 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and high-throughput testing of multilayered supported ionic liquid catalysts for the conversion of CO2 and epoxides into cyclic carbonates†
Paola
Agrigento
ab,
Syed M.
Al-Amsyar
a,
Benjamin
Sorée
c,
Masoumeh
Taherimehr
a,
Michelangelo
Gruttadauria
*b,
Carmela
Aprile
*c and
Paolo P.
Pescarmona
*a
aCentre for Surface Chemistry Catalysis, University of Leuven (KU Leuven), Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: paolo.pescarmona@biw.kuleuven.be; Tel: +32 16 321592
bDipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche (STEBICEF), Sezione di Chimica, University of Palermo, Viale delle Scienze s/n, Ed. 17, I-90128 Palermo, Italy. E-mail: michelangelo.gruttadauria@unipa.it
cUnit of Nanomaterial Chemistry (CNano), Department of Chemistry, University of Namur (UNAMUR), Rue de Bruxelles 61, 5000 Namur, Belgium. E-mail: carmela.aprile@unamur.be; Tel: +32 81 724514
First published on 16th January 2014
Abstract
Multilayered covalently supported ionic liquid phase (mlc-SILP) materials were synthesised by grafting different bis-vinylimidazolium salts on thiol-functionalised silica. These materials, which contain a cross-linked oligomeric network of imidazolium units, were characterised and tested as catalysts for the reaction of carbon dioxide with various epoxides to produce cyclic carbonates. The materials prepared by supporting a bis-imidazolium iodide salt with xylene or octane as a linker between the imidazolium units were identified as the most active catalysts and displayed high turnover numbers and improved productivity compared to known supported ionic liquid catalysts. The most promising mlc-SILP catalysts were further studied to tune the reaction conditions towards optimum catalytic performance and to investigate their versatility with different substrates and their reusability. The rapid and parallel screening of the catalysts was efficiently carried out by means of high-throughput (HT) experimentation.
Introduction
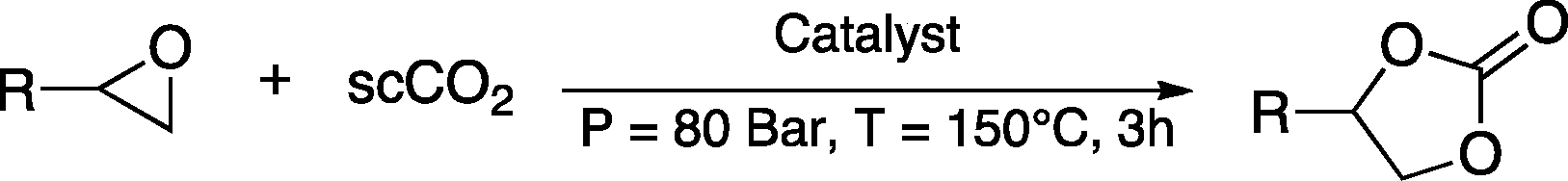
Carbon dioxide is generated in large amounts as the end product of fuel combustion related to human activities and is considered an environmental threat being one of the main greenhouse gases.1 The widespread availability of CO2 stimulated its use as a renewable, inexpensive and non-toxic carbon source for the synthesis of useful chemical products.2 For these features, carbon dioxide represents an attractive green alternative to toxic reactants such as phosgene, isocyanates or carbon monoxide. However, the use of CO2 as feedstock requires scientific and technological progress concerning its separation and conversion. Particularly, the challenge in the conversion of CO2 stems from the high thermodynamic stability of this molecule. This limitation can be overcome by reacting carbon dioxide with high free energy molecules, among which epoxides have been widely studied.3 The reaction of CO2 with epoxides can generate two types of products: cyclic carbonates and polycarbonates, both of which can find relevant applications. This is an attractive reaction in terms of green chemistry and atom efficiency, because CO2 reacts with epoxides without generating any side-product. Various homogeneous and heterogeneous catalysts have been developed to promote this reaction.3–5 Typically, the catalyst contains a nucleophilic species that is able to attack and open the epoxide ring, thus facilitating the addition of CO2 (Scheme 1). State-of-the-art homogeneous catalytic systems for this reaction also include a Lewis acid species that activates the epoxide towards ring opening by the nucleophile.3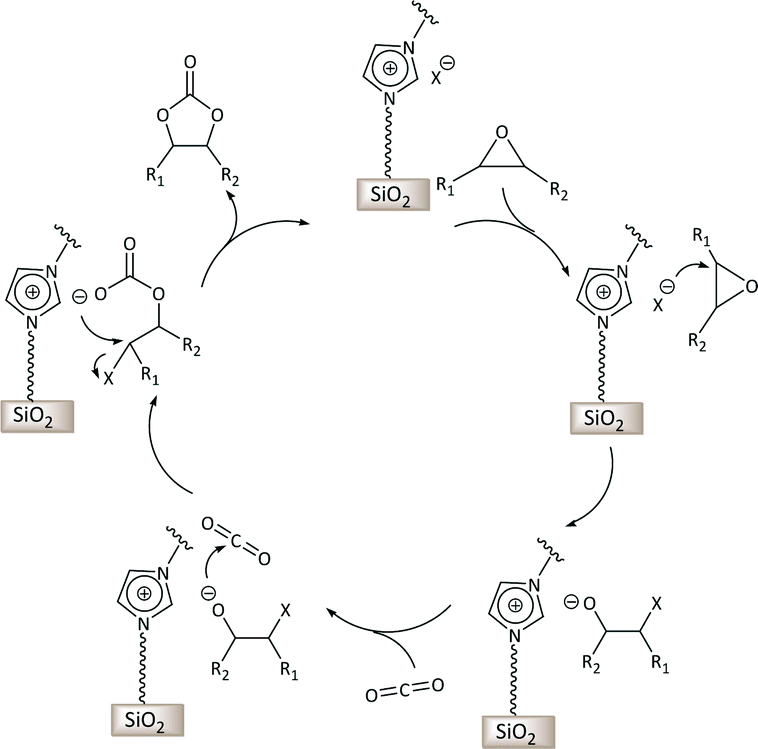 | ||
| Scheme 1 Proposed mechanism for the synthesis of cyclic carbonates via reaction of CO2 with epoxides catalysed by supported imidazolium-based ionic liquids. X− is a halide that acts as a nucleophile. | ||
With the final aim of an industrial application of the reaction of CO2 with epoxides, research effort should be dedicated to the development of suitable heterogeneous catalysts, which can be easily separated from the reaction products and reused in consecutive runs. Supported ionic liquids with halide counterions acting as nucleophiles have been reported as a promising class of metal-free heterogeneous catalysts for this reaction.6,7
The most efficient approach for the immobilisation of ionic liquids on a support is based on covalent grafting, which grants high stability and thus good recyclability of the catalyst. Usually, these catalysts are tested in a solvent-free medium and often with CO2 under supercritical conditions (scCO2), which displays high solubility in various epoxides. Since these supported ionic liquid catalysts are devoid of metal centres that could act as Lewis acids, relatively high temperature (typically ≥100 °C) is required in order to achieve good conversions in the reaction between CO2 and epoxides. This promotes the formation of cyclic carbonates, which are the thermodynamically most favourable products.3 Cyclic carbonates are a class of versatile compounds, finding a broad range of applications as aprotic polar solvents, as electrolytes in secondary batteries, and as intermediates in the production of polymers and fine chemicals.8
Recently, we reported the synthesis of a new class of multilayered covalently supported ionic liquid phase (mlc-SILP) catalysts.9 These materials present a much higher loading of the catalytically active imidazolium salt compared to conventional supported ionic liquids consisting of a covalently anchored monolayer (Fig. 1). The multilayered ionic liquid phase can be generated by grafting a bis-vinylimidazolium halide salt on a suitable support, typically a high surface area silica or a polymer functionalised with groups that allow anchoring of the ionic liquid (e.g. thiol groups, –SH). The two terminal vinyl groups of the bis-vinylimidazolium salt can react not only with the functionalised surface of the support but also with each other through a self-addition reaction of the double bonds; this allows creation of the desired material presenting a cross-linked network of imidazolium units covalently anchored on the support (Fig. 2). The mlc-SILP material prepared by supporting a bromide bis-imidazolium salt on high surface area silica (either amorphous or ordered mesoporous SBA-15) was identified as the best catalyst for the synthesis of cyclic carbonates, displaying very good activity and improved productivity compared to known supported ionic liquid catalysts.9
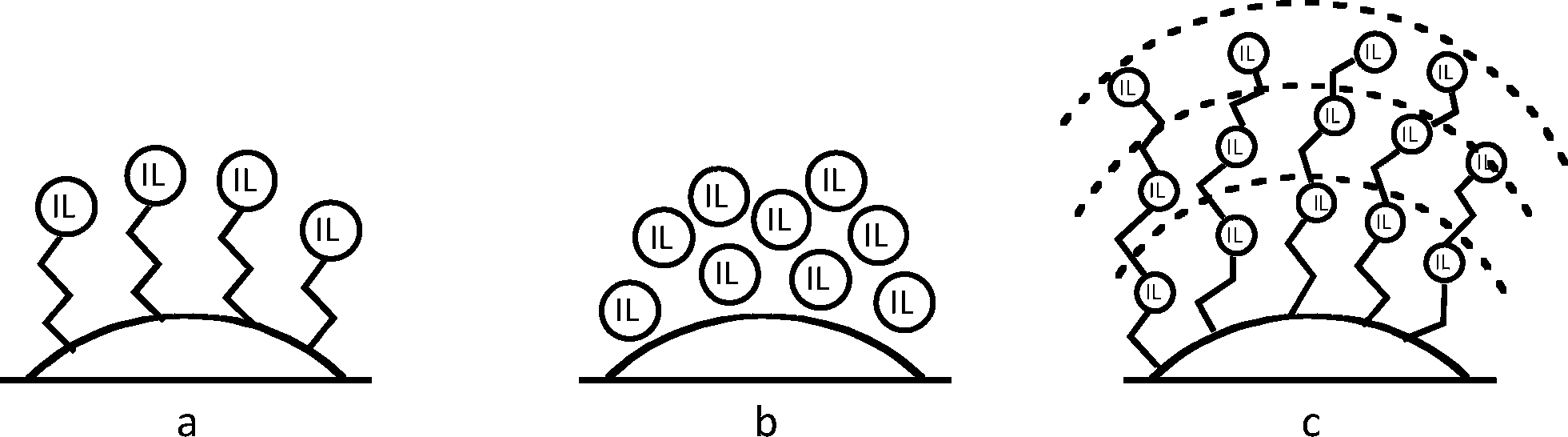 | ||
| Fig. 1 Schematic representation of: (a) covalently anchored monolayer of ionic liquid; (b) adsorbed multilayer of ionic liquid; (c) covalently anchored multilayer of ionic liquid. The latter material combines the asset of a stable, covalent grafting (as in a) with the high loading of ionic liquid of a multilayered material (as in b). | ||
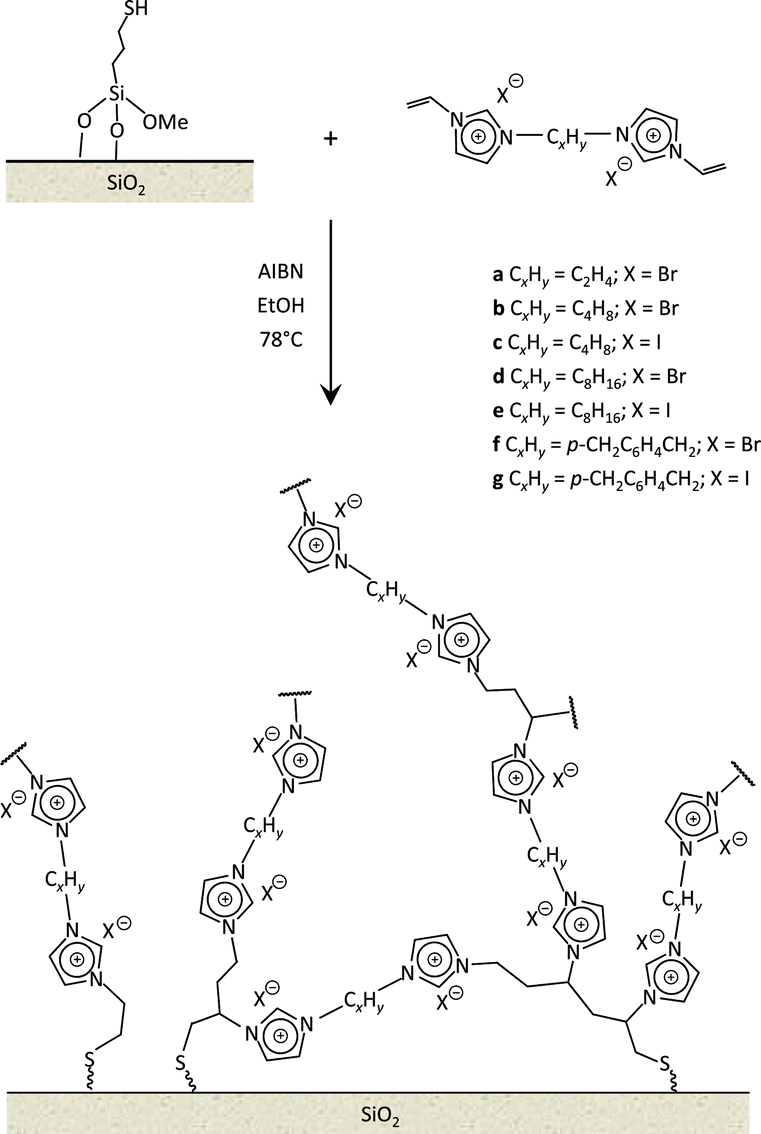 | ||
| Fig. 2 Schematic representation of the synthesis of multilayered supported ionic liquid phase materials from bis-vinylimidazolium salts with different organic linkers (a–g). The wavy lines in the scheme symbolise further extensions of the supported ionic liquid phase. | ||
In this work, the potential of mlc-SILP as heterogeneous catalysts for the reaction of CO2 with epoxides is further explored. A series of mlc-SILP materials was prepared using bis-vinylimidazolium salts with different organic linkers between the two imidazolium units (Fig. 2) and two types of halides as counterions (X = Br or I). Thiol-functionalised silica was employed as the support.9 These materials were characterised and tested as catalysts for the reaction of carbon dioxide with various epoxides. A rapid and efficient parallel screening of the series of mlc-SILP catalysts was carried out using a tailored high-throughput (HT) unit for the study of reactions under supercritical CO2.9–11
Results and discussion
Multilayered covalently supported ionic liquid phase (mlc-SILP) materials are a recently developed class of materials that is finding promising applications in heterogeneous catalysis.9,12 A previous study from our groups showed that mlc-SILPs prepared from bis-imidazolium chloride or bromide with xylene as a linker between the imidazolium units are active catalysts for the reaction of CO2 with epoxides, reaching higher productivity compared to monolayered supported ionic liquids. Here, we present the synthesis, characterisation and catalytic application of a series of new mlc-SILP materials. The multilayered covalently supported ionic liquids were prepared using a series of bis-imidazolium salts, having different lengths and bulkiness of the organic linker connecting the two imidazolium units (Fig. 2). The distance between the two imidazolium units is expected to have a relevant influence on the catalytic performance of the materials, as the attack on the epoxide by the nucleophile is likely to occur in the proximity of the imidazolium units (Scheme 1). Too short a distance between two connected imidazolium units might prevent simultaneous reaction at both of them, while too large a distance would imply a lower loading of active sites per gram of catalyst. Moreover, the chemical nature of the linker can influence the approach of CO2 and of the epoxide to the active sites. The second parameter that was varied in the bis-imidazolium salts is the nature of the halide counterion. This is a crucial element, because the halide is the nucleophile that initiates the catalytic reaction (Scheme 1). The catalytic performance is determined by a combination of the nucleophilicity of the halide towards attack on the epoxide, its coordination ability to the imidazolium unit and its leaving ability from the carbonate intermediate.3 A previous study on mlc-SILPs showed that higher catalytic activity was obtained with Br− compared to Cl−.9 This result stimulated us to investigate mlc-SILPs prepared using Br− and I− as halides. Iodide has been often reported to lead to higher catalytic activity compared to bromide,10 though the complexity of the balance between nucleophilicity, coordination ability and leaving ability implies that different catalytic systems show different orders of activity between the halides.3High surface area amorphous silica (entry 1 in Table 1) functionalised with thiol groups was employed as a support for the new mlc-SILP materials. The bis-vinylimidazolium salts were grafted on the silica support through a thiol–ene coupling reaction between the thiol groups of the modified silica and the double bond of the bis-vinylimidazolium salts, in the presence of 2,2′-azobisisobutyronitrile (AIBN) as a radical initiator (Fig. 2). This reaction offers all of the desirable features of a ‘click reaction’, being highly efficient and simple to execute with no side-products, and proceeding relatively rapidly with high yield. Since the bis-vinylimidazolium salt was used in excess relative to the amount of thiol groups (3.62 mol of salt per mol of thiol group), the formation of a cross-linked oligomeric network of imidazolium units through self-addition reaction of the double bonds is also expected. Therefore, this protocol aims at the formation of a multilayered ionic liquid phase and its covalent grafting on the functionalised silica support.
| Entry | mlc-SILP | Imidazolium unitsb (mmol g−1) | IL/–SH molar ratioc | Loading of supported IL (wt.%) | Surface area (m2 g−1) | Cumulative pore volume (cm3 g−1) |
|---|---|---|---|---|---|---|
| a Synthesis of mlc-SILP materials: bis-vinylimidazolium salt (3.62 mol mol-SH−1, 130 mM), thiol-functionalised silica (1.2 mmol-SH g−1), ethanol, AIBN, 78 °C, 20 h. b Each bis-vinylimidazolium salt contains two imidazolium units. c Molar ratio between the amount of bis-vinylimidazolium salt in the obtained mlc-SILP and the amount of potential anchoring sites on the support (i.e. the original amount of –SH groups). d Data taken from ref. 9. | ||||||
| 1 | SiO2 | — | — | — | 750 | 0.7 |
| 2 | SiO2–ethane–Br | 2.7 | 2.3 | 51 | 129 | 0.2 |
| 3 | SiO2–butane–Br | 2.9 | 2.8 | 58 | 146 | 0.2 |
| 4 | SiO2–butane–I | 2.7 | 3.4 | 67 | 134 | 0.3 |
| 5 | SiO2–octane–Br | 2.5 | 2.5 | 58 | 89 | 0.2 |
| 6 | SiO2–octane–I | 2.2 | 2.4 | 61 | 121 | 0.1 |
| 7 | SiO2–p-xylene–Br | 2.8d | 3.5d | 64d | 124d | 0.1d |
| 8 | SiO2–p-xylene–I | 2.0 | 1.8 | 53 | 133 | 0.3 |
All of the mlc-SILP materials were characterised by means of elemental analysis to evaluate the loading of the ionic liquid phase and the molar ratio between the supported ionic liquid and the original amount of –SH groups (Table 1). The employed method allows reaching a high loading of anchored bis-vinylimidazolium salt (51 to 67 wt.%) and high IL/-SH molar ratios. These results demonstrate the general validity of this synthesis method, in which the use of excess bis-vinylimidazolium salt with respect to the thiol groups leads to the formation of the desired multilayered covalently supported ionic liquid phase. The successful formation of the oligomeric network of supported ionic liquids was confirmed by solid-state 13C NMR spectroscopy (Fig. 3); the absence of the signal corresponding to the vinyl group of the bis-vinylimidazolium salts (at δ = 110 ppm) is an indication that the terminal double bonds reacted completely, either with the thiol groups of the support, or with the vinyl groups of other bis-vinylimidazolium salts. The characteristic signals of the carbon atoms of the imidazolium ring are observed at δ = 121–124 and 136 ppm, whereas the aliphatic carbon atoms resonate in the range 27–60 ppm. These data indicate that the main structure of the bis-vinylimidazolium salts is not affected by the grafting process. The weak signal at δ = 12 ppm is due to the carbon atoms connected to the silicon atoms of the functionalised silica.
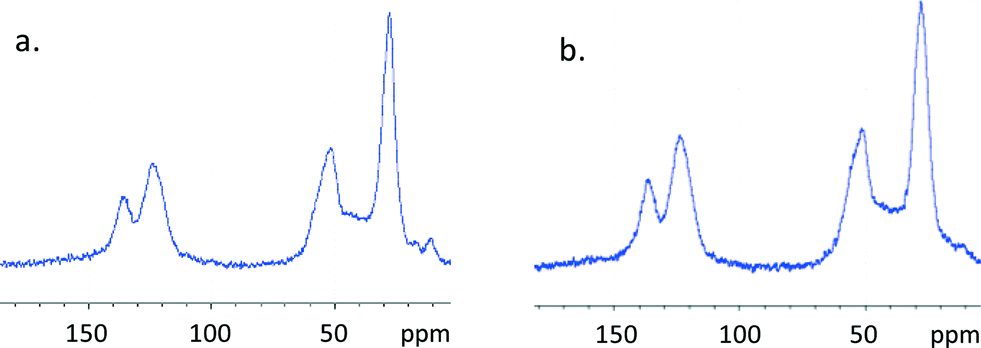 | ||
| Fig. 3 Solid state MAS 13C NMR spectra of SiO2–octane–Br (a) and SiO2–octane–I (b). | ||
The textural properties of the mlc-SILP materials were studied by N2 adsorption/desorption measurements (Table 1). In line with previous reports, a substantial decrease in surface area and cumulative pore volume was observed after grafting of the ionic liquid phase.9 This dramatic change in the textural properties compared to those of the parent silica further confirms the successful grafting of the bis-vinylimidazolium salts on the support.
Although the characterisation data clearly prove the formation of the desired mlc-SILP with all of the employed bis-vinylimidazolium salts, they do not allow identifying the exact structure of the oligomeric network of supported ionic liquids, which is likely to be disordered (see Fig. 2 for a possible representation).
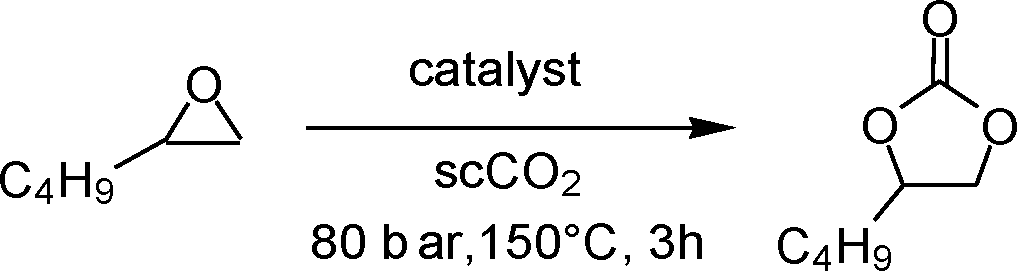
The multilayered covalently supported ionic liquid materials were tested as catalysts for the synthesis of cyclic carbonates via the reaction of epoxides with carbon dioxide under solvent-free conditions and with CO2 under supercritical conditions (i.e. at a pressure above 73.8 bar and a temperature above 31 °C). All of the reactions were carried out in a high-throughput unit that enables a rapid and reliable parallel testing of the series of catalysts in a block consisting of 24 parallel batch reactors.9–11 In order to investigate the versatility of our catalysts, two different epoxides were selected as substrates for the reaction with CO2: an aliphatic terminal epoxide (1,2-epoxyhexane) and an aromatic terminal epoxide (styrene oxide). All of the mlc-SILP catalysts display activity in the conversion of the two epoxides (Table 2), with virtually complete selectivity (>99%) towards the cyclic carbonate product in all cases.13 The lack of activity of the thiol-functionalised silica (entry 9, Table 2) confirms that the observed activity originates from the supported ionic liquid phases.
| Entry | mlc-SILP | Styrene oxide | 1,2-Epoxyhexane | ||||
|---|---|---|---|---|---|---|---|
| Conv.c (%) | Prod.d | TONe | Conv.c (%) | Prod.d | TONe | ||
| a Reaction conditions: epoxide (14.0 mmol), mesitylene (1.40 mmol), 30 mg of catalyst, CO2 (80 bar), 150 °C, 3 h. b Reaction conditions: epoxide (46.7 mmol), mesitylene (4.67 mmol), 100 mg of catalyst, CO2 (80 bar), 125 °C, 3 h. c Conversion and product yields determined by GC, using mesitylene as the internal standard. For selected samples the conversion and yields were confirmed by 1H NMR. d Productivity, defined as gcarbonate gcatalyst−1. e Turnover number, defined as molepoxide converted molhalide−1. | |||||||
| 1 | SiO2–ethane–Br | 48 | 36 | 82 | 20 | 13 | 34 |
| 2 | SiO2–butane–Br | 56 | 43 | 92 | 20 | 14 | 33 |
| 3 | SiO2–butane–I | 66 | 51 | 116 | 29 | 19 | 50 |
| 4 | SiO2–octane–Br | 79 | 60 | 146 | 27 | 18 | 50 |
| 5 | SiO2–octane–I | 85 | 65 | 180 | 37 | 25 | 78 |
| 6 | SiO2–p-xylene–Br | 51 | 39 | 84 | 12 | 8 | 20 |
| 7 | SiO2–p-xylene–I | 99 | 76 | 237 | 45 | 30 | 108 |
| 8 | SiO2–p-xylene–Ib | 37 | 28 | 88 | 21 | 14 | 51 |
| 9 | SiO2–SH | 0 | 0 | 0 | 0 | 0 | 0 |
This set of results shows some clear trends that deserve to be discussed. The mlc-SILP materials with iodide as the counterion display higher activity in terms of conversion, productivity (defined as gcarbonate gcatalyst−1) and turnover number (TON = molepoxide converted molhalide−1) compared to their counterparts with bromide as the counterion (Table 2). This trend is an indication that the leaving ability of the halide plays a role in determining the reaction rate.3 It should be noted that the extent of this effect changes with the nature of the organic linker between the two imidazolium units (for example, compare entries 4 and 5 to entries 6 and 7 in Table 2). A second trend can be observed by comparing the catalytic results as a function of the type of organic linker: the activity gradually increases with the length of the chain passing from ethane to octane, with the trend being more pronounced with styrene oxide as the substrate (compare entries 1, 2 and 4 in Table 2). These results suggest that a better separation between the imidazolium units can prove favourable for simultaneous reaction at both of them. However, the final catalytic performance of each of the mlc-SILP materials is likely to be influenced also by other physicochemical features of the material, which determine how readily the epoxide and the CO2 can access the ionic liquid phase. In this context, the lengthening of the organic linker can increase the affinity of the mlc-SILP for CO2, as suggested by previous studies that evidenced the increase in the solubility of supercritical CO2 in ionic liquids with increasing length of the alkyl chain on the imidazolium cation (from C2 to C8).14 Moreover, the features of the bis-vinylimidazolium salt used as the precursor are expected to influence the way in which the multilayered ionic liquid phase is organised, as reflected by the observed differences in the pore volume of the various mlc-SILPs (Table 1), and this can affect the catalytic performance of the material.
With all of the studied mlc-SILP catalysts, higher epoxide conversions were observed with styrene oxide as compared to 1,2-epoxyhexane (Table 2). This behaviour is different from the generally observed lower conversion with styrene oxide6d,15 and is ascribed to the higher polarity of this substrate, which can favour the interaction with the polar supported ionic liquid phase in which the catalytic reaction takes place.
The best catalysts identified in this set of experiments are SiO2–octane–Br, SiO2–octane–I and SiO2–p-xylene–I, with the latter reaching full conversion of styrene oxide to styrene carbonate under the employed reaction conditions, with a productivity of 76 gcarbonate gcatalyst−1 and TON = 237 (Table 2). The results with these catalysts are markedly superior compared to the previous optimum mlc-SILP catalyst (SiO2–p-xylene–Br),9 which was used in this work as a reference. SiO2–p-xylene–I was also tested at lower temperature (125 °C), achieving lower but still appreciable conversion of the two epoxides with the same complete selectivity towards the cyclic carbonate product (entry 8 in Table 2). The three most promising mlc-SILP catalysts identified in the first set of tests were further employed to study the effects of the amount of the catalyst and the CO2 pressure employed in the reaction, their versatility with different substrates and their reusability.
The effect of the amount of the catalyst employed in the reaction of styrene oxide with CO2 was studied using the two mlc-SILPs prepared with octane as the linker, SiO2–octane–Br and SiO2–octane–I. The higher activity of the catalyst with iodide was confirmed with each of the three tested amounts of the catalyst (Table 3). Remarkably, the degree of conversion of styrene oxide over SiO2–octane–I decreases only moderately upon halving the amount of the catalyst, and upon decreasing it to one quarter of the original loading, an appreciable conversion of 45% is achieved, with excellent productivity (138 gcarbonate gcatalyst−1) and TON (381 molepoxide converted molhalide−1) after 3 h of reaction. Complete selectivity towards styrene carbonate was observed in all cases. These results underline the assets of the mlc-SILP catalysts, which can be employed in rather low amounts due to the high loading of the catalytically active phase (Table 1).
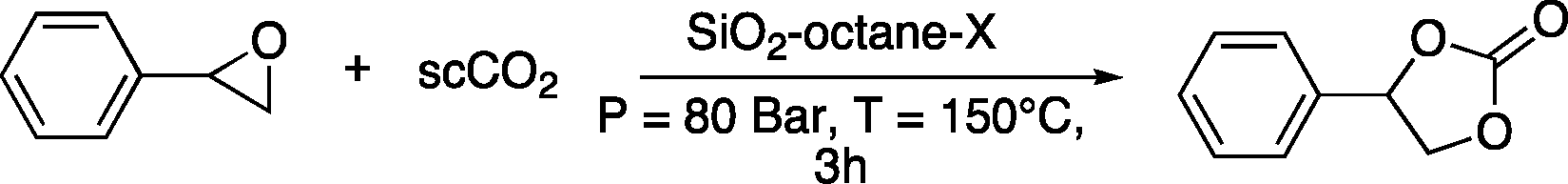
| Entry | Catalyst amount (g) | Halide (X) | Conv.b (%) | Prod.c | TONd | Sel.cyclic carbonate (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: epoxide (14.0 mmol), mesitylene (1.40 mmol), catalyst, CO2 (80 bar), 150 °C, 3 h. b Conversion and product yields determined by GC, using mesitylene as the internal standard. c Productivity, defined as gcarbonate gcatalyst−1. d Turnover number, defined as molepoxide converted molhalide−1. | ||||||
| 1 | 0.030 | I | 85 | 65 | 180 | >99 |
| 2 | 0.016 | I | 72 | 103 | 286 | >99 |
| 3 | 0.008 | I | 45 | 138 | 381 | >99 |
| 4 | 0.030 | Br | 79 | 60 | 145 | >99 |
| 5 | 0.016 | Br | 50 | 72 | 174 | >99 |
| 6 | 0.008 | Br | 24 | 72 | 173 | >99 |
The pressure of carbon dioxide at which the reaction is performed is known to influence the degree of conversion of the epoxide.9,16 Two fluid phases are present under the reaction conditions employed in the first set of tests reported here, and only a fraction of the CO2 present in the reactor dissolves in the bottom phase containing the epoxide.9 Ideally, the CO2 pressure should be high enough to reach a sufficiently high concentration of CO2 dissolved in the phase where the reaction occurs, but not too high to avoid excessive dilution of the epoxide. A previous study with mlc-SILP catalysts showed that higher carbonate yields are obtained with a CO2 pressure of 80 bar compared to 100 bar.9 Here, this parameter was tuned by investigating the catalytic activity of SiO2–p-xylene–I in the reaction of 1,2-epoxyhexane with CO2 at 40, 60 and 80 bar. An intermediate pressure of 60 bar of CO2 (i.e. below the supercritical point) provided the highest epoxide conversion with the employed amount of epoxide and catalyst (Fig. 4), though the differences in the conversion obtained in the three experiments are not major.
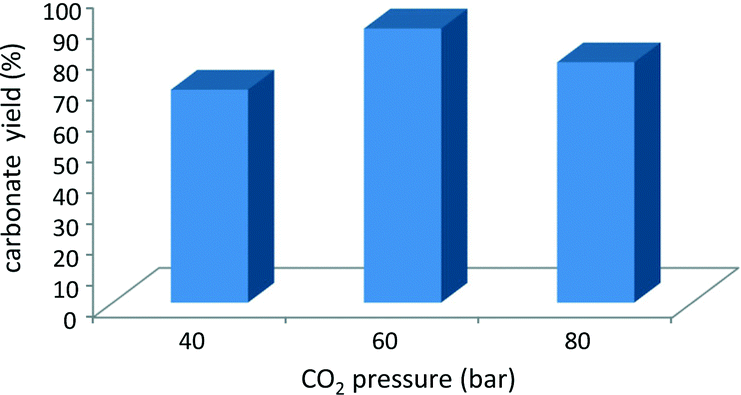 | ||
| Fig. 4 Effect of CO2 pressure on the cycloaddition to 1,2-epoxyhexane over SiO2–p-xylene–I. Reaction conditions: epoxide (23.4 mmol), mesitylene (2.34 mmol), catalyst (50 mg), CO2 (80 bar), 150 °C, 3 h. Conversion and product yields were determined by GC, using mesitylene as the internal standard. These experiments were performed in a batch reactor with two borosilicate windows that enable the direct visualisation of the phases during the reaction.17 | ||
The versatility of the mlc-SILP catalysts in the synthesis of cyclic carbonates from different epoxides and CO2 was tested using SiO2–octane–I (Table 4). Both terminal and internal epoxides were selected as substrates. Internal epoxides are generally more difficult to convert to cyclic carbonates due to the higher steric congestion around the epoxide ring, which can hinder the nucleophilic attack by the halide.3 Moreover, the formation of cyclic carbonates from cyclic epoxides such as cyclohexene oxide and the seldom investigated cyclopentene oxide is further limited by the geometric strain of the five-membered cyclic carbonate ring imposed by the adjacent cyclohexane or cyclopentane ring. In line with these argumentations, higher carbonate yields were observed with the terminal epoxides, while cyclohexene oxide and cyclopentene oxide showed lower reactivity, leading to only low carbonate yields (Table 4). The selectivity towards the cyclic carbonate product was excellent with all substrates. Only the cis-isomer of the cyclic carbonate was observed using cyclohexene oxide and cyclopentene oxide as substrates.11,18
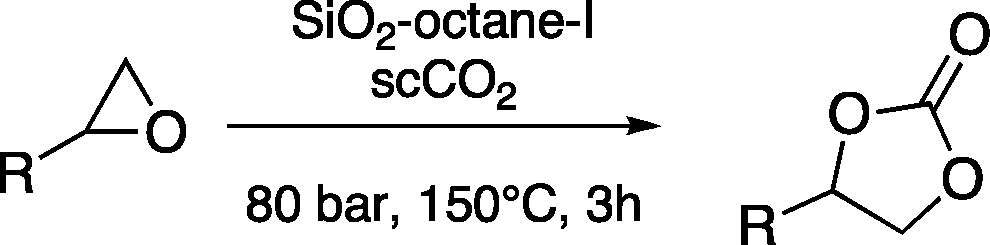
| Entry | Epoxide | Carbonate | Yieldcarbonate (%) | Prod.d | TONe |
|---|---|---|---|---|---|
| a Reaction conditions: epoxide (14.02 mmol), mesitylene (1.40 mmol), 30 mg of catalyst, CO2 (80 bar), 150 °C, 3 h. b Conversion and product yields determined by 1H NMR, using mesitylene as the internal standard. c Conversion determined by GC, using mesitylene as the internal standard. d Productivity, defined as gcarbonate gcatalyst−1. e Turnover number, defined as molepoxide converted molhalide−1. | |||||
| 1 |
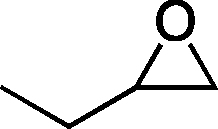
|
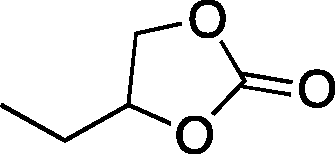
|
32b | 18 | 69 |
| 2 |
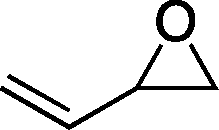
|
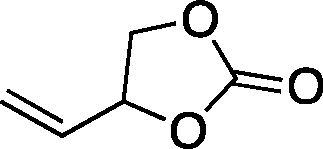
|
67b | 36 | 143 |
| 3 |
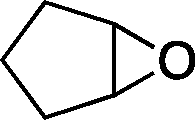
|
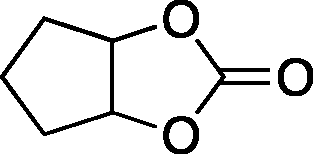
|
5b | 3 | 10 |
| 4 |
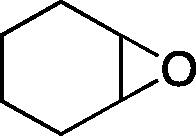
|
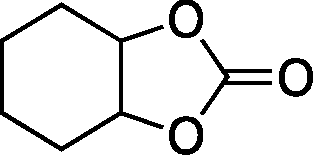
|
10c | 6 | 20 |
The reusability of a selected mlc-SILP catalyst, SiO2–octane–Br, was investigated in consecutive runs of the reaction of styrene oxide with CO2. The tests were carried out with a much lower amount of catalyst relative to the epoxide as compared to the experiments in Table 2, in order to achieve intermediate conversion. These conditions are suitable for monitoring possible changes in the catalytic activity. Mlc-SILP catalysts are not prone to leaching, but have been reported to present a variation in activity upon recycling.9 In line with previous reports, the catalytic activity increased until the third run and then gradually decreased to achieve a similar activity in the fifth as in the first run (Fig. 5). This behaviour has been ascribed to modifications (plasticisation) in the oligomeric network of supported ionic liquids induced by CO2.9
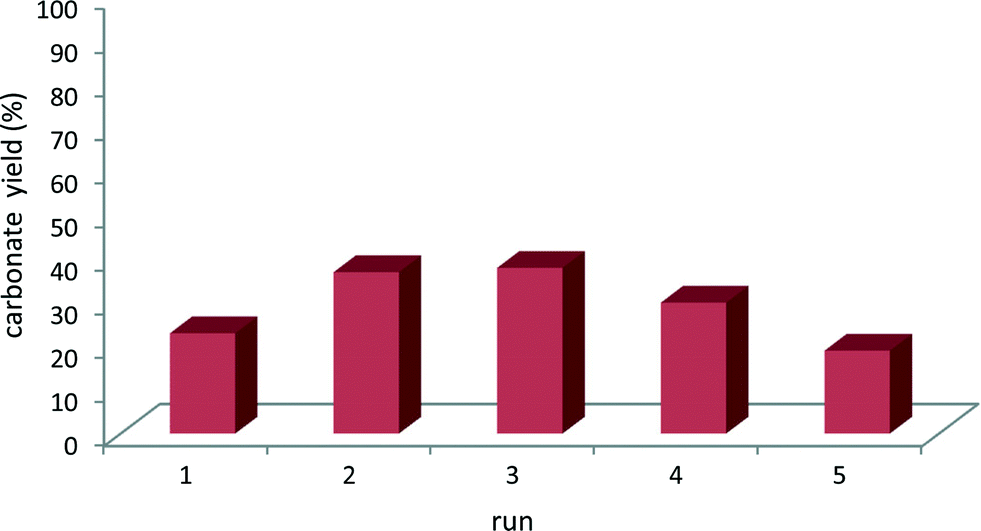 | ||
| Fig. 5 Recycling tests of SiO2–octane–Br as the catalyst for the synthesis of styrene carbonate from CO2 and styrene oxide. Reaction conditions: styrene oxide (174 mmol), mesitylene (17.4 mmol), 75 mg of the catalyst, CO2 (80 bar), 150 °C, 3 h. Conversion and product yields were determined by 1H NMR, using mesitylene as the internal standard. | ||
The mlc-SILP materials prepared by grafting and cross-linking the bis-vinylimidazolium salts on a thiol-functionalised silica support are obtained in the form of a light-yellow powder. The samples typically contain a fraction of polymer-looking slabs with size in the millimetre-range. These flat particles probably originate from a fraction of the bis-vinylimidazolium salt that polymerised without reacting with the support, thus generating unanchored polymeric species. The formation of these slabs was particularly evident for sample SiO2–octane–I. The possible effect of these slabs on the catalytic activity of the material was studied by separating them using a 200 μm analytical sieve and by testing the remaining fine powder as a catalyst in the reaction of CO2 with 1,2-epoxyhexane. The sieved catalyst displays much higher catalytic activity compared to the as-synthesised SiO2–octane–I (entries 1 and 2 in Table 5), indicating that the presence of the large particles is detrimental for the catalytic activity of the material. This suggests that the support plays an important role in the catalytic performance of the multilayered ionic liquid phase. In order to investigate further this hypothesis, a material was prepared using the same conditions as in the synthesis of SiO2–octane–I, but without the silica support. The polymerisation of the bis-vinylimidazolium salt was complete, as indicated by the absence of the signal at 110 ppm in the 13C NMR spectrum (Fig. S1 in the ESI†). The obtained material (octane–I) displays significantly lower catalytic activity compared to SiO2–octane–I in the reaction of CO2 with 1,2-epoxyhexane (Table 5 – the activity difference is particularly evident when comparing the TONs), confirming the importance of the support in creating an accessible and thus catalytically active ionic liquid phase. However, it should be noted that the polymeric octane–I material does not consist exclusively of large particles, and the finer material obtained by sieving octane–I with a 200 μm analytical sieve displays slightly higher activity (entries 3 and 4 in Table 5), though still clearly lower compared to SiO2–octane–I. This set of experiments underlines the complexity of the prepared mlc-SILP materials, which mainly consist of the desired, highly active supported ionic liquid phase, but also contain a fraction of unsupported polymeric ionic liquid phase, which displays decreasing activity as the size of the particles increases. Future studies should aim at optimising the synthesis method to minimise the formation of the polymeric slabs, thus leading to further improvements in the catalytic performance of the mlc-SILP materials.
| Entry | Catalyst | Conv.b (%) | Prod.c | TONd | Sel.cyclic carbonate (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 1,2-epoxyhexane (14.0 mmol), mesitylene (1.40 mmol), catalyst (30 mg), CO2 (80 bar), 150 °C, 3 h. b Conversion and product yields determined by GC, using mesitylene as the internal standard. c Productivity, defined as gcarbonate gcatalyst−1. d Turnover number, defined as molepoxide converted molhalide−1. | |||||
| 1 | SiO2–octane–I | 37 | 25 | 78 | >99 |
| 2 | SiO2–octane–I (sieved) | 61 | 41 | n.d. | >99 |
| 3 | Octane–I | 13 | 9 | 17 | >99 |
| 4 | Octane–I (sieved) | 17 | 12 | 23 | >99 |
Conclusions
Multilayered covalently supported ionic liquid phase (mlc-SILP) materials were synthesised using a protocol involving covalent grafting of bis-vinylimidazolium salts on a thiol-functionalised silica support and cross-linking between the bis-vinylimidazolium units. The method proved versatile and allowed the preparation of a series of mlc-SILP materials with different organic linkers between the imidazolium units and with two types of halides as counterions. The mlc-SILP materials are active heterogeneous catalysts for the reaction of carbon dioxide with various epoxides to yield cyclic carbonates. The most promising catalysts achieved excellent turnover number and productivity compared to the state-of-the-art supported ionic liquid catalysts for the conversion of CO2 and epoxides into cyclic carbonates (see Table S1 in the ESI† for a comparison with selected supported ionic liquid catalysts).6,9,16,19Experimental section
Spectroscopic and analytical methods
1H NMR and 13C NMR spectra of the bis-vinylimidazolium salts were recorded on a Bruker 300 MHz spectrometer. The chemical shifts for 1H and 13C are given in ppm relative to the residual signal of the solvent (CD3OD). The following abbreviations are used to indicate the multiplicity: s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); bs (broad signal). Solid-state 13C CP MAS NMR spectra of the mlc-SILP materials were recorded on an Agilent 400 MHz spectrometer. The samples were packed in zirconia rotors spinning at a frequency of 15 kHz. Tetramethylsilane was employed as a chemical shift reference. Carbon, nitrogen and sulphur contents were determined by combustion analysis using a Thermo Finnigan Flash EA 1112. FT-IR spectra were obtained using a Shimadzu FTIR 8300 infrared spectrophotometer. Melting points were determined using a Kofler hot plate. The specific surface areas (BET method) and the cumulative pore volumes (BJH method) of the silica-based materials, before and after supporting the ionic liquid phase, were determined from the N2 adsorption/desorption isotherms measured at −196 °C using a Micromeritics Tristar 3000 instrument.9Analysis of the reaction mixture at the end of the catalytic test was performed by gas chromatography (GC) on a Trace GC Ultra from Interscience (RTX-5 column, 5 m, 0.1 mm). The temperature profile during the analysis was: 50 °C for 0.50 min, from 50 to 250 °C at 150 °C min−1, 0.5 min at 250 °C. When necessary, the identity of the products was determined by gas chromatography-mass spectrometry (GC-MS) on an Agilent 6890N gas chromatograph (WCOT fused silica column, 30 m, 0.25 mm) coupled to an Agilent 5973 MSD mass spectrometer. Additionally (or alternatively), the conversion and product yields were determined by 1H NMR spectroscopy carried out on a 300 MHz spectrometer (7.0 T). 32 scans were accumulated with a recycle delay of 1 s. The pulse length was 8.0 ms and the power level was 0.0 dB. The samples were prepared by adding around 1 mL of CDCl3 to an aliquot of the reaction solution at the end of the catalytic test. Cyclic carbonates are known compounds and showed spectroscopic data in agreement with their structures.10,11,18
Synthesis of the bis-vinylimidazolium salts
1,4-Bis(iodomethyl)benzene (1g) was prepared according to a procedure reported in the literature.20 Bis-vinylimidazolium salts 2b and 2f were prepared following previously reported methods and showed spectroscopic and analytical data in agreement with those in the literature.9,12c All of the other bis-vinylimidazolium salts are reported here for the first time. Their synthesis procedures were similar to those previously reported (Scheme 2).9,12c In a typical synthesis, a solution of the selected compound 1 (0.01 mol) and 1-vinylimidazole (0.021 mol) in toluene (10 mL, in the case of compounds 2f–g) or chloroform (5 mL, in the case of compound 2a–e) was heated under stirring for 24 h in an oil bath at 90 °C (for toluene) or at 50 °C (for chloroform). After cooling down to room temperature, the mixture was filtered and the residue was washed several times with diethyl ether. Finally, the solid product was dried at 40 °C.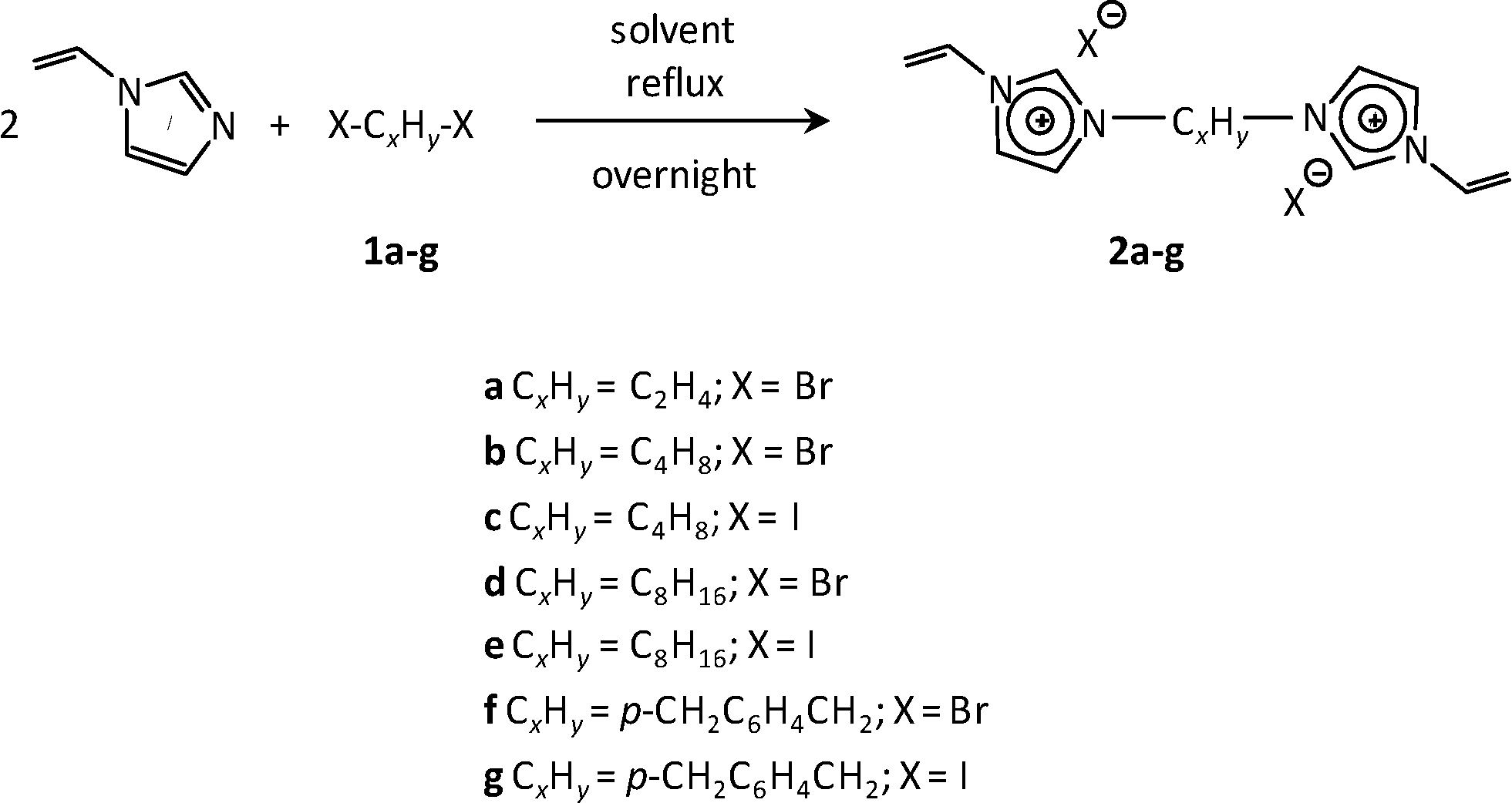 | ||
| Scheme 2 Synthesis of bis-vinylimidazolium salts. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.98 (2H, dd, J = 18.9 and 3.6 Hz, trans-CHCH2), 7.31 (2H, dd, J = 10.4 and 18.8 Hz, CHCH2), 7.85 (2H, d, J = 2.2 Hz, 4-H or 5-H), 8.09 (2H, d, J = 2.3 Hz, 4-H or 5-H), 9.54 (2H, s, 2-H). 13C NMR (CD3OD): δ (ppm) = 50.0, 110.8, 121.4, 124.6, 129.8, 130.9, 137.4. [Found: C, 38.3; H, 4.3; Br, 42.5; N, 14.9; C12H16Br2N4 requires C, 38.3; H, 4.4; Br, 42.5; N, 14.9].
CH2), 5.98 (2H, dd, J = 18.9 and 3.6 Hz, trans-CHCH2), 7.31 (2H, dd, J = 10.4 and 18.8 Hz, CHCH2), 7.85 (2H, d, J = 2.2 Hz, 4-H or 5-H), 8.09 (2H, d, J = 2.3 Hz, 4-H or 5-H), 9.54 (2H, s, 2-H). 13C NMR (CD3OD): δ (ppm) = 50.0, 110.8, 121.4, 124.6, 129.8, 130.9, 137.4. [Found: C, 38.3; H, 4.3; Br, 42.5; N, 14.9; C12H16Br2N4 requires C, 38.3; H, 4.4; Br, 42.5; N, 14.9]. CH2), 5.97 (2H, dd, J = 15.6 and 2.7 Hz, trans-CHCH2), 7.30 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.86 (2H, d, J = 1.5 Hz, 4-H or 5-H), 8.02 (2H, d, J = 1.5 Hz, 4-H or 5-H), 9.48 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.7, 50.4, 110.1, 120.9, 124.5, 129.8, 136.5; IR (nujol)
CH2), 5.97 (2H, dd, J = 15.6 and 2.7 Hz, trans-CHCH2), 7.30 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.86 (2H, d, J = 1.5 Hz, 4-H or 5-H), 8.02 (2H, d, J = 1.5 Hz, 4-H or 5-H), 9.48 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.7, 50.4, 110.1, 120.9, 124.5, 129.8, 136.5; IR (nujol) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max (cm−1): 921, 1154, 1377, 1458, 1550, 2854, 2922. [Found: C, 33.8; H, 4.1; I, 50.9; N, 11.3; C14H20I2N4 requires C, 33.5; H, 4.5; I, 50.3; N, 11.2].
max (cm−1): 921, 1154, 1377, 1458, 1550, 2854, 2922. [Found: C, 33.8; H, 4.1; I, 50.9; N, 11.3; C14H20I2N4 requires C, 33.5; H, 4.5; I, 50.3; N, 11.2].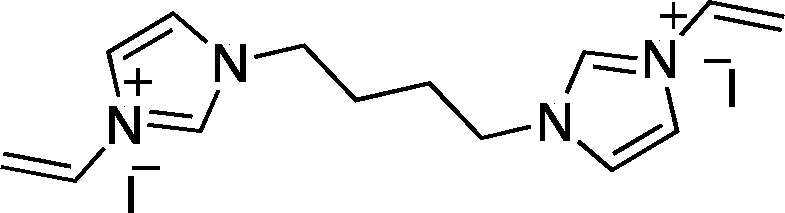 CH2), 5.96 (2H, dd, J = 15.9 and 2.7 Hz, trans-CHCH2), 7.29 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.82 (2H, d, J = 1.8 Hz, 4-H or 5-H), 8.03 (2H, d, J = 1.8 Hz, 4-H or 5-H), 9.44 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.1, 29.7, 30.9, 51.2, 109.9, 120.7, 124.5, 129.8, 136.5. [Found: C, 46.9; H, 6.1; Br, 34.7; N, 12.2; C18H28Br2N4 requires C, 46.8; H, 6.2; Br, 34.5; N, 12.1].
CH2), 5.96 (2H, dd, J = 15.9 and 2.7 Hz, trans-CHCH2), 7.29 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.82 (2H, d, J = 1.8 Hz, 4-H or 5-H), 8.03 (2H, d, J = 1.8 Hz, 4-H or 5-H), 9.44 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.1, 29.7, 30.9, 51.2, 109.9, 120.7, 124.5, 129.8, 136.5. [Found: C, 46.9; H, 6.1; Br, 34.7; N, 12.2; C18H28Br2N4 requires C, 46.8; H, 6.2; Br, 34.5; N, 12.1].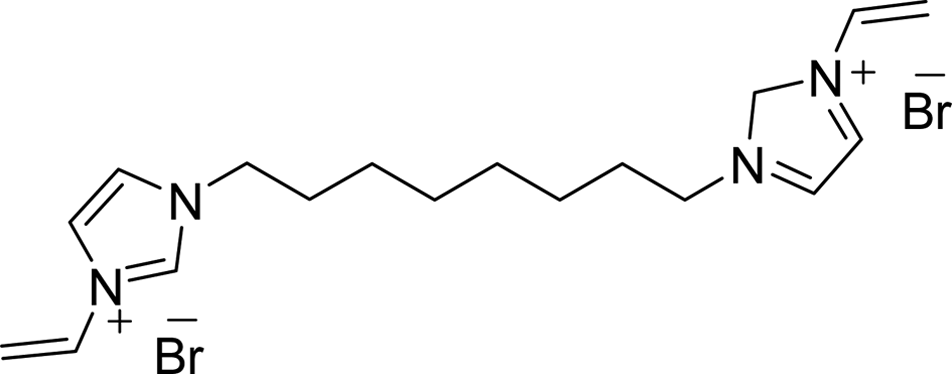 CH2), 5.93 (2H, dd, J = 15.6 and 2.4 Hz, trans-CHCH2), 7.26 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.79 (2H, d, J = 1.2 Hz, 4-H or 5-H), 8.00 (2H, d, J = 1.5 Hz, 4-H or 5-H), 9.44 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.1, 29.8, 30.9, 51.3, 110.0, 120.8, 124.6, 129.9, 136.2. [Found: C, 39.1; H, 5.2; I, 45.8; N, 10.1; C18H28I2N4 requires C, 39.3; H, 5.4; I, 45.1; N, 10.8].
CH2), 5.93 (2H, dd, J = 15.6 and 2.4 Hz, trans-CHCH2), 7.26 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.79 (2H, d, J = 1.2 Hz, 4-H or 5-H), 8.00 (2H, d, J = 1.5 Hz, 4-H or 5-H), 9.44 (2H, s, 2-H); 13C NMR (CD3OD): δ (ppm) = 27.1, 29.8, 30.9, 51.3, 110.0, 120.8, 124.6, 129.9, 136.2. [Found: C, 39.1; H, 5.2; I, 45.8; N, 10.1; C18H28I2N4 requires C, 39.3; H, 5.4; I, 45.1; N, 10.8].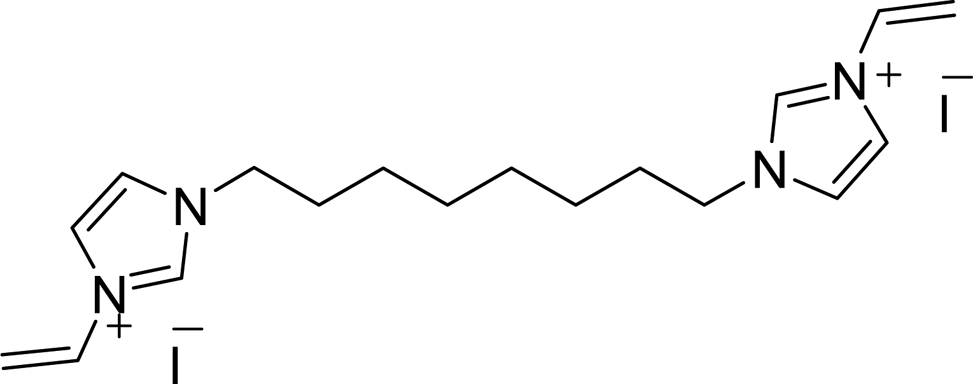 CH2), 5.52 (4H, s, CH2Ph), 5.95 (2H, dd, J = 15.6 and 2.7 Hz, trans-CHCH2), 7.29 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.60 (4H, s, Ph), 7.77 (2H, d, J = 1.8 Hz, 4-H or 5-H), 8.05 (2H, d, J = 1.8 Hz, 4-H or 5-H), 9.46 (2H, s, 2-H). 13C NMR (CD3OD): δ (ppm) = 53.9, 110.3, 121.1, 124.4, 129.8, 128.8, 130.9, 136.1. [Found: C, 39.6; H, 3.7; I, 46.5; N, 10.3; C18H20I2N4 requires C, 39.4; H, 3.5; I, 46.4; N, 10.2].
CH2), 5.52 (4H, s, CH2Ph), 5.95 (2H, dd, J = 15.6 and 2.7 Hz, trans-CHCH2), 7.29 (2H, dd, J = 15.6 and 8.7 Hz, CHCH2), 7.60 (4H, s, Ph), 7.77 (2H, d, J = 1.8 Hz, 4-H or 5-H), 8.05 (2H, d, J = 1.8 Hz, 4-H or 5-H), 9.46 (2H, s, 2-H). 13C NMR (CD3OD): δ (ppm) = 53.9, 110.3, 121.1, 124.4, 129.8, 128.8, 130.9, 136.1. [Found: C, 39.6; H, 3.7; I, 46.5; N, 10.3; C18H20I2N4 requires C, 39.4; H, 3.5; I, 46.4; N, 10.2].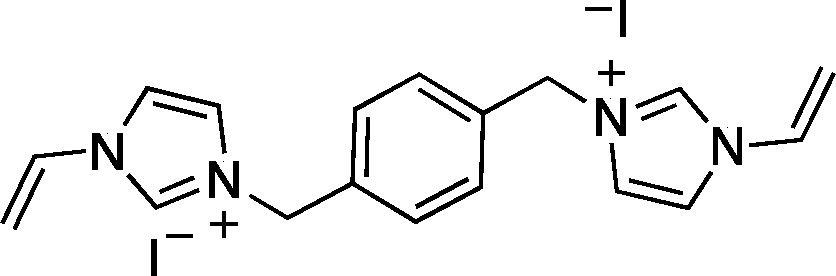
Synthesis of thiol-functionalised silica
Thiol-functionalised silica was prepared using Sigma–Aldrich SiO2 (403563) as the silica source. 1.5 g of silica gel was heated overnight at 150 °C in a vacuum line, in order to remove physically adsorbed water. Then, the solid was reacted with 3-(mercaptopropyl)-trimethoxysilane (2 mL) in 10 mL toluene. The mixture was heated under reflux in an argon atmosphere for 24 h. After cooling to room temperature, the material was isolated by filtration and washed with methanol (50 mL) and diethyl ether (50 mL). The solid was dried under reduced pressure to give the final material (1.9 g). The loading of thiol groups was determined by means of elemental analysis of sulphur (1.2 mmol g−1).Synthesis of mlc-SILP materials 1a–g
The mlc-SILP materials were synthesised using excess bis-imidazolium salt with respect to the –SH groups: 3.62 mol of salt per mol of –SH. In a typical synthesis, the thiol-functionalised silica, a bis-imidazolium salt, 2,2′-azobisisobutyronitrile (AIBN, 60 mg) and ethanol were placed in a three-necked round bottom flask. The reactions were carried out employing a concentration of 130 mM for all of the salts, except for the case of SiO2–p-xylene–I in which the diiodide salt was used as a 40 mM solution due to its lower solubility in ethanol. The suspension was flushed by bubbling argon for 10 min. The flask was heated to 78 °C to favour the dissolution of the bis-imidazolium salt. The reaction mixture was magnetically stirred under argon for 20 h. After cooling down to room temperature, the solid was filtered and washed with hot methanol and diethyl ether and then dried in an oven at 40 °C overnight.Synthesis of the cross-linked polymeric ionic liquid phase (octane–I)
The material was prepared by radical polymerisation. Bis-imidazolium diiodide salt 2e (1.09 mmol, 130 mM), AIBN (0.16 mmol) and ethanol were placed in a three-necked round bottom flask. The reaction conditions were identical to those employed for the synthesis of mlc-SILPs (vide supra).Catalytic tests
The catalytic tests were performed in a high-throughput block consisting of 24 batch reactors under individual magnetic stirring, except for the tests to study the effect of CO2 pressure and the recycling tests, which were carried out in a visualisation batch reactor equipped with two borosilicate windows to allow monitoring of the phases present in the reaction mixture.9–11 Both the 24-reactors block and the visualisation reactor are part of a custom made high-throughput unit (Integrated Lab Solutions, ILS) for performing reactions in (supercritical) carbon dioxide. The pressurisation of the reactors is achieved by means of two ISCO pumps. The 24 parallel batch reactors are pressurised simultaneously with CO2, while check valves protect against back-flow. Risks of overpressure are avoided by automated depressurisation protocols and by the presence of rupture disks. The temperature of the 24-reactors block is regulated using a Huber Thermostat.In a typical experiment, the epoxide, the catalyst and mesitylene (as the GC and NMR internal standard) were introduced in an open glass vial, which was then placed into the selected batch reactor. The reaction block, either the 24-reactors block or the visualisation reactor, was closed and the lines and the reactor were purged with N2 for 10 min. For a typical test carried under 80 bar of CO2, each reactor was pressurised under 40 bar of CO2 while keeping the block at room temperature. Then, the temperature was increased to 150 °C with a rate of 10 °C min−1. During this process, the pressure inside the reactor increased to 60 bar. Once a stable pressure value was reached, the reactor was filled with CO2 to the selected pressure (80 bar). The reactor was kept under the selected conditions for 3 h with a stirring speed of 900 rpm. Then, the stirring was stopped and the reactors were cooled down. Depressurisation was started when the carbon dioxide was not anymore under supercritical conditions.9 The system was allowed to depressurise until the reactors were at room temperature with an internal pressure <2 bar.9 Once the depressurisation was complete, the reactor block was opened and the glass vials were removed and centrifuged at 2000 rpm for 3 min to separate the solid catalyst from the reaction mixture. The supernatant solution was sampled and then diluted with toluene or with ethyl acetate and analysed by GC, or diluted with CDCl3 and analysed by 1H NMR (see Fig. S2 and S3 in the ESI† for representative chromatograms and NMR spectra). The assignment of GC peaks due to new products was performed by gas chromatography-mass spectrometry analysis (GC-MS). 1H NMR was also employed to confirm the absence of the ionic liquid in the solution and to determine the ratio of carbonate to epoxide, which was used to calculate the GC response factor for compounds that are not available commercially.
Selected catalytic tests were performed in duplicate, and the results did not show significant deviations between the two tests. For these experiments, the average of the conversions is reported.
Recycling experiments were performed by removing the supernatant solution from the solid catalyst after the centrifugation step (see above). The solid was washed with toluene (twice) and ethanol (once). After each addition of solvent, the vial containing the suspension was placed in an ultrasonic water bath for about 30 min to favour the removal of reaction residues, followed by centrifugation. Finally, the catalyst was dried in an oven at 85 °C overnight and ground before being used for the next experiment.
Acknowledgements
PA, SMA, MT and PPP acknowledge sponsorship in the frame of a START1 research program. PA and MG acknowledge the University of Palermo and COST Action CM0905 ORCA.Notes and references
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 CAS.
- (a) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC; (b) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS PubMed; (c) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed.
- (a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC; (b) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS PubMed.
- (a) J. Peng and Y. Deng, New J. Chem., 2001, 25, 639 RSC; (b) B. Ochiai, T. Iwamoto and T. Endo, Green Chem., 2006, 8, 138 RSC; (c) D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, J. Mol. Catal. A: Chem., 2013, 378, 326 CrossRef; (d) W. Cheng, X. Chen, J. Sun, J. Wang and S. Zhang, Catal. Today, 2013, 200, 117 CrossRef CAS.
- F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322 CrossRef CAS PubMed.
- (a) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663 CrossRef CAS; (b) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (c) W. Dongfang, G. Yali, G. Shu and X. Yinghao, Ind. Eng. Chem. Res., 2013, 52, 1216 CrossRef; (d) I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255 CrossRef CAS; (e) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS PubMed.
- C. Aprile, F. Giacalone, P. Agrigento, L. F. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2008, 4, 1830 CrossRef PubMed.
- M. Taherimehr, A. Decortes, S. M. Al-Amsyar, W. Lueangchaichaweng, C. J. Whiteoak, E. C. Escudero-Adán, A. W. Kleij and P. P. Pescarmona, Catal. Sci. Technol., 2012, 2, 2231 CAS.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083 RSC.
- (a) P. D. Stevens, J. Fan, H. M. R. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085 CrossRef CAS PubMed; (b) A. Taher, J.-B. Kim, J.-Y. Jung, W.-S. Ahn and M.-J. Jin, Synlett, 2009, 2477 CAS; (c) M. Gruttadauria, L. F. Liotta, A. M. P. Salvo, F. Giacalone, V. La Parola, C. Aprile and R. Noto, Adv. Synth. Catal., 2011, 353, 2119 CrossRef CAS.
- Diols might in principle form as side-products during the catalytic tests through the hydrolysis of the epoxides with adventitious water. However, no diol was observed in any of the tests reported in this work. The suitability of the GC for the analysis of diols was confirmed by injecting standard solutions containing these compounds..
- (a) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896 RSC; (b) D.-W. Park, N.-Y. Mun, K.-H. Kim, I. Kim and S.-W. Park, Catal. Today, 2006, 115, 130 CrossRef CAS.
- C. J. Whiteoak, E. Martin, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469 CrossRef CAS.
- (a) Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS PubMed; (b) W.-L. Dai, L. Chen, S. F. Yin, W.-H. Li, Y.-Y. Zhang, S. L. Luo and C. T. Au, Catal. Lett., 2010, 137, 74 CrossRef CAS; (c) J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167 CrossRef CAS; (d) T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664 RSC.
- The higher conversion of 1,2-epoxyhexane observed at 80 bar compared to the test performed at the same pressure in the 24-reactors block (entry 7 in Table 2) is ascribed to the different volume of epoxide used in the two tests and to the more efficient stirring and larger volume of the visualisation reactor, which implies that at the same CO2 pressure the ratio CO2 to epoxide was not equal in the two experiments..
- (a) B. Gabriele, R. Mancuso, G. Salerno, L. Veltri, M. Costa and A. Dibenedetto, ChemSusChem, 2008, 4, 1778 CrossRef PubMed; (b) C. Beattie, M. North, P. Villuendas and C. Young, J. Org. Chem., 2013, 78, 419 CrossRef CAS PubMed.
- (a) S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584 RSC; (b) W.-L. Dai, L. Chen, S. F. Yin, S. L. Luo and C. T. Au, Catal. Lett., 2010, 135, 295 CrossRef CAS; (c) W.-L Dai, B. Jin, S.-L. Luo, S.-F. Yin, X.-B. Luo and C.-T. Au, J. CO2 Util., 2013, 3–4, 7 CrossRef CAS; (d) J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS.
- T. Kida, A. Kikuzawa, H. Higashimoto, Y. Nakatsuji and M. Akashi, Tetrahedron, 2005, 61, 5763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GC and NMR data and a table with comparison between the catalytic performance of the mlc-SILP materials and other supported ionic liquids. See DOI: 10.1039/c3cy01000g |
| This journal is © The Royal Society of Chemistry 2014 |