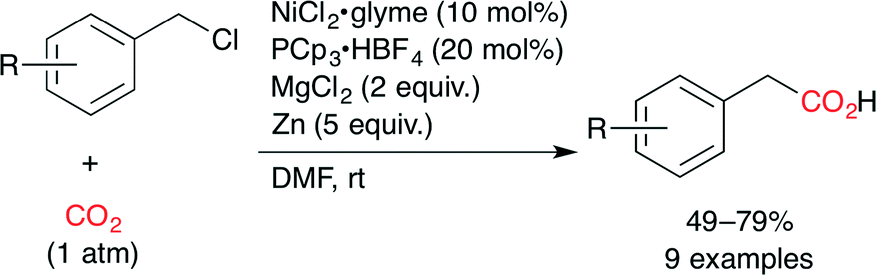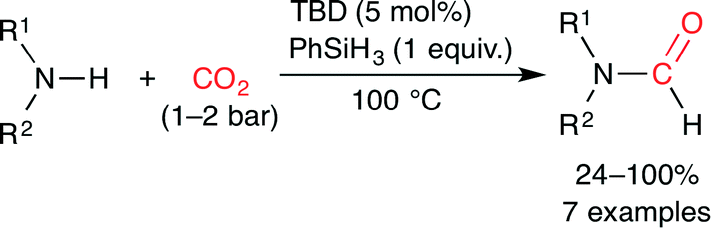DOI:
10.1039/C3CY00993A
(Perspective)
Catal. Sci. Technol., 2014,
4, 1482-1497
Recent progress in catalytic conversions of carbon dioxide
Received
30th November 2013
, Accepted 14th December 2013
First published on 12th February 2014
Abstract
Chemical fixation of carbon dioxide (CO2), which is an inexpensive and renewable carbon source, is becoming more and more important. The development of both new reactions and new catalysts is needed to overcome the kinetic and thermodynamic stability of CO2. Organic and metal catalysts with unique and excellent activity and selectivity have been developed for various chemical conversions of CO2. In this perspective, we provide an overview of the recent progress in this field, classifying it into several categories, where each research is concisely summarized one by one using a single reaction scheme, a representative catalyst structure, and/or a catalytic cycle.

Chihiro Maeda
| Chihiro Maeda was born in Kanagawa, Japan in 1983. He received his bachelor's degree from Kyoto University in 2005, and he obtained his master's degree in 2007 and doctor's degree in 2010 at Kyoto University under the supervision of Professor Atsuhiro Osuka. In 2010, he started his academic career at Keio University as an Assistant Professor. In 2013, he moved to Okayama University to join the Tadashi Ema research group. His main research topic is the synthesis and application of novel porphyrin derivatives. |

Yuki Miyazaki
| Yuki Miyazaki was born in Hyogo, Japan in 1986. He received his bachelor's degree from Okayama University in 2009, and he obtained his master's degree in 2010 and doctor's degree in 2013 at Okayama University under the supervision of Professor Tadashi Ema. His subject during the doctor course was “Metalloporphyrin catalysts for synthesis of cyclic carbonate from CO2 and epoxide”. |

Tadashi Ema
| Tadashi Ema was born in Gifu, Japan in 1966. He received his bachelor's degree from Kyoto University in 1989, and he obtained his master's degree in 1991 and doctor's degree in 1994 at Kyoto University under the supervision of Professor Hisanobu Ogoshi. He was appointed as an Assistant Professor at Okayama University in 1994 and was promoted to an Associate Professor in 2002 and a Full Professor in 2012. His research interest is the design and synthesis of unique molecular catalysts. |
1. Introduction
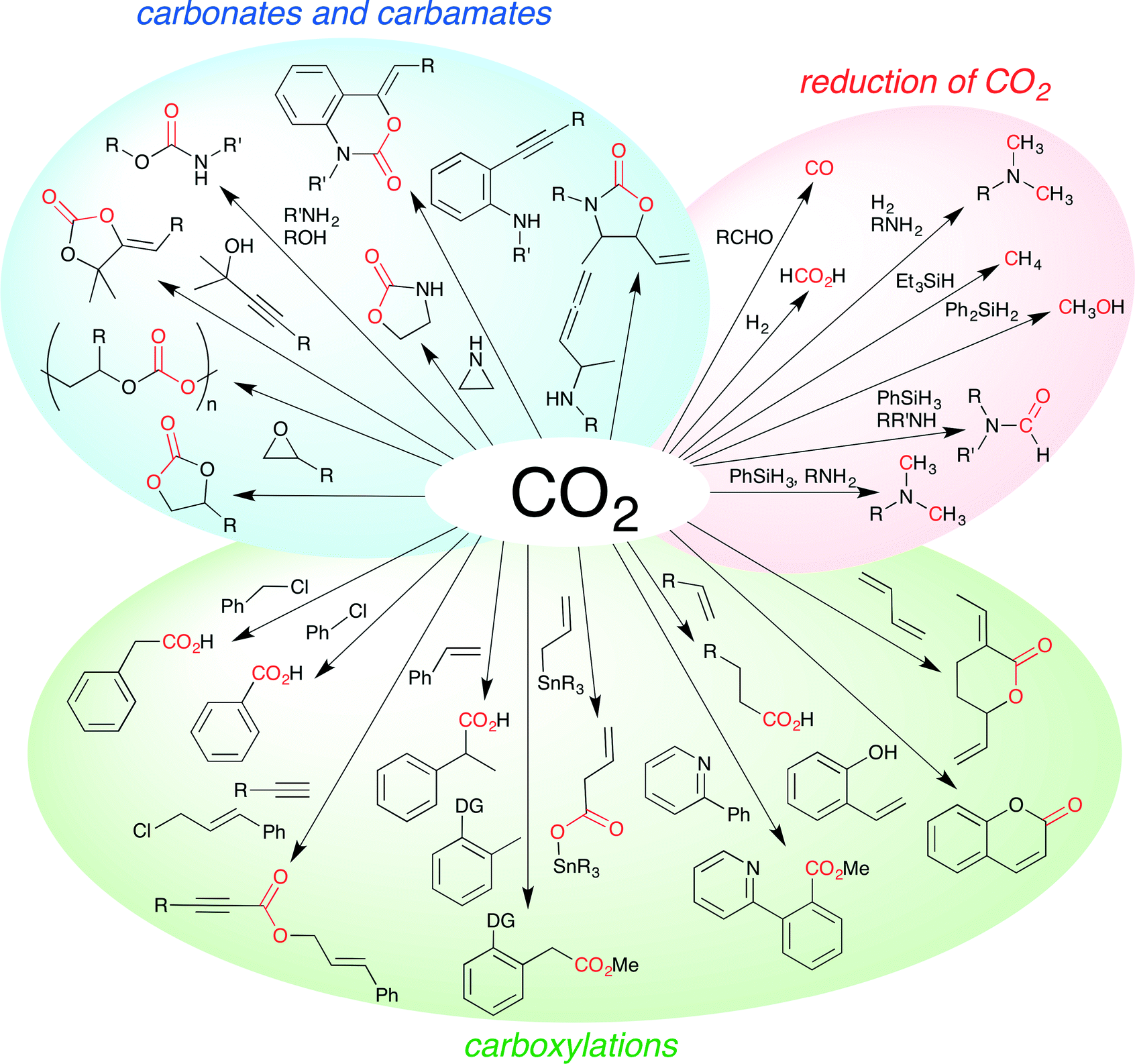
Carbon dioxide (CO2) is the most abundant renewable carbon source in nature. Chemical fixation of CO2 is therefore one of the most important subjects in organic synthesis, and much effort has been devoted to this particular subject.1 Various methods for CO2 fixation have been developed despite the low reactivity and narrow synthetic scope of CO2. Although the Kolbe–Schmitt reaction and carboxylation reactions using Grignard reagents are well-known chemical fixations of CO2, these non-catalytic reactions have some limitations. On the other hand, catalytic reactions are considered to be essential for widening and deepening the synthetic utility of CO2. Both reactions and catalysts need to be developed for this purpose. A large number of inorganic, organic, and metal catalysts have been developed for various chemical conversions of CO2. The achievements over the past five years are particularly noteworthy. The synthesis of cyclic carbonates or polycarbonates from CO2 and epoxides, carboxylation reactions with CO2, reduction of CO2, and other fascinating reactions have been developed and studied extensively and intensively (Fig. 1). In this perspective, we highlight the latest progress in this field, classifying it into several categories. Each research is summarized concisely using a single reaction scheme and/or a representative catalyst structure even if several derivatives of substrates and catalysts were reported. In some cases, a catalytic cycle is added to help the readers understand the mechanistic aspect of the reaction and to promote further development of this research field.
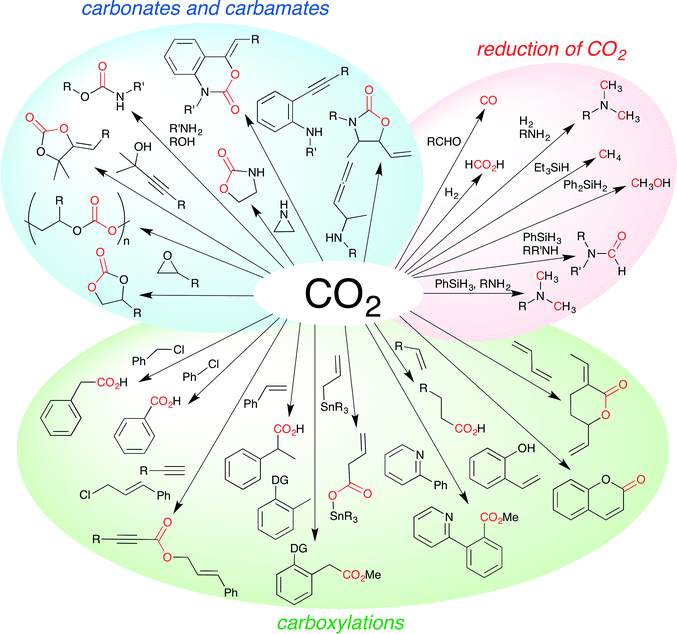 |
| | Fig. 1 Examples of chemical fixation of CO2. | |
2. The synthesis of cyclic carbonates from carbon dioxide and epoxides
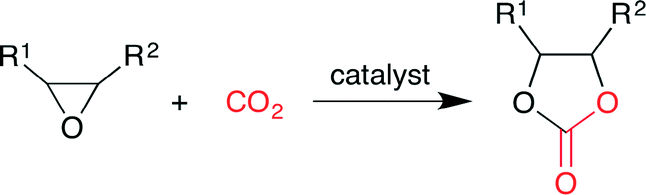
Cyclic carbonates have been widely used as (i) raw materials for polycarbonates, (ii) electrolytes in lithium-ion secondary batteries, (iii) aprotic polar solvents, and (iv) fuel additives. Although cyclic carbonates have been synthesized by the phosgene method, this method has drawbacks, such as the use of highly toxic gas (phosgene), the formation of hydrogen chloride (byproduct) and the generation of waste water containing dichloromethane (solvent) and salts. Although the phosgene method can produce cyclic carbonates cost-effectively on a large scale, the development of environmentally benign methods is required, such as the catalytic method using CO2 and epoxides under mild conditions (Scheme 1).
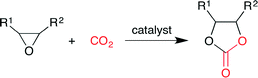 |
| | Scheme 1 The synthesis of cyclic carbonates from epoxides and CO2. | |
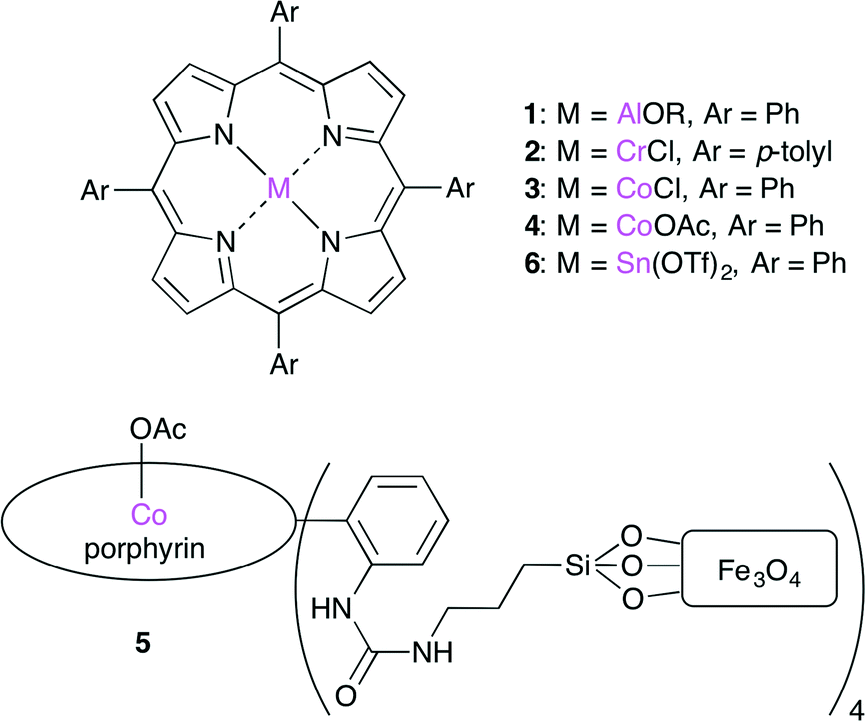
Metalloporphyrin complexes are efficient catalysts (Fig. 2). The Lewis acidic metal center can activate epoxides by coordination. A co-catalyst may be necessary for the nucleophilic ring-opening of the epoxide. Since the pioneering studies by Inoue,2 several metalloporphyrin complexes such as 1–6, having Al,2 Cr,3 Co,4–6 or Sn7 as a metal center, have been developed.
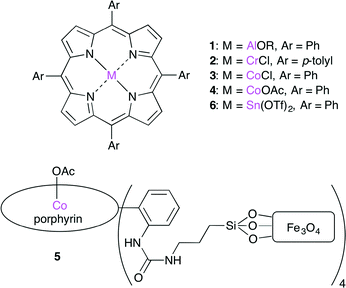 |
| | Fig. 2 Metalloporphyrin catalysts 1–6. | |
Recently, Co porphyrin complex 4 was reported by Jing and co-workers.5 The screening of central metals, axial ligands and co-catalysts indicated that a combination of 4 and phenyltrimethylammonium tribromide (PTAT) showed the highest catalytic activity under mild conditions. The Co porphyrin complex immobilized on magnetic nanoparticles 5 was also developed.6 This heterogeneous catalyst could be recycled by using a magnet up to 16 times with >90% conversion with a low catalyst loading (0.14 mol%).
Tangestaninejad, Moghadam and co-workers have studied Sn porphyrin complex 6.7 The electron-withdrawing axial ligand made the SnIV center more electron-deficient, which led to a strong coordination of the epoxide and an improvement of the catalytic activity. The quaternary ammonium cation in the co-catalyst was found to be important for the enhancement of the nucleophilicity of the counter anion. The reaction is proposed to be accelerated by the activation of both the epoxide and CO2 at the Lewis acidic SnIV center.
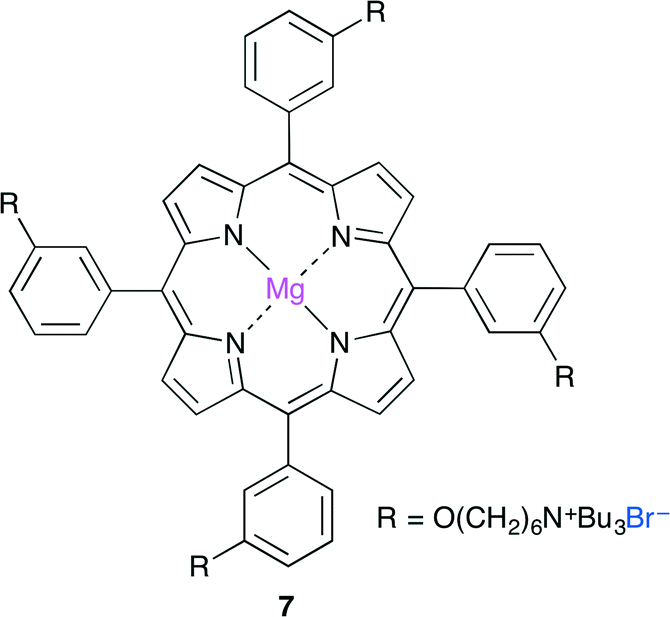
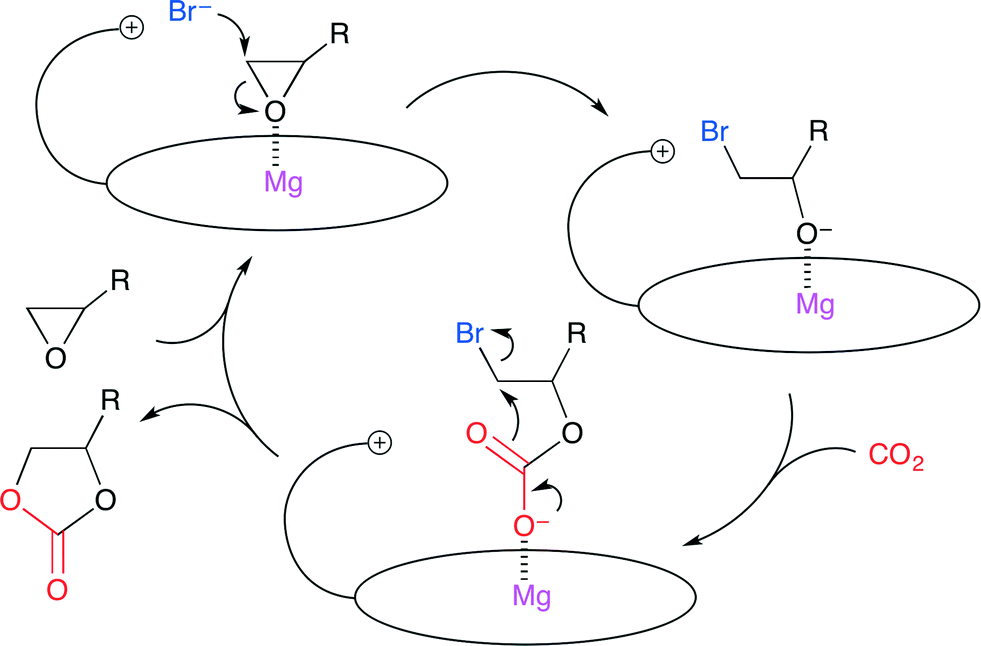
More recently, Ema and co-workers reported a highly active bifunctional catalyst 7 (Fig. 3).8 The Mg center and Br− ion in 7 acted as a Lewis acid and a nucleophile, respectively. As shown in Scheme 2, this reaction is proposed to be initiated by nucleophilic attack by the Br− ion on the less hindered side of the epoxide activated by coordination to the Mg center. The oxyanion generated by ring-opening then attacks CO2 to give a CO2 adduct. The subsequent cyclization gives the product. The use of 0.0008 mol% of 7 at 120 °C resulted in 83% conversion of the epoxide into cyclic carbonate with a TON of 103![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The authors also developed an immobilized version of this bifunctional catalyst. Zn porphyrin was immobilized on biogenous iron oxide via four quaternary ammonium bromide groups.9 This organic–inorganic hybrid catalyst showed a high activity even after recycling ten times without the loss of catalytic activity.
000. The authors also developed an immobilized version of this bifunctional catalyst. Zn porphyrin was immobilized on biogenous iron oxide via four quaternary ammonium bromide groups.9 This organic–inorganic hybrid catalyst showed a high activity even after recycling ten times without the loss of catalytic activity.
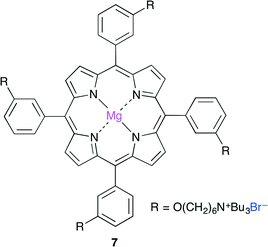 |
| | Fig. 3 Bifunctional catalyst 7. | |
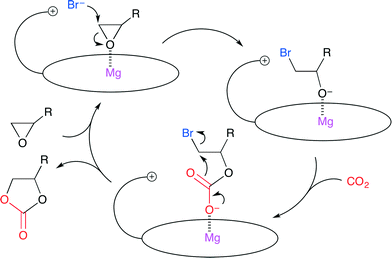 |
| | Scheme 2 A plausible catalytic cycle via the cooperative activation of epoxide with a bifunctional catalyst. | |
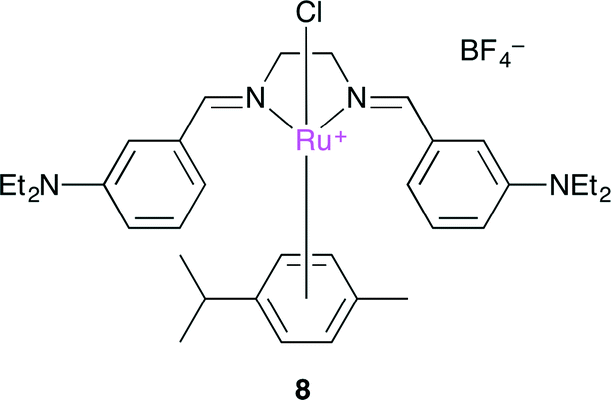
Ulusoy and co-workers developed a cationic diimine–RuII complex 8 (Fig. 4).108 is very attractive due to the easy preparation and chemical stability, and cyclic carbonate could be produced with a low catalyst loading (0.1 mol%). Cyclic voltammetric measurements of several RuII diimine complexes indicated that 8 had the lowest oxidation potential. This result might be attributed to the electron-donating substituents (NEt2) in 8.
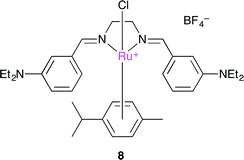 |
| | Fig. 4 Cationic diimine–RuII complex 8. | |
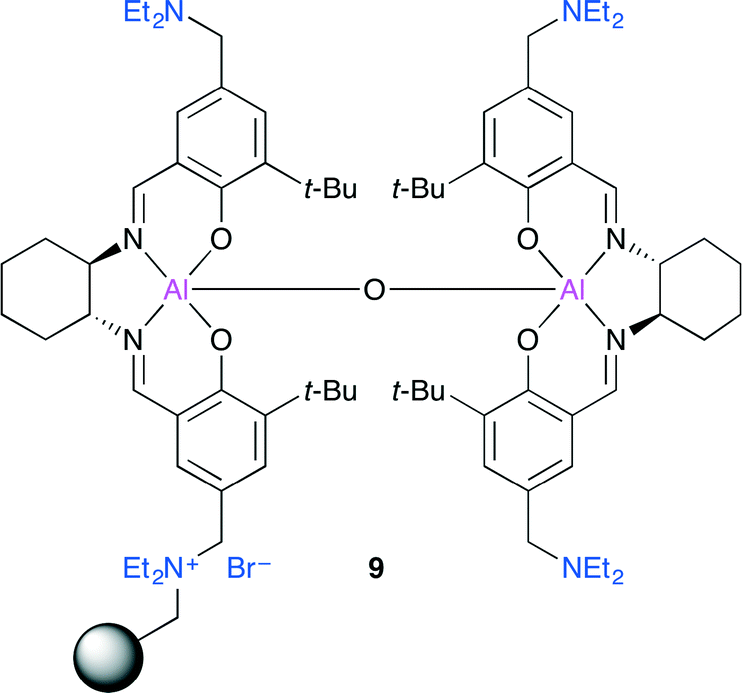
North and co-workers investigated dimeric Al(salen) complexes (Fig. 5).11 Bimetallic complex 9 immobilized on the Merrifield resin via an ammonium bromide group showed a high catalytic activity, and 9 was the first heterogeneous catalyst capable of showing catalytic activity at atmospheric pressure of CO2 at room temperature. The application of 9 to a continuous flow reactor system may be feasible. Although catalyst 9 contains only a single ammonium bromide, 9 showed the highest catalytic activity, which indicated that the amino groups in 9 played an important role in activating CO2.
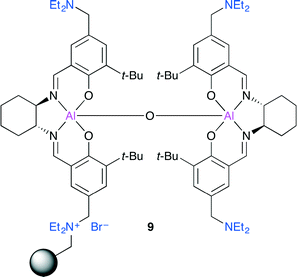 |
| | Fig. 5 Immobilized dimeric Al(salen) complex 9. | |
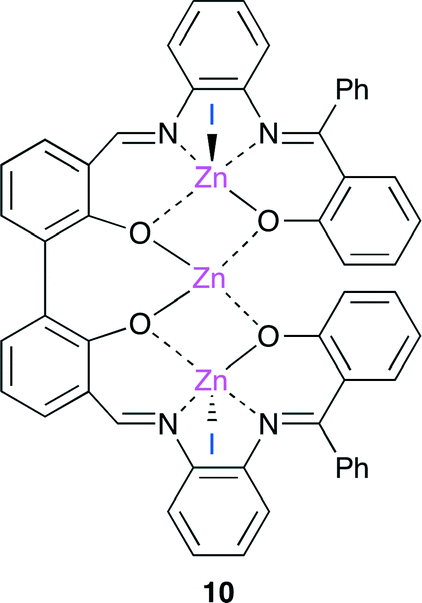
Kleij and co-workers reported a trinuclear bifunctional catalyst 10 derived from a tetraoxo bis-Zn(salphen) complex (Fig. 6).12 The coordinating I− ion and central Zn atom behave as a nucleophile and a Lewis acid, respectively. The proposed catalytic cycle is as follows. (i) The dissociation of one or two I− ion occurs. (ii) The epoxide coordinates to the central Zn atom of 10. (iii) The I− ion attacks the coordinated substrate to form a Zn–alkoxide intermediate. (iv) CO2 insertion gives a linear carbonate structure. (v) The cyclic carbonate is produced through the ring-closure by the intramolecular nucleophilic attack. In addition, catalyst 10 can be reused up to five times without the loss of catalytic activity.
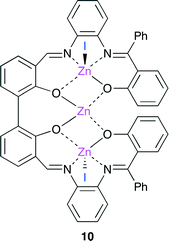 |
| | Fig. 6 Trinuclear bifunctional catalyst 10. | |
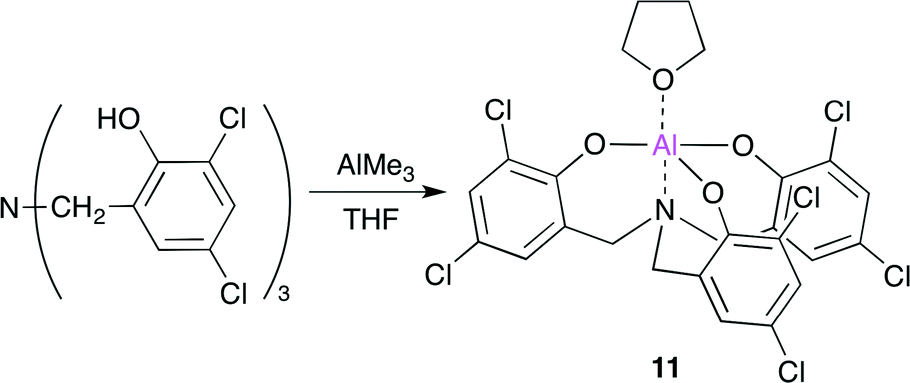
They also reported an Al complex 11 based on an amino triphenolate ligand (Scheme 3).13 By using 11 and tetrabutylammonium iodide (TBAI) as a co-catalyst (100 equiv. of 11) at 90 °C, a high TOF (36000 h−1) and TON (112000) were achieved. The catalytic system also displays a wide substrate scope and functionality tolerance.
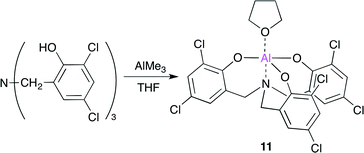 |
| | Scheme 3 The synthesis of Al complex 11. | |
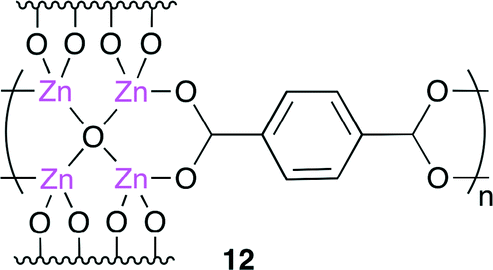
Recently, metal–organic frameworks (MOFs) have received much attention from the viewpoint of gas storage, product separation, magnetism and catalysis. Han and co-workers developed a heterogeneous binary catalyst system for cyclic carbonate synthesis.14 They conducted the reaction using MOF-5 [Zn4O(BDC)3 (BDC = benzene-1,4-dicarboxylate)] 12 (Fig. 7), which was prepared by the copolymerization of a Zn4O cluster with BDC, in the presence of tetrabutylammonium bromide. Although most heterogeneous catalysts need a high reaction temperature (>100 °C) and co-solvent, this catalytic system could produce cyclic carbonate in a high yield at atmospheric CO2 pressure at 50 °C. The effect of the CO2 pressure on the catalytic activity was investigated. A high catalytic activity was exerted at 1–6 MPa, and the yield of cyclic carbonate decreased with an increase in the pressure from 6 to 12 MPa. This is because propylene oxide was dispersed in CO2, which decreased the reaction rate.
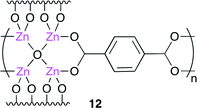 |
| | Fig. 7 The structure of MOF 12. | |
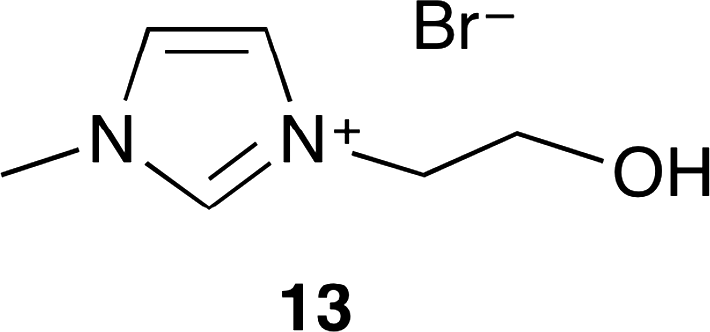
Ionic liquid 13 with the hydroxy group was developed by Zhang and co-workers (Fig. 8).15 The catalytic activities of several ionic liquids were compared with quaternary onium salts and urea. 13 showed the highest catalytic activity, selectivity and reusability without any co-catalyst and co-solvent. This is due to the hydrogen bond of the hydroxy group that activates epoxide efficiently. Moreover, investigations on the solvent effect indicated that protic solvents such as water and methanol tended to be more suitable than aprotic solvents such as dimethylcarbonate and DMF.
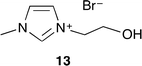 |
| | Fig. 8 Ionic liquid 13. | |
On the basis of this observation, they postulated that water should be a good medium for the synthesis of cyclic carbonates.16 A small amount of water was added to the reaction mixture, and various metal halides, ionic liquids and onium salts were screened. As a result, butyltriphenylphosphonium iodide (PPh3BuI) was found to be the most efficient catalyst. The effect of the amount of water was then investigated in detail. The addition of 0.33 mol% of water remarkably accelerated the reaction to give a maximal product yield. The TOF value was four-fold higher than that in the absence of water. A further increase in the amount of water decreased the yield and selectivity because of the side reaction giving 1,2-propylene glycol. Other solvents with or without hydroxy groups were also tested. Water was the best solvent for PPh3BuI, suggesting that protic solvents with hydroxy groups are good promoters to improve the catalytic activity of the catalyst.
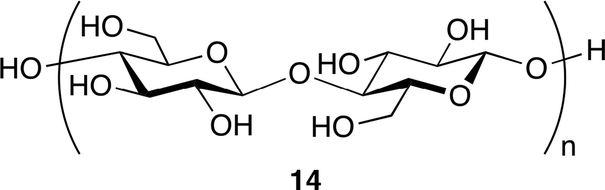
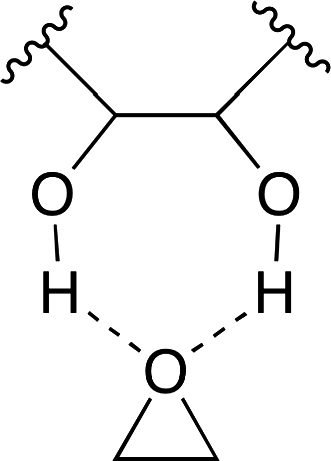
A binary catalyst system composed of cellulose 14 and KI was reported by Jiang, Han and co-workers (Fig. 9).17 They envisioned that cellulose, which is abundant in nature, might be able to activate epoxide by the hydrogen bond, and tested cellulose and various alcohols for catalytic activity. The cellulose–KI combination was found to be very active, stable, and recyclable. Based on the results that other 1,2-diols also showed a high catalytic activity, the two adjacent hydroxy groups of cellulose or 1,2-diols seem to activate epoxide via double hydrogen bonds (Fig. 10).
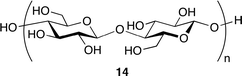 |
| | Fig. 9 The structure of cellulose. | |
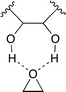 |
| | Fig. 10 Hydrogen bonding between the diol and epoxide. | |
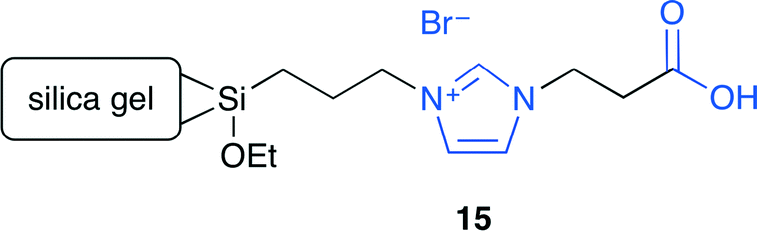
Park and co-workers developed a carboxylic acid-functionalized ionic liquid, which was immobilized on silica gel.18 A series of ionic liquids with or without the carboxyl group and different halide anions were prepared. As a result of screening, 15 showed a high catalytic activity and selectivity (Fig. 11). The immobilized ionic liquids with other functional groups such as the hydroxy group exhibited a lower catalytic activity. The non-functionalized ionic liquid increased the conversion rate but with a lower selectivity in the presence of a small amount of water. In contrast, when a small amount of acetic acid was added to the non-functionalized ionic liquid, the reaction became much faster, yielding a much higher conversion and selectivity. Therefore, the carboxyl group of 15 improved the catalytic performance probably by hydrogen bonding with the epoxide.
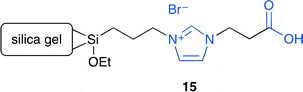 |
| | Fig. 11 Immobilized ionic liquid 15. | |
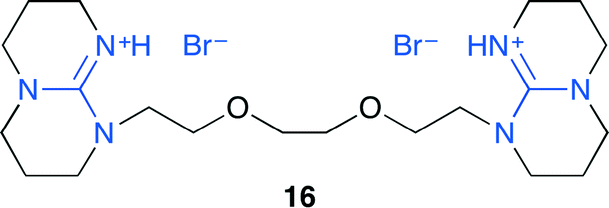
He and co-workers developed ionic liquid 16 with strong Lewis basic 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), which could be reused at least five times (Fig. 12).19 The reaction intermediates were investigated by using in situ FT-IR spectroscopy. 16 can activate CO2 and epoxide through carbamic acid formation and the hydrogen bond, respectively. Moreover, 16 could easily convert ethylene carbonate produced from ethylene oxide into dimethyl carbonate by treating with methanol in one pot.
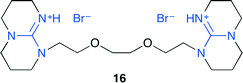 |
| | Fig. 12 Ionic liquid 16. | |
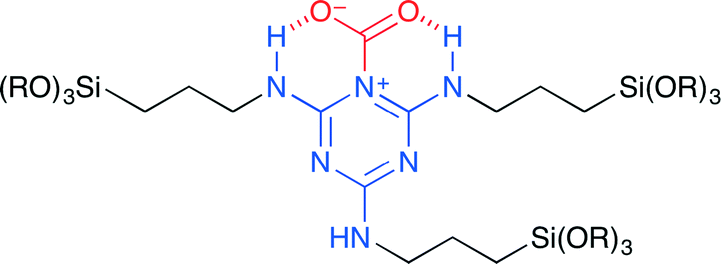
Park and co-workers synthesized periodic mesoporous organosilica (PMO) 17 from N2,N4,N6-tris(3-(triethoxysilyl)propyl)-1,3,5-triazine-2,4,6-triamine (TBTS).20 This is the first application of the nitrogen-rich PMO to the coupling reaction of CO2 with epoxides. The SEM and TEM measurements revealed that TBTS-PMO had a uniform hexagonal plate morphology with short channels, which were perpendicularly arranged to the platelets. CO2 may be adsorbed at the TBTS-PMO surface to form pseudo-cyclic carbamate (Fig. 13). The cooperative action of the triazine moiety and the amino NH group seems to play an important role in the activation of CO2. DFT calculations supported this proposal.
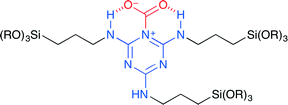 |
| | Fig. 13 A proposed mode of CO2 activation of 17. | |
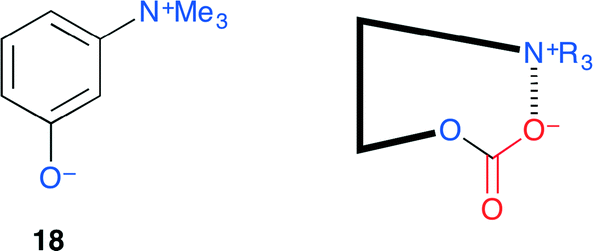
Sakai and co-workers proposed an ammonium betaine framework for the activation of CO2 to give a reactive intermediate that may be useful for various organic transformations (Fig. 14).21 The aryloxide anion has both nucleophilic and leaving abilities, both of which are essential features for catalysis, and the quaternary ammonium cation may stabilize the anion formed by the nucleophilic addition to CO2. Among eight candidates, 18 was found to be the most active. A metal-free, halogen-free and solvent-free organocatalytic reaction was achieved with 1 mol% catalyst loading.
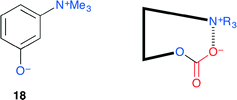 |
| | Fig. 14 Activation of CO2 by ammonium betaine 18. | |
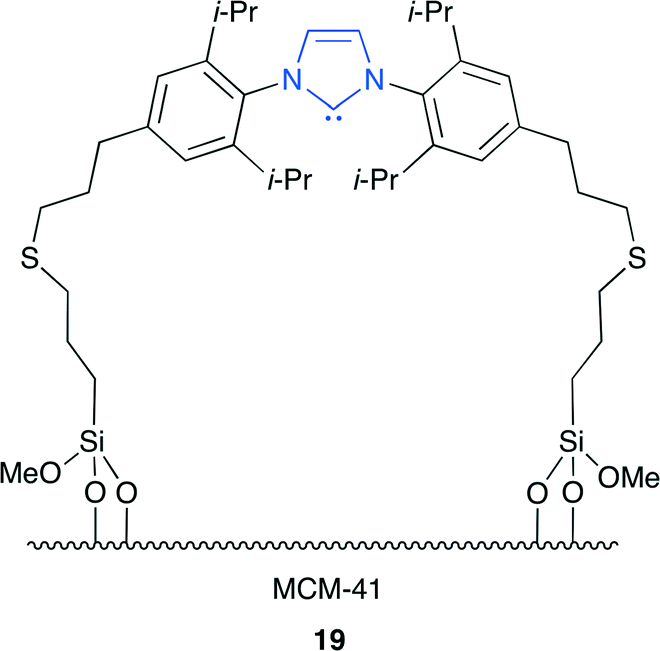
Lu and co-workers reported the N-heterocyclic carbene (NHC)-immobilized MCM-41 catalyst 19 (Fig. 15).22 It is well known that NHC can activate CO2 by forming a stable imidazolium–carboxylate (NHC–CO2) adduct. This is the efficient heterogeneous catalyst with high catalytic activity and reusability without metal and halogen. The in situ diffuse reflectance infrared fourier transform spectroscopy (DRIFTS) and thermogravimetric analysis (TGA) were used to investigate the reversible CO2 capture–release behavior of 19. 19 is an excellent CO2 capture agent because it can form an NHC–CO2 adduct at a relatively low temperature and can decarboxylate upon heating.
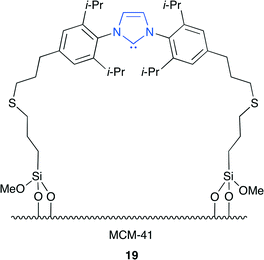 |
| | Fig. 15 Immobilized NHC 19. | |
3. The synthesis of polycarbonates
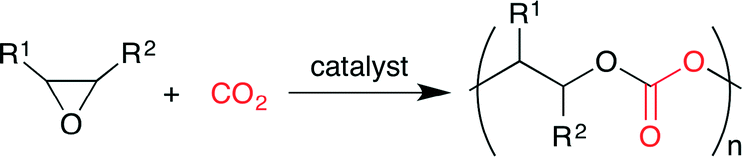
Synthetic CO2-based polymers with high CO2 content are expected to find wide applications in green and sustainable society. Polycarbonates can be synthesized directly from CO2 and epoxides (Scheme 4). Usually, mild reaction conditions are required to obtain uniformly distributed polycarbonates with high carbonate linkage. Under harsh reaction conditions, a polymer chain is shortened by the back-biting mechanism at the polymer terminus to give cyclic carbonate. Therefore, highly active catalysts are required for the copolymerization of epoxides and CO2.
 |
| | Scheme 4 The synthesis of polycarbonates from epoxides and CO2. | |
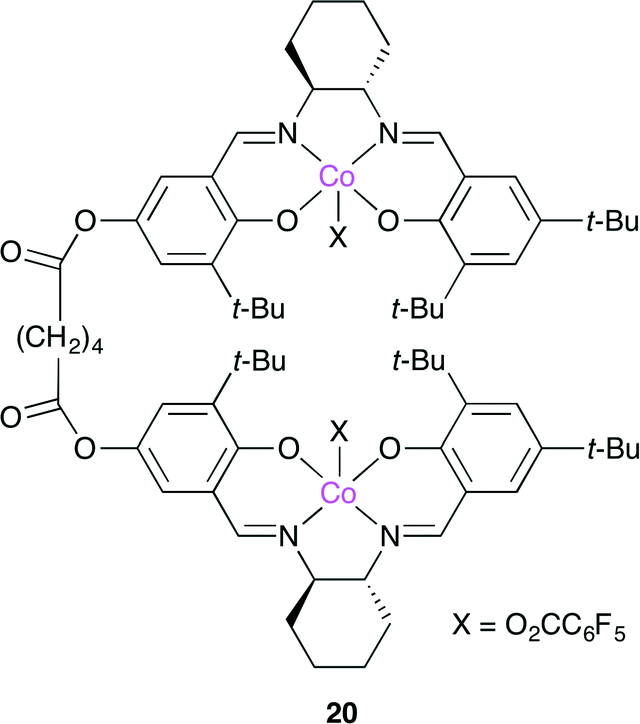
Nozaki and co-workers developed bimetallic catalyst 20, which is a dimer of a Co(salen) complex (Fig. 16).23 Epoxide is activated by one metal center in 20, whereas the alkoxide anion or carbonate anion of the polymer chain is coordinated to another metal center. These activated species might promote CO2 insertion or nucleophilic attack to the epoxide, followed by the inter-nuclear migration. By repeating this process, polycarbonate is produced. Among several catalysts with various linker lengths, 20 was the most active probably because this bimetallic mechanism took place most efficiently.
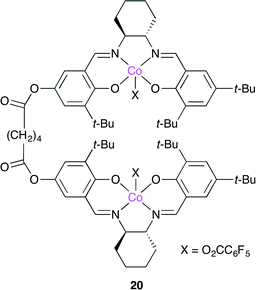 |
| | Fig. 16 Dimeric Co(salen) complex 20. | |
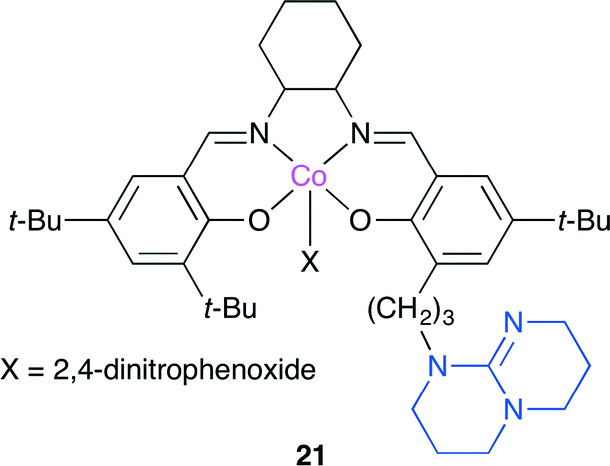
Lu, Darensbourg and co-workers developed a highly active salen-type catalyst 21 bearing TBD (Fig. 17).24 Although it is usually difficult to selectively copolymerize epoxide containing electron-withdrawing substituents, such as styrene oxide and epichlorohydrin, they succeeded in producing polycarbonate from epichlorohydrin with >99% polymer selectivity and >99% carbonate linkages by using 21 at 0 °C. Based on the mass spectra and X-ray crystal structures of reaction intermediates together with kinetic studies using in situ FT-IR spectroscopy, a catalytic cycle was also proposed for a binary catalyst system.
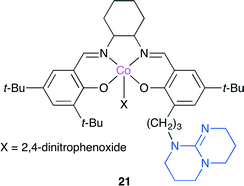 |
| | Fig. 17 Bifunctional Co(salen) catalyst 21. | |
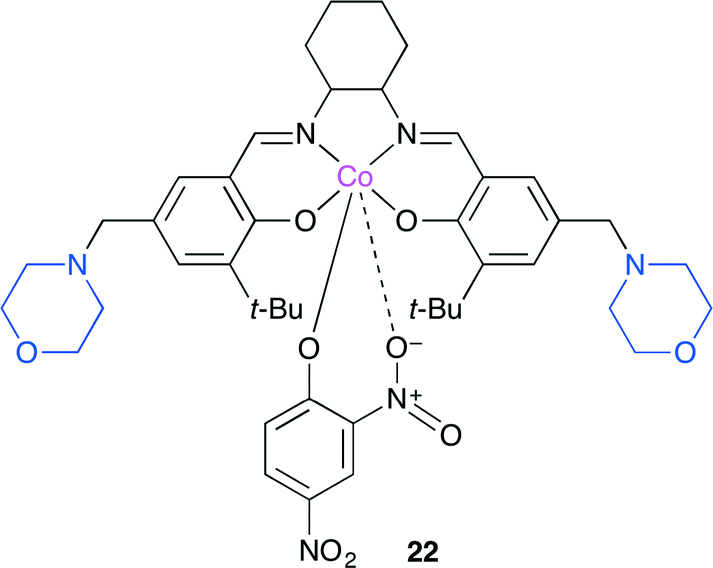
Niu and co-worker reported a new bifunctional Co(salen)-type catalyst with two Lewis basic units 22 (Fig. 18).25 The copolymerization of CO2 with propylene oxide selectively produced the alternating copolymer. In addition, the terpolymerization of CO2, propylene oxide and cyclohexene oxide with a high cyclohexene carbonate content was also realized without the formation of cyclic carbonates or ether linkages. The glass transition temperature (Tg) of the terpolymer obtained was adjustable between 40 and 118 °C by controlling the relative ratios of propylene oxide and cyclohexene oxide.
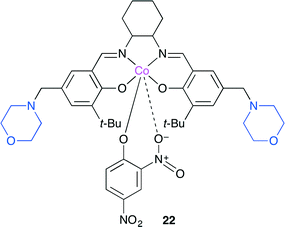 |
| | Fig. 18 Bifunctional Co(salen) catalyst 22. | |
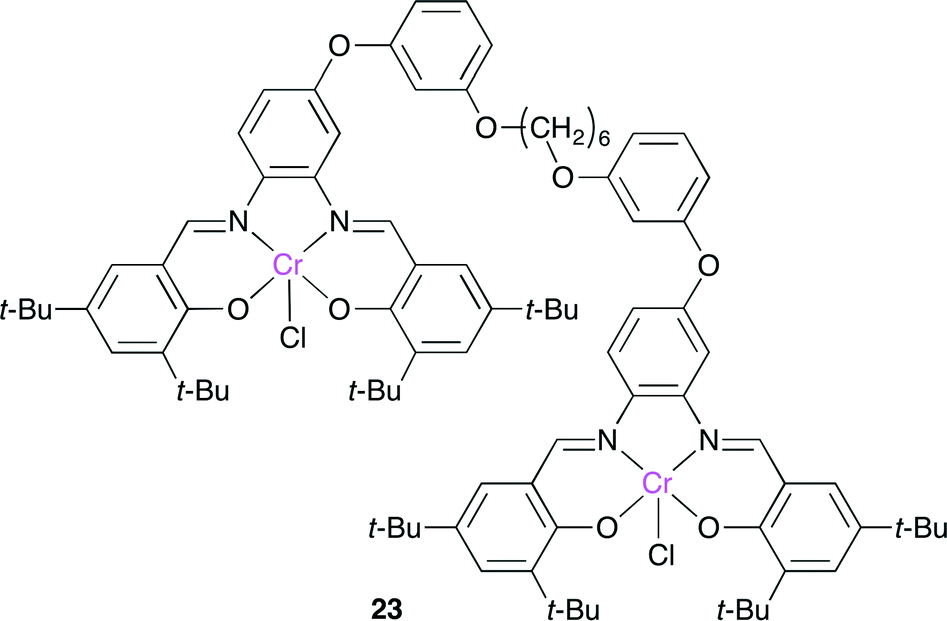
Rieger and co-workers reported a bimetallic Cr(salphen) catalyst 23, which has two salphen units bridged by a flexible alkyl chain (Fig. 19).26 Although there are a few reports on dimeric salphen catalysts, they have a rigid bridging spacer with a long metal–metal distance. The copolymerization of propylene oxide and CO2 with 23 was tolerant to dilution, giving a copolymer of a high molecular weight with a high selectivity, whereas the much slower copolymerization with a lower selectivity proceeded with the monomeric Cr(salphen).
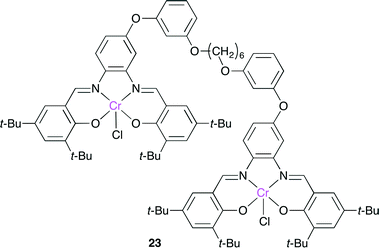 |
| | Fig. 19 Bimetallic Cr(salphen) catalyst 23. | |
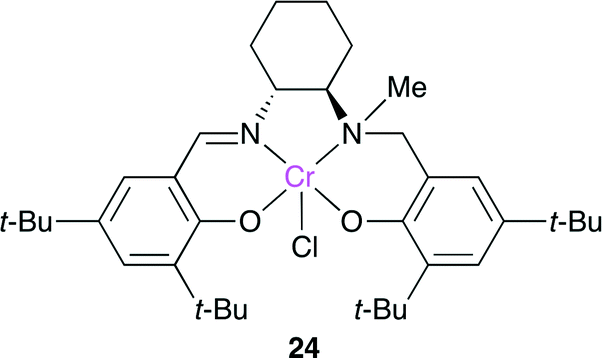
The salalen-type catalyst 24 was developed by Nozaki and co-workers (Fig. 20).27 Salalen is a reduced structure of salen, and the electronic and steric properties and flexibility of salalen are different from those of salen. 24 with [PPN]Cl ([PPN]+ = bis(triphenylphosphoranylidene)iminium) as a co-catalyst showed a high catalytic performance for the copolymerization of cyclohexene oxide and CO2 (TOF = 100 h−1) at the atmospheric pressure of CO2, which is the highest of the reported catalysts.
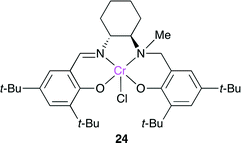 |
| | Fig. 20 Cr(salalen) catalyst 24. | |
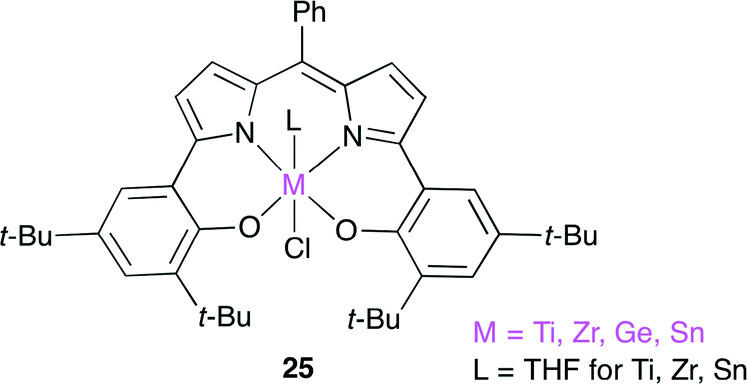
Furthermore, they reported a new catalyst 25 for the copolymerization of epoxides and CO2 (Fig. 21).28 Complexes 25 are composed of a tetravalent metal ion such as Ti, Zr, Ge and Sn, a trianionic [ONNO]-tetradentate ligand, and a monoanionic axial ligand. This is the first example of tetravalent metal complexes used as catalysts for the copolymerization. The Ti and Ge complexes were found to give the alternating copolymer with high carbonate linkage (TOF up to 76 h−1). It is attractive to use the less toxic metals such as Ti and Ge, and further development is expected.
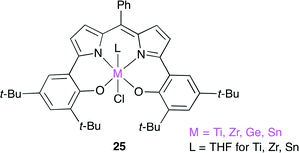 |
| | Fig. 21 Tetravalent metal complexes 25. | |
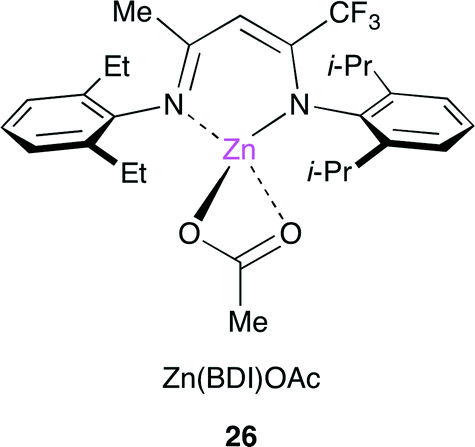
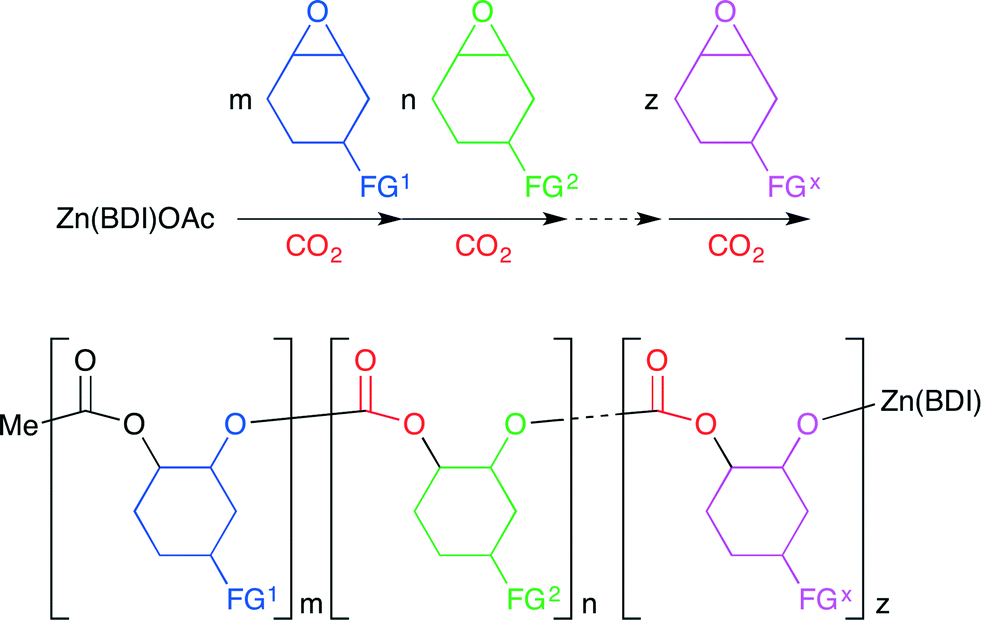
Coates and co-workers reported a method for the living multiblock copolymerization catalyzed by Zn(BDI) complex 26 (BDI = β-diiminate) (Fig. 22).29 Various cyclohexene oxide derivatives containing protected alcohol, lipophilic, hydrophilic or fluorophilic functional groups could be copolymerized sequentially with CO2 by 26 without any co-catalyst. Various block copolymers containing different carbonate units in a single chain were synthesized with good control of block sequence and length in one pot (Scheme 5).
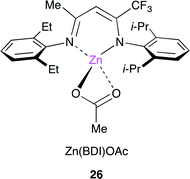 |
| | Fig. 22 Zn(BDI) complex 26. | |
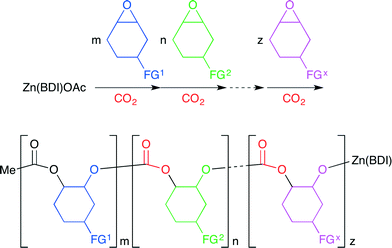 |
| | Scheme 5 The preparation of a multiblock cyclohexene carbonate copolymer. | |
4. Carboxylations with CO2
In addition to the ring-opening reactions of epoxides with CO2, much effort has been made to develop catalytic conversions of CO2 into other useful compounds. In many cases, researchers search for new reactions as well as new catalysts. In this section, catalytic carboxylation reactions with C–C bond formation are highlighted.
4.1 Carboxylation of aryl and benzyl halides
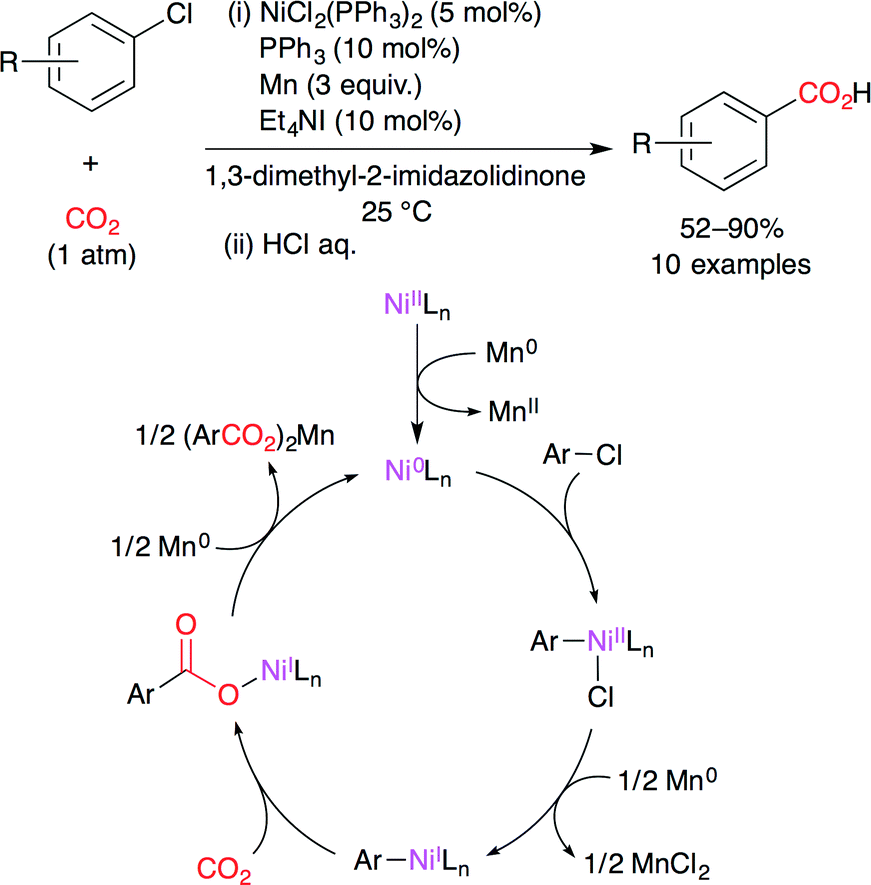
Tsuji and co-workers developed the nickel-catalyzed carboxylation of aryl and alkenyl chlorides with CO2 (1 atm) at room temperature (Scheme 6).30 This new catalytic system is also highly active for aryl bromides, tosylates and triflates. A proposed reaction mechanism is shown in Scheme 6. The NiII complex is reduced to a Ni0 species upon treatment with Mn0. The subsequent oxidative addition of aryl chloride gives a NiII intermediate. NiII is then reduced by the Mn0/Et4NI system to afford a NiI intermediate. This nucleophilic NiI species reacts with CO2 to give the carboxylatonickel intermediate, which is reduced by Mn0 to give the corresponding manganese carboxylate with the regeneration of the Ni0 catalyst.
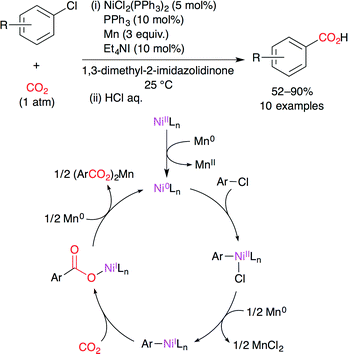 |
| | Scheme 6 The Ni-catalyzed carboxylation of aryl chlorides with CO2. | |
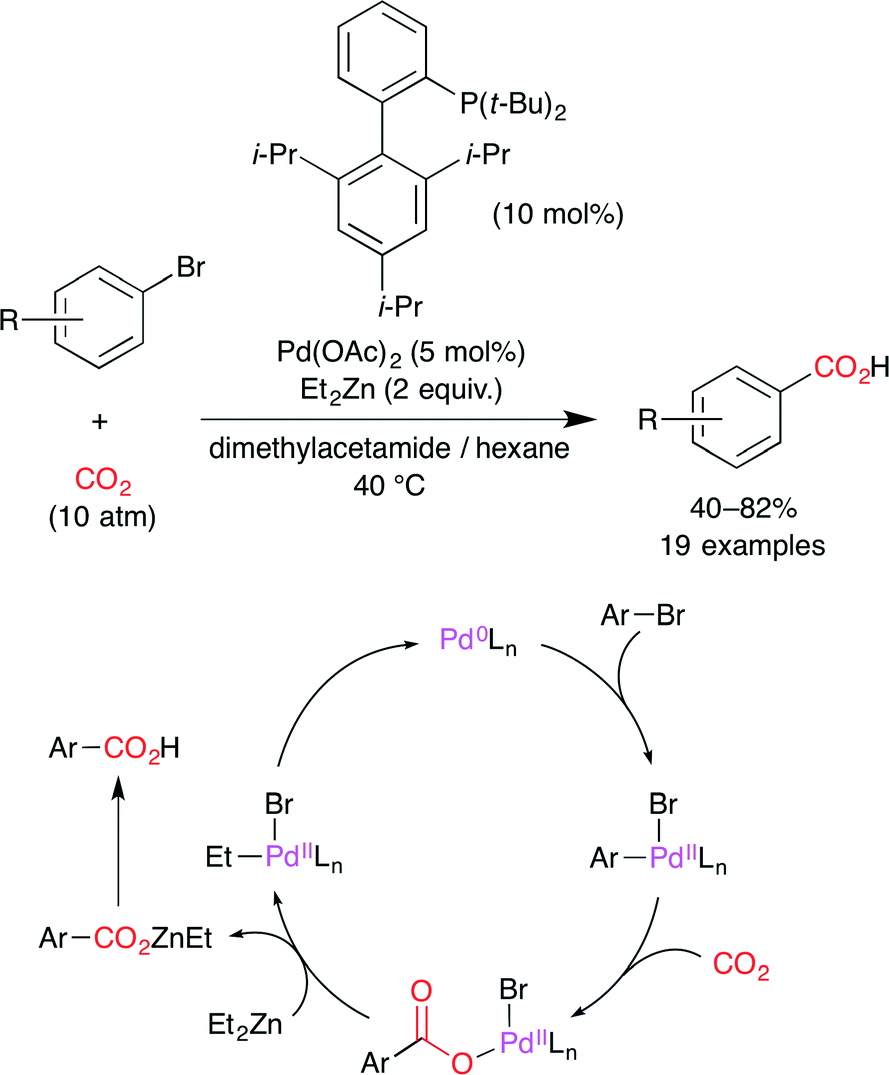
Martín and co-worker reported the Pd0-catalyzed carboxylation of aryl bromides with CO2 (Scheme 7).31 The proposed reaction mechanism is shown in Scheme 7. The oxidative addition of aryl bromide to a Pd0 complex forms a PdII–aryl species, and CO2 insertion gives the Pd–carboxylate intermediate. Subsequent transmetalation with Et2Zn delivers the Zn–carboxylate with the regeneration of the Pd0 catalyst.
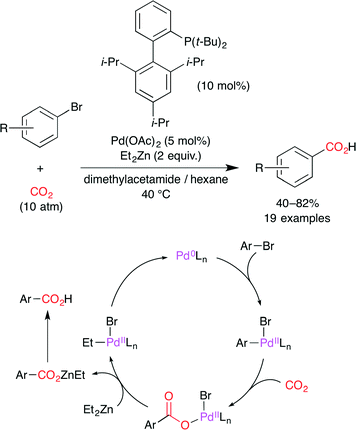 |
| | Scheme 7 The Pd-catalyzed carboxylation of aryl bromides with CO2. | |
They also reported the Ni-catalyzed carboxylation of benzyl chlorides with CO2 (Scheme 8).32 A combination of NiCl2·glyme, PCp3·HBF4 (Cp = cyclopentyl), MgCl2 and Zn dust was found to provide phenylacetic acid derivatives in good yields at atmospheric CO2 pressure at room temperature. Secondary and tertiary alkyl bromides also underwent the carboxylation under similar reaction conditions in moderate yields.
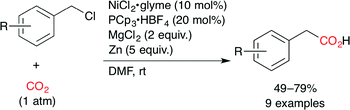 |
| | Scheme 8 The Ni-catalyzed carboxylation of benzyl chlorides with CO2. | |
4.2 Hydrocarboxylation
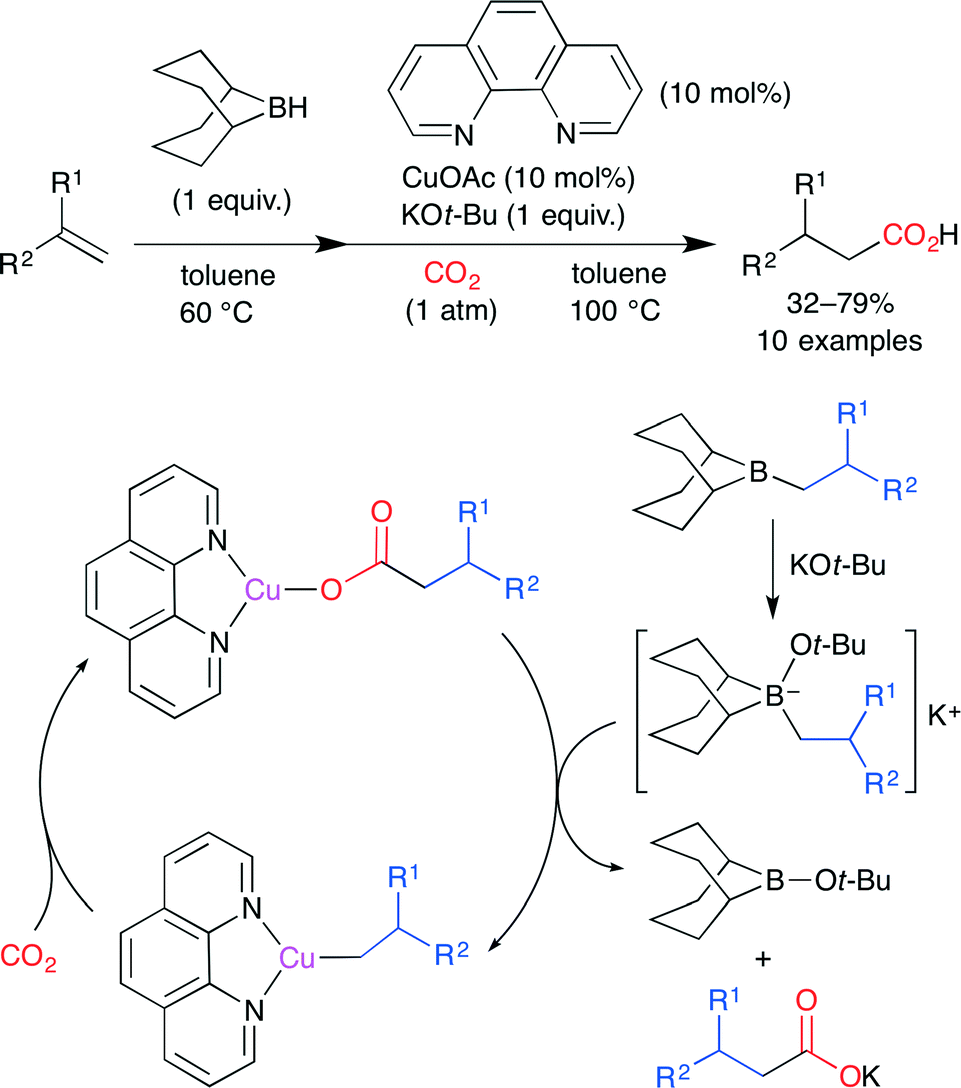
Ohmiya, Sawamura and co-worker developed the reductive carboxylation of terminal alkenes by using a Cu(phen) complex (Scheme 9).33 Reactive alkylborane is produced by the hydroboration of alkene with 9-borabicyclo[3.3.1]nonane (9-BBN), and tetravalent borate is formed by the stoichiometric reaction of alkylborane with KOt-Bu. Subsequent B–Cu transmetalation between the Cu(phen) complex and alkylborate gives an alkylcopper(I) species. Finally, CO2 insertion takes place to afford the carboxylate copper complex. This is an attractive method because of easily accessible alkylborane and inexpensive Cu(phen).
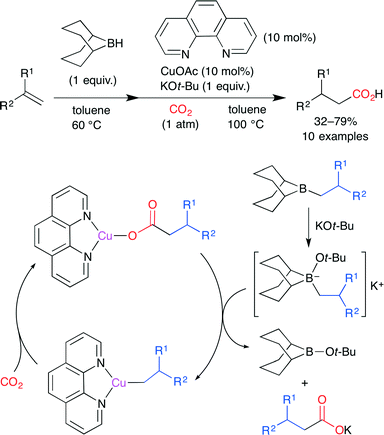 |
| | Scheme 9 The Cu-catalyzed carboxylation of alkylboranes with CO2. | |
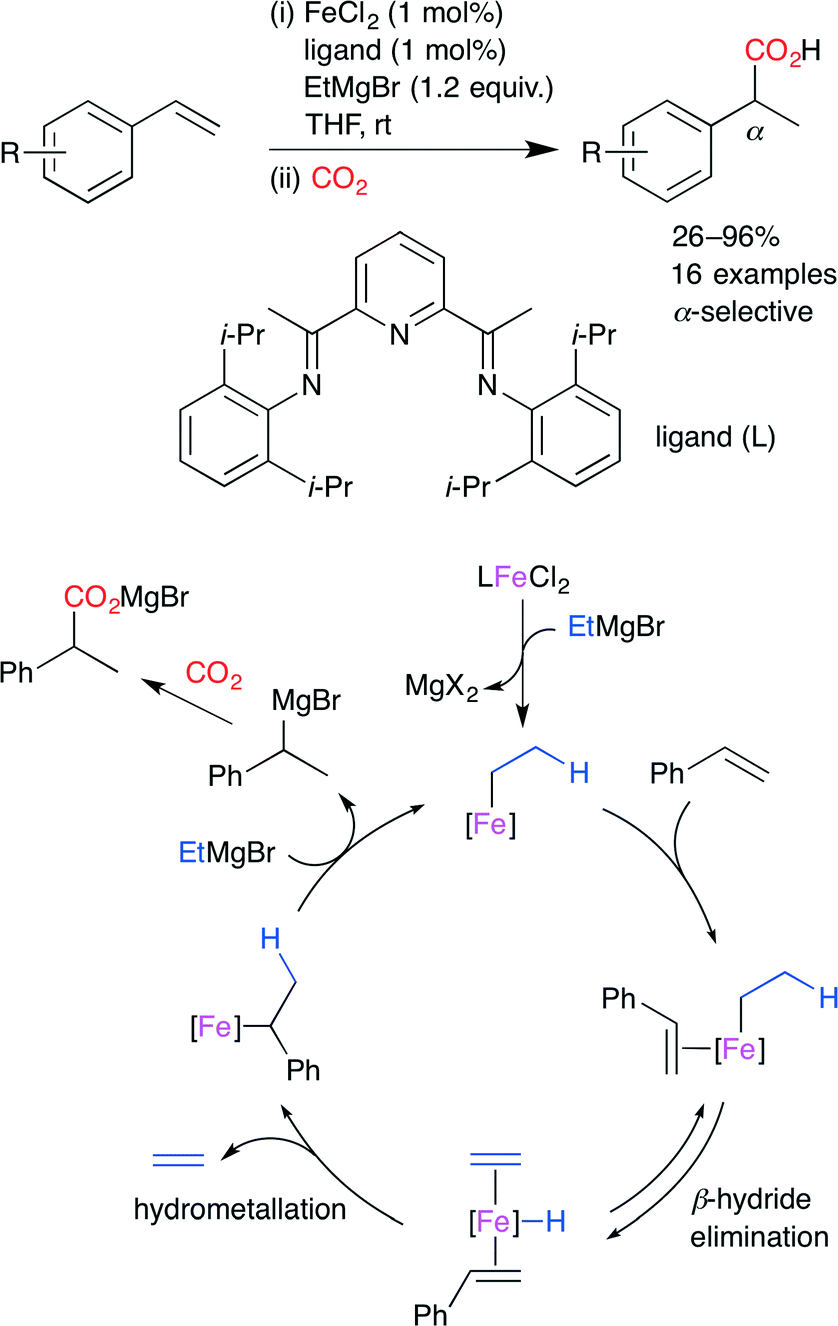
Thomas and co-worker developed the Fe-catalyzed hydrocarboxylation of styrene derivatives by using a highly active bench-stable FeII precatalyst (Scheme 10).34 Using 1 mol% FeCl2, bis(imino)pyridine-based ligand (1 mol%), and EtMgBr (1.2 equiv.), a variety of styrene derivatives were transformed into the corresponding α-aryl carboxylic acids with high regioselectivity at atmospheric CO2 pressure. The catalyst was found to be equally active with a loading of 0.1 mol%. The proposed mechanism is shown in Scheme 10. The alkylation of the iron precatalyst with EtMgBr and the coordination of styrene give an organometallic complex, which undergoes β-hydride elimination to give an iron hydride complex. The hydrometalation of styrene and the subsequent transmetalation with EtMgBr releases the hydromagnesiated intermediate with the regeneration of the initial complex. Finally, the hydromagnesiated intermediate reacts with CO2 to form the carboxylated product.
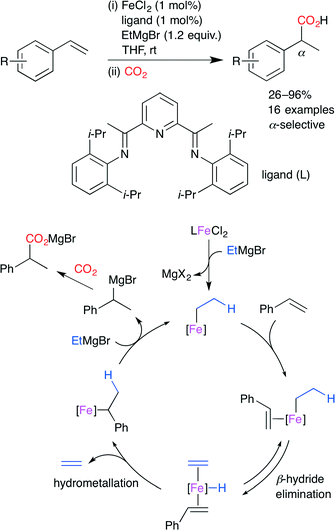 |
| | Scheme 10 The Fe-catalyzed regioselective hydrocarboxylation of styrene derivatives with CO2. | |
4.3 Carboxylation via the C–H bond activation
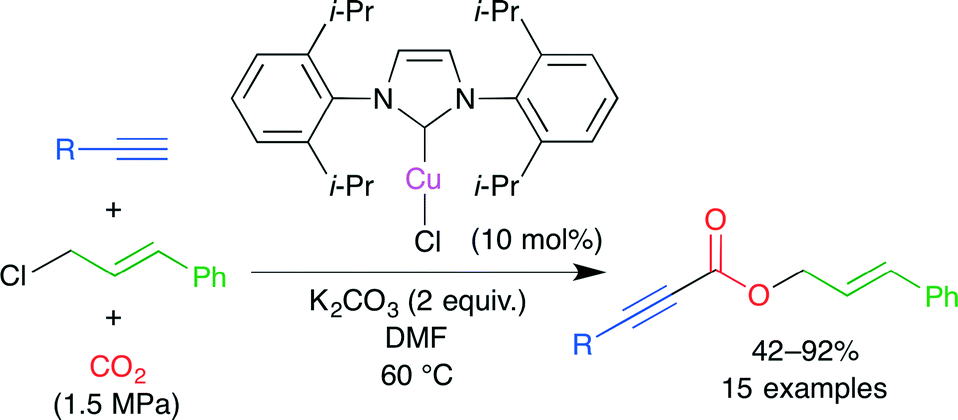
In a transition metal-catalyzed carboxylation, an active carbon–metal bond is initially formed. In the presence of alkyl halide, however, a selective carboxylative coupling is difficult because the cross-coupling byproduct is easily formed from this active species and alkyl halide. Zhang, Lu and co-workers reported the carboxylative three-component coupling of terminal alkynes, allylic chlorides and CO2 using an NHC–CuI complex (Scheme 11).35 In this particular case, the use of less reactive allylic chloride suppressed the cross-coupling side reaction, leading to a high selectivity. Carboxylic acid esters were obtained directly from the reaction mixture. The catalyst could be easily recovered in a high yield by chromatographic separation.
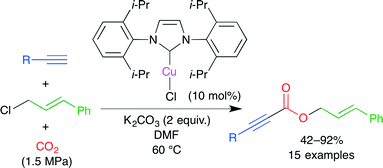 |
| | Scheme 11 The Cu-catalyzed carboxylative coupling of terminal alkynes, allylic chlorides and CO2. | |
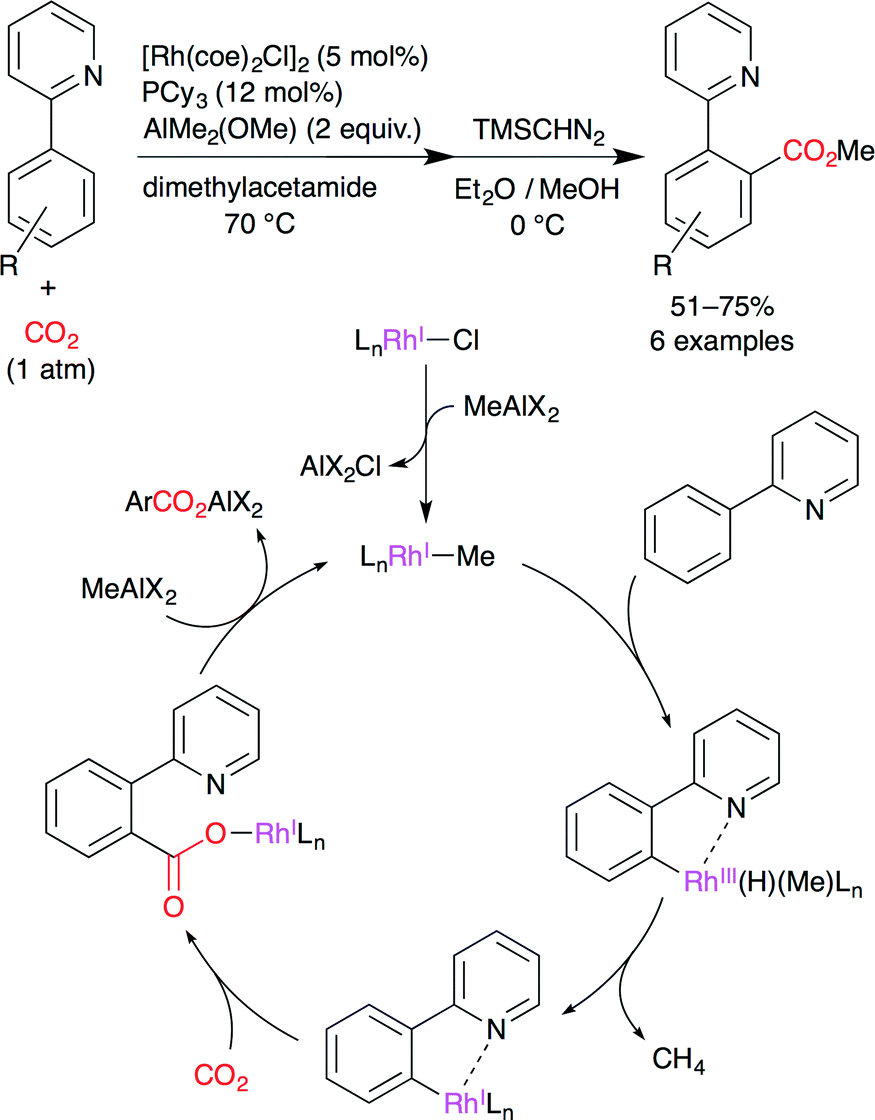
Iwasawa and co-workers succeeded in the development of RhI-catalyzed direct carboxylation of aromatic compounds via chelation-assisted C–H bond activation (Scheme 12).36 It is well known that RhI complexes are efficient catalysts for C–H activation and that an arylrhodium(I) species has sufficient nucleophilicity. A proposed catalytic cycle is shown in Scheme 12. (i) A methylrhodium(I) species is generated by the reaction of RhI chloride with the methylaluminum reagent. (ii) C–H bond activation occurs at the o-position by the chelation effect of the pyridyl group. (iii) The reductive elimination of methane gives the arylrhodium(I) species. (iv) The nucleophilic carboxylation of arylrhodium(I) affords the rhodium carboxylate. (v) Transmetalation with the methylaluminum reagent gives the aluminum carboxylate, regenerating the initial methylrhodium(I) species. The pyrazole ring can also be used as a directing group.
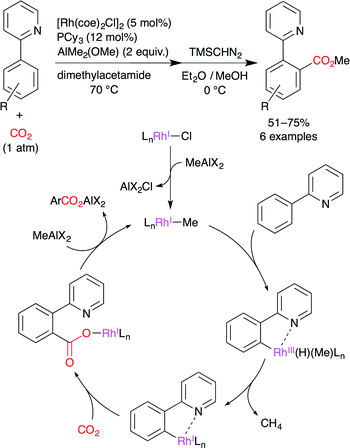 |
| | Scheme 12 The Rh-catalyzed direct carboxylation of arenes with CO2. | |
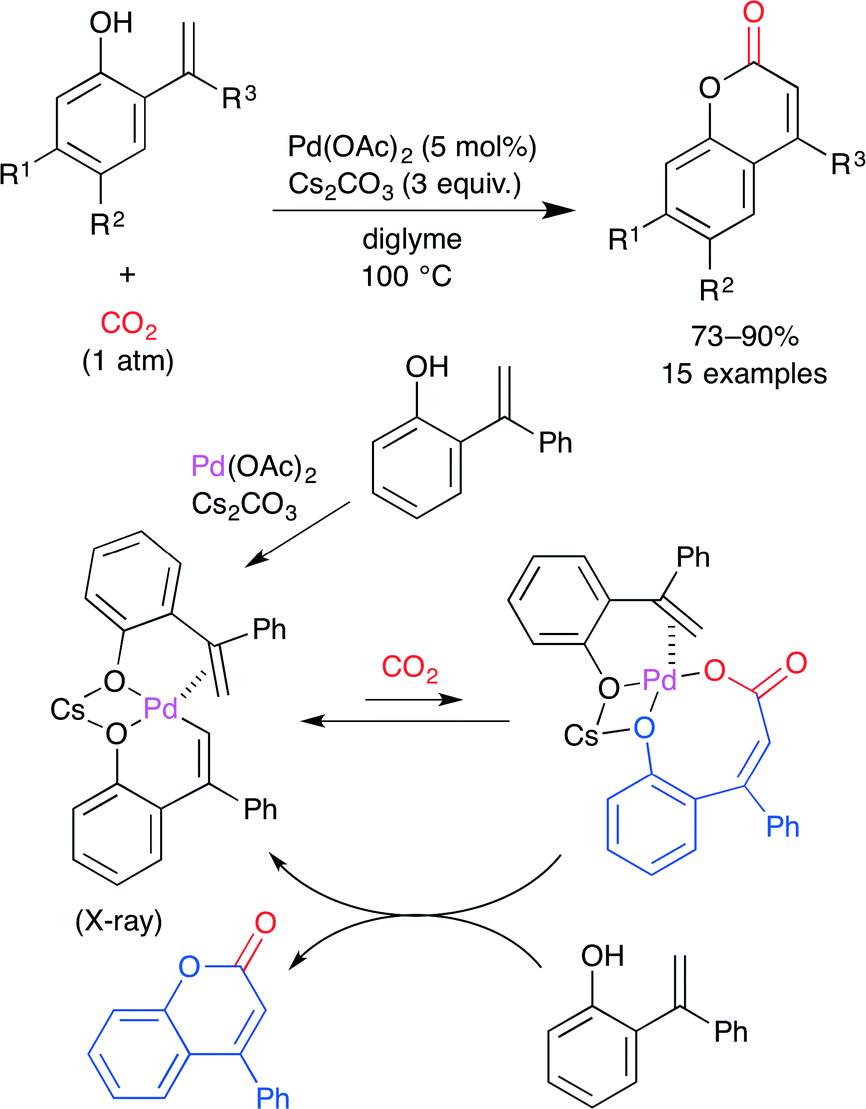
This group also reported the PdII-catalyzed direct carboxylation of the alkenyl C–H bond with CO2 (Scheme 13).37 The treatment of 2-hydroxystyrenes with Pd(OAc)2 and Cs2CO3 under atmospheric CO2 pressure produced coumarins in good yields. The reaction mechanism, which was proposed on the basis of the X-ray crystal structure and reactivity test of the intermediates, is shown in Scheme 13. First, the six-membered alkenyl palladium intermediate is generated by the chelation-assisted alkenyl C–H bond cleavage of 2-hydroxystyrene with Pd(OAc)2 along with the coordination of the second molecule of 2-hydroxystyrene. Subsequently, this alkenyl palladium species undergoes reversible carboxylation to afford a palladium carboxylate intermediate. This is replaced by another molecule of 2-hydroxystyrene to release the product with the regeneration of the six-membered alkenyl palladium intermediate.
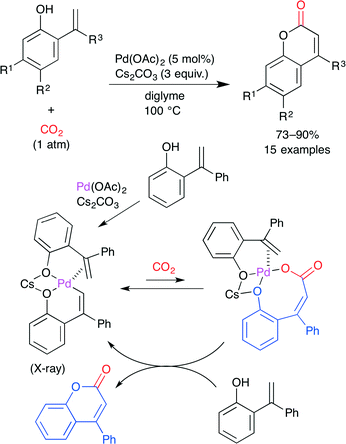 |
| | Scheme 13 The Pd-catalyzed direct carboxylation of 2-hydroxystyrene derivatives with CO2. | |
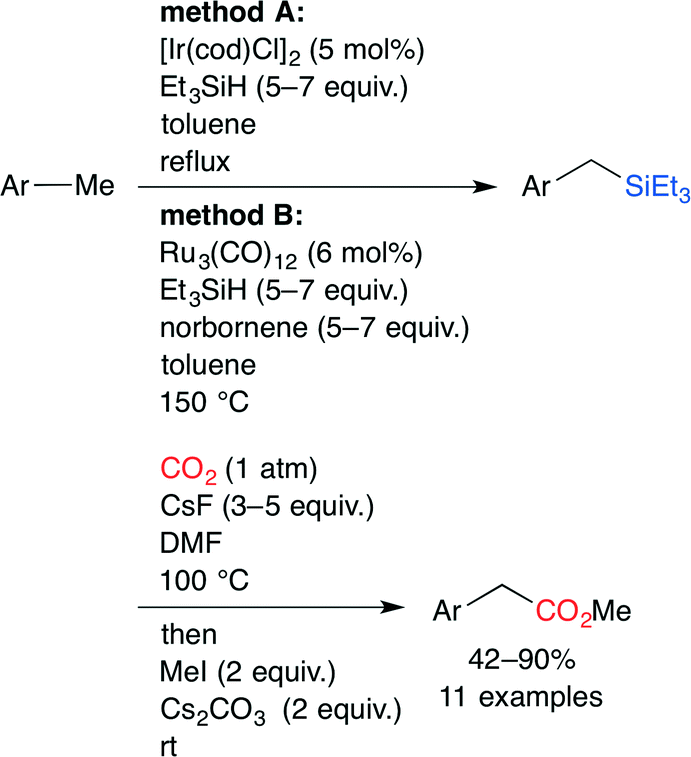
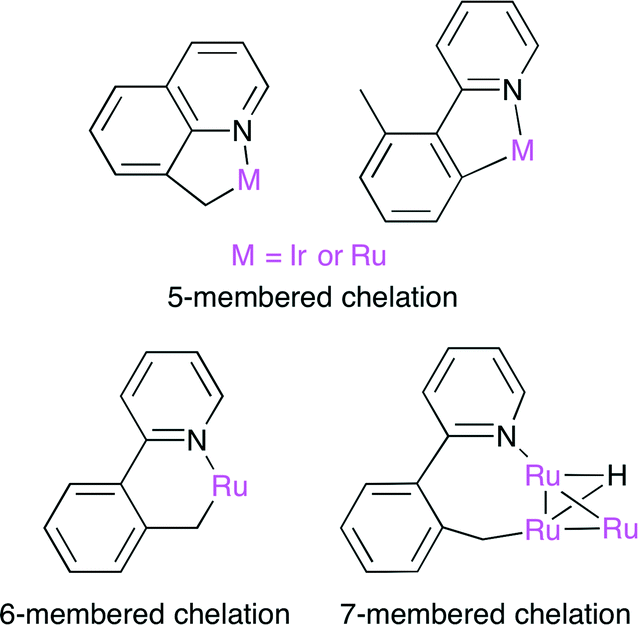
The carboxylation of a C(sp3)–H bond with CO2 is one of the most challenging transformations in current organic chemistry. A novel sequential protocol for the benzylic C(sp3)–H carboxylation via C(sp3)–H silylation catalyzed by IrI or Ru0 complexes was developed by Mita, Sato and co-workers (Scheme 14).38 Because the pyridyl group in the substrate behaves as a directing group, Ir and Ru complexes can form stable 5–7 membered complexes when the benzylic C(sp3)–H bond is selectively activated by these chelation modes (Fig. 23).
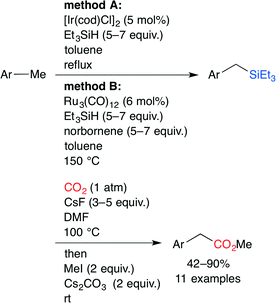 |
| | Scheme 14 Benzylic C(sp3)–H carboxylations. | |
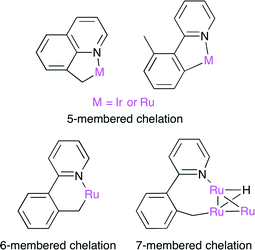 |
| | Fig. 23 The chelation mode between benzylic C(sp3)−H and Ir or Ru. | |
4.4 Other carboxylations
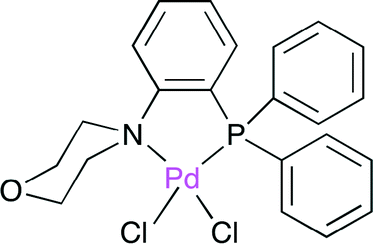
Feng, Bao and co-workers synthesized the PdII complex with the air-stable P,N-bidentate ligand (Fig. 24), where the cyclic secondary amine moiety is linked to the benzene ring of triphenylphosphine.39 The X-ray crystal structure indicated that the soft P and hard N atoms in the ligand are coordinated to the PdII center. The selective synthesis of δ-lactone was effected by the palladium-catalyzed telomerization of 1,3-butadiene with CO2 (Scheme 15). Among several ligands prepared, this P,N-bidentate ligand (Fig. 24) was the most suitable for this synthesis.
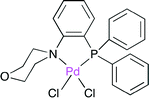 |
| | Fig. 24 The Pd complex with the P,N-bidentate ligand. | |
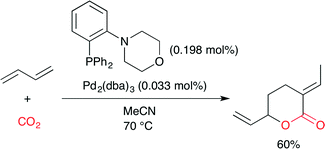 |
| | Scheme 15 The Pd-catalyzed telomerization of 1,3-butadiene with CO2. | |
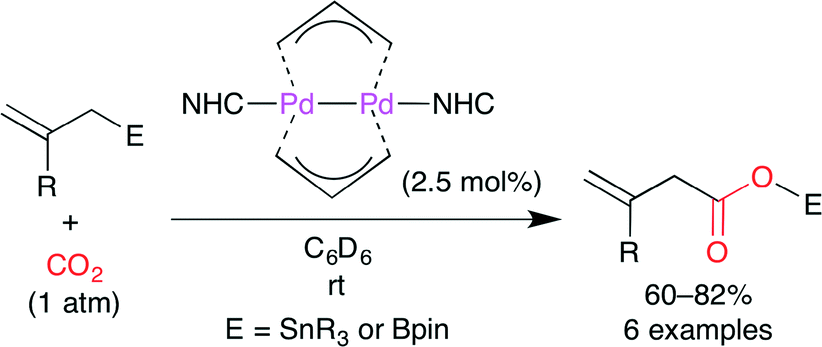
Hazari and co-workers developed a facile synthesis of a PdI-bridging allyl dimer and its use in the catalytic carboxylation of allylstannanes and allylboranes with CO2 (Scheme 16).40 The allyl-bridged PdI dimer is attractive because of a metal–metal bond, two bridging allyl ligands, and a terminal ligand (NHC). This catalyst had been derived from (η1-allyl)(η3-allyl)Pd(L) complex, which is air-, moisture-, and temperature-sensitive. The authors found a more efficient synthetic method, which is a direct reaction of Pd(allyl)2 with NHCs. Bridging allyl ligands are nucleophilic like η1-allyl ligands rather than electrophilic like η3-allyl ligands. The insertion of CO2 followed by transmetallation gives the product.
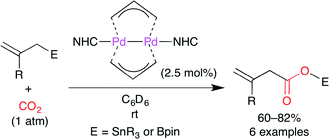 |
| | Scheme 16 The carboxylation of allylstannanes and allylboranes with CO2. | |
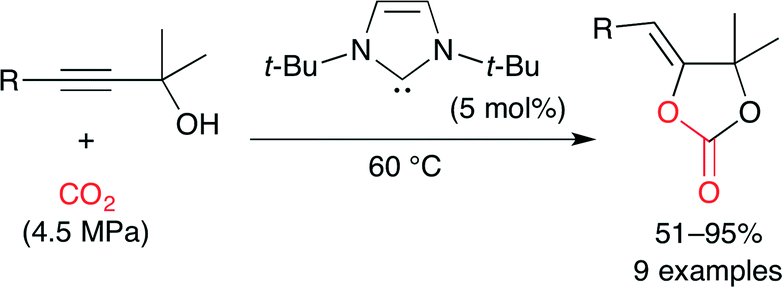
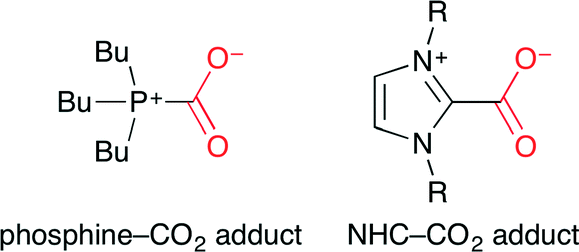
Ikariya and co-workers reported that NHCs and the corresponding CO2 adducts act as effective catalysts for carbonate synthesis under relatively mild reaction conditions (Scheme 17).41 NHCs are expected to exhibit similar chemical properties to tributylphosphine (Bu3P) which forms zwitterionic CO2 adducts (Fig. 25) to promote the nucleophilic addition of CO2 to electrophiles. This method can easily synthesize hindered internal cyclic carbonates.
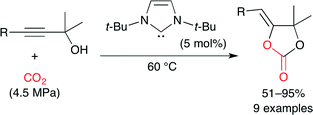 |
| | Scheme 17 The carboxylative cyclization of propargylic alcohol and CO2. | |
 |
| | Fig. 25 Zwitterions of phosphine–CO2 and NHC–CO2 adducts. | |
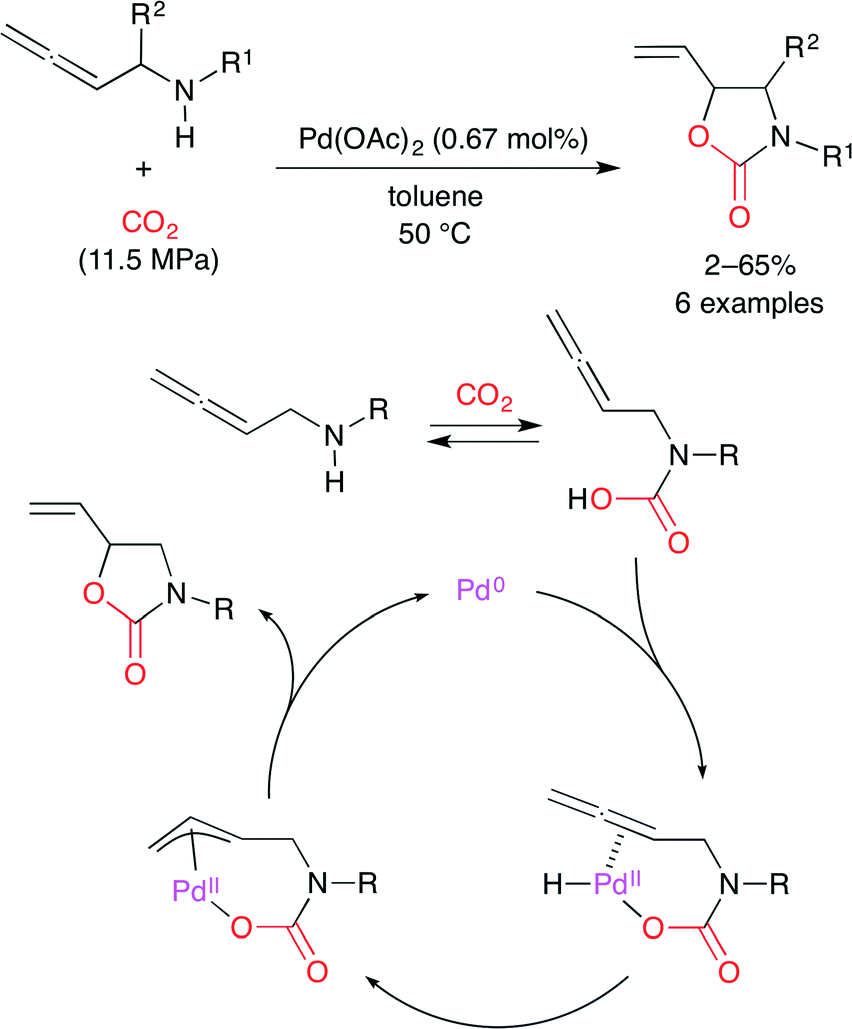
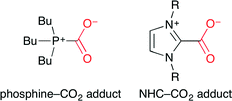
They have also demonstrated the Pd-catalyzed carboxylative cyclization of α-allenyl amines to give oxazolidinones (Scheme 18).42 A possible catalytic mechanism is as follows. (i) The oxidative addition of carbamic acid generated gives a hydridopalladium species. (ii) The allene moiety then inserts into the Pd–H bond to give an η3-allylpalladium intermediate. (iii) The reductive elimination gives the cyclization product. This is the first example of the Pd-catalyzed cyclic urethane synthesis via the intramolecular addition of carbamic acid. This direct addition system is an environmentally benign process giving no wastes.
 |
| | Scheme 18 The carboxylative cyclization of allenic amines. | |
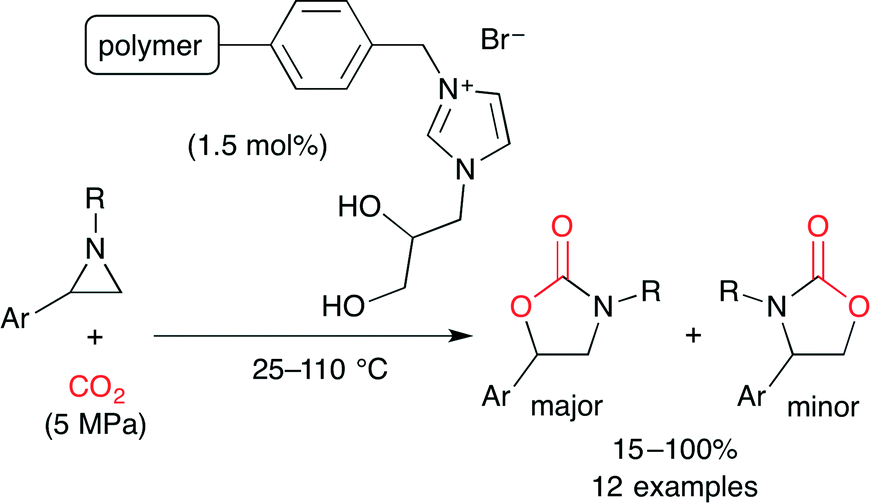
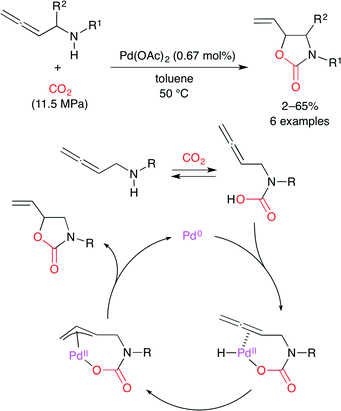
Bhanage and co-workers used polymer-supported diol-functionalized ionic liquids as a catalyst for the coupling of aziridine and CO2 (Scheme 19).43 Hydrogen bonding between the aziridine and the vicinal hydroxy groups in the ionic liquid activates the aziridine ring. The Br− anion selectively attacks the α-position of the phenyl group to ring-open. The CO2 insertion followed by cyclization affords the product. The catalyst was readily recovered by simple filtration, and the catalytic activity was retained after four-time reuse.
 |
| | Scheme 19 The carboxylation of aziridine with CO2. | |
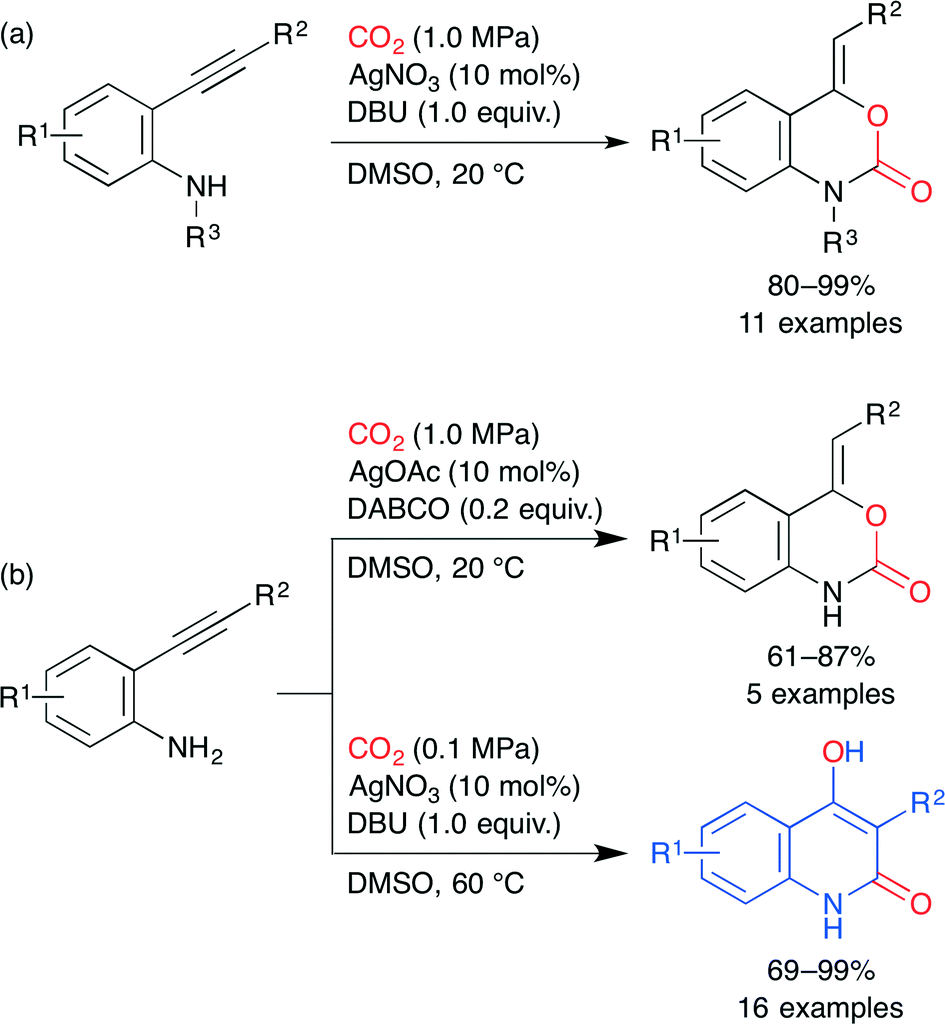
The benzoxazin-2-one and 4-hydroxyquinolin-2(1H)-one skeletons are seen in several drugs. Ag-catalyzed incorporation of CO2 into o-alkynylanilines was developed by Yamada and co-workers. By using AgNO3 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under CO2, secondary o-alkynylanilines were converted into the corresponding benzoxazin-2-ones (Scheme 20a).44 Primary o-alkynylanilines also underwent the same reaction by using 1,4-diazabicyclo[2.2.2]octane (DABCO) instead of DBU. Surprisingly, when DBU was used for primary o-alkynylanilines, 4-hydroxyquinolin-2(1H)-ones were produced in excellent yields (Scheme 20b).45 An intramolecular rearrangement involving the generation of isocyanate from benzoxazin-2-one formed in situ is proposed. In both cases, the AgI ion activates the C–C triple bond most efficiently to promote the cyclization of a carbamate intermediate.
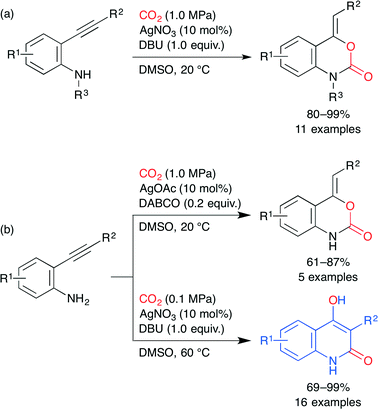 |
| | Scheme 20 The Ag-catalyzed incorporation of CO2 into o-alkynylanilines. | |
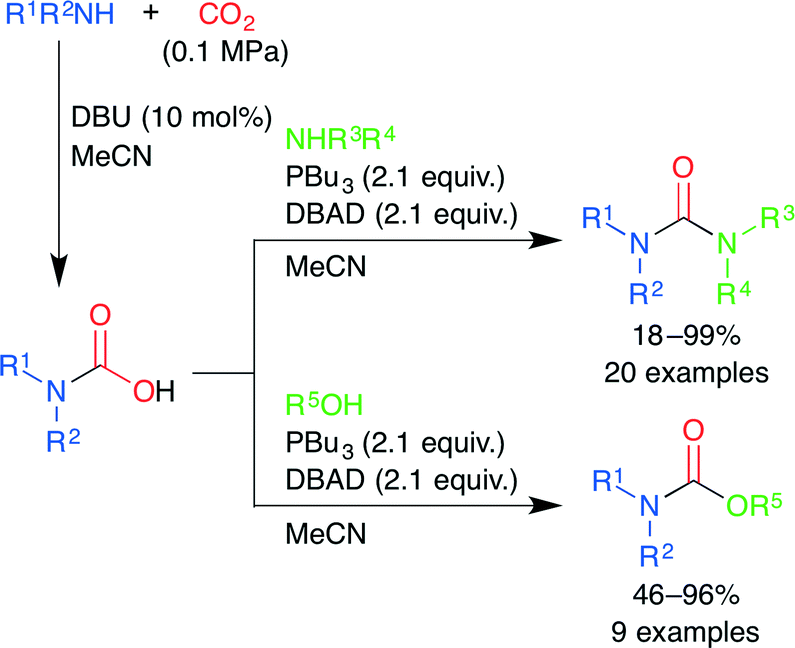
A mild and efficient synthesis of ureas and carbamates via carbamic acids generated by the DBU-catalyzed reaction of amines and CO2 was developed by Peterson and co-workers (Scheme 21).46 The carbamic acids generated from primary amines were dehydrated under Mitsunobu conditions to give isocyanates, which reacted with amines and alcohols to yield the corresponding ureas and carbamates, respectively. On the other hand, the carbamic acids from secondary amines react with activated alcohols by the SN2 mechanism. Various unsymmetrical di- and trisubstituted ureas and carbamates could be easily constructed. This parallel process may be useful for the synthesis of various building blocks containing urea or carbamate.
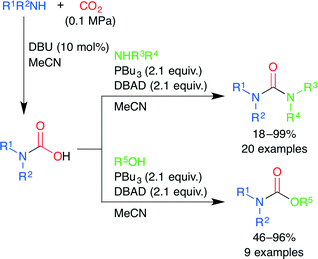 |
| | Scheme 21 The synthesis of ureas and carbamates via carbamic acids generated by the DBU-catalyzed reactions of amines and CO2. | |
5. The reduction of CO2
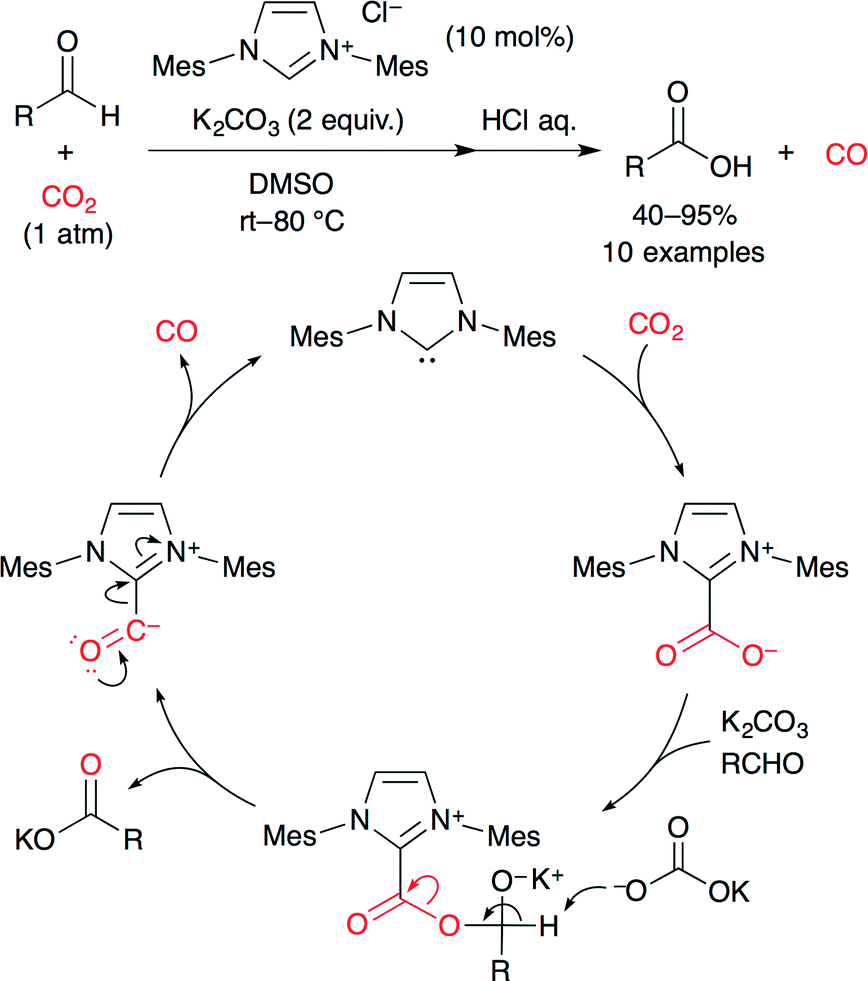
The reduction of CO2 can produce carbon monoxide (CO), formic acid, methanol, methane, and so on. Recently, catalytic reductions of CO2 have been studied actively. For example, Zhang and co-worker achieved the NHC-catalyzed reduction of CO2 to CO using aromatic aldehydes as reductants (Scheme 22).47 Originally, they had expected that carbonate and/or anhydride would be obtained from the reaction between cinnamaldehyde and imidazolium–carboxylate (NHC–CO2), but actually, CO2 behaved as an oxidant. A catalytic cycle is shown in Scheme 22. (i) NHC reacts with CO2 to form NHC–CO2. (ii) The carboxylate attacks the formyl group, generating a possible intermediate. (iii) Deprotonation results in the NHC–CO complex and a carboxylate salt via the C–O bond splitting. (iv) The NHC–CO complex releases CO with the regeneration of the NHC.
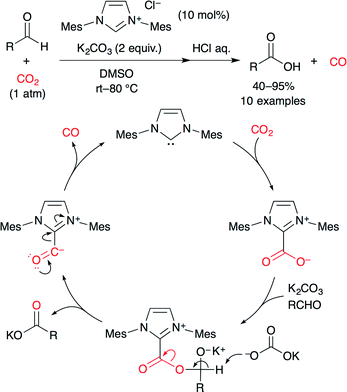 |
| | Scheme 22 The NHC-catalyzed reduction of CO2. | |
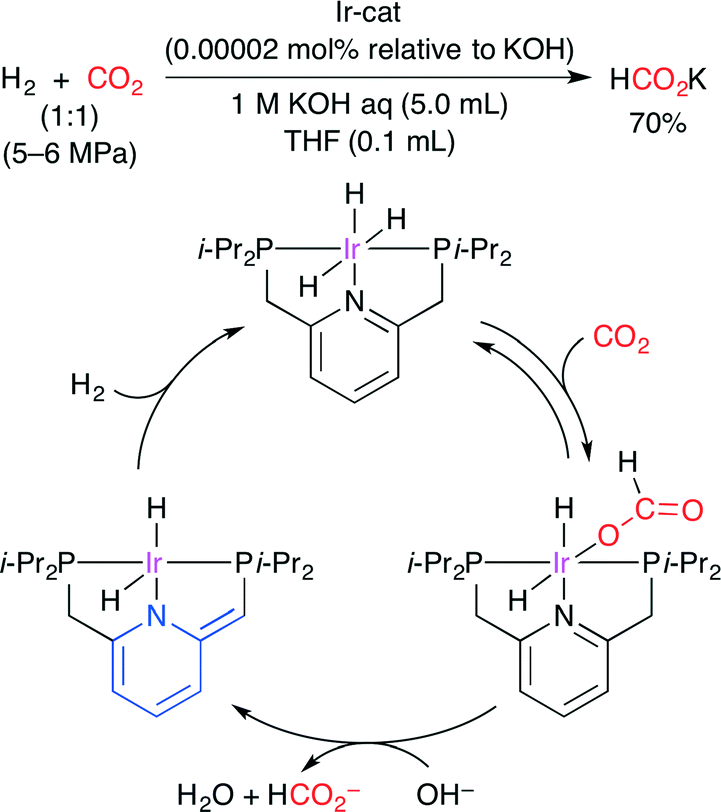
The catalytic hydrogenation of CO2 to formate was reported by Nozaki and co-workers (Scheme 23).48 By using a PNP-ligated IrIII trihydride complex, potassium formate was formed from CO2 in aqueous potassium hydroxide with an extremely high TON (3500000) at 120 °C and TOF (150000 h−1) at 200 °C. A plausible mechanism is shown in Scheme 23. (i) CO2 is inserted into the IrIII trihydride complex. (ii) Deprotonative dearomatization promotes the dissociation of the formate ligand. (iii) Hydrogenation regenerates the initial trihydride complex.
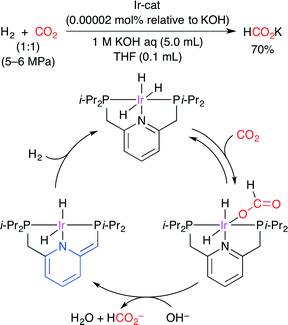 |
| | Scheme 23 The Ir-catalyzed hydrogenation of CO2. | |
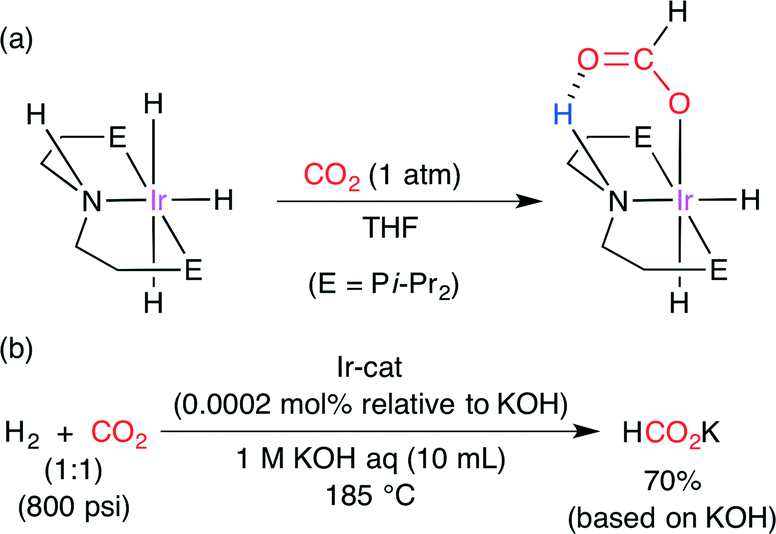
The catalytic hydrogenation of CO2 was further studied by Hazari and co-workers.49 Computational calculations on several Ir complexes indicated that CO2 insertion is trans-influenced and that hydride is the best trans-influence ligand. In addition, they found a promising IrIII trihydride complex exhibiting a deviation from prediction (Scheme 24a). It has a hydrogen-bond donor in the secondary coordination sphere, and CO2 insertion seems to be favored by hydrogen bonding, which was indeed supported by DFT calculations. The transition-state stabilization by hydrogen bonding was calculated to be −4.59 kcal mol−1. The CO2-inserted IrIII complex in Scheme 24a was characterized by NMR, IR and X-ray diffraction analysis. This CO2-inserted IrIII complex, which is air-stable and water-soluble, produced potassium formate from CO2 and H2 in aqueous KOH at 185 °C in up to 70% yield with a high TON of 348000 (Scheme 24b).
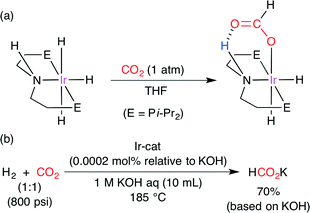 |
| | Scheme 24 (a) The insertion of CO2 into the Ir-trihydride complex. (b) The Ir-catalyzed hydrogenation of CO2. | |
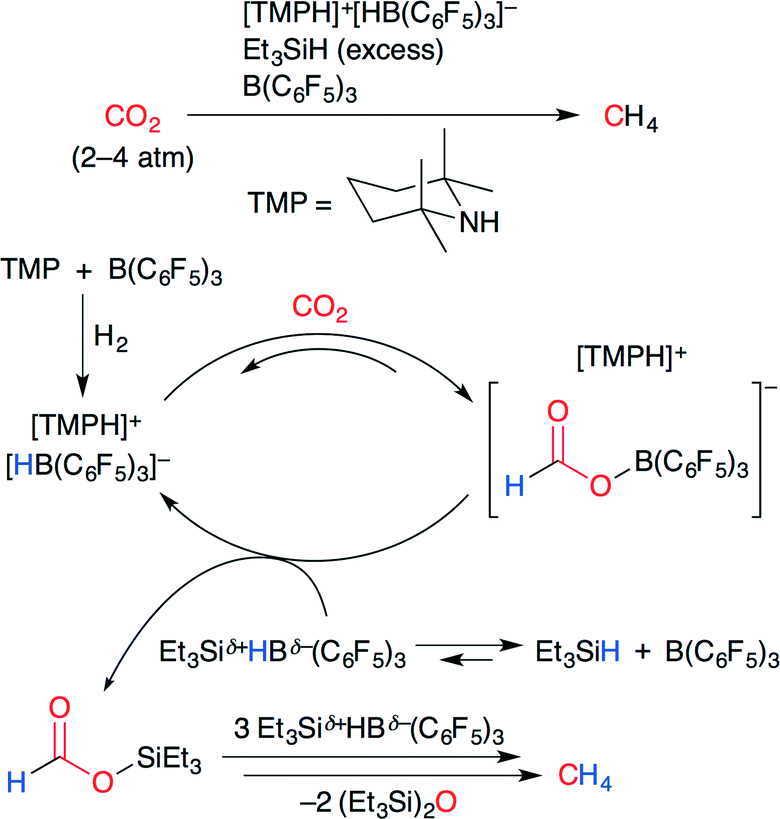
The catalytic conversion of CO2 into methane using a frustrated Lewis pair (FLP) was developed by Piers and co-workers (Scheme 25).50 A FLP consists of a bulky Lewis acid and a bulky Lewis base which cannot be quenched by each other because of steric hindrance. As shown in Scheme 25, the ammonium hydridoborate ion pair [TMPH]+[HB(C6F5)3]−, which was prepared by the treatment of a FLP (TMP/B(C6F5)3) with H2, reacted with CO2 to give a formatoborate. The subsequent silylation of formatoborate with Et3SiH and B(C6F5)3 afforded formatosilane, which was ultimately reduced to methane by further hydrosilylation.
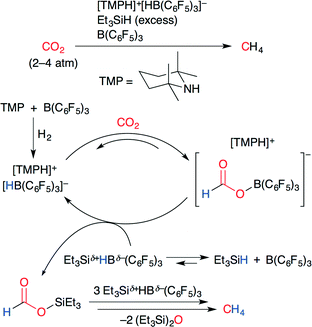 |
| | Scheme 25 The FLP-catalyzed deoxygenative hydrosilylation of CO2. | |
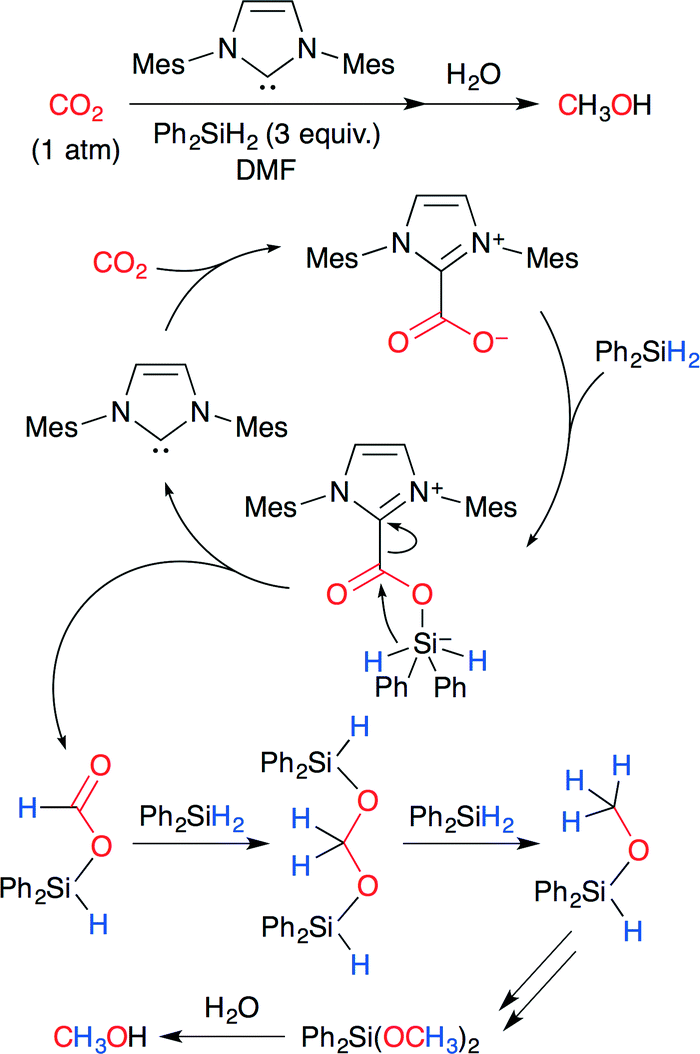
Zhang, Ying and co-worker developed the NHC-catalyzed reduction of CO2 to methanol (Scheme 26).51 It is known that NHCs behave as nucleophiles for CO2 to form imidazolium carboxylates (NHC–CO2 adducts). The reaction is proposed to proceed in the following way. The carboxylate moiety of the NHC–CO2 adduct attacks the silane center, and simultaneous hydride transfer forms formoxysilane. The formoxysilane reacts with free hydrosilanes in the presence of NHC. Methanol is produced by further reduction and workup. NHC could be reduced to 0.05 mol%, and the TON reached 1840. This is the first example of hydrosilylation of CO2 using an organocatalyst.
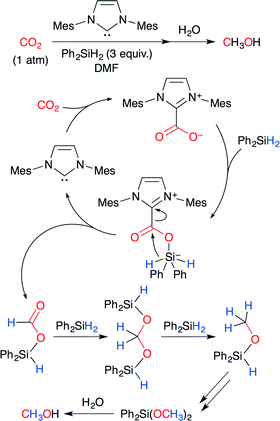 |
| | Scheme 26 The NHC-catalyzed hydrosilylation of CO2. | |
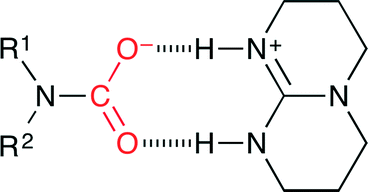
Cantat and co-workers reported organocatalytic reductive formylation of amines with CO2 (Scheme 27).52 The three-component reaction of CO2, amine and PhSiH3 in the presence of TBD (5 mol%) afforded formamide. As a result of the screening of base catalysts, TBD showed the best performance, which is due to the formation of the carbamate salt (Fig. 26). The hydride transfer from silicon to carbon is facilitated by the hydrogen bonds between the carbamate anion and the TBDH+ cation. Secondary amines were more reactive than primary amines, and aliphatic amines were more reactive than aromatic amines, which indicates that the activity was mainly governed by the basicity of the amines.
 |
| | Scheme 27 The TBD-catalyzed reductive formylation of amines with CO2. | |
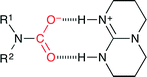 |
| | Fig. 26 The formation of the carbamate salt with TBD. | |
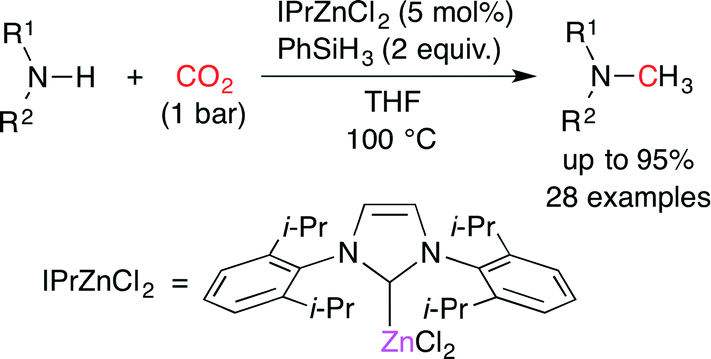
Cantat and co-workers found that ZnII salts are effective for promoting the reactions of amines with CO2 and hydrosilanes to give N-methylamines. They screened many combinations of ZnII salts and neutral ligands and found that IPrZnCl2 was the best catalyst (Scheme 28).53 PhSiH3 and THF were the best hydride source and solvent, respectively. Various primary and secondary amines were methylated. They proposed a possible reaction pathway. The Zn-catalyzed reductive formylation of amine with CO2 and PhSiH3 would give formamide, which would be further reduced to N-methylamine by the Zn-catalyzed hydrosilylation.
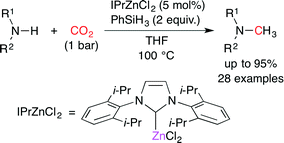 |
| | Scheme 28 The Zn-catalyzed methylation of amines with CO2. | |
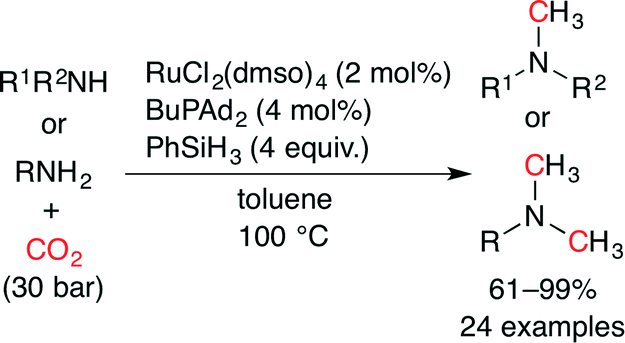
Beller and co-workers independently reported the methylation of amines with CO2 using a Ru catalyst (Scheme 29).54 As a result of the screening of 15 metal precursors including Ru, Rh, Cu and Fe complexes and 12 phosphine and nitrogen ligands, a combination of RuCl2(dmso)4 and BuPAd2 showed the highest catalytic performance. A variety of aromatic and aliphatic, secondary and primary amines were successfully transformed into the desired tertiary amines in good to excellent yields. Notably, functional groups such as ester and nitrile were tolerated under the reaction conditions.
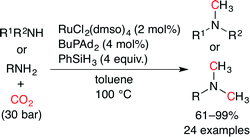 |
| | Scheme 29 The Ru-catalyzed methylation of amines with CO2. | |
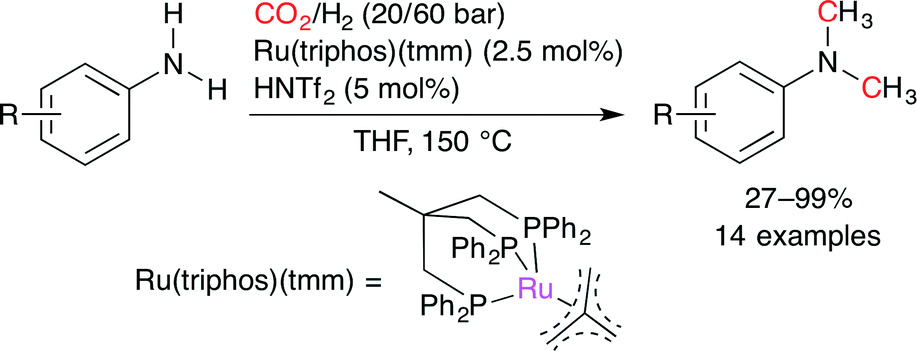
Klankermayer and co-workers disclosed the Ru-catalyzed methylation of primary and secondary aromatic amines with CO2 and H2 (Scheme 30).55 A combination of the triphos-based Ru complex, Ru(triphos)(tmm), with an acid such as trifluoromethanesulfonylimide (HNTf2), methanesulfonic acid (MSA), and p-toluenesulfonic acid (p-TsOH) afforded methylated products in excellent yields. The acid co-catalyst was essential for the reaction.
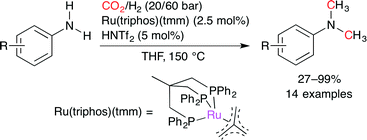 |
| | Scheme 30 The Ru-catalyzed methylation of aromatic amines using CO2 and H2. | |
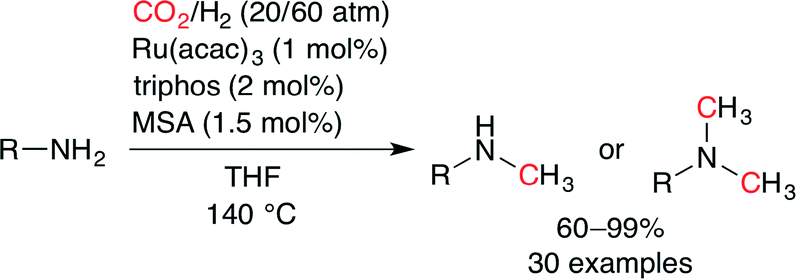
Beller and co-workers also independently succeeded in the Ru-catalyzed methylation of aromatic and aliphatic amines with CO2 and H2 (Scheme 31).56 By using a ruthenium(III) precursor and triphos, the desired methylated amines were obtained in good to excellent yields. Furthermore, selective mono-methylation of primary amines was achieved by tuning the reaction time and catalyst loading. Although aliphatic amines showed a poor reactivity under the standard conditions, the replacement of MSA with LiCl (7.5 mol%) gave satisfactory results. Several control experiments indicated that methylation of amines proceeded predominantly via formamide intermediates.
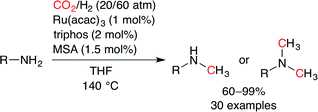 |
| | Scheme 31 A general catalytic methylation of amines. | |
6. Conclusions and outlook
The efficient conversion of CO2 into useful compounds is a longstanding dream of synthetic chemists. Catalysts play a key role in expanding the synthetic potential. Indeed, new reactions and new catalysts have been actively developed over the past several years. In this perspective, we provide an overview of the recent progress. Although the efficiency of CO2 fixation is far below that of enzymatic reactions in photosynthesis, synthetic methods are rich in variation. The coupling reactions of CO2 with epoxides to give cyclic carbonates or polycarbonates are highly atom-efficient, producing no by-products. Some of the catalysts recently developed showed extremely high TON and TOF or a high reusability without the loss of catalytic activity. Catalytic carboxylation reactions involving C–C bond formation are very useful and powerful, and synthetic variation is increasing. Not only C–X bonds but also C–H bonds can be activated by transition metals. The number of new methods for the reduction of CO2 is also increasing rapidly. CO2 can be successfully reduced into CO, formic acid, methanol, methane, formamide, or methylamine using hydrosilane or H2 as a reductant. Organocatalysts as well as metal catalysts are effective for these transformations. The electrochemical or photochemical reduction of CO2 has also been studied, although they are out of range of this perspective.57 Further developments and breakthroughs toward the creation of a green and sustainable society are expected.
Notes and references
- Reviews:
(a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed;
(b) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed;
(c) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC;
(d) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC;
(e) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC;
(f) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS PubMed;
(g) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed;
(h) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC;
(i) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC;
(j) I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS PubMed;
(k) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- W. J. Kruper and D. V. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- R. L. Paddock, Y. Hiyama, J. M. McKay and S. T. Nguyen, Tetrahedron Lett., 2004, 45, 2023–2026 CrossRef CAS PubMed.
- L. Jin, H. Jing, T. Chang, X. Bu, L. Wang and Z. Liu, J. Mol. Catal. A: Chem., 2007, 261, 262–266 CrossRef CAS PubMed.
- D. Bai, Q. Wang, Y. Song, B. Li and H. Jing, Catal. Commun., 2011, 12, 684–688 CrossRef CAS PubMed.
- F. Ahmadi, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and A. R. Khosropour, Inorg. Chem. Commun., 2011, 14, 1489–1493 CrossRef CAS PubMed.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC.
- T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485–2492 RSC.
- M. Ulusoy, E. Çetinkaya and B. Çetinkaya, Appl. Organomet. Chem., 2009, 23, 68–74 CrossRef CAS.
- J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577–2579 RSC.
- M. V. Escárcega-Bobadilla, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem.–Eur. J., 2013, 19, 2641–2648 CrossRef PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS PubMed.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS PubMed.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023–1028 RSC.
- Z.-Z. Yang, Y.-N. Zhao, L.-N. He, J. Gao and Z.-S. Yin, Green Chem., 2012, 14, 519–527 RSC.
- E. A. Prasetyanto, M. B. Ansari, B.-H. Min and S.-E. Park, Catal. Today, 2010, 158, 252–257 CrossRef CAS PubMed.
- Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS PubMed.
- H. Zhou, Y.-M. Wang, W.-Z. Zhang, J.-P. Qu and X.-B. Lu, Green Chem., 2011, 13, 644–650 RSC.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191–15199 CrossRef CAS PubMed.
- H. Li and Y. Niu, Appl. Organomet. Chem., 2011, 25, 424–428 CrossRef CAS.
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367–14369 CrossRef CAS PubMed.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- J. G. Kim, C. D. Cowman, A. M. LaPointe, U. Wiesner and G. W. Coates, Macromolecules, 2011, 44, 1110–1113 CrossRef CAS.
- T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106–9109 CrossRef CAS PubMed.
- A. Correa and R. Martín, J. Am. Chem. Soc., 2009, 131, 15974–15975 CrossRef CAS PubMed.
- T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221–1224 CrossRef PubMed.
- H. Ohmiya, M. Tanabe and M. Sawamura, Org. Lett., 2011, 13, 1086–1088 CrossRef CAS PubMed.
- M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900–11903 CrossRef CAS PubMed.
- W.-Z. Zhang, W.-J. Li, X. Zhang, H. Zhou and X.-B. Lu, Org. Lett., 2010, 12, 4748–4751 CrossRef CAS PubMed.
- H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251–1253 CrossRef CAS PubMed.
- K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954–10957 CrossRef CAS PubMed.
- T. Mita, K. Michigami and Y. Sato, Org. Lett., 2012, 14, 3462–3465 CrossRef CAS PubMed.
- Y. Dai, X. Feng, B. Wang, R. He and M. Bao, J. Organomet. Chem., 2012, 696, 4309–4314 CrossRef CAS PubMed.
- D. P. Hruszkewycz, J. Wu, N. Hazari and C. D. Incarvito, J. Am. Chem. Soc., 2011, 133, 3280–3283 CrossRef CAS PubMed.
- Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194–4197 CrossRef CAS PubMed.
- Y. Kayaki, N. Mori and T. Ikariya, Tetrahedron Lett., 2009, 50, 6491–6493 CrossRef CAS PubMed.
- R. A. Watile, D. B. Bagal, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, J. Mol. Catal. A: Chem., 2011, 351, 196–203 CrossRef CAS PubMed.
- T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848–851 CrossRef CAS PubMed.
- T. Ishida, S. Kikuchi and T. Yamada, Org. Lett., 2013, 15, 3710–3713 CrossRef CAS PubMed.
- S. L. Peterson, S. M. Stucka and C. J. Dinsmore, Org. Lett., 2010, 12, 1340–1343 CrossRef CAS PubMed.
- L. Gu and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 914–915 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef PubMed.
- O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- For the electrochemical or photochemical reduction of CO2, see for example: J. Mao, K. Li and T. Peng, Catal. Sci. Technol., 2013, 3, 2481–2498 CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence