Ligand-free copper(I) oxide nanoparticle-catalysed amination of aryl halides in ionic liquids
Michael T.
Keßler
,
Silas
Robke
,
Sebastian
Sahler
and
Martin H. G.
Prechtl
*
Department of Chemistry, Institute of Inorganic Chemistry, University of Cologne, Greinstraße 6, 50939 Cologne, Germany. E-mail: martin.prechtl@uni-koeln.de; Web: http://catalysis.uni-koeln.de Fax: +49 221 4701788
First published on 8th October 2013
Abstract
In the following, we present a simple and feasible methodology for a C–N coupling reaction using nanoscale Cu2O catalysts incorporated in n-Bu4POAc ionic liquid media. It is shown that a wide range of amines and aryl halides can be coupled selectively in high yields, without the use of ligands or additives (bases) and without precautions against water or air. All catalyses can be carried out with a nanoparticle catalyst loading as low as 5 mol%, based on the used precursor.
Introduction
Cross coupling reactions belong to the most important and synthetically versatile reactions in chemical synthesis. C–N coupling reactions have been known for years and can easily be conducted with (noble) metal catalysts, e.g. palladium or nickel.1,2 Unfortunately, these catalysts are, though used in low loadings, extremely expensive or suffer from the disadvantage of toxicity.3,4 In the last decade, scientific research has focussed on the development of inexpensive and less toxic metals such as iron5–8 and copper.9–11 One promising catalyst turns out to be copper(I) oxide which showed remarkable activity in homogeneous as well as heterogeneous C–N bond formation reactions.12–14 Homogeneously driven copper(I) catalysed C–N bond formation sometimes requires only homeopathic catalyst loadings, but the use of ligands, base or other additives as well as inert conditions are usually necessary.15–17 Heterogeneous C–N cross coupling catalysed by copper(I) requires rather harsh reaction conditions but does not need ligands and shows a good reaction workup.18,19However, up to now nanoscale copper(I) catalysed cross coupling reactions, especially C–N bond formation, have rarely been reported in scientific literature13,20–22or show drawbacks in versatility and catalyst loadings.20,22–25 Furthermore, it would be more convenient to conduct catalysis at temperatures right below the boiling point of water without the necessity for ligands, additives and inert reaction conditions.26 Nonetheless, we want to establish a Cu2O-nanocatalyst which combines the advantages of classical homogeneous and heterogeneous catalyses. On the one hand, Cu(I) nanocatalysts show good yields and high TONs under moderate reaction conditions; on the other hand, excellent reaction workup and low product contamination are secured. Apart from a few publications,20,23 Cu2O nanoparticles (Cu2O-NPs) were considered as rather unreactive and non-versatile catalysts, which was why they seemed to be useless in C–N coupling reactions. As a rather toxic alternative with adequate activity and versatility, CuI is widely employed.21,27
Usually solvents can alter the chemo-physical properties of catalysts.28 Ionic liquids (ILs) are widely known as so-called “designer solvents”. They can act as promoters and activators,29,30 as protecting agents and stabilisers,28,30–35 as reducing agents11,30,35 and certainly as (co-)solvents themselves for molecular and nanoscale species.36–39 Only limited reports about the use and influence of ionic liquids as solvents for nanocatalytic C–N cross coupling reactions are known.40,41 To the best of our knowledge, publications about the incorporation and stabilisation of Cu2O nanoparticles in ionic liquid media and their use for catalytic amination reaction have not been reported yet.
Herein we established a new approach to catalytic C–N couplings with Cu2O-NP in ionic liquid media. With regard to future applications for a recyclable catalytic system, we tried in particular to simplify the reaction process. The influence of the reaction parameters such as reaction time, temperature and catalyst loading as well as the influence of additives, solvents or inert atmosphere were investigated. Finally, we could establish a versatile methodology for the coupling of iodo- and bromobenzenes with a broad scope of amines and ammonia with a remarkable selectivity. It has to be pointed out that the synthesis of the Cu2O nanoparticles proceeds directly in the used ionic liquid from cheap precursors like basic CuCO3 with subsequent C–N coupling reactions. The as-synthesised nanoparticles are not separated or purified in an additional step but can be directly used as catalysts for amination reactions. In contrast to many other homogeneous and heterogeneous catalyses, the presented coupling reaction in ionic liquid medium turns out to have no necessity for ligands, additional base, additives or an inert atmosphere and can even proceed in the presence of water at temperatures below 80 °C.
Synthesis of the catalytic nanoparticle phase
In addition to thermal decomposition,35 laser ablation or sputtering methods,42 reduction with hydrogen or hydrazine solution43,44 using ionic liquid as a reducing agent for the synthesis of Cu2O nanoparticles in terms of reduction of the precursor is a potential alternative. We have recently published a low-temperature synthesis of Cu2O nanoparticles in n-Bu4POAc elsewhere.11 The obtained nanoparticles had an average diameter of 5.5 nm (±1.2 nm), pointing out that basic CuCO3 usually decomposes to CuO at 295 °C.45 In our low-temperature synthesis, CuCO3 is converted to Cu2O-NPs via an “ionic liquid-induced” reduction at 120 °C (Scheme 1).11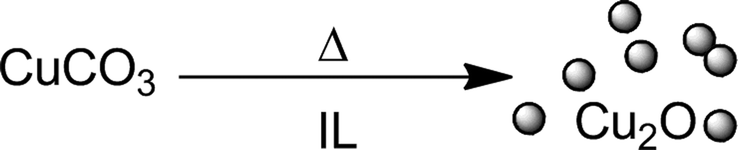 | ||
| Scheme 1 Synthesis of Cu(I) oxide nanoparticles by thermal reduction of copper(II) carbonate in ionic liquid medium. | ||
Amination reactions with ammonia
Amination reactions of iodobenzene and its derivatives with aqueous ammonia solution belong to the most common and most desired reactions in chemical synthesis. Both reactants are inexpensive, easy to handle and readily available. It is well known that Cu(I) in homogeneous46,47 as well as in heterogeneous48,49 catalysis promotes cross coupling reactions.In our case, we were pleased to see that iodobenzene couples smoothly even with ammonia in aqueous solution, in the presence of a ligand-free Cu2O nanocatalyst in n-Bu4POAc (Scheme 2). It is quite uncommon that the yields were satisfactory or high at temperatures of about 75–85 °C within 24 h even in the absence of an additional base (see Table 1, entries 7 and 10–13).
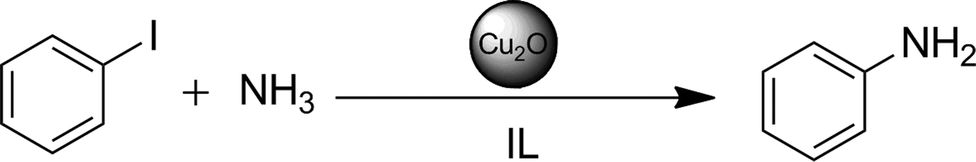 | ||
| Scheme 2 Amination of iodobenzene with aqueous ammonia solution as a test reaction for the activity of the Cu2O NPs in IL. | ||
| Entry | IL | T (°C) | t (h) | Cat.-loadingb (mol%) | Additives | Conv.c (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: 1 mmol iodobenzene, 1 ml NH3 (aq., 20%, 12 mmol), Cu-cat in 1 g IL.a No Cu2O-NPs detectable.b Based on used precursor amount (CuCO3).c Conversions were determined using 1H NMR with hexamethyldisilane as the internal standard. | ||||||
| 1 | bmim NTf2 | 85 | 24 | 100a | 0 | |
| 2 | bmmim NTf2 | 85 | 24 | 100a | 0 | |
| 3 | bmim OAc | 85 | 24 | 100 | 38 | |
| 4 | bmpyrr OAc | 85 | 24 | 100 | <1 | |
| 5 | bmim Cl | 85 | 24 | 100a | 34 | |
| 6 | (n-Bu4P)2SO4 | 85 | 24 | 100 | 0 | |
| 7 | n-Bu4POAc | 85 | 24 | 100 | 61 | |
| 8 | n-Bu4POAc | 85 | 24 | 100 | K2CO3 | 63 |
| 9 | n-Bu4POAc | 85 | 24 | 100 | KI | 59 |
| 10 | n-Bu4POAc | 85 | 16 | 100 | 64 | |
| 11 | n-Bu4POAc | 85 | 16 | 10 | 83 | |
| 12 | n-Bu4POAc | 100 | 24 | 100 | 40 | |
| 13 | n-Bu4POAc | 75 | 16 | 10 | 92 | |
Different ionic liquids as reaction media were investigated by varying the anion as well as the cation to change the polarity of the ionic liquid. Polarity, basicity and acidity, which are crucial attributes for application in catalysis, are summarized in the so-called Kamlet–Taft parameters. We tried other polar ionic liquids with different Kamlet–Taft parameters50 for the synthesis of Cu2O from CuCO3, pointing out that the particles obtained in n-Bu4POAc were the most active. Rather apolar ionic liquids such as 1-butyl-3-methylimidazolium N,N-bistrifluoromethylsulfonylimide (bmim NTf2) and 1-butyl-2,3-dimethylimidazolium N,N-bistrifluoromethylsulfonylimide (bmmim NTf2) are not capable of reducing the Cu(II) species.11 Therefore, we were not surprised that we could not detect any nanoparticles in the ionic liquids. Furthermore, Cu(II) salts do not support the amination of aryl halides very well (Table 1, entries 1 and 2). However, highly polar ionic liquids often suffer from high melting points ((n-Bu4P)2SO4) and seem to be impractical as reaction media (Table 1, entry 6). The obtained reaction mixtures show high viscosities, leading to a rather inhomogeneous intermixture of educts and the catalytic IL-phase. Nevertheless, acetate-based ionic liquids combine both low melting points and the ability to form Cu2O. The catalytic activities of the formed copper(I) species, 1-butyl-3-methylimidazolium acetate (bmim OAc) and butyl-methylpyrrolidinium acetate (bmpyrr OAc), are rather low (Table 1, entries 3 and 4); only the high polarity of the reaction medium seems to support the amination reaction (Table 1, entry 5). Interestingly, only the nanoparticles generated in n-Bu4POAc show good results in the amination of iodobenzene with a yield of about 61% (Table 1, entries 7 and 10–13).
With n-Bu4POAc as our reaction medium of choice, we investigated the influence on the reaction time towards the yield. We observed that the conversion of iodobenzene to aniline increases drastically with time and a maximum conversion could be reached after 16 h (64%) (Fig. 1, Table 1, entry 10).
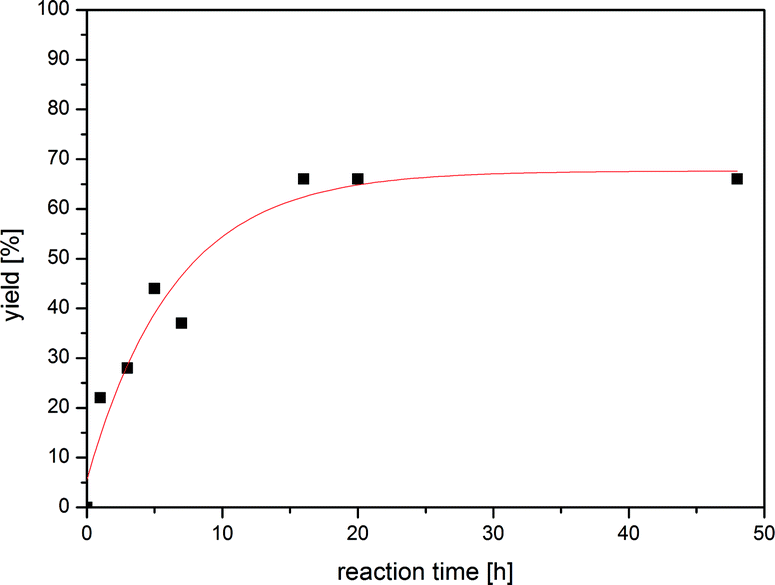 | ||
| Fig. 1 Plot of reaction time and corresponding yield of aniline. The maximum yield is reached within 16 h at 85 °C. | ||
Investigation of the catalyst loading showed that a decrease in the amount of Cu2O raised the yield drastically. The maximum value of about 83% was achieved using only 10 mol% of the catalyst (Table 1, entry 11). The catalyst loading is referred to as the amount of catalyst precursor applied. In fact, due to the high surface/volume ratio of the particles, the amount of real active catalyst is even lower.11,41 Loadings of the catalyst below 10 mol% as well as loadings higher than 10 mol% lowered the yield of aniline. Moreover, with high catalyst loadings, the system's miscibility probably suffers due to the higher viscosity of the liquid phase. Analogous to several other publications dealing with Pd- or Ni-NPs, it is possible that the Cu2O-NPs act as a catalyst reservoir for Cu(I), and a molecular species might act as the active species.32,51 There are different ways of how a molecular species can theoretically be leached from the particle surface as Beletskaya and co-workers have revealed. Usually there is an equilibrium between leaching and the Ostwald-ripening of the particles. In this context, the synonym “nanosalt” is introduced for metal–chalcogen nanoparticles.51 Up to now, the mechanism for the leaching effect has not been intensively investigated, but usually the Ar–X species is believed to generate molecular metal species by oxidative addition.26 During the extraction of the reaction mixture using n-pentane, copper species can be leached out of the catalytic phase and contaminate the final product. In order to investigate the amount of copper in the organic phase, ICP-OES measurements revealed that the extract contains only a maximum of 0.041 μmol (lowest value found: 0.00029 μmol) copper. Based on the utilised amount of catalyst precursor (CuCO3 0.05 mmol), the leached copper amount is equivalent to a maximum of 0.082%, which is remarkably low.
Moreover, the outcome of the amination reaction catalysed by Cu(I) oxide nanoparticles could be improved further by adjusting the reaction temperature. The yields can effectively be increased to an excellent yield of 92% at temperatures as low as 75 °C (Table 1, entry 13). The higher conversions at lower temperatures are most likely related to the low boiling point of ammonia; thus, the concentration in the liquid phase is higher at lower temperature. It is a remarkable fact that a ligand- and additive-free approach shows best results far below the boiling point of water, which makes the reaction also possible in water. Most other ligand-free approaches need temperatures between 100 °C and 135 °C.16 A further increase in the reaction temperature affects the yield of aniline negatively (Fig. 2).
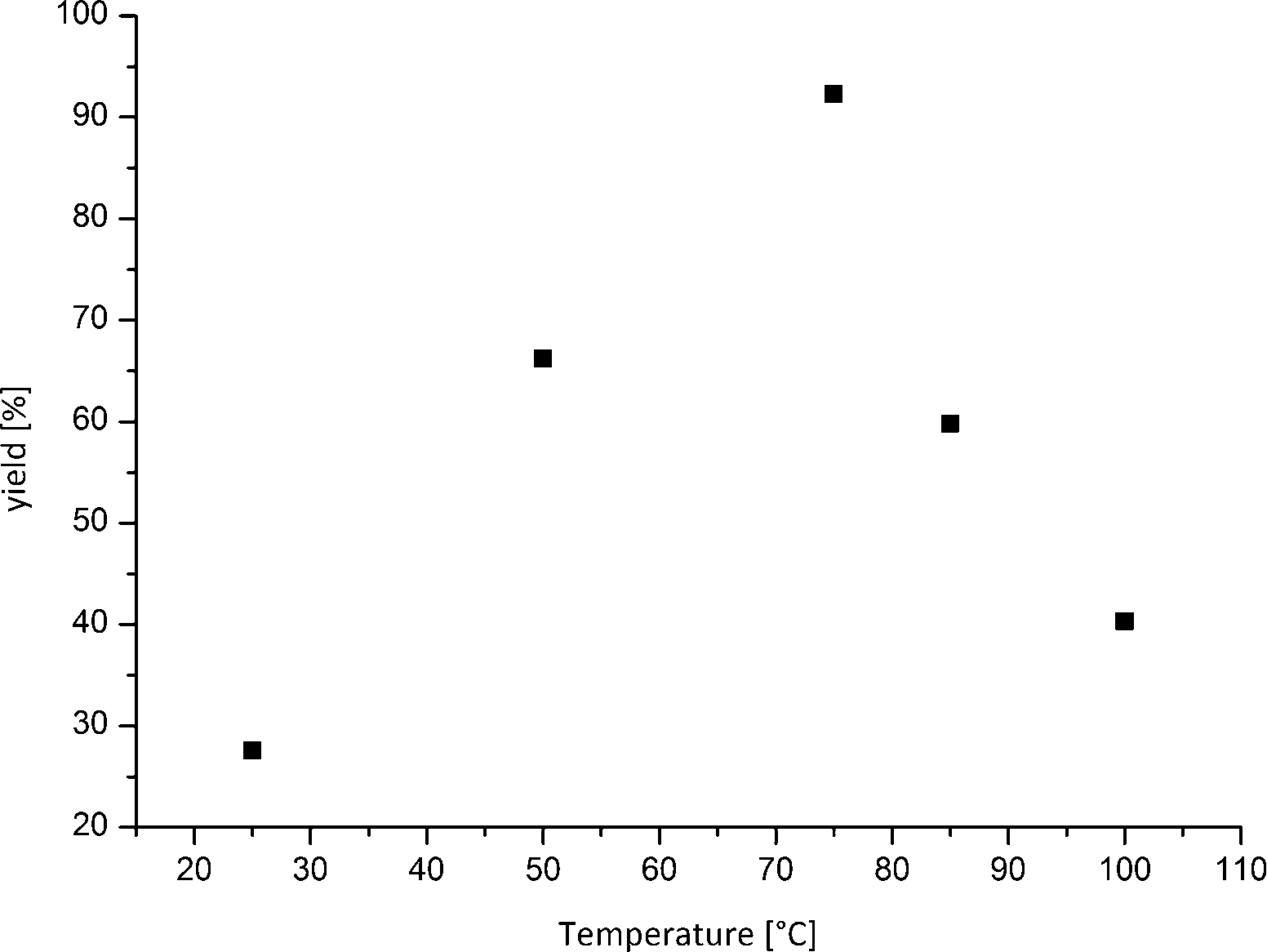 | ||
| Fig. 2 Correlation between reaction temperature and corresponding yield of aniline. The best results could be achieved at 75 °C within 16 h and a catalyst loading of 10 mol%. | ||
The addition of additives like a base (K2CO3) or KI (Table 1, entries 8 and 9) to change the pH-value or to simulate a different copper(I)-species did not improve the yield of aniline measurably. Obviously it is not necessary to neutralise the HX species,16,19 which is produced during the reaction process, by an additional base. On the one hand, the acetate ionic liquid may act as a buffer reagent and keep the pH-value constant, and on the other hand, ammonia can act as a proton scavenger as well.
In comparison to the used copper(I) oxide nanoparticles, we investigated different (bulk) copper salts in oxidation states (I) and (II) (Fig. 3). In contrast to ref. 17, we could not show activity for Cu2+ species in our reaction sequence. All used Cu(II) salts (Cu(NO3)2, CuSO4, CuCl2, Cu(OAc)2 and CuF2) failed completely in catalysing the amination of iodobenzene with ammonia under the same conditions. A competing reaction might be the formation of a stable Cu(NH3)42+ complex which colours the reaction mixture immediately. CuI (as used in other manuscripts)22 and bulk Cu2O applied in ionic liquid media showed very poor activity of <1% and 3% yields, respectively.
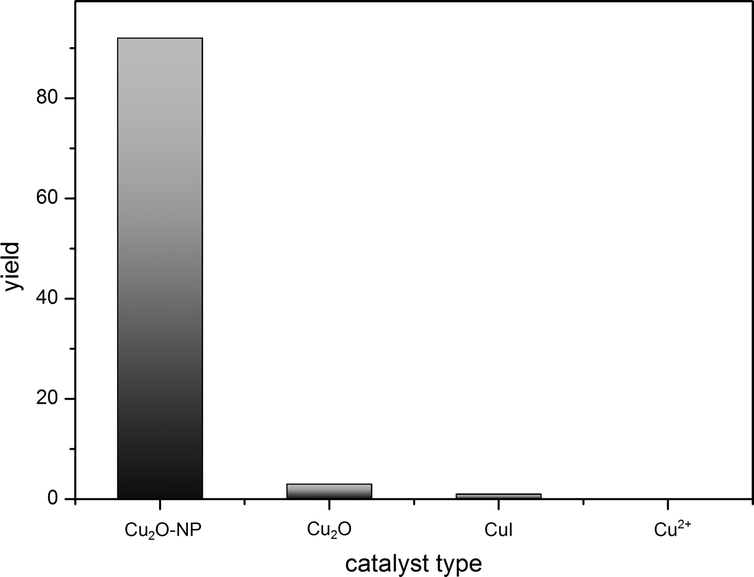 | ||
| Fig. 3 Correlation between yield and copper catalyst for the amination of iodobenzene. | ||
Reactions with primary and secondary amines
In order to show the versatility of our catalytic system, aryl halides were coupled with primary and secondary amines as well. Due to the increasing electron density at the nitrogen atom, there is a distinct tendency for per-N-substitution reactions to give mono-, di- and tri-substituted amines, as well as quaternary ammonium salts in an undesirable product mixture.52 Hence the reaction sequence, the reaction conditions as well as the workup must prevent per-arylation and guarantee the selective synthesis of only one product (Scheme 3).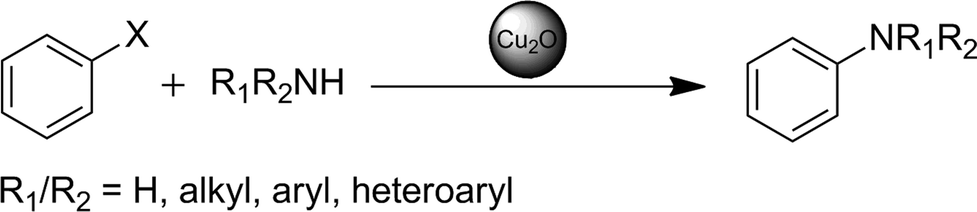 | ||
| Scheme 3 Amination of aryl halides with primary and secondary amines. | ||
As a starting point, N-arylation reactions of iodobenzene with primary amines were chosen with a catalyst loading of 10 mol%. The results of the arylation experiments are listed in Table 2, showing remarkable results concerning yields and catalyst loadings. We were pleased to find out that we could reduce the loading of the copper-catalyst to 5 mol% without a significant loss in activity (Table 2, entry 2).
| Entry | Amine | Cat.-loadingb (mol%) | Conversion (sec. amine)a (%) |
|---|---|---|---|
| Reaction conditions: 1 mmol iodobenzene, 1 ml prim. amine (CH3-NH2 (40 wt%) 12.9 mmol, n-Bu-NH2 10.1 mmol, iso-Bu-NH2 10.6 mmol, cHex-NH2 8.7 mmol, n-Oct-NH2 6.0 mmol, iso-Pr-NH2 11.6 mmol, tert-Bu-NH2 9.5 mmol, C6H5-NH2 10.7 mmol), Cu-cat in 1 g (3.1 mmol) IL, reaction temperature: 75 °C, reaction time: 16 h.a Conversions were determined using 1H NMR with hexamethyldisilane as the internal standard.b Based on used precursor amount (CuCO3). No by-products could be detected. | |||
| 1 | CH3-NH2 | 10 | 65 |
| 2 | CH3-NH2 | 5 | 65 |
| 3 | n-Bu-NH2 | 5 | 71 |
| 4 | iso-Bu-NH2 | 5 | 95 |
| 5 | cHex-NH2 | 5 | 77 |
| 6 | n-Oct-NH2 | 5 | 90 |
| 7 | iso-Pr-NH2 | 5 | 99 |
| 8 | tert-Bu-NH2 | 5 | 57 |
| 9 | C6H5-NH2 | 5 | 65 |
The best results could be obtained with linear and branched alkyl amines (65%–99%, Table 2, entries 2–4 and 6–8), followed by cyclic (77%, Table 2, entry 5) and aryl amines (65%, Table 2, entry 9). The low yields of the coupling between iodobenzene and methyl amine can be explained by the rather low electron density (e.g. compared to n-butyl amine). No further arylation of the desired secondary amine was detectable, which might be in correlation with the high excess of primary amine in the reaction mixture. A similar screening with secondary amines is shown in Table 3.
| Entry | Amine | Cat.-loadingb (mol%) | Conversion (tert. amine)a (%) |
|---|---|---|---|
| Reaction conditions: 1 mmol iodobenzene, 1 ml sec. amine (Et2NH 9.6 mmol, n-Bu2NH 5.9 mmol, piperidine 10.1 mmol, Ph2NH 6.9 mmol, morpholine 11.5 mmol, L-Prolin 11.7 mmol), Cu-cat in 1 g (3.1 mmol) IL, reaction temperature: 75 °C, reaction time: 16 h.a Conversions were determined using 1H NMR with hexamethyldisilane as the internal standard.b Based on used precursor amount (CuCO3). No by-products could be detected. | |||
| 1 | Et2NH | 5 | 65 |
| 2 | n-Bu2NH | 5 | 16 |
| 3 | Piperidine | 5 | 99 |
| 4 | Ph2NH | 5 | 0 |
| 5 | Morpholine | 5 | 99 |
| 6 | L-Prolin | 5 | 33 |
In summary, the conversions of the coupling between secondary amines and aryl halides are slightly lower in comparison to the conversions of the coupling between primary amines and aryl halides. This might be due to emerging steric effects. The starting materials (secondary amines) and products (tertiary amines) are much bulkier and thus slightly disfavoured in the transmetallation steps. Diethylamine gave a satisfactory yield of 65% (Table 3, entry 1). There was on the contrary no conversion detectable when diphenylamine was used as the amination reagent (Table 3, entry 4). Nevertheless, there are also some examples which verify the versatility of this method with good to excellent results (e.g. morpholine and piperidine, both 99%, Table 3, entries 3 and 5). These heterocyclic compounds are less bulky than common secondary alkyl amines due to their fixed ring structure. No by-products, such as perarylated ammonium derivatives, could be detected in the reaction mixture.
Screening of different aryl halides
We were also able to show that our catalytic system was capable of coupling different substituted bromo- and iodoaryl derivatives with both ammonia and amines. The reaction tolerates functionalities like OH-, alkyl or methoxy groups on the aromatic ring. Although the yields are rather low in some cases, the reaction tolerates some common functional groups. Again, steric effects caused by substituents in the vicinity of the halide (Table 4, entries 6, 7, and 9) seem to hamper the amination completely. Aryl chlorides do not undergo amination reaction at temperatures as low as 75 °C (Table 4, entry 4).| Entry | Aryl–X | Amine | Cat.-loadingb (mol%) | Conversiona (%) |
|---|---|---|---|---|
| Reaction conditions: 1 mmol aryl halide, 1 ml piperidine (10.1 mmol), diethylamine (9.6 mmol) or ammonia solution (20%, 12 mmol), Cu-cat in 1 g (3.1 mmol) IL, reaction temperature: 75 °C, reaction time: 16 h.a Conversions were determined using 1H NMR with hexamethyldisilane as the internal standard.b Based on used precursor amount (CuCO3). No by-products could be detected. | ||||
| 1 | C6H5Br | NH3 | 10 | 12 |
| 2 | 3-Iodoanisole | NH3 | 5 | 92 |
| 3 | 4-Iodotoluene | NH3 | 10 | 79 |
| 4 | 1,2-Dichlorobenzene | Et2NH | 5 | 0 |
| 5 | C6H5Br | Et2NH | 5 | 10 |
| 6 | 2-Iodophenol | Et2NH | 5 | 0 |
| 7 | 2-Iodoanisole | Et2NH | 5 | 0 |
| 8 | 3-Iodoanisole | Et2NH | 5 | 24 |
| 9 | 2-Iodotoluene | Et2NH | 5 | 0 |
| 10 | 4-Iodotoluene | Et2NH | 5 | 33 |
| 11 | 3-Iodoanisole | Piperidine | 5 | 93 |
| 12 | 4-Iodotoluene | Piperidine | 5 | 85 |
Conclusions
In conclusion, we established an efficient C–N coupling reaction catalysed by Cu2O nanoparticles. Therefore we consciously avoided rather toxic catalyst materials, for example, CuI. We could show the versatility of this method in about 30 examples. The coupling of aryl halides with ammonia or primary or secondary amines gave exclusively the corresponding primary, secondary or tertiary amines in satisfactory to excellent yields and selectivity. The nanoparticle catalyst was synthesised in n-Bu4POAc – a polar ionic liquid with low melting point – which acted as a reaction medium, reductant and stabilising agent. Additionally the as-prepared catalyst did not require any workup and could subsequently be used for the amination reaction. The catalyst loading was as low as 5 mol%, and the whole reaction as well as the workup requires neither inert conditions nor any additional ligands, base or further additives. In sum, this protocol shows benefits in versatility, selectivity, effectivity and preparative demand which clearly distinguishes it from previous publications. Our future work will focus on a recyclable catalytic system.Experimental
General
An aqueous solution of n-Bu4POH (40 wt%) and basic CuCO3, as well as all other copper salts and LiNTf2, was obtained from ABCR Chemicals®. 1-chlorobutane, 1,2-dimethylimidazole and methylimidazole, and all primary and secondary amines, as well as all aryl halides, were purchased from Sigma Aldrich®. Sulphuric acid, acetic acid, aqueous NH3 solution (20%), K2CO3 and KI were obtained from the chemical stock of the institute. All chemicals were used without further purification, and all reactions were conducted under non-inert conditions. 1H-NMR and 31P-NMR spectra were recorded using a Bruker® AVANCE II 300 spectrometer at 298 K (300.1 MHz, external standard tetramethylsilane (TMS)). The obtained Cu2O nanoparticles were analysed using a powder X-ray diffractometer (STOE®-STADI MP, Cu-Kα irradiation, λ = 1.540598 Å) and a TEM Phillips® EM 420, 120 kV as described in ref. 11. The ionic liquid n-Bu4POAc and the Cu2O nanoparticles were prepared according to our previous report.11 All other ILs were synthesized according to literature-known methods.30,33,34,53,54 All ICP-OES measurements (Cu leaching) were performed on an AMETEK Spectro Arcos equipped with an ESI SC4-DX autosampler and run using the Spectro Smart Analyser Vision software. Two identical reaction samples were prepared for the amination of iodobenzene with ammonia. The reaction mixtures were extracted with n-pentane according to the procedure for amination reactions (see above). n-Pentane was completely removed under reduced pressure, and the residue was digested in 2 ml of 65% HNO3 at 90 °C for 3 h to give a clear yellow solution. The crude solution was allowed to cool to room-temperature and diluted to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 1:100 (sample 1), as well as to 1:50 and 1:500 (sample 2). Every probe was measured three times and correlated with a reference material and a blank probe. The characteristic spectral lines at 224.7 nm and 324.7 nm were used to determine the amount of leached copper-catalyst.
:50 and 1:100 (sample 1), as well as to 1:50 and 1:500 (sample 2). Every probe was measured three times and correlated with a reference material and a blank probe. The characteristic spectral lines at 224.7 nm and 324.7 nm were used to determine the amount of leached copper-catalyst.
Ionic liquid synthesis11
The synthesis of n-Bu4POAc is adapted from ref. 11 and modified to our preparative demands. In a 100 ml round bottom flask, 50 ml of 40 wt% tetrabutylphosphonium hydroxide solution (72.4 mmol; n-Bu4POH) is mixed with 4.12 ml (72.4 mmol) of 99% acetic acid under vigorous stirring. After further stirring for 25 min, the residual water is removed under reduced pressure at 60 °C. The resulting ionic liquid is subsequently dried under reduced pressure for at least 72 h leaving a colourless hygroscopic and waxy solid behind. Yield, 20.8 g (95%). 1H-NMR (300 MHz, rt, CDCl3): δ (ppm) = 2.07–2.18m (8H), 1.48–1.58q (8H), 1.36–1.48q (8H), 0.86–0.93t (12H). 31P-NMR (60 MHz, rt, CDCl3): δ (ppm) = 33.25.Nanoparticle synthesis11
The Cu2O nanoparticles were synthesised in an oven-dried 25 ml crimp top vial equipped with a butyl-rubber septum and a glass stirring bar. 0.05 mmol (11 mg) of basic CuCO3 was suspended in 1 g (3.1 mmol) IL and heated to 120 °C for 12 h while stirring at 500 rpm in a vial holder. The resulting catalytic phase was a brownish red dispersion of Cu2O nanoparticles in IL. The particles in IL could be easily dispersed in acetone, ethanol or isopropanol.Procedure for amination reactions
The as-prepared nanoparticle dispersion was allowed to cool down to room temperature in the 25 ml crimp-top vial. Then 1 mmol aryl halide and 1 ml of amine or ammonia solution (NH3 (20 wt%) 12 mmol, CH3-NH2 (40 wt%) 12.9 mmol, n-Bu-NH2 10.1 mmol, iso-Bu-NH2 10.6 mmol, cHex-NH2 8.7 mmol, n-Oct-NH2 6.0 mmol, iso-Pr-NH2 11.6 mmol, tert-Bu-NH2 9.5 mmol, C6H5-NH2 10.7 mmol, Et2NH 9.6 mmol, n-Bu2NH 5.9 mmol, piperidine 10.1 mmol, Ph2NH 6.9 mmol, morpholine 11.5 mmol and L-Prolin 11.7 mmol) were added with vigorous stirring. The vial was closed again and heated up to 75 °C for 16 h. The reaction mixture was cooled and extracted with n-pentane (3 × 5 ml). The organic phases were combined, and the solvent was evaporated under reduced pressure. The residue was analysed using 1H-NMR techniques (internal standard: hexamethyldisilane, 0.1 mmol, 20 μl) and compared with literature data.Acknowledgements
We acknowledge the Ministerium für Innovation, Wissenschaft und Forschung NRW (MIWF-NRW) for financial support (M. H. G. Prechtl). Furthermore, the Evonik® Foundation is acknowledged for a scholarship (M. T. Keßler) and the Robert-Lösch-Foundation for a project grant. J.-H. Choi is acknowledged for helpful discussion. For access to ICP-OES analysis, we thank M. Staubwasser and J. Scheld (UoC, Inst. of Geology).Notes and references
- M. H. Ali and S. L. Buchwald, J. Org. Chem., 2001, 66, 2560–2565 CrossRef CAS PubMed.
- S. Nagao, T. Matsumoto, Y. Koga and K. Matsubara, Chem. Lett., 2011, 40, 1036–1038 CrossRef CAS.
- G. K. Bielmyer, C. Decarlo, C. Morris and T. Carrigan, Environ. Toxicol. Chem., 2013, 32, 1354–1359 CrossRef CAS PubMed.
- E. Patel, C. Lynch, V. Ruff and M. Reynolds, Toxicol. Appl. Pharmacol., 2012, 258, 367–375 CrossRef CAS PubMed.
- S. Gulak and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2012, 51, 1357–1361 CrossRef PubMed.
- M. Mayer, A. Welther and A. Jacobi von Wangelin, ChemCatChem, 2011, 3, 1567–1571 CrossRef CAS.
- A. Welther, M. Bauer, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 1088–1093 CrossRef CAS.
- A. Welther and A. Jacobi von Wangelin, Curr. Org. Chem., 2013, 17, 326–335 CrossRef CAS.
- P. P. Arquilliere, C. C. Santini, P. H. Haumesser and M. Aouine, ECS Trans., 2011, 35, 11–16 CrossRef CAS PubMed.
- P. Arquilliere, P. H. Haumesser and C. C. Santini, Microelectron. Eng., 2012, 92, 149–151 CrossRef CAS PubMed.
- M. T. Kessler, C. Gedig, S. Sahler, P. Wand, S. Robke and M. H. G. Prechtl, Catal. Sci. Technol., 2013, 3, 992–1001 CAS.
- R. T. Gephart, D. L. Huang, M. J. B. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488–6492 CrossRef CAS PubMed.
- S. J. Ahmadi, S. Sadjadi, M. Hosseinpour and M. Abdollahi, Monatsh. Chem., 2011, 142, 801–806 CrossRef CAS.
- P. J. Ji, J. H. Atherton and M. I. Page, J. Org. Chem., 2012, 77, 7471–7478 CrossRef CAS PubMed.
- S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586–5587 CrossRef CAS PubMed.
- Z.-J. Liu, J.-P. Vors, E. R. F. Gesing and C. Bolm, Green Chem., 2011, 13, 42–45 RSC.
- P.-F. Larsson, A. Correa, M. Carril, P.-O. Norrby and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5691–5693 CrossRef CAS PubMed.
- J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971–9983 CrossRef CAS PubMed.
- A. Correa and C. Bolm, Adv. Synth. Catal., 2007, 349, 2673–2676 CrossRef CAS.
- S. Uk Son, I. Kyu Park, J. Park and T. Hyeon, Chem. Commun., 2004, 778–779 RSC.
- H. J. Xu, Y. F. Liang, Z. Y. Cai, H. X. Qi, C. Y. Yang and Y. S. Feng, J. Org. Chem., 2011, 76, 2296–2300 CrossRef CAS PubMed.
- B. C. Ranu, R. Dey, T. Chatterjee and S. Ahammed, ChemSusChem, 2012, 5, 22–44 CrossRef CAS PubMed.
- S. Jammi, S. Krishnamoorthy, P. Saha, D. S. Kundu, S. Sakthivel, M. A. Ali, R. Paul and T. Punniyamurthy, Synlett, 2009, 3323–3327 CAS.
- B. X. Tang, S. M. Guo, M. B. Zhang and J. H. Li, Synthesis-Stuttgart, 2008, 1707–1716 CAS.
- M. Kidwai, S. Bhardwaj and S. Poddar, Beilstein J. Org. Chem., 2010, 6, 35 CrossRef PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753–7808 CrossRef CAS.
- G. Lefevre, G. Franc, C. Adamo, A. Jutand and I. Ciofini, Organometallics, 2012, 31, 914–920 CrossRef CAS.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, J. Mol. Catal. A: Chem., 2009, 313, 74–78 CrossRef CAS PubMed.
- C. C. Cassol, A. P. Umpierre, G. Machado, S. I. Wolke and J. Dupont, J. Am. Chem. Soc., 2005, 127, 3298–3299 CrossRef CAS PubMed.
- M. H. G. Prechtl, P. S. Campbell, J. D. Scholten, G. B. Fraser, G. Machado, C. C. Santini, J. Dupont and Y. Chauvin, Nanoscale, 2010, 2, 2601–2606 RSC.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- C. S. Consorti, F. R. Flores and J. Dupont, J. Am. Chem. Soc., 2005, 127, 12054–12065 CrossRef CAS PubMed.
- C. C. Cassol, G. Ebeling, B. Ferrera and J. Dupont, Adv. Synth. Catal., 2006, 348, 243–248 CrossRef CAS.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS PubMed.
- R. Venkatesan, M. H. G. Prechtl, J. D. Scholten, R. P. Pezzi, G. Machado and J. Dupont, J. Mater. Chem., 2011, 21, 3030–3036 RSC.
- V. Calo, A. Nacci, A. Monopoli and P. Cotugno, Angew. Chem., Int. Ed., 2009, 48, 6101–6103 CrossRef CAS PubMed.
- V. Calo, A. Nacci, A. Monopoli and P. Cotugno, Chem.–Eur. J., 2009, 15, 1272–1279 CrossRef CAS PubMed.
- C. Amiens, B. Chaudret, M. Respaud and P. Lecante, Actual. Chim., 2005, 19–27 CAS.
- L. M. Lacroix, S. Lachaize, F. Hue, C. Gatel, T. Blon, R. P. Tan, J. Carrey, B. Warot-Fonrose and B. Chaudret, Nano Lett., 2012, 12, 3245–3250 CrossRef CAS PubMed.
- C. T. Yang, Y. Fu, Y. B. Huang, J. Yi, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 7398–7401 CrossRef CAS PubMed.
- I. Geukens, J. Fransaer and D. E. De Vos, ChemCatChem, 2011, 3, 1431–1434 CrossRef CAS.
- H. Wender, L. F. de Oliveira, P. Migowski, A. F. Feil, E. Lissner, M. H. G. Prechtl, S. R. Teixeira and J. Dupont, J. Phys. Chem. C, 2010, 114, 11764–11768 CAS.
- M. Ranjbar, S. Fardindoost, S. M. Mahdavi, A. I. Zad and N. Tahmasebi G, Sol. Energy Mater. Sol. Cells, 2011, 95, 2335–2340 CrossRef CAS PubMed.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441–3461 CrossRef CAS PubMed.
- H. Henmi, T. Hirayama, N. Mizutani and M. Kato, Thermochim. Acta, 1985, 96, 145–153 CrossRef CAS.
- S. M. Islam, S. Mondal, P. Mondal, A. S. Roy, K. Tuhina and M. Mobarok, Inorg. Chem. Commun., 2011, 14, 1352–1357 CrossRef CAS PubMed.
- Y. Zou, H. Lin, P. A. Maggard and A. Deiters, Eur. J. Inorg. Chem., 2011, 4154–4159 CAS.
- P. Liu, P. H. Li and L. Wang, Synth. Commun., 2012, 42, 2595–2605 CrossRef CAS.
- X. Qi, L. Zhou, X. Jiang, H. Fan, H. Fu and H. Chen, Chin. J. Catal., 2012, 33, 1877–1882 CrossRef CAS.
- P. G. Jessop, D. A. Jessop, D. B. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
- G. Verardo, A. G. Giumanini and P. Strazzolini, Synth. Commun., 1994, 24, 609–627 CrossRef CAS.
- D. B. Zhao, Z. F. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876–15882 CrossRef CAS PubMed.
- Y. G. Cui, I. Biondi, M. Chaubey, X. Yang, Z. F. Fei, R. Scopelliti, C. G. Hartinger, Y. D. Li, C. Chiappe and P. J. Dyson, Phys. Chem. Chem. Phys., 2010, 12, 1834–1841 RSC.
| This journal is © The Royal Society of Chemistry 2014 |