked to hydrogenolysis, although detailed product formation and therefore cleavage pathways were not elucidated. Also, several nickel based heterogeneous systems were used for lignin hydrogenolysis.75a,b These catalytic systems were reported to yield the propylguaiacol and propylsyringol products with good selectivity, without a significant reduction of the aromatics. Furthermore, arenes were obtained in an elegant one-pot solvent-hydrogen transfer strategy using Raney® nickel and zeolites.75c A related reductive cooperative palladium/zinc system was also reported to yield the aromatic products in good yields.76 Catechols were obtained in a high yield using heterogeneous copper-doped porous metal oxides.77
3.2 Oxidative approaches for bond cleavage in lignin and model compounds
Oxidation is an important strategy for the depolymerisation of lignin. Organisms, like white-rot fungi, degrade lignin through oxidative mechanisms, typically using manganese containing enzymes.58 For the commercial production of vanillin from lignin, selective aerobic oxidative cleavage is performed using copper.57 Many simple oxidants can be utilised, however, due to over-oxidation, a selective product formation while maintaining high conversion values is challenging. The oxidative cleavage of lignin and lignin model compounds has been studied, especially in the chemistry of bleaching, and for a comprehensive overview of all the different catalysts, we refer readers to the following reviews.9,78Here, we will discuss a few recent reports that highlight the potential of soluble transition metal/ligand complexes for the selective depolymerisation of lignin towards useful chemicals and how the choice of catalyst influences the products obtained.
Thorn and coworkers studied the use of vanadium catalysts for selective lignin bond cleavage.79 Initial mechanistic studies, using a simple diol model system, showed that cleavage can be achieved under aerobic conditions. They isolated a V(III) species, indicating that the reaction occurs via a two electron pathway. Other model substrates, containing a diol-moiety, could also be subjected to oxidative cleavage and it was shown that phenyl substituents facilitate the reactions.80 Toste and Son reported a very efficient β–O–4 bond cleavage, using catalyst [V1] (Scheme 12).81 Other vanadium catalysts have been reported for similar selective bond cleavage reactions of lignin model compounds.82 Vanadium catalysts can induce an oxidative bond scission at different locations, as was demonstrated using a 13C labelled dimeric lignin model compound.83 The vanadium catalyst [V2] cleaved the benzylic C–C bond, to yield 2,6-dimethyloxybenzoquinone and the corresponding aldehyde. In contrast, with the vanadium catalyst [V1], a selective bond cleavage of the ethereal C–O bond took place. Mechanistic studies using deuterium labelled model compounds were also provided. Additionally, it was shown that benzylic C–C bond cleavage using the catalyst [V2] is restricted to phenolic substrates. A cobalt based oxidation catalyst was reported to oxidise both aromatic rings to benzoquinones in similar dimeric lignin models.84
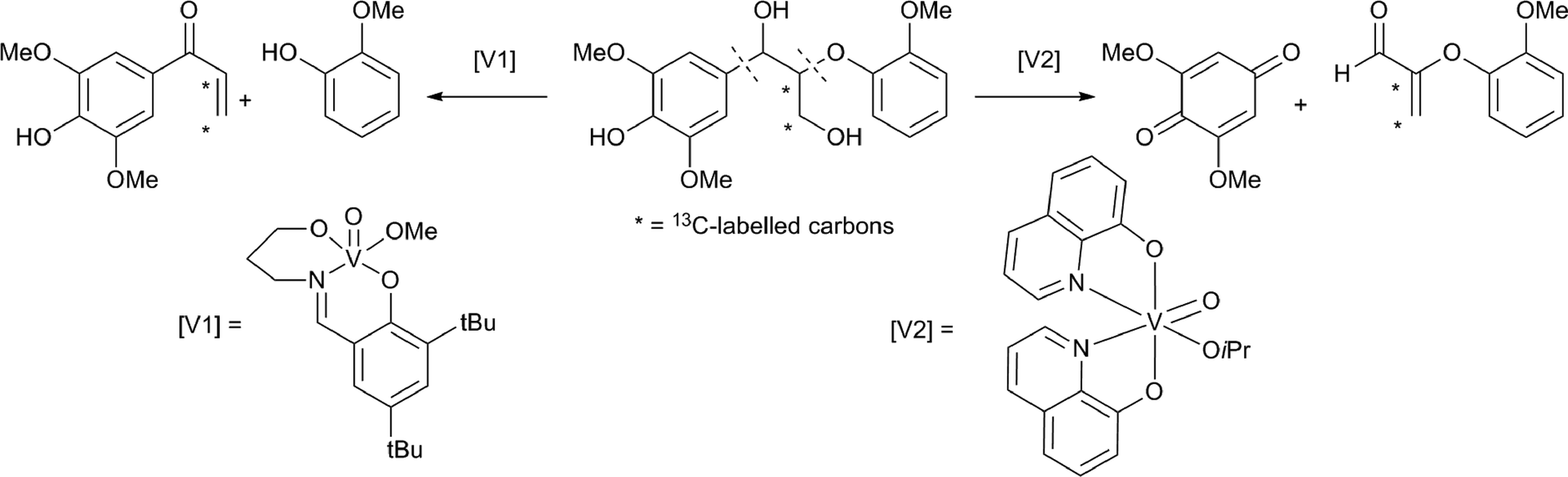 | ||
| Scheme 12 Examples of site selective oxidative cleavage of a β–O–4 motif containing lignin model, using two different vanadium based catalysts.81,83 | ||
Stahl and coworkers screened a range of catalysts for the selective oxidation of either the primary (1°) γ-alcohol or the secondary (2°) α-alcohol of a lignin model compound (Scheme 13).85 Copper/TEMPO derived oxidation catalysts showed good primary alcohol selectivity, while palladium, iron and chromium catalysts selectively oxidized the secondary alcohol. Copper/TEMPO catalysts have previously been reported for selective primary alcohol to aldehyde oxidation.86 In addition, it was shown that when the primary alcohol was oxidised to the corresponding aldehyde, a subsequent retro-aldol cleavage of the β–O–4 motif took place. On the other hand, oxidation of the secondary alcohol can lead to the corresponding dehydration product (Scheme 13). Additionally, it was demonstrated that a catalytic amount of AcNH-TEMPO combined with inorganic acids was a very effective and selective catalyst for secondary alcohol oxidation for this and many similar model compounds. A study by Hanson et al. compared the copper/TEMPO catalyst system to several vanadium based oxidants.87 They found that products were associated with similar selectivity patterns, with the copper system preferring oxidation of the primary alcohol, followed by oxidative cleavage, whilst the vanadium catalysts induced mostly secondary alcohol oxidation, followed by dehydration and further oxidation reactions.
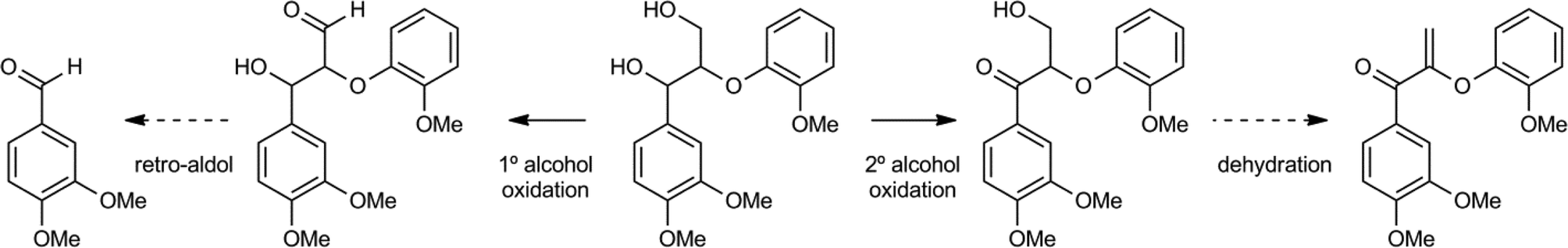 | ||
| Scheme 13 Two distinct oxidation pathways of a β–O–4 containing lignin model.86 | ||
Several of the oxidative systems discussed above have been tested on real lignin. The group of Toste applied [V1] on various organosolv lignin samples obtained from Miscanthus giganteus.88 They demonstrated, using GPC and 1H–13C correlation NMR spectroscopy, that dioxasolv and acetosolv lignin could be effectively depolymerised by β–O–4 bond cleavage. Additionally, they confirmed, by GC-MS, that vanillin, syringic acid, syringaldehyde, 4-hydroxybenzaldehyde, vanillic acid, 3-hydroxy-1-(4-hydroxy-3-methoxyphenyl)propan-1-one, 3-hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)propan-1-one and 4-hydroxybenzoic acid were formed as the main products, in the given respective order of quantity. The catalyst [V2] was also tested and showed a similar depolymerisation activity. Combining [V1] and [V2], a C–C and a C–O bond cleavage catalyst, did not significantly improve the efficiency of lignin degradation. Ethanosolv lignin was not depolymerised, which was explained by the etherification of the free benzylic hydroxyl groups during the acid pre-treatment. The cobalt-Schiff base catalyst developed by Bozell et al. was tested in the oxidative cleavage of the organosolv fraction of tulip poplar, under oxygen pressure.84 As in the case of the lignin model compounds, the benzoquinone products were detected by 1H-NMR. Stahl and co-workers demonstrated that TEMPO/HNO3/HCl selectively oxidised secondary alcohols in aspen lignin.85 All of these examples show a remarkable correlation between the studies using model compound and real lignin samples in terms of reaction pathways and product distribution. Furthermore, Chen et al. reported the selective cleavage of the 5–5′ linkage in lignin, from pine-kraft-AQ pulp, using dinuclear manganese catalysts and hydrogen peroxide under relatively mild conditions.89
4. Oils and fats
The use of triglycerides from biomass is an ancient practice and oil and fats are arguably still the most utilised renewable feedstocks in the chemical industry. In addition to the recent interest for biodiesel production, glycerol and the fatty acids or methyl esters thereof, obtained after ester hydrolysis, have many applications in for example, the production of soaps, lubricants, coatings, plastics and food additives.4c,90 Recently, much research has been focused on the production of monomers for plastics and biodegradable polymers using fatty acid based substrates.91 Therefore, the specific and selective chemical transformation of fatty acids have become an increasingly important area in homogeneous catalysis.Animal and plant fats contain many differing fatty acid units, attached to glycerol. Among these, unsaturated fatty acids are of most importance for use as renewable resources, because the double bond provides a convenient handle for further chemical valorisation. Some important examples are shown in Fig. 5. Generally, a first step towards the utilisation of triglycerides is the trans-esterification or hydrolysis of the ester, and indeed many procedures exist, involving acidic and basic procedures.1b,c
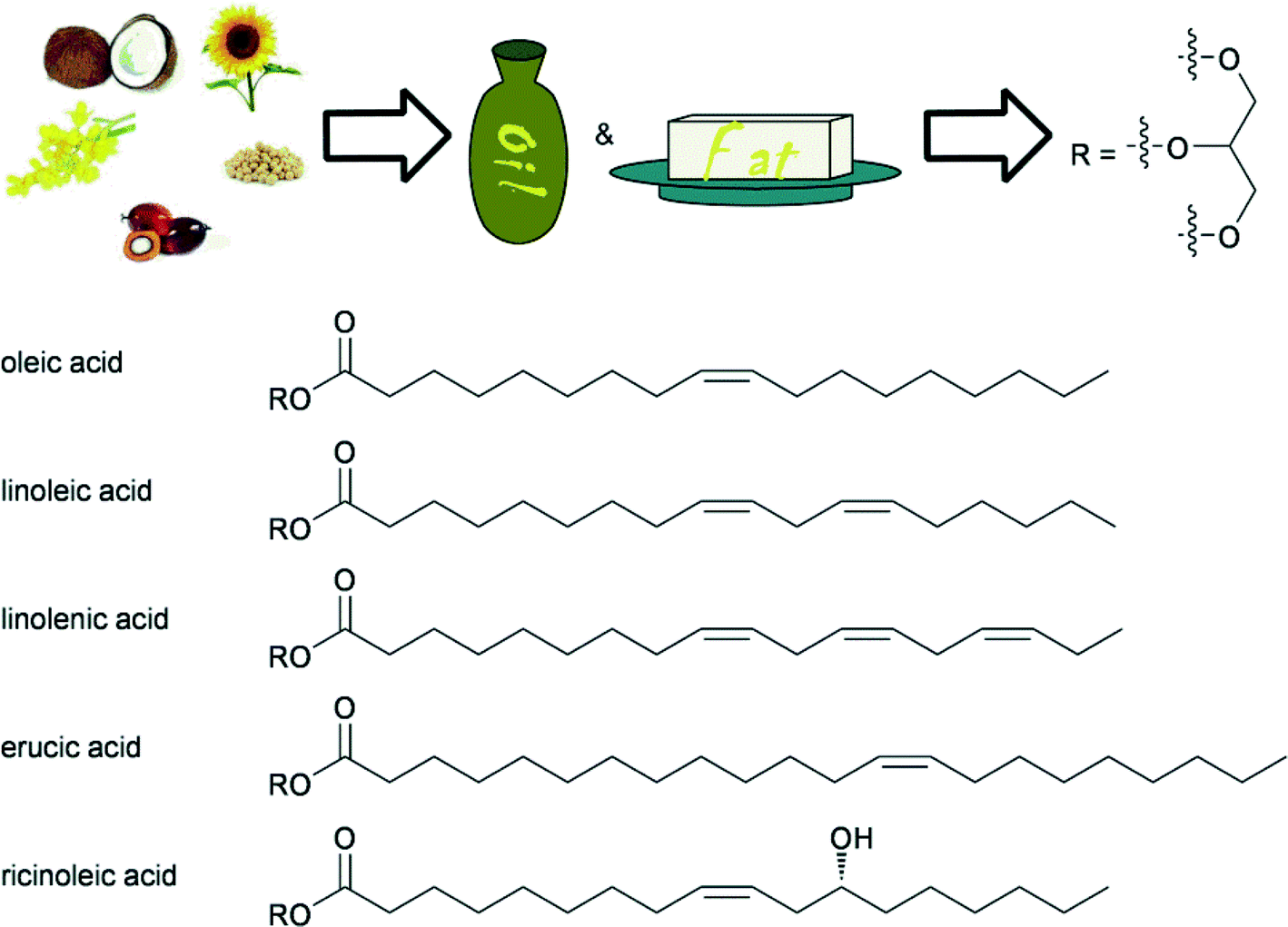 | ||
| Fig. 5 Typical fatty acids as part of triglycerides used in industrial processes as feedstocks, obtained from various types of oils. | ||
It is important to mention that triglyceride trans-esterification, or hydrolysis, releases glycerol as an unavoidable side-product, which has thus become another important target in the field of biomass valorisation.92 Many important small molecule products can be produced from glycerol.4c For example, 1,2- and 1,3-propanediol can be obtained through dehydroxylation, using heterogeneous catalysts. Homogeneous catalysts for the valorisation of glycerol do also exist. Recent examples of the applications of homogeneous catalysts are glycerol oxidation to dihydroxyacetone, performed with remarkable activity and selectivity, using a palladium phenanthroline complex (Scheme 14a)25 and etherification of glycerol using Lewis acids, especially Bi(OTf)3 (Scheme 14b).93 Another example is the direct desymmetrisation of glycerol using chiral catalysts, to obtain chiral building blocks.94
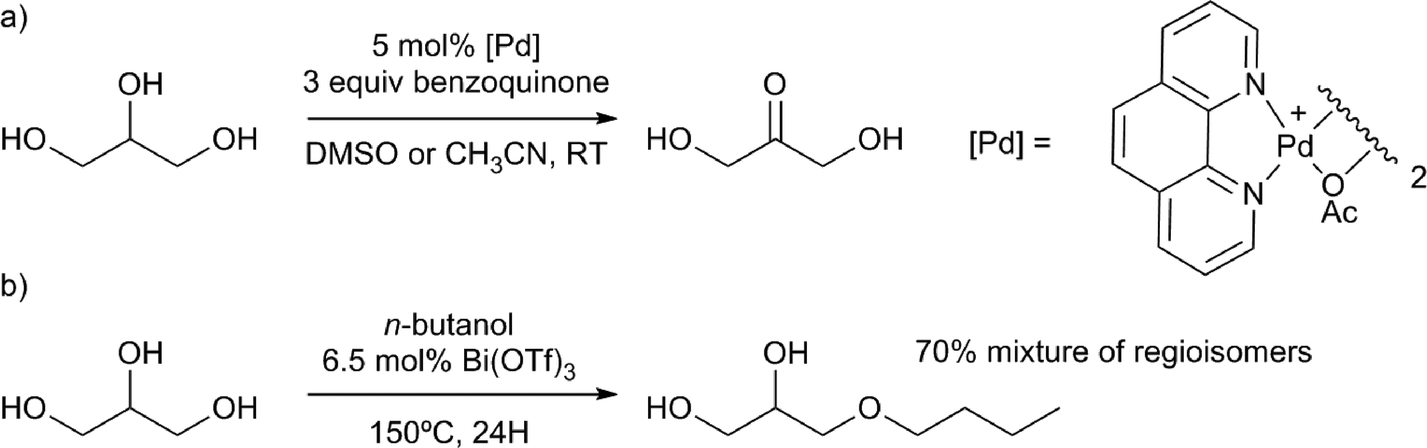 | ||
| Scheme 14 Valorisation of glycerol by a) selective palladium catalysed oxidation to dihydroxyacetone25 and b) Lewis acid catalysed etherification.93 | ||
In addition to valorising fatty acids and glycerol separately, the direct conversion of triglycerides can also be performed. For example, the groups of Larock and Meier showed, in a series of papers, how polymers can be obtained from vegetable oils as starting materials, by applying different metathesis strategies.95 Below, an overview of the different selective transformations of fatty acids using homogeneous catalysts is given, that have already been developed or show promise for further development for the valorisation of vegetable and animal fats and oils.
4.1 Double bond modification of fatty acids
The double bonds present in unsaturated fatty acids are the most obvious handle, and offer many possibilities for chemical modification (Scheme 15). These involve known methodologies, which encompass the addition of diverse functionalities across the double bond, increasing the functional diversity of obtained products. In many cases, these reactions are preceded by fast double bond isomerisation, allowing highly selective terminal or 3-functionalisation.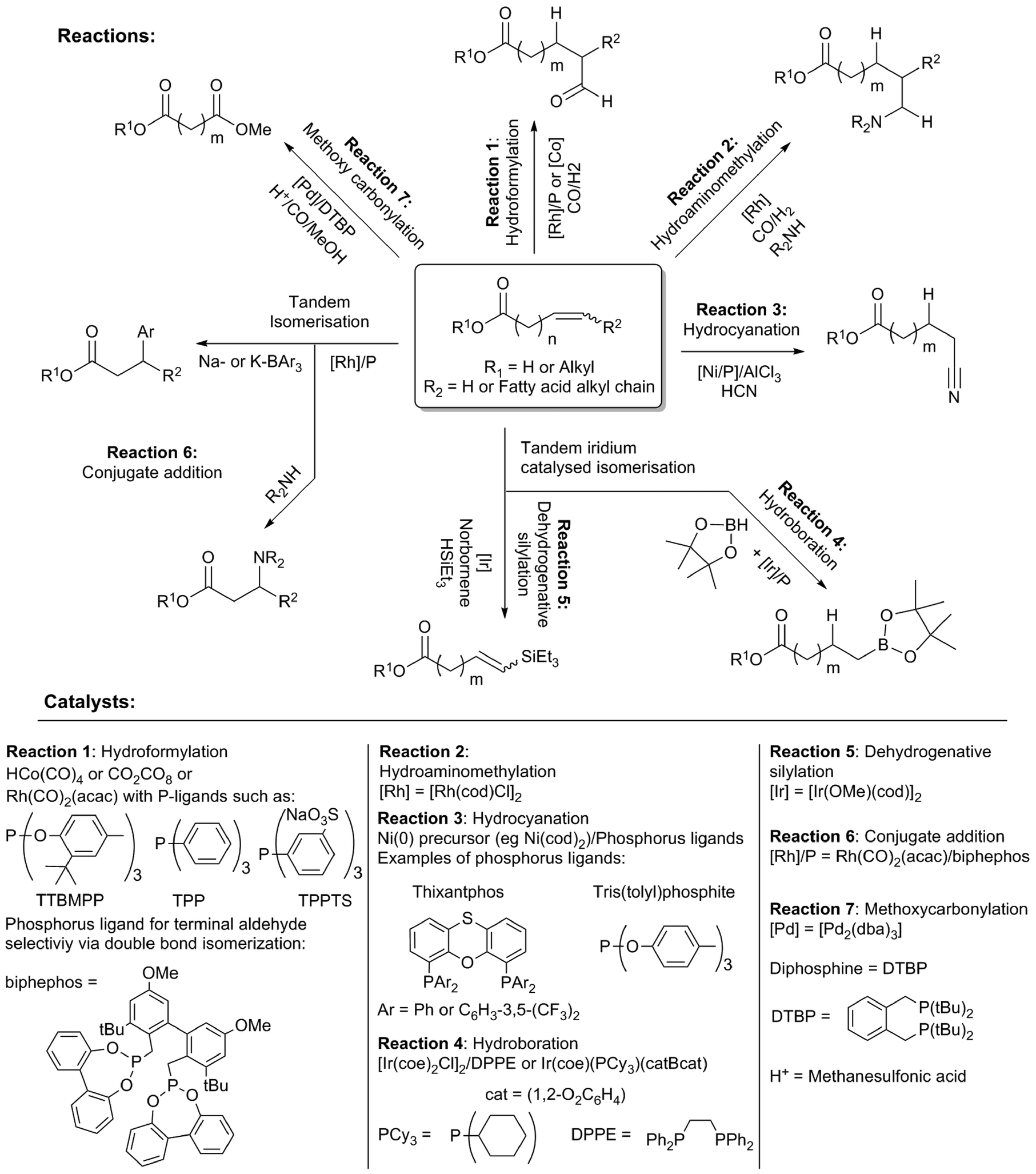 | ||
| Scheme 15 Strategies for double bond modification in unsaturated fatty acids/esters and homogeneous transition metal catalysts used. | ||
Hydroformylation is an important reaction that can be used to yield formyl-functionalised fatty acids (Scheme 15, Reaction 1). Rhodium catalysts are considered superior to cobalt systems in terms of selectivity and activity.96 Activity, selectivity for aldehydes and the extent of isomerisation can be modulated by the choice of ligand. For example, bulky phosphite ligands, like tris(2-tert-butyl-4-methylphenyl)phosphite (TTBMPP, reported rates up to 400–500 mol mol−1 h−1 for methyl oleate and methyl linoleate) outperforming cheaper triphenylphosphine (TPP),97 which again was reported to be the superior to (PhO)3P (2074 mol mol−1 h−1 compared to 1254 mol mol−1 h−1 respectively, in the hydroformylation of soybean oil).98 Alternatively, a catalyst system capable of concomitant double bond isomerisation can provide high selectivity towards the terminal aldehyde product.99 Hydroformylation products can be hydrogenated to hydroxylated fatty acids for material synthesis.96a,100 Recently, a biphasic catalyst system using Rh/TPPTS, combined with a phase transfer catalyst was shown to provide a high conversion in the hydroformylation of methyl oleate and also double unsaturated substrates like methyl linoleate and methyl linolenate.101 An amine functionality can be introduced in one reaction step by hydraminomethylation with [Rh(cod)Cl]2 (Scheme 15, Reaction 2).102
Nickel-based catalysts, combined with diphosphine or (di)phosphite ligands, were reported for the hydrocyanation of ω-unsaturated fatty acid esters.103 (Scheme 15, Reaction 3) In addition, there is extensive patent literature concerning the hydrocyanation of fatty acids, fatty acid esters and oils.104 Thixantphos diphosphine ligands induced similar yields, but were outperformed by the well-known tris(tolyl)phosphite ligands.
The hydroboration of ω-unsaturated fatty acid esters was carried out with iridium–phosphine catalysts, providing the terminal hydrobonated products in >90% yield (Scheme 15, Reaction 4).105 A tandem double bond isomerisation and hydroboration of methyl oleate could be achieved using the same catalyst system, although only with moderate yields (around 50%).106 Similar, iridium-based catalyst have been used for the tandem isomerisation and dehydrogenative trialkylsilylation of the double bond (Scheme 15, Reaction 5).107 Moreover, functional groups were regioselectively introduced at the 3-position by rhodium catalysed isomerisation, followed by conjugate addition using carbon and nitrogen nucleophiles (Scheme 15, Reaction 6).108 The yield was highly dependent on the nucleophile and the fatty acid ester derivative.
The direct, highly selective introduction of an ester functional group into the terminal position of internally unsaturated fatty acid methyl esters was reported by Cole-Hamilton et al. by methoxycarbonylation (Scheme 15, Reaction 7).109 They applied the palladium–phosphine (DTBP) catalyst that was developed by Lucite for their methyl methacrylate process.110 The catalyst causes a rapid double bond isomerisation, providing a terminal ester group.111 In addition, by the use of fatty acids that contain multiple unsaturations, branched ester groups can be obtained, although with a low yield.112 Methoxycarbonylation was applied to vegetable oil obtained from a supermarket.113 Also, triglyceride esters could be subjected to methanolysis in tandem using the same catalyst, leading directly to diester compounds. The diesters can be converted to diols with LiAlH4 or catalytic hydrogenation, using the Milstein catalyst or ruthenium/triphos.113,114 The diester compounds obtained from methoxycarbonylation were found to be excellent monomers for the synthesis of new polymers.115
4.2 Double bond conjugation, isomerisation and selective transfer hydrogenation
The selective isomerisation of the double bonds in fatty acids can lead to valuable precursors for further functionalisation. In addition to the rhodium, iridium and palladium isomerisation catalysts mentioned above, Fe(CO)5 was also found to be an active isomerisation catalyst.108,116 Similarly, silver was utilised for lactone formation.117 Also, the use of [Pd(μ-Br)tBu3P]2 leads to isomerisation and can be combined with a metathesis catalyst to create specific unsaturated product distributions.118Valuable products from fatty acids, obtained from various types of natural oils, are conjugated linoleic acids (CLAs). CLAs have many applications, as food additives or starting materials for the production of polymers, coatings and paints in industry.119 Various methods exist, which provide CLAs from unconjugated fatty acids, using alkaline processes, biocatalysts, photocatalysts and metal catalysts.119a In the food industry homogeneous transition metal complexes are usually avoided, however, recent advances in the field of bioplastics led to a renewed interest in selective transition metal catalyst systems. Using mild reaction conditions, many metal precursors catalyse the conversion of linoleic acid, or methyl linoleate, to CLAs, although tin based promoters have to be employed for an efficient conversion (Scheme 16).119b,120 RhCl(PPh3)3 and [RhCl(C8H14)2]2 stood out as the more efficient catalysts for this reaction, producing the desired CLAs in over 95% selectivity. Several good separation and recycling procedures were reported, using, for example, biphasic systems.121
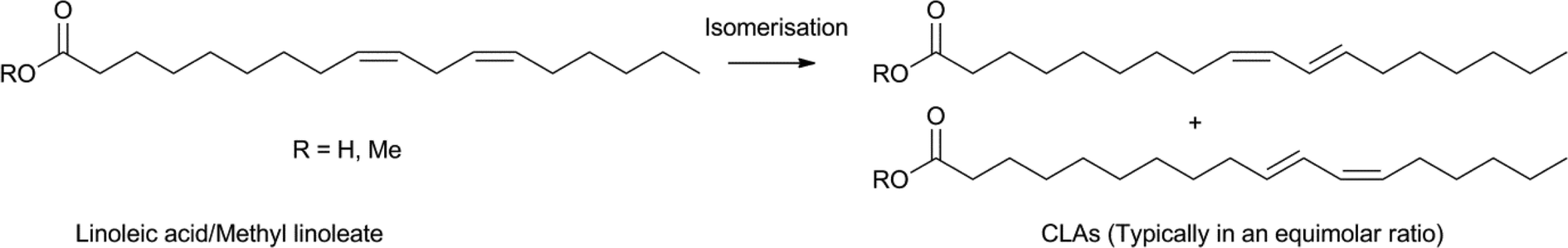 | ||
| Scheme 16 Isomerisation of linoleic acid or methyl linoleate CLAs. | ||
Recently, the selective conversion of polyenes to monoenes, using a ruthenium(III) chloride catalyst, under transfer hydrogenation conditions was reported.122 The selective transfer hydrogenation was achieved when the substrate contained an aromatic group. It was proposed that the aromatic group acts as a ligand for the ruthenium, after reduction of the ruthenium(III) halide precursors, followed by the formation of allyl complexes of the fatty acid chains. A ruthenium N-heterocyclic carbene complex [RuCl2(p-cymene)IMES] was applied as the catalyst to form 12-oxostearate from methyl-ricinoleate.123 Although a hydrogen borrowing mechanism was suggested, this reaction could also involve isomerisation of the double bond, as was shown by DSM, for the formation of methyl 12-oxo-stearate from ricinoleic acid ester using the Lucite catalyst.124
4.3 Double bond metathesis
As a result of significant progress in the field of olefin metathesis, with efficient and selective catalysts readily accessible, the ruthenium catalysed double bond metathesis of unsaturated fatty acids has become another important tool for fatty acid valorisation.91b,125 Metathesis is most commonly applied to transform fat products into monomers for polymerisation reactions. A plethora of recent examples and applications can be found in the literature.118,126 The self-metathesis of methyl oleate, to yield the C18 diester and the C18 alkene, is commercially exploited by Elevance.127 Also, cross-metathesis with, for example, ethylene or methyl acrylate has been applied for this purpose (Scheme 17a). Polyene monomers and norbornene modified monomers derived from fatty acids have also been polymerised using acyclic diene metathesis, or ring opening metathesis (Scheme 17b).128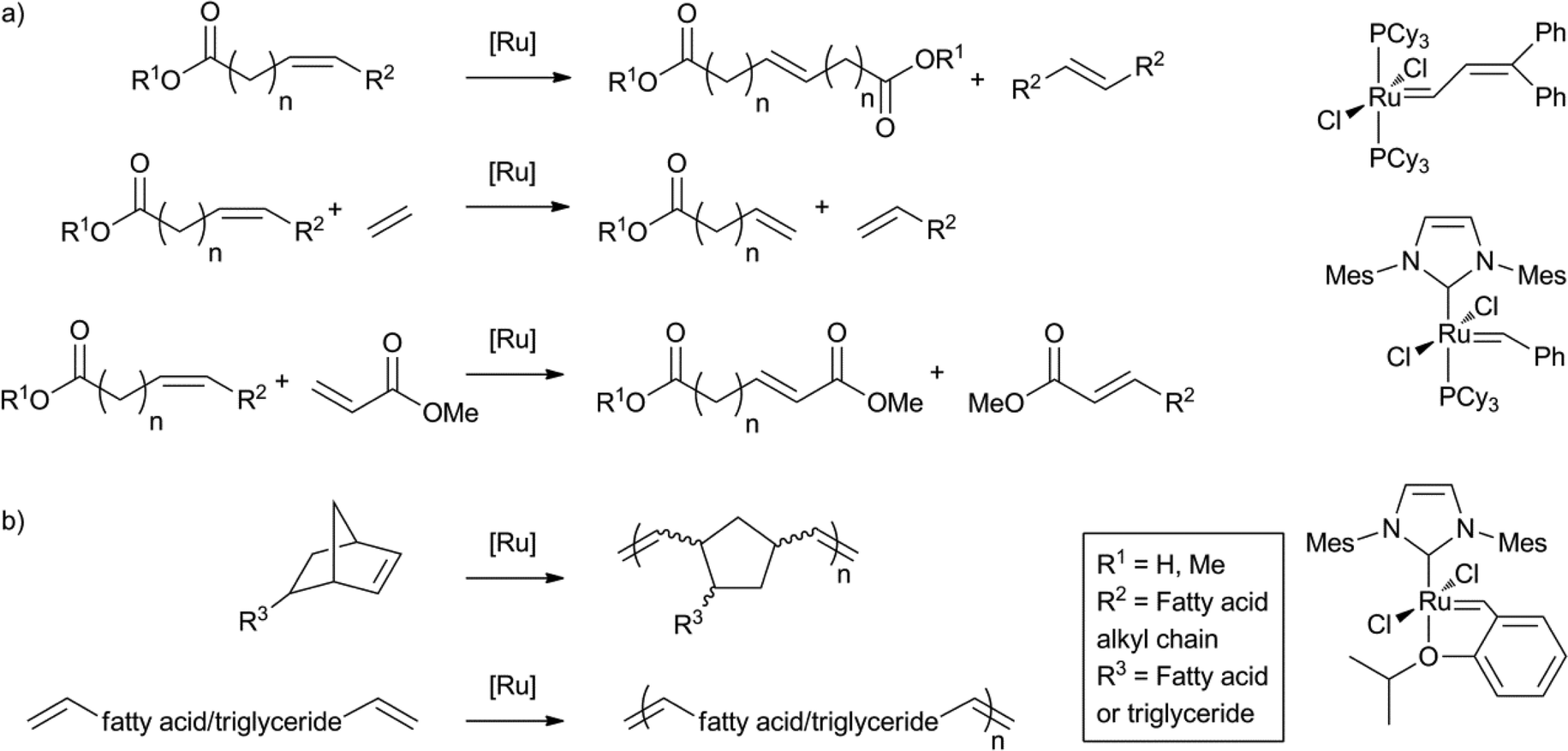 | ||
| Scheme 17 Application of ruthenium catalysed double bond metathesis for a) monomer synthesis and b) polymerisation. | ||
Recently, Z-selective molybdenum metathesis catalysts were reported.129 Furthermore, a ruthenium based Z-selective catalyst system for the homodimerisation of methyl 10-undecenenoate was demonstrated by Grubbs et al. with excellent yields and selectivity (Scheme 18a).130 Of the catalysts applied, ones bearing Ar = 2,6-dimethyl-4-methoxybenzene and 2,6-dimethyl-4-chlorobenzene provided products with the highest isolated yields (>90%). The most active complex (TON close to 1000, Scheme 18b) was used in the synthesis of several insect pheromones, from fatty acid derived starting materials.131 The Z-selective fatty acid type intermediates were obtained in excellent isolated yields from cross-metathesis reactions of alcohols, like oleyl alcohol, or 11-eicosenol with 1-butene, 1-pentene, 1-hexene, 1-heptene, 1-decene and 1,4-hexadiene establishing this methodology as a very useful synthetic tool for the synthesis of Z-alkene containing products from fatty acid derived substrates.
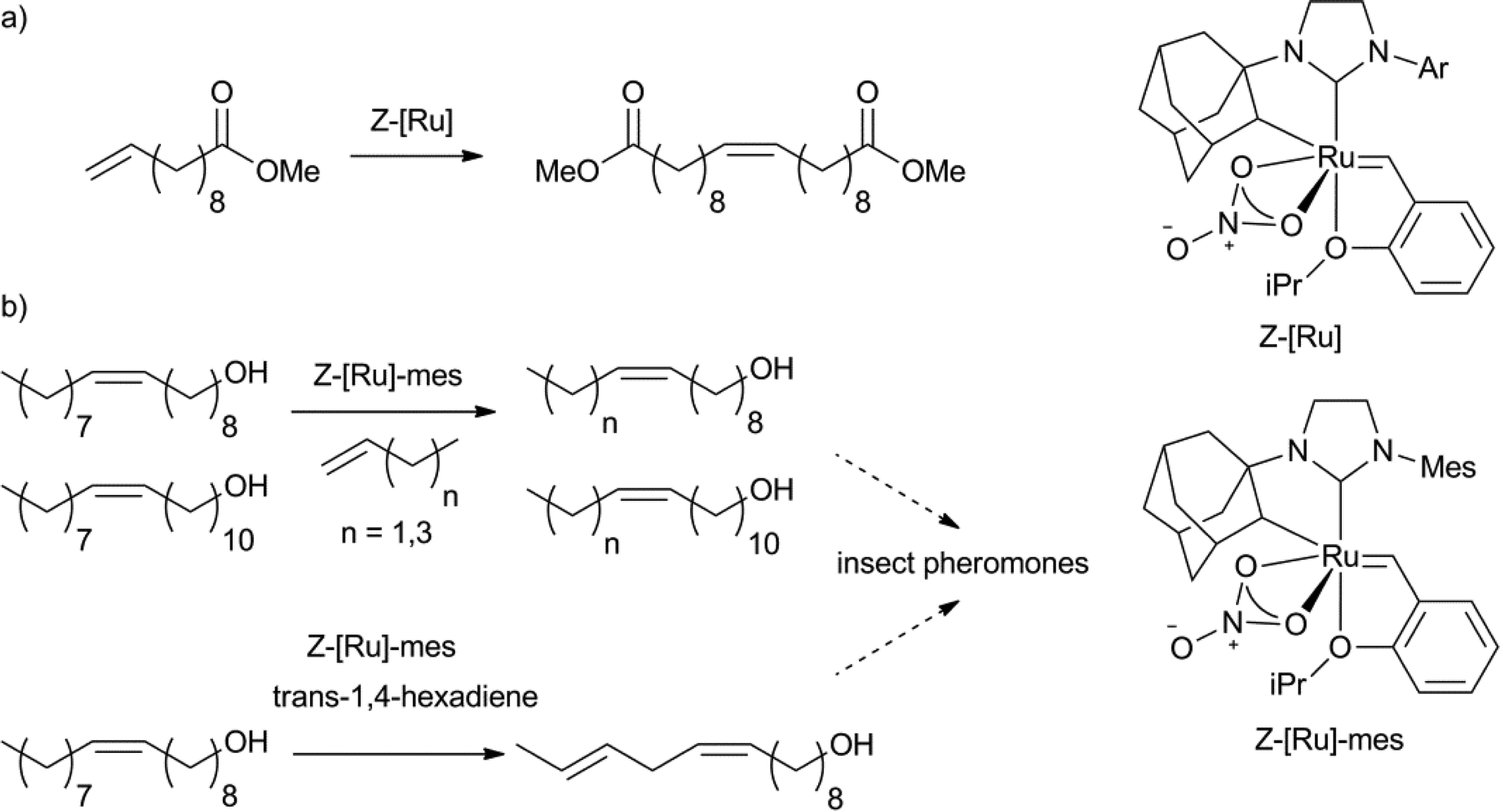 | ||
| Scheme 18 Examples of Z-selective metathesis of a) methyl 10-undecenoate to obtain the Z-homodimer130 and b) oleyl alcohol and 11-eicosenol as substrates for the synthesis of Z-alkene containing insect pheromones.131 | ||
4.4 Oxidative cleavage, oxidation and epoxidation
Oxidation is another extensively explored research area and many known procedures have been applied for the oxidation, oxidative cleavage or epoxidation of fatty acids.1c,132,13b For example, ketones can be obtained via a palladium catalysed Hoechst–Wacker oxidation and selective, metal-free methods have since been developed.132a,133Epoxidation or hydroxylation of the olefin functionality provides new synthetic platforms for further modification. The epoxides themselves are utilised for the production of plastics, in which reactions are usually carried out using organic reagents or heterogeneous catalysts.132b,134 However, recently, interesting and more selective homogenous catalysts have been discovered. For example, the enantioselective epoxidation of fatty acids can be achieved using the Sharpless–Katzuki epoxidation catalyst (Scheme 19).135 The product proved to be a valuable precursor for the enantioselective synthesis of several fluorine containing and other pharmaceuticals compounds.134,136 A Sharpless-type asymmetric dihydroxylation has also been reported.132a,136
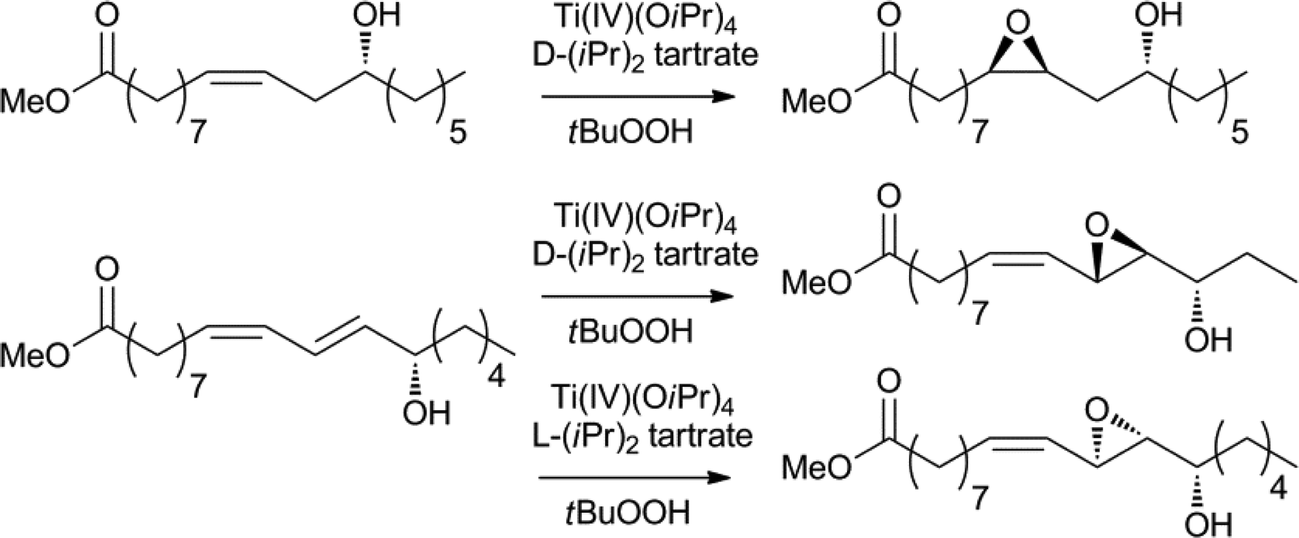 | ||
| Scheme 19 Enantioselective epoxidation of fatty acid derivatives.135,136 | ||
Aldehyde products are typically hard to obtain in a selective fashion and the outcome of these reactions is particularly sensitive to variable reaction parameters. Very recently, the group of Klein Gebbink reported a highly aldehyde selective, one-pot oxidative cleavage of several unsaturated fatty acids and esters (Scheme 20).137 The catalytic system is based on Fe(OTf)2, with a mixture of diastereoisomers of the bpbp ligand, combined with the sequential addition of hydrogen peroxide, sulfuric acid and sodium periodate. One-pot methods for the oxidative cleavage of unsaturated fatty acids, yielding carboxylic acids, were also reported.138
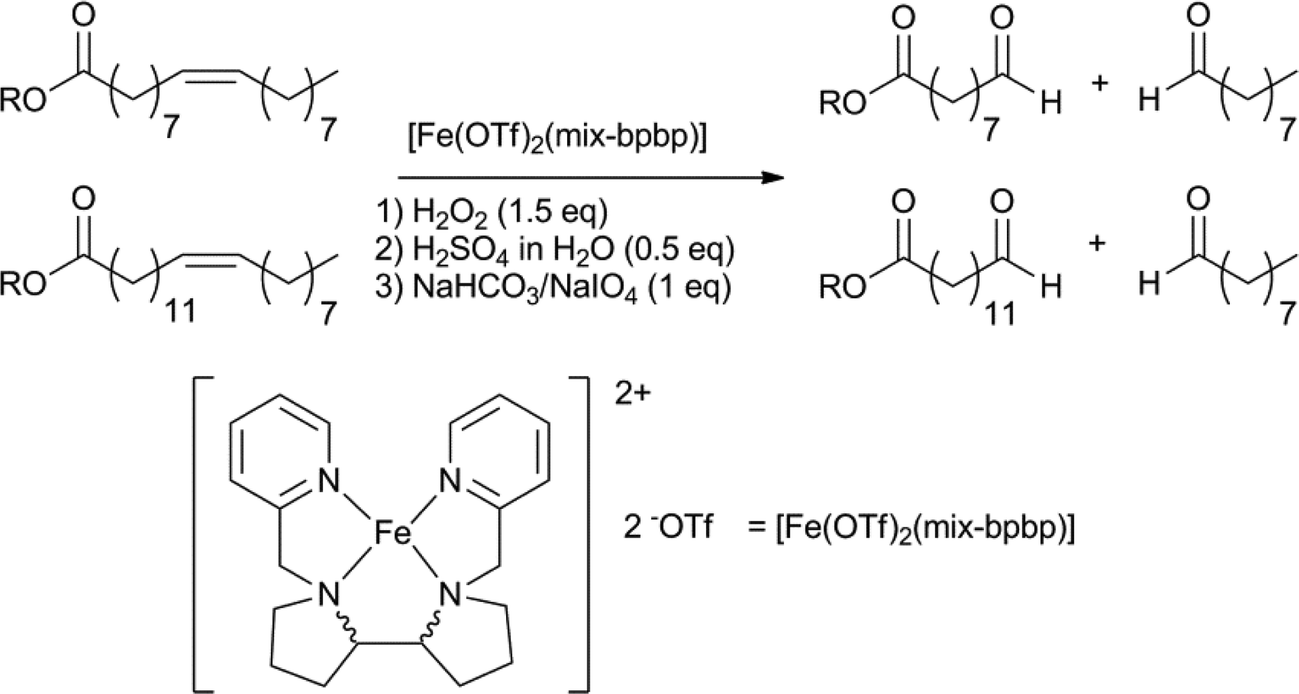 | ||
| Scheme 20 One-pot selective iron catalysed oxidative cleavage of fatty acids, to give aldehyde products.137 | ||
5. Conclusions and future prospects
5.1 (Hemi)cellulose
Very few reactions have been performed on cellulose or lignocellulose using soluble transition metal catalysts. The only, notorious, exception being the direct dehydration of cellulose and lignocellulose to HMF, under the influence of acid and CrCl2, which serves to isomerise glucose to fructose. Also, the reactions on sugars are relatively scarce, although recently increasing. Hydrogenation is still best performed using heterogeneous catalysts, although some very active, water-soluble homogeneous catalysts were developed, that merit further investigation, in particular with regard to their longevity. The selective oxidation of unprotected sugars is an important step towards the protection group free conversions of sugars, an area that was previously solely the domain of biocatalysis, using enzymes.Probably the most important contribution of homogeneous catalysis towards the conversion of sugar-based building blocks lies in the one carbon extension using CO-chemistry, key steps in the conversion of levulinic acid to adipic acid and other monomers for polymers. These processes may well find applications in the coming decade.
5.2 Lignin
Interesting approaches have been shown and detailed for the selective cleavage of lignin model compounds, using homogeneous catalysts. These new methods are a significant advance towards the selective depolymerisation of lignin, into processable aromatic subunits, which might, in the future be utilised for the production of aromatic platform chemicals. Reductive approaches, especially those that are redox neutral, show much promise; but further research should focus on strategies that reduce the extent of the functionality found in lignin. More progress was demonstrated in the application of oxidative approaches on real lignin samples. However, these generally led to products with an increased level of functionality, compared to the ones obtained via a reductive cleavage.This raises a key question regarding lignin valorisation: what exactly are the aromatic compounds that should be targeted and can suitable lignin-derived platform chemicals be identified?2a,55 Lignin, compared to sugars or other biomass resources, is much less uniform in structure, and more challenging to degrade, thus, significant new insight will be needed to tackle this challenge. It is possible that lignin derived building blocks will find application both in the “emerging”, as well as “drop-in” strategies, depending on the reaction methodology and extent of the functionality of the products. To this end, the selective depolymerisation of lignin, as well as the high-yield production of defined aromatic compounds needs to be addressed. It will be important for further progress in this field that the focus shifts away from simple lignin model compounds, to more sophisticated lignin models, or lignin samples and that the feedstock compatibility is taken into account in the earlier stages of catalyst development. The development of homogeneous catalysts for the selective cleavage of lignin, for the production of aromatic platform chemicals, is still somewhat in its infancy, but there are many promising indications that this strategy will become viable in the future.
5.3 Fatty acids and oils
This overview shows many well established and promising methodologies for the selective transformation of fatty acids, using homogeneous catalysts. These strategies allow for an arsenal of products to be obtained from common fatty acids, like oleic acid. Additionally, the direct transformation of fatty acid mixtures, as well as triglycerides themselves, allows for a more versatile product spectrum. The direct valorisation of vegetable oil mixtures, to obtain starting materials for the production of new bio-based polymers, is highly desired. Overall, the valorisation of fatty acids and triglycerides is at an advanced stage, as is demonstrated by several commercial applications.5.4 Role of homogeneous catalysts
It is clear, from the above discussion, that there are a number of areas where homogeneous catalysis may well be the method of choice for the conversion of biomass or platform chemicals, as a result of a unique reactivity or selectivity, including the enantioselective transformation of biomass related substrates. Most of the applications described in this Perspective are a proof-of-principle, with the conversion and selectivity values demonstrated on a laboratory scale, and in most publications rather low turnover numbers are reported. However, for applications in bulk chemicals, this parameter is tremendously important. The Achilles heel of homogeneous catalysts is not their problematic separation from the products, as many people think; but rather their instability. The development of successful, industrially applicable processes very much relies on the ingenuity of chemists in finding catalysts with high turnover numbers, either by increasing the rate, or by improving the catalyst stability. This is indeed the strong point of homogeneous catalysis: many parameters can be changed in the catalyst, as well as in the reaction conditions, leading to an infinite number of possible solutions.In all the bulk chemical processes, the catalysts are recycled. In most cases this is done by distilling off the product from the catalyst. If the catalyst is unstable during distillation, a number of other separation techniques can be used. Two-phase catalysis, in which the catalyst is contained in one phase and the starting materials and product in the other phase, is already practised on a large scale. Organic–aqueous biphasic systems are an obvious choice, especially in connection with biomass conversion, but combinations of very non-polar with very polar organic solvents can also be used. So far, the use of fluorous solvents and ionic liquids has not been applied, industrially. This is largely because of the high costs. The use of membranes, to separate the catalyst from the product stream, has already been developed, industrially. This method would also seem to be highly applicable to this field. Catalyst immobilisation is, from an industrial perspective, not seen as a useful approach, in view of the instability of the catalyst. In fine chemicals production, catalysts are rarely recycled. Here, also, sufficient turnover numbers need to be achieved, but in view of the higher costs of fine chemicals, these may be much lower, in the order of 500–20000.
Many creative solutions, as well as new, large-scale productions utilising renewables, based on homogeneous catalysis are expected in the years to come.
Acknowledgements
The authors gratefully acknowledge financial support from the European Commission (SuBiCat Initial Training Network, Call FP7-PEOPLE-2013-ITN, grant no. 607044).Notes and references
- (a) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS PubMed; (b) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed; (c) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (d) P. N. R. Vennestøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502 CrossRef PubMed.
- (a) Top value-Added Chemicals from Biomass Vol. II—Results of Screening for Potential Candidates from Biorefinery Lignin, ed. J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, U. S. Department of Energy (DOE) by the Pacific Northwest National Laboratory, Richland, WA, US, 2007, PNNL-16983 Search PubMed; (b) Top Value-Added Chemicals from Biomass Vol. I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, ed. T. Werpy and G. Petersen, U. S. Department of Energy (DOE) by the National Renewable Energy Laboratory a DOE national Laboratory, 2004 Search PubMed.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- (a) J. H. Clark, R. Luque and A. Matharu, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 183 CrossRef CAS PubMed; (b) P. Gallezot, ChemSusChem, 2008, 1, 734 CrossRef CAS PubMed; (c) M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827 CrossRef CAS PubMed; (d) N. Yan and P. J. Dyson, Curr. Opin. Chem. Eng., 2013, 2, 178 CrossRef; (e) A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2012, 2, 1487 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- (a) J. C. Hicks, J. Phys. Chem. Lett., 2011, 2, 2280 CrossRef CAS; (b) S. Dutta, ChemSusChem, 2012, 5, 2125 CrossRef CAS PubMed.
- (a) H. Li, F. Chang, Y. Zhang, D. Hu, L. Jin, B. Song and S. Yang, Current Catalysis, 2012, 1, 221 CrossRef CAS; (b) H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, John Wiley & Sons, Inc., Hoboken, NJ, US, 2nd edn, 2006 Search PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis Understanding the Art, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2004 Search PubMed.
- (a) Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and Gandini, 2008, Elsevier, Amsterdam Search PubMed; (b) A. Gandini, Macromolecules, 2008, 41, 9491 CrossRef CAS.
- R.-J. van Putten, J. C. Van der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- K.-I. Seri, Y. Inoue and H. Ishida, Chem. Lett., 2000, 22 CrossRef CAS.
- C. B. Rasrendra, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, Top. Catal., 2010, 53, 1241 CrossRef CAS.
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4, 2141 Search PubMed.
- F.-F. Wang, C.-L. Liu and W.-S. Dong, Green Chem., 2013, 15, 2091 RSC.
- S. Kolarić and V. Šunjić, J. Mol. Catal. A: Chem., 1996, 110, 189 CrossRef.
- S. Rajagopal, S. Vancheesan, J. Rajaram and J. C. Kuriacose, J. Mol. Catal., 1992, 75, 199 CrossRef CAS.
- G. Descotes, J. Sabadie and D. Sinou, Tetrahedron Lett., 1978, 19, 3351 CrossRef.
- C. Ramstadius, A. M. Träff, P. Krumlinde, J.-E. Bäckvall and I. Cumpstey, Eur. J. Org. Chem., 2011, 4455 CrossRef CAS.
- M. Trincado, K. Kühlein and H. Grützmacher, Chem.–Eur. J., 2011, 17, 11905 CrossRef CAS PubMed.
- C. Wang, A. Pettman, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548 CrossRef CAS PubMed.
- R. M. Painter, D. M. Pearson and R. M. Waymouth, Angew. Chem., Int. Ed., 2010, 49, 9456 CrossRef CAS PubMed.
- M. Jäger, M. Hartmann, J. G. de Vries and A. J. Minnaard, Angew. Chem., Int. Ed., 2013, 52, 7809 CrossRef PubMed.
- R. Nygaard Monrad and R. Madsen, J. Org. Chem., 2007, 72, 9782 CrossRef CAS PubMed.
- D. J. Hayes, S. W. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries-Industrial Processes and Products: Status Quo and Future Directions Vol 1, ed. B. Kamm, P. R. Gruber and M. Kamm, John Wiley & Sons, Weinheim, Germany, 2006, pp. 139–163 Search PubMed.
- A. M. Raspolli Galletti, C. Antonetti, V. De Luise, D. Licursi and N. Nassi o Di Nasso, BioResources, 2012, 7, 1824 CrossRef.
- (a) H. D. Hoving, A. de Rijke, G. M. C. Wagemans, R. F. M. J. Parton and K. Babic, WO Pat., WO2013/034763, 2013 Search PubMed; (b) R. F. M. J. Parton, M. P. W. M. Rijkers and J. A. Kroon, US Pat., US2012/0330040, 2012 Search PubMed.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed.
- (a) K. Osakada, T. Ikariya and S. Yoshikawa, J. Organomet. Chem., 1982, 231, 79 CrossRef CAS; (b) H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS; (c) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed.
- W. Li, J.-H. Xie, H. Lin and Q.-L. Zhou, Green Chem., 2012, 14, 2388–2390 RSC.
- J. Deng, Y. Wang, T. Pan, Q. Xu, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163 CrossRef CAS PubMed.
- F. Joo, Z. Tóth and M. T. Beck, Inorg. Chim. Acta, 1977, 25, L61 CrossRef CAS.
- M. Chalid, A. A. Broekhuis and H. J. Heeres, J. Mol. Catal. A: Chem., 2011, 341, 14 CrossRef CAS.
- J. M. Tukacs, D. Király, A. Strádi, G. Novodarszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057 RSC.
- C. Delhomme, L.-A. Schaper, M. Zhang-Preße, G. Raudaschl-Sieber, D. Weuster-Botz and F. E. Kühn, J. Organomet. Chem., 2013, 724, 297 CrossRef CAS.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247 RSC.
- L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529 CrossRef CAS PubMed.
- L. Qi and I. T. Horvath, ACS Catal., 2012, 2, 2247 CrossRef CAS.
- N. J. Wise and J. M. J. Williams, Tetrahedron Lett., 2007, 48, 3639 CrossRef CAS.
- Y. Wei, C. Wang, X. Jiang, D. Xue, J. Li and J. Xiao, Chem. Commun., 2013, 49, 5408 RSC.
- J. G. de Vries, N. Sereinig, E. W. M. van de Vondervoort and M. C. C. Janssen, WO Pat., WO2012/131027, 2012 Search PubMed.
- P. K. Wong, C. Li, L. Stubbs, M. van Meurs, D. G. Anak Kumbang, S. C. Y. Lim and E. Drent, WO Pat., WO2012/134397, 2012 Search PubMed.
- (a) H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS PubMed; (b) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed.
- (a) G. Papadogianakis, L. Maat and R. A. Sheldon, J. Chem. Soc., Chem. Commun., 1994, 2659 RSC; (b) C. M. Nicklaus, A. J. Minnaard, B. L. Feringa and J. G. de Vries, ChemSusChem, 2013, 6, 1631 CrossRef CAS PubMed.
- F. M. A. Geilen, T. vom Stein, B. Engendahl, S. Winterle, M. A. Liauw, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2011, 50, 6831 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083 CrossRef CAS PubMed.
- C. M. Nicklaus, P. H. Phua, T. Buntara, S. Noel, H. J. Heeres and J. G. de Vries, Adv. Synth. Catal., 2013, 355, 2839 CrossRef CAS.
- G. Flèche and M. Huchette, Starch/Staerke, 1986, 38, 26 CrossRef.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199 CrossRef CAS PubMed.
- S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS PubMed.
- D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905 CrossRef CAS.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227 CrossRef CAS PubMed.
- (a) J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Martina, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29 CrossRef CAS; (b) G. Brunow and K. Lundquist, Functional Groups and Bonding Patterns in Lignin (including the Lignin-Carbohydrate Complexes), in Lignin and Lignans, CRC Press, Boca Raton, FL (USA), 2011, pp. 167–299 Search PubMed; (c) M. Y. Balakshin, E. A. Capanema and H.-M. Chang, Recent Advances in the Isolation and Analysis of Lignins and Lignin-Carbohydrate Complexes, in Characterization of Lignocellulosic Materials, ed. T. Q. Hu, Blackwell publishing Ltd, Oxford, UK, 2009, p. 148 Search PubMed; (d) S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846 CrossRef CAS.
- H. R. Bjørsvik and L. Liguori, Org. Process Res. Dev., 2002, 6, 279 CrossRef.
- C. A. Gasser, G. Hommes, A. Schäffer and P. F.-X. Corvini, Appl. Microbiol. Biotechnol., 2012, 95, 1115 CrossRef CAS PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090 CrossRef CAS PubMed; H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869 Search PubMed.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554 CrossRef CAS PubMed.
- S. C. Chmely, S. Kim, P. N. Ciesielski, G. Jiménez-Osés, R. S. Paton and G. T. Beckham, ACS Catal., 2013, 3, 963–974 CrossRef CAS.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093 RSC.
- T. vom Stein, T. Weigand, C. Merkens, J. Klankenmayer and W. Leitner, ChemCatChem, 2013, 5, 439 CrossRef CAS.
- D. Weickmann and B. Plietker, ChemCatChem, 2013, 5, 2170 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed.
- P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480 CrossRef CAS PubMed.
- B. S. Samant and G. W. Kabalka, Chem. Commun., 2012, 48, 8658 RSC.
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640 RSC.
- A. C. Atesin, N. A. Ray, P. C. Stair and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 14682 CrossRef CAS PubMed.
- R. S. Assary, A. C. Atesin, Z. Li, L. A. Curtiss and T. J. Marks, ACS Catal., 2013, 3, 1908 CrossRef CAS.
- A. Wu, B. O. Patrick and B. R. James, Inorg. Chem. Commun., 2012, 24, 11 CrossRef CAS.
- M. Nagy, K. David, G. J. P. Britovsek and A. J. Ragauskas, Holzforschung, 2009, 63, 513 CrossRef CAS.
- (a) Q. Song, F. Wang and J. Xu, Chem. Commun., 2012, 48, 7019 RSC; (b) Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 RSC; (c) X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499 CrossRef CAS PubMed.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttämaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806 RSC.
- K. Barta, G. Warner, E. Beach and P. T. Anastas, Green Chem., 2014, 16, 191 RSC.
- S. R. Collinson and W. Thielemans, Coord. Chem. Rev., 2010, 254, 1854 CrossRef CAS.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, A. D. Sutton and D. L. Thorn, J. Am. Chem. Soc., 2009, 131, 428 CrossRef CAS PubMed.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611 CrossRef CAS PubMed.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791 CrossRef CAS PubMed.
- G. Zhang, B. L. Scott, R. Wu, L. A. Silks and S. K. Hanson, Inorg. Chem., 2012, 51, 7354 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410 CrossRef CAS PubMed.
- B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415 CrossRef CAS PubMed.
- (a) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC; (b) P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805 CrossRef CAS; (c) J. M. Hoover, J. E. Stevens and S. S. Stahl, Nat. Protoc., 2012, 7, 1161 CrossRef CAS PubMed.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794 CrossRef CAS.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369 CrossRef CAS.
- C.-L. Chen, E. A. Capanema and H. S. Gracz, J. Agric. Food Chem., 2003, 51, 1932 CrossRef CAS PubMed.
- (a) U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854 CrossRef CAS PubMed; (b) A. Behr, A. Westfechtel and J. Pérez Gomes, Chem. Eng. Technol., 2008, 31, 700 CrossRef CAS.
- (a) G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Polymers, 2010, 2, 440 CrossRef CAS; (b) M. Desroches, M. Escouvois, R. Auvergne, S. Caillol and B. Boutevin, Polym. Rev., 2012, 52, 38 CrossRef CAS; (c) Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13 RSC.
- F. Liu, K. De Oliviera Vigier, M. Pera-Titus, Y. Pouilloux, J.-M. Clacens, F. Decampo and F. Jérome, Green Chem., 2013, 15, 901 RSC.
- Z. X. Giustra and K. L. Tan, Chem. Commun., 2013, 49, 4370 RSC.
- (a) Q. Tian and R. C. Larock, J. Am. Oil Chem. Soc., 2002, 79, 479 CrossRef CAS; (b) T. C. Mauldin, K. Haman, X. Sheng, P. Henna, R. C. Larock and M. R. Kessler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6851 CrossRef CAS; (c) P. H. Henna and R. C. Larock, Macromol. Mater. Eng., 2007, 292, 1201 CrossRef CAS; (d) P. H. Henna and R. C. Larock, J. Appl. Polym. Sci., 2009, 112, 1788–1797 CrossRef CAS; (e) P. H. Henna, M. R. Kessler and R. C. Larock, Macromol. Mater. Eng., 2008, 293, 979 CrossRef CAS; (f) Y. Xia, Y. Lu and R. C. Larock, Polymer, 2010, 51, 53 CrossRef CAS; (g) P. A. Fokou and M. A. R. Meier, Macromol. Rapid Commun., 2008, 29, 1620 CrossRef CAS; (h) U. Biermann, J. O. Metzger and M. A. R. Meier, Macromol. Chem. Phys., 2010, 211, 854 CrossRef CAS.
- (a) E. H. Pryde, J. Am. Oil Chem. Soc., 1984, 61, 419 CrossRef CAS; (b) A. Guo, D. Demydov, W. Zhang and Z. S. Petrovic, J. Polym. Environ., 2002, 10, 49 CrossRef CAS.
- K. F. Muilwijk, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Oil Chem. Soc., 1997, 74, 223 CrossRef CAS.
- P. Kandanarachchi, A. Guo and Z. Petrovic, J. Mol. Catal. A: Chem., 2002, 184, 65 CrossRef CAS.
- A. Behr, D. Obst and A. Westfechtel, Eur. J. Lipid Sci. Technol., 2005, 107, 213 CrossRef CAS.
- A. Guo, W. Zhang and Z. S. Petrovic, J. Mater. Sci., 2006, 41, 4914–4920 CrossRef CAS.
- J. Boulanger, A. Ponchel, H. Bricout, F. Hapiot and E. Monflier, Eur. J. Lipid Sci. Technol., 2012, 114, 1439 CrossRef CAS.
- A. Behr, M. Fiene, C. Buß and P. Eilbracht, Eur. J. Lipid Sci. Technol., 2000, 102, 467 CrossRef CAS.
- W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. Commun., 1997, 1521 RSC.
- (a) D. Bourgeois, P. Marion and C. Rosier, US Pat., US20040122251A1, 2004 Search PubMed; (b) A. Allgeier and C. Lenges, US Pat., US20050059737A1, 2005 Search PubMed.
- K. Y. Ghebreyessus and R. J. Angelici, Organometallics, 2006, 25, 3040 CrossRef CAS.
- G. M. Lee, C. M. Vogels, A. Decken and S. A. Westcott, Eur. J. Inorg. Chem., 2011, 2433 CrossRef CAS.
- T. Huber, D. Firlbeck and H. M. Riepl, J. Organomet. Chem., 2013, 744, 144 CrossRef CAS.
- D. M. Ohlmann, L. J. Gooßen and M. Dierker, Chem.–Eur. J., 2011, 17, 9508 CrossRef CAS PubMed.
- C. Jiménez-Rodriguez, G. R. Eastham and D. J. Cole-Hamilton, Inorg. Chem. Commun., 2005, 8, 878 CrossRef.
- (a) W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. Lan Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC; (b) G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609 RSC.
- P. Roesle, C. J. Dürr, H. M. Möller, L. Cavallo, L. Caporaso and S. Mecking, J. Am. Chem. Soc., 2012, 134, 17696 CrossRef CAS PubMed.
- M. R. L. Furst, T. Seidensticker and D. J. Cole-Hamilton, Green Chem., 2013, 15, 1218 RSC.
- M. R. L. Furst, R. Le Goff, D. Quinzler, S. Mecking, C. H. Botting and D. J. Cole-Hamilton, Green Chem., 2012, 14, 472 RSC.
- G. Walther, J. Deutsch, A. Martin, F.-E. Baumann, D. Fridag, R. Franke and A. Köckritz, ChemSusChem, 2011, 4, 1052 CrossRef CAS PubMed.
- (a) D. Quinzler and S. Mecking, Angew. Chem., Int. Ed., 2010, 49, 4306 CrossRef CAS PubMed; (b) F. Stempfle, D. Quinzler, I. Heckler and S. Mecking, Macromolecules, 2011, 44, 4159 CrossRef CAS; (c) D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2010, 49, 8564 CrossRef CAS PubMed.
- K.-C. Shih and R. J. Angelici, J. Org. Chem., 1996, 61, 7784 CrossRef CAS PubMed.
- L. J. Gooßen, D. M. Ohlmann and M. Dierker, Green Chem., 2010, 12, 197 RSC.
- D. M. Ohlmann, N. Tschauder, J.-P. Stockis, K. Gooßen, M. Dierker and L. J. Gooßen, J. Am. Chem. Soc., 2012, 134, 13716 CrossRef CAS PubMed.
- (a) A. Philippaerts, S. Goossens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2011, 4, 684 CrossRef CAS PubMed; (b) D. D. Andjelkovic, B. Min, D. Ahn and R. C. Larock, J. Agric. Food Chem., 2006, 54, 9535 CrossRef CAS PubMed.
- A. Behr, H. Witte and Z. Bayrak, Eur. J. Lipid Sci. Technol., 2013, 115, 721 CrossRef CAS.
- (a) R. L. Quirino and R. C. Larock, J. Am. Oil Chem. Soc., 2012, 89, 1113 CrossRef CAS; (b) C. S. Consorti, G. L. P. Aydos, G. Ebeling and J. Dupont, Appl. Catal., A, 2009, 371, 114 CrossRef CAS.
- S. Perdiau, S. Harder, H. J. Heeres and J. G. de Vries, ChemSusChem, 2012, 5, 2427 CrossRef PubMed.
- S. Shahane, C. Fischmeister and C. Bruneau, Catal. Sci. Technol., 2012, 2, 1425 CAS.
- N. Sereinig, M. C. C. Janssen and J. G. de Vries, WO Pat., WO 2013/013990A1, 2013 Search PubMed.
- M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1073 CrossRef CAS.
- (a) L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837 CrossRef CAS; (b) C, Vilela, A. J. D. Silvestre and M. A. R. Meier, Macromol. Chem. Phys., 2012, 213, 2220 CrossRef CAS; (c) X. Miao, R. Malacea, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2911 RSC; (d) M. Sacristán, J. C. Ronda, M. Galià and V. Cádiz, J. Appl. Polym. Sci., 2011, 122, 1649 CrossRef.
- T. W. Abraham, H. Kaido, C. W. Lee, R. L. Pederson, Y. Schrodi and M. J. Tupy, US Pat., US20090264672A1, 2009 Search PubMed.
- (a) Y. Xia and R. C. Larock, Polymer, 2010, 51, 2508 CrossRef CAS; (b) U. Biermann, J. O. Metzger and M. A. R. Meier, Macromol. Chem. Phys., 2010, 211, 854–862 CrossRef CAS.
- (a) I. Ibrahem, M. Yu, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3844 CrossRef CAS PubMed; (b) M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962 CrossRef CAS PubMed; (c) S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461 CrossRef CAS PubMed.
- B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693 CrossRef CAS PubMed.
- M. B. Herbert, V. M. Marx, R. L. Pederson and R. H. Grubbs, Angew. Chem., 2013, 125, 328 CrossRef.
- (a) A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2008, 110, 812 CrossRef; (b) M. Guidotti, R. Psaro, M. Sgobba and N. Ravasio, Catalytic Processes for the Selective Epoxidation of Fatty Acids: More Environmentally Benign Routes, in Catalysis for Renewables: From Feedstock to Energy Production, 2007, ch. 12, pp. 257–272 Search PubMed.
- I. Hermans, K. Janssen, B. Moens, A. Philippaerts, B. van Berlo, J. Peeters, P. A. Jacobs and B. F. Sels, Adv. Synth. Catal., 2007, 349, 1604 CrossRef CAS.
- S. G. Tan and W. S. Chow, Polym.-Plast. Technol. Eng., 2010, 49, 1581 CrossRef CAS.
- (a) T. A. Foglia, P. E. Sonnet, A. Nunez and R. L. Dudley, J. Am. Oil Chem. Soc., 1998, 75, 601 CrossRef CAS; (b) M. Nor Omar, H. A. Moynihan and R. J. Hamilton, Eur. J. Lipid Sci. Technol., 2003, 105, 43 CrossRef.
- M. Nor bin Omar, N. N. M. Nor, H. Moynihan and R. Hamilton, Biotechnology, 2007, 6, 283 CrossRef CAS.
- P. Spannring, V. Yazerski, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. Klein Gebbink, Chem.–Eur. J., 2013, 19, 15012 CrossRef CAS PubMed.
- (a) P. Spannring, I. Prat, M. Costas, M. Lutz, P. C. A. Bruijnincx, B. Weckhuysen and B. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 708 RSC; (b) P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, RSC Adv., 2013, 3, 6606 RSC.
| This journal is © The Royal Society of Chemistry 2014 |