 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the surface modifications following the mechanochemical preparation of a Ag/Al2O3 selective catalytic reduction catalyst
Kathryn
Ralphs
a,
Carmine
D'Agostino
b,
Robbie
Burch
a,
Sarayute
Chansai
a,
Lynn F.
Gladden
*b,
Christopher
Hardacre
*a,
Stuart L.
James
*a,
Jonathan
Mitchell
b and
Sarah F. R.
Taylor
a
aCentre for the Theory and Application of Catalysis, CenTACat, School of Chemistry and Chemical Engineering, Queen's University, Belfast, BT9 5AG, UK. E-mail: c.hardacre@qub.ac.uk; s.james@qub.ac.uk
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Pembroke Street, Cambridge CB2 3RA, UK. E-mail: lfg1@cam.ac.uk
First published on 3rd December 2013
Abstract
The surface modification of a mechanochemically prepared Ag/Al2O3 catalyst compared with catalysts prepared by standard wet impregnated methods has been probed using two-dimensional T1–T2 NMR correlations, H2O temperature programmed desorption (TPD) and DRIFTS. The catalysts were examined for the selective catalytic reduction of NOx using n-octane in the presence and absence of H2. Higher activities were observed for the ball milled catalysts irrespective of whether H2 was added. This higher activity is thought to be related to the increased affinity of the catalyst surface towards the hydrocarbon relative to water, following mechanochemical preparation, resulting in higher concentrations of the hydrocarbon and lower concentrations of water at the surface. DRIFTS experiments demonstrated that surface isocyanate was formed significantly quicker and had a higher surface concentration in the case of the ball milled catalyst which has been correlated with the stronger interaction of the n-octane with the surface. This increased interaction may also be the cause of the reduced activation barrier measured for this catalyst compared with the wet impregnated system. The decreased interaction of water with the surface on ball milling is thought to reduce the effect of site blocking whilst still providing a sufficiently high surface concentration of water to enable effective hydrolysis of the isocyanate to form ammonia and, thereafter, N2.
1. Introduction
Recently the trend in lean-burn NOx reduction for mobile applications has been towards ammonia Selective Catalytic Reduction (SCR), or NOx Storage and Reduction (NSR), or a combination of SCR and NSR. Nevertheless, selective catalytic reduction using an on-board hydrocarbon fuel, Hydrocarbon Selective Catalytic Reduction (HC-SCR), would offer significant benefits if catalysts with sufficient low temperature activity could be developed. Silver-based catalysts appear to be the most promising but are relatively inactive at temperatures below about 350 °C.1–18 Various modifications to the standard wet impregnation preparation technique have been investigated with a view to improving the activity of alumina-supported Ag catalysts. For example, co-precipitation,10 sol–gel processes11–15 and hydrolysis16–18 methods have been reported but, to date, the highest activity catalysts prepared by these methods are produced via wet impregnation from aqueous solutions using AgNO3 as the Ag precursor. In addition, the effect of the addition of hydrogen has been extensively studied.19–29 A strong increase in the activity of the catalyst was observed on addition of hydrogen, for example Burch and co-workers19,20 observed a decrease in the light off temperature for a wet impregnated Ag/Al2O3 catalyst from 410 to 195 °C.Mechanochemical treatment of solid materials by ball milling and related techniques has a long history in the context of inorganic materials synthesis,30–34 and has recently been investigated for organic35 and metal–organic synthesis.36 Its use in preparing heterogeneous catalysts has recently been reviewed and showed significant increases in activity could be achieved for a wide range of reactions including deNOx, CO oxidation and VOC removal.37 Recently, we showed38 that Ag/Al2O3 catalysts could be prepared simply by ball milling alumina and a Ag precursor, for example Ag2O or Ag metal. These materials were highly active for the HC-SCR of NOx even in the low temperature range, showing light-off temperatures (i.e., temperature at which the conversion is 50%) below 300 °C, with the catalyst prepared using silver oxide resulting in a light-off temperature of 240 °C. The increased activity was not correlated to an increase in the BET surface. In addition, transmission electron microscopy and UV-Vis studies of the impregnated and ball milled catalysts revealed a similar particle size distribution of the silver.38
In this paper, the mechanochemically prepared Ag/Al2O3 catalyst was examined for the HC-SCR of NOx in the presence of hydrogen to assess whether the activity could be further improved. In addition, in situ diffuse reflectance infra-red spectroscopy (DRIFTS) and NMR have been used to compare the changes in the surface interactions due to the mechanochemical processing. T1–T2 NMR two-dimensional correlations have been used39 to investigate surface interactions of two important species involved in the SCR reaction, n-octane and water, within Al2O3 supports and Ag/Al2O3 catalysts prepared by standard wet impregnation and by the ball milling method. NMR relaxometry has been widely used to characterise surface interactions in porous materials40–43 and has been very recently applied to heterogeneous catalysts44,45 and molecular dynamics.46 In particular, the use of the T1/T2 ratio has the advantage of being independent of surface-to-volume ratio of the pores.47 Therefore, such a ratio will only be dependent on the surface properties and not upon the geometrical characteristics of the pore structure. Hence, it allows us to compare materials with different pore structures and gives us a direct correlation with the surface interactions and how the ball milling process affects these. This is because the interactions are independent of surface-to-volume ratio. In addition, this makes the method also more robust to effects deriving from a different “filling” of the pore volume by different liquids. However, we anticipate that in the present work, the pores are small (order of few to tens of nanometers) in all samples and so the observed relaxation times are surface-dominated in all cases. If the volume of liquid dropped below monolayer coverage, then we might see a change due to different volume within the pore space, but this is not relevant to the saturated systems used here.
The aim of this work is to gain a better understanding how the ball milling process can improve the SCR activity and can change the NMR relaxation properties of the n-octane and water chemical species within the catalysts, from which it is possible to infer the strength of surface interaction with the catalytic surface and hence gain further insights into the understanding of the catalytic performance.
2. Experimental
2.1 Catalyst preparation
Ag/Al2O3 catalyst, denoted as Ag2O_BM, was prepared by ball milling using Ag2O as the Ag precursor. The Ag2O precursor (0.0438 g) (99.99%, Sigma Aldrich) and 2.00 g of as received γ-Al2O3 (Grace-Davison, BET surface area = 261 m2 g−1) were well-mixed using a spatula before being placed into a 500 cm3 sintered aluminium oxide grinding jar with seven 10 mm diameter sintered alumina grinding balls. Milling was performed in a Retsch PM100 Planetary Ball Mill at a rotation speed of 150 rpm for 1 h. A standard Ag/Al2O3 catalyst, denoted as Ag_STD, was prepared by wet impregnation using 0.022 M AgNO3 solution to obtain a silver loading of 2 wt% on the same γ-Al2O3 as used for the ball milled sample. The catalyst was then dried in an oven overnight at 100 °C. The resulting Ag2O_BM and Ag_STD catalysts were calcined at 650 °C for 2 h. To prepare the ball milled alumina blank, the same procedure was used without the silver oxide present. The Ag content of the catalysts was determined by the inductively coupled plasma method and determined to be approximately 2 wt% Ag. The BET specific surface areas of the catalysts were 235 m2 g−1 and 258 m2 g−1 for Ag2O_BM and Ag_STD, respectively. As reported previously, for both catalysts under reaction conditions, the silver was found to be in the form of both isolated Ag+ ions and Agnδ+ (n ≤ 8) clusters using diffuse reflectance UV-vis spectroscopy.382.2 Catalytic activity tests
Typically, the catalytic activity tests over Ag/Al2O3 catalysts were carried out in a fixed-bed flow reactor system, consisting of a quartz reactor tube. The catalysts were held in place between plugs of quartz wool and a K-type thermocouple was placed in the centre of the catalyst bed. Each of the gases in the feed system was controlled individually by mass flow controllers, while n-octane and water vapour were introduced to the system by means of separate saturators with Ar as a carrier gas. The n-C8H18 saturator was placed in an ice/water bath and the H2O saturator temperature was controlled using a thermostatic bath. All the lines following the water saturator were trace heated to prevent condensation.The feed gas stream consisted of 720 ppm NO; 4340 ppm n-C8H18 (as C1); 4.3% O2, 7.2% H2O (when added); 7.2% CO2; 0.72% H2 and Ar balance was introduced to the reactor which was heated from 150 to 600 °C and then back down to 150 °C stepwise at 50 °C intervals dwelling at each temperature for 40 min in order to obtain steady state conditions. The total gas flow rate was 276 cm3 min−1 over 276 mg of catalyst which was sieved to obtain particle size of 250–450 μm. The space velocity for all catalytic tests was 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm3 g−1 h−1 (calculated using the total gas flow rate divided by the amount of the catalyst used in the activity test). The inlet and outlet NOx concentrations were determined by a Signal 4000VM series chemiluminescence detector. The oxidation of n-octane was measured online using a Bruker Tensor 27 IR spectrometer, fitted with a gas cell of volume 190 cm3.
000 cm3 g−1 h−1 (calculated using the total gas flow rate divided by the amount of the catalyst used in the activity test). The inlet and outlet NOx concentrations were determined by a Signal 4000VM series chemiluminescence detector. The oxidation of n-octane was measured online using a Bruker Tensor 27 IR spectrometer, fitted with a gas cell of volume 190 cm3.
All the activity data was measured during the decreasing temperature ramp. Using this method, the activity remained constant once the desired temperature had been reached and the conversions were calculated from an average of outlet NOx readings at each temperature. In contrast, measurements made on ramping the temperature up showed that steady state was reached after 15–20 min although it should be noted that the values between the two ramps once steady state had been reached only showed small variations. The time to reach steady state on increasing the temperature was associated with a dip in the NOx concentration once the temperature had been reached; however, this is not due to reaction but adsorption of the NOx.
2.3 NMR relaxometry
Four different samples were used for the experiments namely as received γ-Al2O3 (Al2O3_STD), ball milled γ-Al2O3 (Al2O3_BM), wet impregnated 2% Ag/γ-Al2O3 (Ag_STD) and ball milled 2% Ag/γ-Al2O3 (Ag2O_BM). NMR experiments were performed in a Bruker DMX 300 operating at a 1H frequency of 300.13 MHz. NMR relaxation experiments were performed using a T1–T2 pulse sequence, which comprises a saturation recovery measurement to encode T1 (using a comb of 90° pulses) followed by a Carr–Purcell Meiboom–Gill (CPMG) echo train of 180° pulses to encode T2. The sequence is schematically shown in Fig. 1. The T1 recovery interval, tdelay, was varied logarithmically between 1 ms and 5 s in 32 steps. The echo spacing between the 180° pulses of the CPMG was set to 250 μs. More details of the pulse sequence used are reported elsewhere.39,41,48 | ||
| Fig. 1 T 1–T2 NMR pulse sequence. The thin and thick vertical bars represent 90° and 180° radiofrequency (RF) pulses, respectively. T1 relaxation is encoded in the variable time tdelay. T2 relaxation is encoded in the train of n 180° pulses. A single data point is acquired at the centre of each echo time, τ. The cross-hatched patterns (HG) represent homospoil magnetic field gradients. | ||
Samples were prepared by soaking the catalytic material in n-octane or water for at least 24 h. The wet catalyst was then removed from the liquid, placed on a pre-soaked filter paper (with the liquid n-octane or water) in order to remove excess external liquid and finally placed into the NMR tube. To ensure a saturated atmosphere in the NMR tube, hence minimising errors due to evaporation of volatile liquids, a small amount of pure liquid was placed onto absorbed filter paper, which was then placed in the cap of the NMR tube. All the NMR measurements were performed at atmospheric pressure and 20 °C using 5 mm NMR glass tubes.
2.4 H2O temperature-programmed desorption (TPD)
The H2O-TPD measurements were performed to investigate the adsorption properties of water on the supports (Al2O3_STD and Al2O3_BM) and the Ag catalysts (Ag_STD and Ag2O_BM). In each of the experiments, approximately 50 mg of fresh sample was used and pre-treated in Ar at a flow rate of 100 cm3 min−1 for 30 min at 300 °C. The catalyst was then cooled down to 50 °C where a gas mixture of 0.5% H2O–1% Kr/Ar with the same flow rate was introduced for one hour. Water vapour was introduced to the system by means of a saturator with Ar flow as a carrier gas. The H2O saturator temperature was carefully controlled at 10 °C using a Grant™ GD120 thermostatic bath. Following H2O adsorption, the catalyst was purged by 1% Kr/Ar flow at the total rate of 100 cm3 min−1 for 30 min to remove any weakly bound H2O. Finally, the temperature was ramped at a rate of 10 °C min−1 up to 700 °C under 1% Kr/Ar flow. Kr was used as an internal standard for quantification. The desorbed H2O was monitored by a Hiden quadrupole mass spectrometer.2.5 DRIFTS analysis
In situ DRIFTS measurements were performed using a Bruker Vertex 70 FTIR spectrometer equipped with a liquid N2-cooled detector. In a typical experiment, 25 mg of catalyst was placed in a ceramic crucible in the DRIFTS cell.Prior to the experiments, the catalyst was pre-treated by heating in 5% O2/Ar with a total flow rate of 100 cm3 min−1 up to 300 °C for 1 h and then cooled down in flowing Ar to 250 °C. The IR spectrum of the Ag/Al2O3 catalyst at 250 °C under flowing Ar was taken as a background. Two 4-way VICI valves were installed to allow us to switch between two gas mixtures. The DRIFTS was used to examine the relative surface concentrations of isocyanate on each catalyst. Therefore, each catalyst was exposed to C8H18 + O2 gas feed at 250 °C for 30 min and followed by a flowing NO + O2 feed for 10 min. In all cases, the in situ DRIFTS spectra were recorded with a resolution of 4 cm−1 and with an accumulation of 16 scans every 10 s. The DRIFTS spectra were analyzed by the OPUS software. The concentrations of the reactants used were 720 ppm NO + 4.3% O2 and 4340 ppm (as C1) n-C8H18 + 4.3% O2 and Ar balance. The total flow rate was 100 cm3 min−1.
3. Results and discussion
Fig. 2 shows a comparison of the NOx conversion of the most active ball-milled Ag catalyst (Ag2O_BM) with that of a conventional wet impregnated Ag/Al2O3 catalyst (Ag_STD) in the presence and absence of H2. For the purpose of comparison, we take as the reference point the light-off temperature which we define as the temperature for 50% NOx conversion. Fig. 2 shows that the ball-milled catalyst has a light-off temperature of only about 240 °C whereas the wet impregnated catalyst has a light-off temperature of about 370 °C in the absence of hydrogen, in agreement with previously reported results.38 In the presence of 0.72% hydrogen, the light-off temperature was reduced from 240 °C (without H2) to 165 °C (with H2) for Ag2O_BM and from 370 °C (without H2) to 180 °C (with H2) for Ag_STD. In addition, 100% NOx conversion was obtained at 220 °C and 298 °C for the ball milled and wet impregnated catalysts, respectively, compared with ~300 °C for the ball milled catalyst in the absence of hydrogen and 80% conversion at ~450 °C for the Ag_STD catalyst. Although a wider range of operational temperatures is observed for Ag_STD than Ag2O_BM, the activity of the ball milled catalyst is significant as these results show the lowest temperature activity for n-octane-SCR reported up to date, albeit in the presence of hydrogen.19,20 The effect of hydrogen is consistent with previous studies on HC-SCR over silver catalysts.8,21-23 It is now generally accepted that the role of the hydrogen is to modify the chemical reactions rather than to significantly change the nature of the silver under reaction conditions. | ||
| Fig. 2 NOx conversion as a function of catalyst temperature for the SCR of NOx reaction with n-octane over Ag_STD (●) and Ag2O_BM (♦) catalysts in the absence (solid) and the presence (dashed) of H2. Feed stream: 720 ppm NO, 4340 ppm (as C1) n-C8H18, 4.3% O2, 7.2% CO2, 7.2% H2O, 0.72% H2 and Ar balance. Space velocity: 60000 cm3 g−1 h−1. | ||
Moreover, the SCR activity with n-octane over Ag catalysts in the absence of water was investigated and presented in Fig. 3. For both Ag_STD and Ag2O_BM catalysts, it is clearly seen that the conversion of NOx was suppressed when there was no water added into the SCR feed. This is consistent with previous work by Shimizu et al.17 This has been attributed to the possibility that, with water in the feed, the deposition of carbonaceous species such as carboxylates and carbonates is reduced.17 However, it is also noted that water is necessary for the hydrolysis of NCO species formed during the reaction to form nitrogen. These have been reported to be important intermediates in the SCR reaction over Ag catalysts and thus the presence of water is critical in the reaction mechanism.8,23
 | ||
| Fig. 3 NOx conversion as a function of catalyst temperature for the SCR of NOx reaction with n-octane over Ag_STD (●) and Ag2O_BM (♦) catalysts in the presence (solid) and the absence (dashed) of H2O. Feed stream: 720 ppm NO, 4340 ppm (as C1) n-C8H18, 4.3% O2, 7.2% CO2, 7.2% H2O (when added), and Ar balance. Space velocity: 60000 cm3 g−1 h−1. | ||
It is well known that both the activation of the hydrocarbon and presence of water are critical for the HC-SCR of NOx over Ag based catalysts,1,3,8,17,49,50 therefore, due to the fact that there is a significant difference in NOx conversion profile and operational temperature window for the Ag2O_BM and Ag_STD catalysts, the interactions of n-octane and water were examined on each catalyst using two-dimensional T1–T2 NMR.
The two-dimensional T1–T2 NMR correlation maps for the pure Al2O3 samples are shown in Fig. 4 in the presence of water or n-octane. In all cases, a single peak is observed, with water giving rise to broader peaks due to the greater uncertainty involved in fitting the exponential decay function for short T2 relaxation times. The position of the dashed diagonal in each plot is determined from the maximum peak intensity and corresponds to the T1/T2 ratio. This ratio is considered to be an indicator of the strength of interaction between the liquid and the solid surface. An increase in the magnitude of T1/T2 indicates an increase in the strength of the surface interaction of a given molecular species with the surface.41 Water in Al2O3_STD exhibits T1/T2 = 63, whereas in Al2O3_BM it gives T1/T2 = 57. The values for n-octane are T1/T2 = 31 in Al2O3_STD and T1/T2 = 38 in Al2O3_BM. The significant difference between the T1/T2 ratios for water and those for n-octane clearly indicate that water has a stronger interaction with the alumina surface compared with n-octane. This result is expected given the polarity and ability of water to form hydrogen bonds with the surface hydroxyl groups of the Al2O3. Comparing the two alumina samples, small but significant changes are observed for each liquid. Water has a lower strength of interaction with Al2O3_BM compared with Al2O3_STD; conversely, n-octane has a higher strength of interaction with Al2O3_BM. Although these differences in the T1/T2 ratios are not large, they are reproducible and greater than the error associated with determining the individual T1/T2 ratios from the correlation plots (see Fig. 6, error bars). Therefore, we infer that the ball milling process changes the properties, specifically the affinity of the Al2O3 surface towards water and n-octane.
 | ||
| Fig. 4 T 1/T2 of: (a) water in Al2O3_STD; (b) water in Al2O3_BM; (c) n-octane in Al2O3_STD; (d) n-octane in Al2O3_BM. | ||
The T1–T2 correlations for water or n-octane in the Ag/Al2O3 catalysts are shown in Fig. 5. The ball milling decreases the T1/T2 of water within the catalyst: water in Ag_STD gives T1/T2 = 47, and in Ag2O_BM gives T1/T2 = 35. Conversely, the ratio for n-octane increases with ball milling from T1/T2 = 32 in Ag_STD to T1/T2 = 38 in Ag2O_BM. This trend in T1/T2 observed for the catalysts is similar to that observed for the Al2O3 supports.
 | ||
| Fig. 5 T 1/T2 of: (a) water in Ag_STD; (b) water in Ag2O_BM; (c) n-octane in Ag_STD; (d) n-octane in Ag2O_BM. | ||
The T1/T2 ratios are summarised in Fig. 6. The difference in the T1/T2 values between water and n-octane for the different samples can be seen in terms of competitive interaction strength. In the context of the catalytic data presented here it is the observation that the reduction in the interaction strength of water compared with n-octane with the ball milled sample compared with the conventionally prepared sample that is of particular significance.
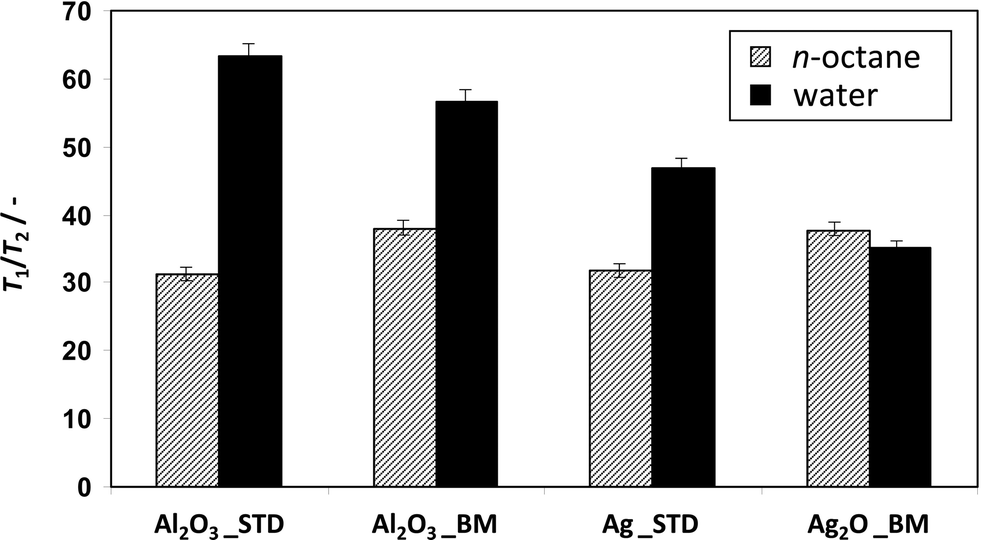 | ||
| Fig. 6 Summary of T1/T2 of water and n-octane in Al2O3 and Ag/Al2O3 samples. Error bars are also reported. | ||
It is noted that paramagnetic species might affect both T1 and T2 relaxation times. However, these oxides are known to be free of paramagnetic species. Even in the case of traces of paramagnetic impurities, these would be the same in all samples and, therefore, would affect the relaxation times in a similar manner for both n-octane and water. Hence, this cannot be the determining factor in the differences in T1/T2 observed. Indeed, the ball mill used in this work has sintered alumina grinding balls, which excludes the possibility of contamination from introducing, for example, paramagnetic iron particles during the ball milling process, which could, for instance, be present if steel balls were used in the milling process.
In addition to NMR study, the interaction of water on the Al2O3 supports and Ag catalysts was probed using temperature programmed desorption. The concentration profiles of desorbed H2O as a function of catalyst temperature are illustrated in Fig. 7. Fig. 7a shows that there is a difference in the desorption profiles and the amount of desorbed H2O between Al2O3_STD and Al2O3_BM. The concentration of H2O on Al2O3_STD peaks at about 135 °C whereas Al2O3_BM sample shows two desorption features at 112 and 215 °C. The calculated amount of desorbed H2O is 457.0 and 343.7 μmol gcat−1 for Al2O3_STD and Al2O3_BM, respectively. For the Ag_STD and Ag2O_BM catalysts, similar profiles for the desorbed H2O were observed, in which water starts to desorb at 55 °C and peaks at about 130 °C before gradually declining as the temperature was raised up to 700 °C. The amount of H2O desorbed is found to be 653.3 and 443.5 μmol gcat−1 for Ag_STD and Ag2O_BM samples, respectively. As shown in Fig. 7, the amount of water desorbed on the ball-milled support and ball-milled Ag catalyst is significantly lower than desorbed from the received support and wet-impregnated Ag catalyst. It should be noted that, whilst a small drop in the BET surface area is found for the ball milled catalysts compared with the as received alumina and the wet impregnated prepared silver catalyst of ~10%38 and will contribute to the lower amounts of water desorbed, the extent of the decrease found in the TPD is much higher. Therefore, a smaller number of adsorption sites for the water is found for the ball-milled samples compared with the standard catalyst/support.
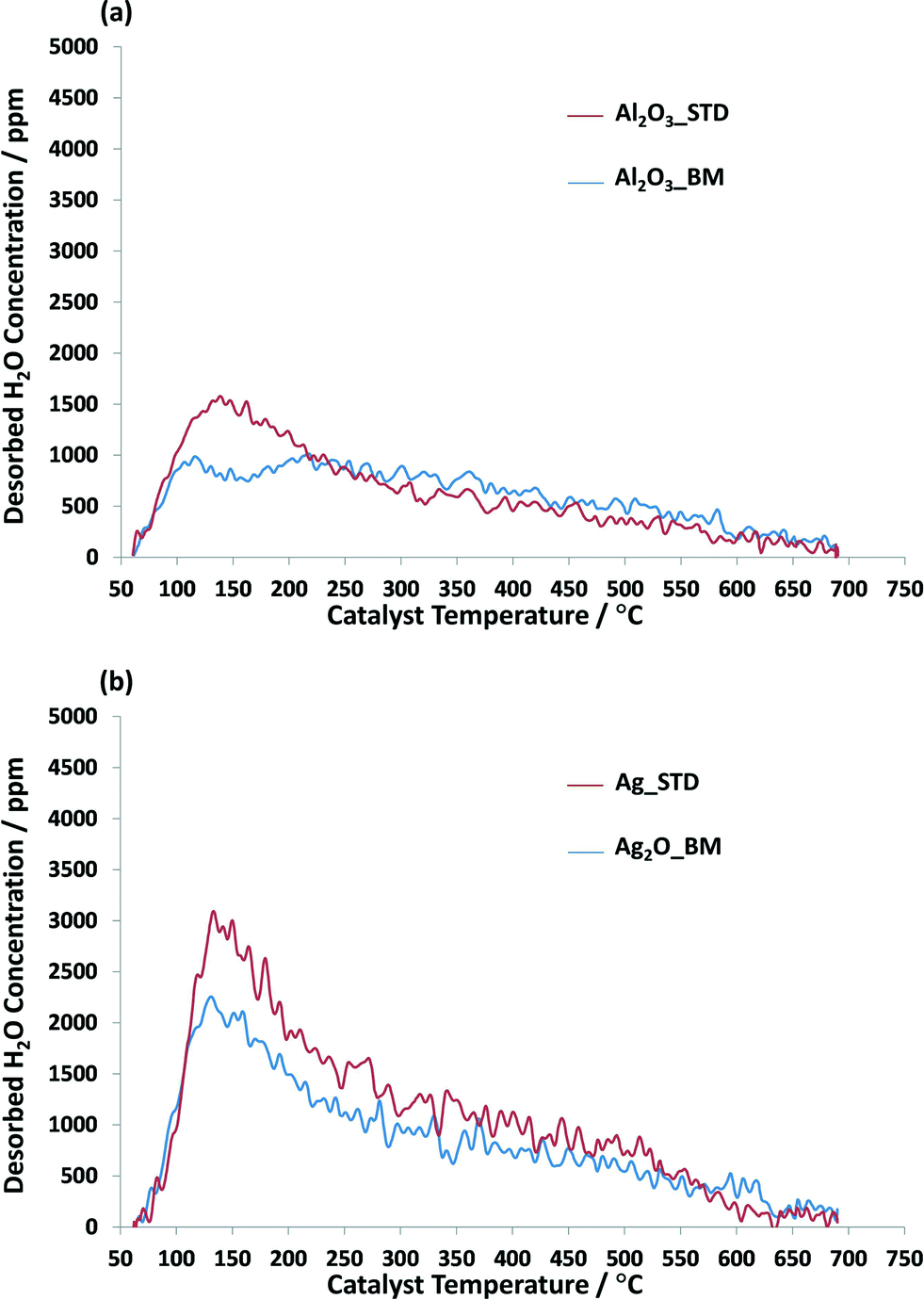 | ||
| Fig. 7 TPD profiles of H2O desorption on (a) Al2O3 supports and (b) Ag/Al2O3 catalysts. | ||
The ball milling is, therefore, inducing surface changes of the samples as observed by both TPD and NMR measurements. It is known that the absolute relaxation times depend on the density of adsorption sites, which is manifested as a weak dependence in T1/T2.51 However, this sensitivity of T1/T2 to adsorption site density can only be detected when the surface chemistry of the system being studied is the same. For example, in previous work on water in porous plasters, small changes in T1/T2 were related to variations in the number of surface sites; this interpretation was appropriate as the type of surface adsorption sites did not change between samples.40 In this work, the TPD results presented suggests that the ball milling process is decreasing the number of adsorption sites for water molecules, hence in the discussion made above, this may be expected to affect the T1/T2 values for water, which are lower on the ball milled samples. However, a quantitative analysis is not sufficient to explain the NMR data. Indeed, NMR relaxation results also show that for the ball milled samples there is a consistent increase in the T1/T2 values for the hydrocarbon, which implies also qualitative changes of adsorption sites. This suggests that the ball milling changes the properties of some of the adsorption sites, which results in a lower affinity for water and higher affinity for the hydrocarbon. In summary, there is a quantitative but also a qualitative change of the surface due to ball milling, which increases the affinity of the surface towards the hydrocarbon relative to water. As shown below, the DRIFTS results and the activation barrier for n-octane oxidation are also in line with such findings.
It has to be highlighted that the details of such changes at this stage are not yet fully elucidated and more work is required to obtain more detailed insights. However, the current results show evidence that such changes are occurring, which support the hypothesis made in previous work.38
It has to be noted that such NMR relaxation measurements aims at characterizing the surface affinity of the materials for the different species and are carried out at room temperature. It is clear that reaction conditions are different in terms of temperature and complexity of all the reaction species involved. NMR relaxation measurements under such conditions would be challenging and not necessarily feasible. However, the measurements reported here offer more reliability, are easier to interpret and yet give a good indication on surface properties of those materials; hence, they can be used to help understand the catalytic data, especially when combined with the other methods used in this work.
To correlate with the NMR measurements, in situ DRIFTS was used to examine the relative amounts and rates of formation of isocyanate as a function of preparation method. It is now widely accepted2,5-8,12,13,21–29,52–55 that the surface NCO species is only observed when the catalyst is active and that it is probably a key intermediate in the HC-SCR reaction on Ag/Al2O3 catalysts. In order to examine the NCO formation on each catalyst (Ag2O_BM and Ag_STD), both Ag catalysts were exposed to C8H18 + O2 for 30 min at 250 °C and the species monitored using DRIFTS. This allowed the active/activated CxHyOz species to be adsorbed on the catalyst. Subsequently, a NO + O2 mixture was fed over the pre-treated Ag catalysts. It should be noted that these experiments were conducted in the absence of water to prevent the fast hydrolysis of surface NCO. The results are shown in Fig. 8.
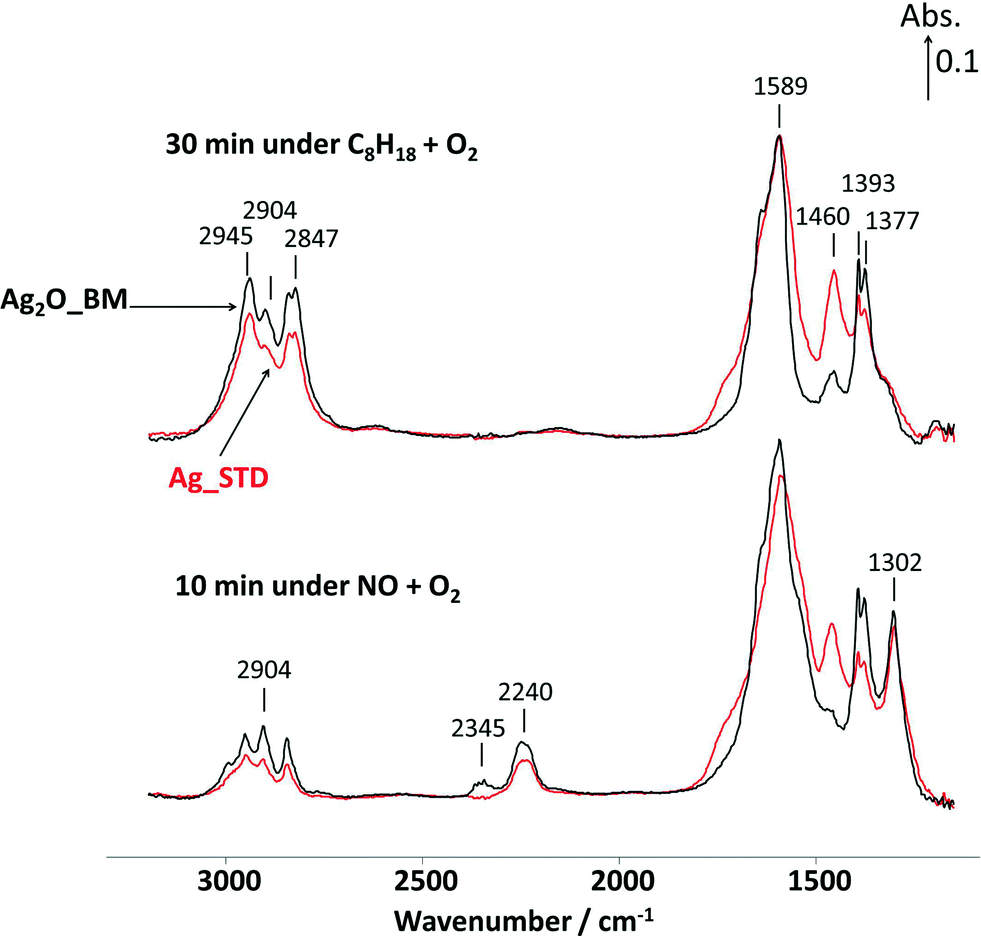 | ||
| Fig. 8 In situ DRIFT spectra on Ag catalysts recorded at 250 °C under C8H18 + O2 and NO + O2 conditions at 250 °C. Feed conditions: 720 ppm NO, 4340 ppm (as C1) n-C8H18, 4.3% O2, and Ar balance. | ||
From Fig. 8, after 30 min reaction under C8H18 + O2 conditions at 250 °C, a number of overlapping IR bands are observed below 1800 cm−1 due to the partial oxidation of n-octane. These species include carboxylates, acetates and formates.5,12,13,21,22,24–29,52 IR bands in the –CH– stretching region between 3100 and 2800 cm−1 were also observed and assigned to n-octane in the gas phase as well as adsorbed hydrocarbon species.3,12,13,21,22,52 When replacing the C8H18 + O2 with the NO + O2 feed, it is observed that the IR bands between 3100 and 2800 cm−1 decreased significantly and the NCO was formed shown by the band at 2240 cm−1 from the reaction between partially oxidized CxHyOz and NOx. The appearance of the IR band at 1302 cm−1 is due to NOx adsorption under NO + O2 gas feed. In comparison with Ag_STD, the DRIFTS spectra observed on Ag2O_BM sample revealed that there was a small peak observed at 2345 cm−1, attributed to both adsorbed and gas phase CO2 being formed from oxidation of n-octane.
Fig. 9 shows the dynamic change in the concentration of the NCO species on Ag2O_BM and Ag_STD catalysts after switching in the NO + O2 feed. It is clearly seen that the integrated area of the NCO band on Ag2O_BM is significantly higher than that found over the Ag_STD catalyst over the entire time of the experiment. In addition, the initial rate of NCO formation on Ag2O_BM is faster than on Ag_STD.
 | ||
| Fig. 9 Relative NCO intensity to the highest integrated peak area of NCO corresponding to Fig. 8 obtained from the integrated area of the DRIFT band at 2240 cm−1 under flowing NO + O2 at 250 °C. Prior to the measurement, both Ag_STD (■) and Ag2O_BM (♦) catalysts were pre-treated under C8H18 + O2 for 30 min at 250 °C. Feed conditions: 720 ppm NO, 4340 ppm (as C1) n-C8H18, 4.3% O2, and Ar balance. The lines provide a guide for the eye. | ||
In order to clarify which reactions are affected by the different preparation methods, the activation barriers for the Ag_STD and Ag2O_BM catalysts were measured for both the activation of the hydrocarbon and the reduction of the NOx. Fitting the deNOx reaction rate data in the kinetically controlled zone to the Arrhenius equation provides the pre-exponential factor and the activation energies. The apparent activation energy calculated from the rate of NOx conversion (<15% conversion) for NOx reduction on Ag2O_BM and Ag_STD were found to be 52.7 (±3.5) and 64.2 (±3.4) kJ mol−1, respectively. These may be compared with the barriers determined by Shimizu et al.18 for the SCR of NOx with n-hexane on 2 wt% Ag/Al2O3 catalyst of 67 kJ mol−1 for the NO reduction. This increase in barrier is consistent with the decreased activity shown using n-hexane versus n-octane due to the increased stability of the shorter hydrocarbon with respect to oxidation. In addition, the activation barrier for n-octane conversion (<15% conversion) was also calculated and found to be 45.7 (±3.8) and 57.1 (±4.5) kJ mol−1 for Ag2O_BM and Ag_STD, respectively. The apparent activation barriers determined indicate that the ball milled catalyst can activate the hydrocarbon more easily than for the wet impregnated sample; however, as the barrier for both catalysts is lower than found for the NOx reduction, it indicates that a subsequent step, for example the interaction of the NOx with the activated hydrocarbon to form NCO may be rate determining.
The NMR, together with the TPD results, show a decreased interaction of water and an increased interaction of the hydrocarbon with the surface which may result in the decreased activation barrier for n-octane activation and will increase the surface concentration of hydrocarbon. Whilst this is important as the precursor to the formation of NCO an increase in the n-octane interaction alone would not necessarily lead to higher NOx activity. The presence of water is also important as it provides a hydrolysis route for the decomposition of the isocyanate intermediate to form ammonia and carbon dioxide and subsequently nitrogen.8,23,52–55 It should be noted that, at the high water concentrations employed herein, although the hydrolysis step is important to complete the catalytic cycle and form N2, it is unlikely that this step is rate determining. Water can, however, also act as a poison for the reaction if present at too high a concentration by site blocking preventing the adsorption of both n-octane and NO.17,49,50 In the case of the ball milled catalyst, the interaction of n-octane is found to be increased relative to the wet impregnated catalyst whereas the opposite is true in the case of water. This is consistent with the surface of the ball milled catalyst increasing the rate of NCO formation, due to increased n-octane at the surface, whilst maintaining sufficient surface concentration of water for efficient hydrolysis. In the case of the wet impregnated catalyst both the n-octane concentration will be lower and the water concentration higher leading to reduced NCO formation and potentially increased site blocking by water hence the lower activity observed.
4. Conclusions
The work presented, herein, highlights that highly active Ag catalysts for NOx reduction by hydrocarbons on Ag/Al2O3 catalysts can be prepared by ball milling Ag2O with Al2O3 support. When compared with conventionally prepared wet-impregnated catalysts, the ball milled catalyst lowered the reaction temperature for NOx reduction with n-octane by ~130 °C. In addition, in the presence of H2, the lowest NOx reduction temperature was observed, to date. Two-dimensional T1–T2 NMR correlation experiments and water TPD measurements coupled with in situ DRIFTS analysis has indicated that the active sites on the ball milled Ag catalyst increase the formation of the intermediate NCO species by increasing the hydrophobicity of both the support and catalyst. In the case of the ball milled catalyst, the interaction of water is reduced whilst the n-octane is enhanced which provides a balance between sufficient water to hydrolyse the NCO and yet not to act as a site blocker for NO or the hydrocarbon. This concentration change coupled with the lower activation barrier for n-octane activation found for the ball milled material may be the cause of the increased activity of this catalyst compared with the wet impregnated material.Acknowledgements
We gratefully acknowledge funding for this work from the Department of Education and Leaning in Northern Ireland and the CASTech grant (EP/G012156/1) from the EPSRC.References
- K. A. Bethke and H. H. Kung, J. Catal., 1997, 172, 93 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283 CrossRef CAS.
- A. Iglesias-Juez, A. B. Hungría, A. Martínez-Arias, A. Fuerte, M. Fernándezcía, J. A. Anderson, J. C. Conesa and J. Soria, J. Catal., 2003, 217, 310 CAS.
- E. F. Iliopoulou, A. P. Evdou, A. A. Lemonidou and I. A. Vasalos, Appl. Catal., A, 2004, 274, 179 CrossRef CAS PubMed.
- J. Wang, H. He, Q. Feng, Y. Yu and K. Yoshida, Catal. Today, 2004, 93–94, 783 CrossRef PubMed.
- L.-E. Lindfors, K. Eränen, F. Klingstedt and D. Y. Murzin, Top. Catal., 2004, 28, 185 CrossRef CAS.
- H. He and Y. Yu, Catal. Today, 2005, 100, 37 CrossRef CAS PubMed.
- K. Shimizu and A. Satsuma, Phys. Chem. Chem. Phys., 2006, 8, 2677 RSC.
- V. Demidyak, C. Hardacre, R. Burch, A. Mhadeshwar, D. Norton and D. Hancu, Catal. Today, 2011, 164, 515 CrossRef PubMed.
- X. She and M. Flytzani-Stephanopoulos, J. Catal., 2006, 237, 79 CrossRef CAS PubMed.
- K. Takagi, T. Kobayashi, H. Ohkita, T. Mizushima, N. Kakuta, A. Abe and K. Yoshida, Catal. Today, 1998, 45, 123 CrossRef CAS.
- A. Sultana, M. Haneda, T. Fujitani and H. Hamada, Catal. Lett., 2007, 114, 96 CrossRef CAS PubMed.
- J. Li, Y. Zhu, R. Ke and J. Hao, Appl. Catal., B, 2008, 80, 202 CrossRef CAS PubMed.
- H. Kannisto, H. H. Ingelsten and M. Skoglundh, J. Mol. Catal. A: Chem., 2009, 302, 86 CrossRef CAS PubMed.
- V. I. Pârvulescu, B. Cojocaru, V. Pârvulescu, R. Richards, Z. Li, C. Cadigan, P. Granger, P. Miquel and C. Hardacre, J. Catal., 2010, 272, 92 CrossRef PubMed.
- A. Martínez-Arias, M. Fernández-García, A. Iglesias-Juez, J. A. Anderson, J. C. Conesa and J. Soria, Appl. Catal., B, 2000, 28, 29 CrossRef.
- K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2000, 25, 239 CrossRef CAS.
- K. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151 CrossRef CAS.
- J. P. Breen and R. Burch, Top. Catal., 2006, 39, 53 CrossRef CAS PubMed.
- R. Burch, J. P. Breen, C. J. Hill, B. Krutzsch, B. Konrad, E. Jobson, L. Cider, K. Eränen, F. Klingstedt and L. E. Lindfors, Top. Catal., 2004, 30–31, 19 CrossRef.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2010, 276, 49 CrossRef CAS PubMed.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2011, 281, 98 CrossRef CAS PubMed.
- J. P. Breen, R. Burch, C. Hardacre, C. J. Hill and C. Rioche, J. Catal., 2007, 246, 1 CrossRef CAS PubMed.
- S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2003, 42, 179 CrossRef CAS.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 222, 368 CrossRef CAS PubMed.
- S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma, T. Hattori and T. Kojima, Chem. Eng. Sci., 2007, 62, 5335 CrossRef CAS PubMed.
- A. Satsuma, J. Shibata, A. Wada, Y. Shinizaki and T. Hattori, Stud. Surf. Sci. Catal., 2003, 145, 235 CrossRef CAS.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, Appl. Catal., B, 2004, 54, 137 CrossRef CAS PubMed.
- J. Shibata, K.-I. Shimizu, Y. Takada, A. Shichi, H. Yoshida, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 227, 367 CrossRef CAS PubMed.
- P. Baláž, in Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265 CrossRef CAS PubMed.
- T. Ishida, N. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAS PubMed.
- J. Huang, T. Takei, T. Akita, H. Ohashi and M. Haruta, Appl. Catal., B, 2010, 95, 430 CrossRef CAS PubMed.
- J. Huang, E. Lima, T. Akita, A. Guzmán, C. Qi, T. Takei and M. Haruta, J. Catal., 2011, 278, 8 CrossRef CAS PubMed.
- A. Bruckman, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131 RSC.
- A. Lazuen-Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC.
- K. Ralphs, C. Hardacre and S. L. James, Chem. Soc. Rev., 2013, 42, 7701 RSC.
- U. Kamolphop, S. F. R. Taylor, J. P. Breen, R. Burch, J. J. Delgado, S. Chansai, C. Hardacre, S. Hengrasmee and S. L. James, ACS Catal., 2011, 1, 1257 CrossRef CAS.
- L. F. Gladden and J. Mitchell, New J. Phys., 2011, 13, 035001 CrossRef.
- K. M. Song, J. Mitchell, H. Jaffel and L. F. Gladden, J. Mater. Sci., 2010, 45, 5282 CrossRef CAS.
- D. Weber, J. Mitchell, J. McGregor and L. F. Gladden, J. Phys. Chem. C, 2009, 113, 6610 CAS.
- J. Mitchell, M. D. Hurlimann and E. J. Fordham, J. Magn. Reson., 2009, 200, 198 CrossRef CAS PubMed.
- J. P. Korb, New J. Phys., 2011, 13, 035016 CrossRef.
- C. D'Agostino, G. L. Brett, P. J. Miedziak, D. W. Knight, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem.–Eur. J., 2012, 18, 14426 CrossRef CAS PubMed.
- C. D'Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle , Chem.–Eur. J., 2013, 19, 11725 Search PubMed.
- C. D'Agostino, J. Mitchell, L. F. Gladden and M. D. Mantle, J. Phys. Chem. C, 2012, 116, 8975 Search PubMed.
- P. J. McDonald, J. P. Korb, J. Mitchell and L. Monteilhet, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 011409 Search PubMed.
- P. J. McDonald, J. Mitchell, M. Mulheron, P. S. Aptaker, J. P. Korb and L. Monteilhet, Cem. Concr. Res., 2007, 37, 303 Search PubMed.
- A. Abe, N. Aoyamo, S. Sumiya, N. Kakuta and K. Yoshida, Catal. Lett., 1998, 50, 5 Search PubMed.
- S. Sumiya, M. Saito, H. He, Q. Feng, N. Takezawa and K. Yoshida, Catal. Lett., 1998, 50, 87 Search PubMed.
- I. Foley, S. A. Farooqui and R. L. Kleinberg, J. Magn. Reson., Ser. A, 1996, 123, 95 Search PubMed.
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493 Search PubMed.
- S. Tamm, H. H. Ingelsten and A. E. C. Palmqvist, J. Catal., 2008, 255, 304 Search PubMed.
- K. Eränen, F. Klingstedt, K. Arve, L.-E. Lindfors and D. Y. Murzin, J. Catal., 2004, 227, 328 Search PubMed.
- N. Bion, J. Saussey, M. Haneda and M. Daturi, J. Catal., 2003, 217, 47 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |