 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organosilane oxidation by water catalysed by large gold nanoparticles in a membrane reactor†
Vitaly
Gitis
*ab,
Rolf
Beerthuis
a,
N. Raveendran
Shiju
*a and
Gadi
Rothenberg
a
aVan 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098XH Amsterdam, The Netherlands. E-mail: n.r.shiju@uva.nl; Web: http://hims.uva.nl/hcsc Tel: +31 20 525 6515
bUnit of Environmental Engineering, Ben-Gurion University of the Negev, PO Box 653, Beer-Sheva 84105, Israel. E-mail: gitis@bgu.ac.il; Fax: +972 8 6479397; Tel: +972 8 6479031
First published on 20th May 2014
Abstract
We show that gold nanoparticles catalyse the oxidation of organosilanes using water as oxidant at ambient conditions. Remarkably, monodispersions of small gold particles (3.5 nm diameter) and large ones (6–18 nm diameter) give equally good conversion rates. This is important because separating large nanoparticles is much easier, and can be done using ultrafiltration instead of nanofiltration. We introduce a simple setup, constructed in-house, where the reaction products are extracted through a ceramic membrane under pressure, leaving the gold nanoparticles intact in the vessel. The nominal substrate/catalyst ratios are ca. 1800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with typical TONs of 1500–1600, and TOFs around 800 h−1. But the actual activity of the large nanoparticles is much higher, because most of their gold atoms are “inside”, and therefore unavailable. Control experiments confirm that no gold escapes to the membrane permeate. The role of surface oxygen as a possible co-catalyst is discussed. Considering the ease of product separation and the robustness of the ceramic membrane, this approach opens opportunities for actual applications of gold catalysts in water oxidation reactions.
:1, with typical TONs of 1500–1600, and TOFs around 800 h−1. But the actual activity of the large nanoparticles is much higher, because most of their gold atoms are “inside”, and therefore unavailable. Control experiments confirm that no gold escapes to the membrane permeate. The role of surface oxygen as a possible co-catalyst is discussed. Considering the ease of product separation and the robustness of the ceramic membrane, this approach opens opportunities for actual applications of gold catalysts in water oxidation reactions.
Silicon alcohols (silanols) are widely used in organic synthesis, particularly in cross-coupling reactions, and in industry as synthons for silicon-based polymeric materials.1 Traditionally, they are synthesized by hydrolysis of halosilanes, stoichiometric oxidation of organosilanes, or reactions of siloxanes with alkali reagents.2 However, these are not clean processes. In contrast, the catalytic oxidation of silanes with water is environmentally benign. It produces silanols with a high selectivity and hydrogen gas as the only by-product. Catalysing chemical reactions in water at room temperature is one of the challenges facing chemists today.3 Oxidizing silanes to silanols is usually catalysed by noble metals, such as rhenium, ruthenium, iridium, silver and gold.
Although gold is traditionally considered (and prized) as inert, its nanoparticles (AuNPs) are enjoying a newly acquired status as catalysts for many reactions.4–17 Kaneda and co-workers18 showed that hydroxyapatite-supported gold particles (3.0 ± 0.9 nm) catalysed silane oxidation in water at 80 °C. Using a different approach, Asao et al.19 reported the use of nanoporous gold with pore size around 30 nm received from a gold–silver alloy for this type of reactions. Recently, carbon nanotube-supported AuNPs (AuCNT) were reported as highly active for silane oxidation in THF at room temperature.20 Gold nanoparticles also catalyse selective activation of alkynes, giving fast hydrogenation or hydration reactions,21 as well as catalysts for the selective conversion of biomass into chemicals22 and in fuel cell reactions.23
The particles' size and shape, dictated by the preparation recipes,24 often determine the catalytic performance. Generally, the smaller the particle, the higher the catalytic activity, down to the three-layered Au55 cluster. Indeed, small gold particles (2–5 nm diameter) catalyse a variety of reactions.25–29 Exceptions are few yet notable, such as the unusually large 38 nm AuNPs reported as catalysts for hydrogen peroxide decomposition to hydroxyl radicals, enhancing the chemiluminescence of the luminol–H2O2 system.30
The problem is that although small AuNPs are exciting as catalysts, their size hampers practical application. Gold is too expensive a catalyst to throw away, and separating particles smaller than 2 nm is no mean feat. Acceptable Au separation levels often require repeated purification cycles or a combination of methods, increasing operational costs and decreasing throughput. One way to solve this is by using pressure-driven membrane separation, a relatively new technology. State-of-the-art nanofiltration (NF) membranes can retain even single-nm particles, but these membranes are expensive, delicate, and have low flow rates.28 Alternatively, separation using ultrafiltration (UF) membranes demands significantly lower transmembrane pressures, and the membranes are cheaper and more robust.31 The problem is that UF membranes are only effective for particles 8–10 nm diameter and larger.
In this paper we show that AuNPs as large as 18 nm can catalyze the oxidation of silanes to silanols with water, generating hydrogen as the only by-product (Scheme 1). We demonstrate the effectiveness of these reactions in a dedicated membrane pressure reactor, using a ceramic membrane for separating the product mixture from the catalyst. Using large AuNPs in such a reactor combines the advantages of high conversion rates and good flux values with full retention of the catalysts.
 | ||
| Scheme 1 Oxidation of silanes to silanols with water using Au nanoparticles. | ||
We prepared monodispersed gold aquasols of varying sizes using the citric acid/tannic acid method (Fig. 1 shows TEM images and gold nanoparticle size distribution; more details are given in the ESI†). In a typical reaction (Scheme 1), organosilanes were oxidized in water in presence of gold nanoparticle catalyst under 1 bar N2 pressure. A production of corresponding silanols was accompanied by a formation of hydrogen gas (this was quantified volumetrically, see ESI†). Reactions were run for up to 24 h in a stainless steel high-pressure reactor, equipped with a membrane, constructed in-house. The gold was retained in the reactor by a porous ceramic membrane placed in the bottom, and the product was isolated and determined using 1HNMR and gas chromatography (full details and drawings in the ESI†). The extraction of reaction products through the membrane was possible using only 4–5 bar additional nitrogen pressure.
 | ||
| Fig. 1 Transmission electron micrographs of gold nanoparticle suspensions with different average diameters: 3.1 ± 0.5 nm (I); 4.8 ± 0.5 nm (II); 9.2 ± 0.8 nm (III). Size distribution of gold nanoparticle suspensions A–G, determined using dynamic light scattering (IV). The distribution function analysis is displayed as scattered intensity per particle size: A (3.1 nm), B (4.8 nm), C (9.2 nm), D (18.3 nm), E (27.5 nm), F (37.4 nm) and G (45 nm). | ||
Fig. 2 shows the conversion profiles of dimethylphenylsilane 1a (top) and the yield profiles for dimethylphenylsilanol 2a (bottom) vs. time in the presence of AuNPs of different sizes. The profiles are divided into small particles and large ones (left and right, respectively) for clarity. For comparison, we also performed a control reaction in the absence of any catalyst. These graphs show two important things. First, there is no significant difference in the conversion when using AuNPs up to 18 nm in diameter. This is important because the filtering of larger particles (>10 nm) is much easier, and can be done using standard ultrafiltration technology. Second, there is a marked difference between substrate conversion and product yield in the absence of AuNPs. Practically, this means that the formation of the disiloxane by-product is suppressed in the presence of the catalyst, as the silane is quickly consumed in the oxidation reaction.
 | ||
| Fig. 2 Conversion profiles of dimethylphenylsilane 1a (top graphs) and yield profiles for dimethylphenylsilanol 2a (bottom graphs) in the presence of AuNPs of different diameters (3.1 nm, 4.8 nm, 18.3 nm, 37 nm and 45 nm). Reaction conditions: 2.93 mmol 1a, 103.3 mM acetone, 222 mM water, 0.31 mg catalyst (1.57 × 10−6 mol gold; 0.05% Au relative to substrate) and 296 K. All experiments were performed in duplicates. | ||
Our dead-end membrane reactor enables quick and efficient separation of the AuNPs. This is easily monitored using UV-visible spectroscopy.32,33Fig. 3 shows the spectra of the reaction mixture before and after the membrane. The permeate shows no absorbance, confirming 100% retention of the AuNPs. Conversely, the samples taken from the reaction mixture show the typical gold plasmon peak at 520 nm, which increases as the effective concentration of the particles increases.32
 | ||
| Fig. 3 UV-visible spectra of the reaction mixture at 0, 1, 2 and 4 h for 18 nm AuNPs, compared with the spectrum of the membrane permeate. Note that the increase in the absorbance of the plasmon peak at 520 nm is due to the removal of solvent during the sampling, which increases the concentration of the NPs.34 | ||
The AuNP catalysts exhibit high turnover numbers (TONs) and turnover frequencies (TOFs). Considering that ca. 85% conversion is reached after 2 h (with 3.1 nm particles) using a nominal substrate/catalyst ratio of 1870:1, typical TONs are 1500–1600, with TOFs around 800 h−1. But the actual catalytic activity is much higher, especially for the larger AuNPs, because of their smaller effective surface area. If we consider that the particles are monodistributed and spherical (see DLS and TEM analyses), the effective substrate/catalyst molar ratios are 2500:1 (3 nm), 3100:1 (6 nm), 10600:1 (18 nm) and 25000:1 (45 nm). Note that this does not mean that gold atoms on the surface of large particles are more active than on smaller ones. Rather, we think that only a small number of atoms (or facets) are actually active, and the nice part is that this “window” of activity ranges up to at least 18 nm sized particles.
Following this, we tested the re-using of the gold NPs in this reactor system (all these control experiments were run in duplicate). Here, we first repeated the reaction with 18 nm particles (Fig. 2c), and after 4 h filtered all the liquids through the membrane, and washed the remains with acetone. The acetone was filtered and new reactants and solvent were added, using the same solid catalyst that remained in the vessel. The results showed 50% conversion and 100% selectivity after 2 h. STEM analysis of the reaction mixture shows some agglomeration after the reaction (see Fig. S3, ESI†). However, not all particles were agglomerated, though they were left deliberately in the reaction mixture for 6 h.
We then examined the scope of the reaction, testing various aromatic and aliphatic organosilanes in the membrane reactor (structure 1a–e, Scheme 1). Table 1 shows the conversion, yield, and nominal TON values. Aliphatic silanes (entries 2, 5) were more active than the aromatic ones. In all cases, we observed complete retention of the nanoparticles within the membrane reactor. Moreover, the membrane could be reused several times.
| No | Substrate | Conversion, % | Yield,b % | TON |
|---|---|---|---|---|
| a Reaction conditions: 0.2 g organosilane, 7.6 ml acetone, 10 ml deionized water, 0.31 mg AuNPs (6 nm) catalyst, reaction time 2 h. Total reaction volume 17.8 ml, magnetic stirring at 400 rpm, 296 K. b GC yield, corrected for the presence of an external standard. c Isolated yield. | ||||
| 1 | 1a, (CH3)2SiHC6H5 | 80.3 | 79.5c | 749 |
| 2 | 1b, (C2H5)3SiH | 90.0 | 86.8 | 984 |
| 3 | 1c, (C6H5)2SiHCH3 | 81.1 | 78.2 | 520 |
| 4 | 1d, (C6H5)3SiH | 79.8 | 76.6 | 389 |
| 5 | 1e, [(CH3)2CH]3SiH | 83.4 | 80.8 | 669 |
We may anticipate that the reaction begins with the insertion of the Si–H bond on the Au nanoparticle to generate a silyl-metal hydride intermediate. The silyl-metal hydride intermediate is subsequently attacked by a nucleophile derived from water to generate the silanol (this attack is most likely an SN2 type, and therefore an inversion would be expected, since it would be easier for the oxygen to attack trans to the NP surface). However, this is only a suggestion. In the final step of the cycle, Au–Hδ+ and Osδ−⋯Hδ+ species should react to produce H2, regenerating the catalyst surface (Scheme 2).
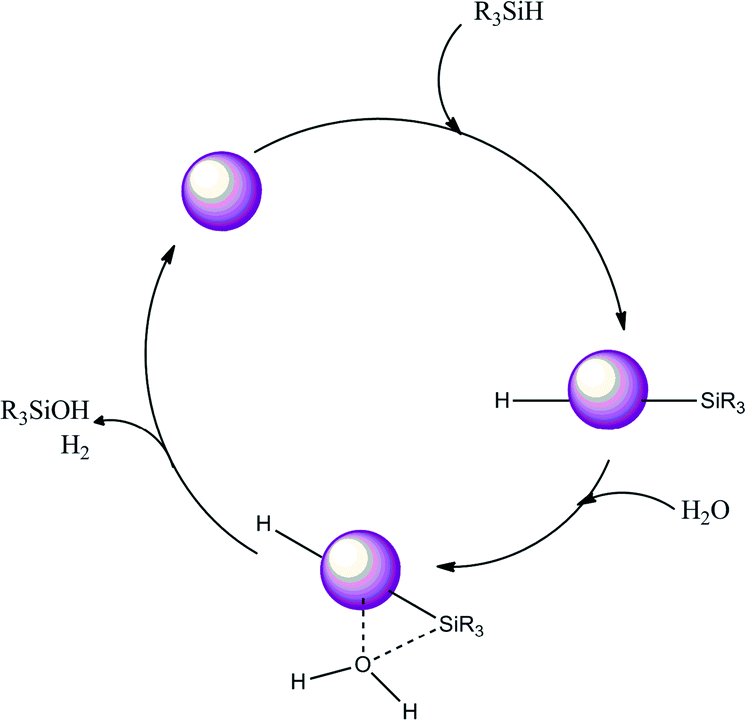 | ||
| Scheme 2 Suggested route for the oxidation of silanes to silanols with water using Au nanoparticles. | ||
The critical step of the reaction is most likely the dissociative activation of water. Our observations show that this step does not depend on the particle size alone below a certain size. Recent surface science studies proposed that an oxygen atom on Au(111) surface (Os) acts as a basic co-catalyst that promotes various organic reactions at low temperatures.35–38 Cooperation of metal NPs as redox sites and the surface oxygen as a basic co-catalyst leading to enhanced reactivity for the dissociation of water was also confirmed on Ru, Rh, Pd, Ir, and Pt surfaces.39 Furthermore, recent theoretical work showed that the reaction barrier for the water dissociation on the Pd(111) surface contaminated with Os (0.34 eV) was significantly lower than that on the clean Pd(111) surface (0.92 eV).40 This is due to the bonding interaction between the Os and a hydrogen atom in H2O at the transition state (Osδ−⋯Hδ+⋯OH). This model was consistent with previous results of the surface science experiments for water dissociation on clean and oxygen precovered Pd surfaces, in which OH groups were detected only on the latter surface.41
Enhanced reactivity due to adsorbed species on Au surface is reported for CO oxidation as well. For the vapor phase CO oxidation, oxide-supported Au particles in the 2–4 nm size range are the most active.42 Adding water vapor can increase the CO oxidation rate substantially, presumably due to the presence of hydroxyl groups on the surface.43,44 Carbon-supported Au catalysts, which were inactive for the vapor-phase CO oxidation up to 373 K, showed excellent CO oxidation rates in the aqueous phase at 300 K.44 Moreover, a 50-fold increase in rate was observed on going from acidic to basic pH, though Au particles used in aqueous-phase studies were larger than 4 nm.45 All these facts show that the adsorbed species play a major role in enhancing the activity even for larger particles. A similar effect may be operating in our case, making larger particles as active as smaller ones.
Conclusions
Water oxidation of silanes to silanols is efficiently catalysed by “large” gold nanoparticles (6–18 nm in diameter). This catalytic oxidation has several advantages, namely using water as a clean oxidant, high activity and selectivity for silanols. Moreover, it works well with a number of silanes with different types of substituents. Combining this reaction with ultrafiltration membrane reactor technology solves the problem of difficult separation of nanoparticles from the reaction mixture, and opens opportunities for bona fide applications.Acknowledgements
V.G. thanks the European Commission for a Marie Curie International Exchange Fellowship. We thank A. Duek for preparing the AuNP suspensions, and Dr. E. Garnett and M. de Goede (FOM Institute AMOLF) for help with the STEM experiments.Notes and references
- Y. Okada, M. Oba, A. Arai, K. Tanaka, K. Nishiyama and W. Ando, Inorg. Chem., 2010, 49, 383–385 CrossRef CAS PubMed.
- W. Li, A. Wang, X. Yang, Y. Huang and T. Zhang, Chem. Commun., 2012, 48, 9183–9185 RSC.
- K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Baumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. J. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS PubMed.
- G. J. Hutchings, M. Brust and H. Schmidbaur, Chem. Soc. Rev., 2008, 37, 1759–1765 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- T. Takei, T. Akita, I. Nakamura, T. Fujitani, M. Okumura, K. Okazaki, J. H. Huang, T. Ishida and M. Haruta, in Adv. Catal., ed. B. C. Gates and F. C. Jentoft, Elsevier Academic Press Inc, San Diego, 2012, vol. 55, pp. 1–126 Search PubMed.
- A. V. Biradar and T. Asefa, Appl. Catal., A, 2012, 435, 19–26 CrossRef PubMed.
- S. Das and T. Asefa, Top. Catal., 2012, 55, 587–594 CrossRef CAS.
- A. F. Lee, S. F. J. Hackett, G. J. Hutchings, S. Lizzit, J. Naughton and K. Wilson, Catal. Today, 2009, 145, 251–257 CrossRef CAS PubMed.
- N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS PubMed.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- C. T. Campbell, J. C. Sharp, Y. X. Yao, E. M. Karp and T. L. Silbaugh, Faraday Discuss., 2011, 152, 227–239 RSC.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 5302–5304 RSC.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. W. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093–10095 CrossRef CAS PubMed.
- J. John, E. Gravel, A. Hagège, H. Li, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533 CrossRef CAS PubMed.
- M. Garcia-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Perez-Ramirez and N. Lopez, ChemPhysChem, 2008, 9, 1624–1629 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- V. Mazumder, Y. Lee and S. H. Sun, Adv. Funct. Mater., 2010, 20, 1224–1231 CrossRef CAS.
- A. V. Gaikwad, P. Verschuren, S. Kinge, G. Rothenberg and E. Eiser, Phys. Chem. Chem. Phys., 2008, 10, 951–956 RSC.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- A. Tsoukala, L. Peeva, A. G. Livingston and H. R. Bjorsvik, ChemSusChem, 2012, 5, 188–193 CrossRef CAS PubMed.
- L. Wen, J. K. Fu, P. Y. Gu, B. X. Yao, Z. H. Lin and J. Z. Zhou, Appl. Catal., B, 2008, 79, 402–409 CrossRef CAS PubMed.
- Z. F. Zhang, H. Cui, C. Z. Lai and L. J. Liu, Anal. Chem., 2005, 77, 3324–3329 CrossRef CAS PubMed.
- A. V. Gaikwad, V. Boffa, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2008, 47, 5407–5410 CrossRef CAS PubMed.
- A. V. Gaikwad and G. Rothenberg, Phys. Chem. Chem. Phys., 2006, 8, 3669–3675 RSC.
- J. Wang, H. F. M. Boelens, M. B. Thathagar and G. Rothenberg, ChemPhysChem, 2004, 5, 93–98 CrossRef CAS PubMed.
- A. V. Gaikwad, P. Verschuren, E. Eiser and G. Rothenberg, J. Phys. Chem. B, 2006, 110, 17437–17443 CrossRef CAS PubMed.
- K. M. Kosuda, A. Wittstock, C. M. Friend and M. Baeumer, Angew. Chem., Int. Ed., 2012, 51, 1698–1701 CrossRef CAS PubMed.
- C. G. F. Siler, B. Xu, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2012, 134, 12604–12610 CrossRef CAS PubMed.
- J. L. Gong and C. B. Mullins, Acc. Chem. Res., 2009, 42, 1063 CrossRef CAS PubMed.
- B. Xu, L. Zhou, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2010, 49, 343 Search PubMed.
- A. Shavorskiy, M. J. Gladys and G. Held, Phys. Chem. Chem. Phys., 2008, 10, 6150–6159 RSC.
- Y. Cao and Z.-X. Chen, Surf. Sci., 2006, 600, 4572–4583 CrossRef CAS PubMed.
- M. Wolf, S. Nettesheim, J. M. White, E. Hasselbrink and G. Ertl, J. Chem. Phys., 1990, 92, 1509 CrossRef CAS PubMed.
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed.
- M. Date, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129 CrossRef CAS PubMed.
- W. C. Ketchie, Y.-L. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94–101 CrossRef CAS PubMed.
- W. C. Ketchie, M. Murayama and R. J. Davis, Top. Catal., 2007, 44, 307–317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures for the synthesis of the gold nanoparticles and the catalytic oxidation of silanes, DLS data for the various nanoparticles, photo and schematic diagram of the experimental setup and SEM image of the membrane. See DOI: 10.1039/c3cy00506b |
| This journal is © The Royal Society of Chemistry 2014 |