Glycol metalloporphyrin derivatives in solution or immobilized on LDH and silica: synthesis, characterization and catalytic features in oxidation reactions†
Kelly A. D. F.
Castro
ab,
Mário M. Q.
Simões
b,
M. Graça P. M. S.
Neves
*b,
José A. S.
Cavaleiro
b,
Fernando
Wypych
a and
Shirley
Nakagaki
*a
aLaboratório de Química Bioinorgânica e Catálise; Universidade Federal do Paraná (UFPR), CP 19081, CEP 81531-990, Curitiba, Paraná, Brazil. E-mail: shirleyn@ufpr.br
bDepartment of Chemistry and QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: gneves@ua.pt
First published on 23rd October 2013
Abstract
We reacted [meso-tetrakis(pentafluorophenyl)porphyrin] with ethylene glycol to obtain porphyrins substituted with one to four hydroxyalkyloxy groups at position 4 of the meso-aryl groups. We then inserted iron(III) or manganese(III) into the mono- and tetra-substituted free-base derivatives, which afforded the corresponding metalloporphyrin complexes. Next, we immobilized the iron(III) and manganese(III) porphyrins on two supports: layered double hydroxide (LDH) and silica (synthesized by the sol–gel process). We characterized the resulting solids using powder X-ray diffraction (PXRD), UV–vis electronic spectroscopy, and electron paramagnetic resonance (EPR), and we investigated the catalytic activity of the materials in both homogeneous and heterogeneous media. The metalloporphyrins provided good catalytic conversions in (Z)-cyclooctene oxidation. As for cyclohexane oxidation, the catalysts were selective for the alcohol instead of the ketone. In the case of heterogeneous catalysis, the immobilized metalloporphyrins furnished slightly lower yields as compared with homogeneous catalysis. However, these solids presented a major advantage: reusability. Indeed, these solid catalysts retained their activity for at least three cycles in the case of (Z)-cyclooctene oxidation.
Introduction
In recent years, various scientists have developed a large number of systems based on synthetic metallomacrocycles to model cytochrome P-450.1–8 Within this class of compounds, porphyrins and analogues have deserved special attention. These versatile derivatives find application in different areas such as catalysis,1–13 medicine,14,15 electronics,16 solar cells,17,18 and sensors,19,20 among others.21 In the field of catalysis, metalloporphyrins have proven to be able to effectively oxidize hydrocarbons and other organic compounds under mild conditions.3,22–36Many studies have shown that structurally modifying metalloporphyrins3,5,37 improves catalytic performance in homogeneous medium. This is a consequence of the stereo-electronic effect that substituents exert on the metalloporphyrin macrocycle: (1) they increase the lifetime and reactivity of the active catalytic species in solution, and (2) they inhibit the formation of dimeric species and the oxidative self-destruction process that inactivates the catalyst.5
Extensive investigations have shown that halogenated metalloporphyrins are more resistant to degradation via free-radical attack than those containing electron-donating substituents. Metalloporphyrins containing electron-withdrawing substituents also efficiently catalyze the oxygenation of hydrocarbons and a wide variety of other organic compounds.3,5,12,38,39
In this context, the free-base ligand [5,10,15,20-tetrakis(pentafluorophenyl) porphyrin], [H2(TPFPP)], constitutes a surprisingly versatile compound, mainly for catalytic purposes.40–44 It consists of an important platform to prepare new porphyrin ligands because it can undergo many types of structural modification via nucleophilic substitution of the p-fluorine atoms.43,44 Chemically modifying [H2(TPFPP)] or the corresponding metal complexes45 with appropriate nucleophiles allows for their functionalization with substituents that will make them potentially applicable in the photodynamic therapy (PDT) of several diseases.44,46,47 In addition, by choosing the right substituents, one can immobilize the metalloporphyrin on different supports, creating new materials for heterogeneous catalysis.11–13,21–25
In general, metalloporphyrins containing bulky or electronegative substituents at any position of the meso-aryl groups present high catalytic efficiency, resulting in good product yields during the oxidation of many substrates.12,13,25,26,48–51 However, the use of these catalysts in homogeneous medium is limited because it is not possible to recover or reuse the catalyst, not to mention that parallel reactions sometimes deactivate the catalytic species.3,5
Therefore, researchers have searched for strategies to immobilize metalloporphyrins on different inorganic supports, so as to enable catalyst recovery and reuse and to prevent catalytic species deactivation.11–13,24–26,34,35,48,52 The immobilization procedure and the support may also lead to a more selective catalysis: the combination of the porphyrin complex and the support may create structures with cavities and pores, culminating in unusual selectivity, such as size- and shape-selectivity.53,54
Studies have shown that metalloporphyrin immobilization on rigid supports, such as silica gel, inhibits dimer formation because it suppresses the interactions between the metalloporphyrin complexes fixed on the support surface.24
In the last decade, silica preparation by sol–gel process emerged as a very useful technique to immobilize different compounds, including catalytically active metalloporphyrins. The sol–gel method produces high-purity solids under mild conditions and at low cost. One possible sol–gel route is the hydrolytic route: polymerization of silicon alkoxides [e.g., tetraethyl orthosilicate (TEOS) (Si(OC2H5)4] affords an inorganic solid bearing siloxane (Si–O–Si) and silanol groups (Si–OH). This process is conducted in the presence of a solvent, usually an alcohol, and an acid or base catalyst.55
Layered double hydroxides (LDH) are another example of inorganic supports; they consist of synthetic lamellar compounds with a structure derived from natural brucite. In the LDH structure, magnesium ions are octahedrally coordinated to hydroxyl groups.56 These octahedra are interconnected, resulting in “two-dimensional” layers that interact via van der Waals forces. Because LDH have divalent and trivalent ions, they contain positively charged layers of metal hydroxides stacked along the basal direction. Intercalation of hydrated anions ensures total neutralization, so the general formula of an LDH can be represented as [M1−xIIIMxII(OH)2]x+[Ax/nn−·yH2O]x−.26,56,57
In this paper, we report on the synthesis of metalloporphyrins bearing appended ethylene glycol units. We aimed to obtain complexes displaying the appropriate structure to interact with solid supports such as LDH and silica (obtained by the sol–gel process). We evaluated the catalytic activity of the synthesized complexes in (Z)-cyclooctene and cyclohexane oxidation in both homogeneous and heterogeneous media. We also describe the recovery and reuse of the heterogeneous catalysts.
Experimental
All the chemicals used in this study were purchased from Aldrich, Sigma, or Merck and were of analytical grade. Iodosylbenzene (PhIO) was synthesized through the hydrolysis of iodosylbenzene diacetate.58 The resulting solid was carefully dried under reduced pressure and kept at 5 °C. Mg/Al LDH at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and intercalated with nitrate anions was prepared as described previously.26,59 Porphyrin P1, used as platform for further functionalization, was prepared according to the literature.60
:1 and intercalated with nitrate anions was prepared as described previously.26,59 Porphyrin P1, used as platform for further functionalization, was prepared according to the literature.60
Synthesis of the free-base porphyrins P2–P6 from the reaction of ethylene glycol with porphyrin P1 (see Fig. 1 for abbreviations)
:1) mixture as the eluent. After solvent evaporation, P1 was recrystallized in dichloromethane–petroleum ether in 11% yield. Molecular formula: C44H10F20N4; 1H NMR δH, ppm (CDCl3): −2.92 (broad s, 2H, NH) and 8.12 (s, 8H, H-β); 19F NMR δF, ppm (CDCl3): −160.00 (8F, dd, J = 25.4 and 8.5 Hz, Ar o-F), −174.7 (4F, t, J = 21.2 Hz, Ar p-F), and −184.79 to −184.97 (8F, m, Ar m-F); UV–vis λmax, nm (CH2Cl2): 410, 504, 539, 582, and 648; MS (m/z): 975 [M]+.
:1 v/v) as the mobile phase. The first fraction was identified as porphyrin P2 (61% yield): m.p. >300 °C; 1H NMR δH, ppm (CD3OD): 9.10 (broad s, 8H, H-β), 4.10 (t, J = 4.5 Hz, 2H, CH2), and 4.70 (t, J = 4.5 Hz, 2H, CH2) (the signal relative to the NH protons was not observed due to their replacement by deuterium from deuterated methanol); 19F NMR δF, ppm (CD3OD): −190.13 (dt, J = 7.5 and 21.4, 6F, F-meta), −184.33 (dd, J = 7.7 and 21.4, 2F, F-meta), −180.54 (t, J = 21.4, 3F, F-para), −167.34 (dd, J = 7.7 and 21.5, 2F, F-ortho), and −165.30 (m, 6F, F-ortho); UV–vis (CH2Cl2) λmax, nm (logε): 412 (5.20), 506 (4.08), 582 (3.90), and 656 (2.95); HRMS (FAB+) m/z: calculated for C46H16F19N4O2 (M + H)+: 1017.096, found: 1017.0964 (Fig. S1, S6, and S11 – ESI†).
:1 v/v) was the mobile phase. P2, P3, P4, P5, and P6 were crystallized in methanol. The first fraction was identified as porphyrin P2 (31.5 mg, 6% yield). The second fraction was identified as porphyrin P3 (8% yield) with the following characteristics: m.p. >300 °C; 1H NMR δH, ppm (CD3OD): 9.10 (broad s, 8H, H-β), 4.10 (t, J = 4.5, 2H, CH2), and 4.70 (t, J = 4.5, 2H, CH2); 19F NMR δF, ppm (CD3OD): −188.64 (dt, J = 7.3 and 21.3, 4F, F-meta), −182.81 (dd, J = 7.9 and 22.1, 4F, F-meta), −179.08 (t, J = 21.3, 2F, F-para), −165.79 (dd, J = 7.5 and 22.1, 4F, F-ortho), and −163.74 (dd, J = 7.7 and 21.3, 4F, F-ortho); UV–vis (CH2Cl2) λmax, nm (logε): 412 (5.45), 506 (4.10), 582 (3.61), and 654 (2.94). HRMS (FAB+) m/z: calculated for C48H20F18N4O4 (M + H)+: 1059.1231, found: 1059.12507 (Fig. S2, S7, and S11 – ESI†).
The third fraction was identified as porphyrin P4 (14% yield) and presented the following characteristics: m.p. >300 °C; 1H NMR δH, ppm: 9.10 (broad s, 8H, H-β), 4.10 (t, J = 4.4, 2H, CH2), and 4.70 (t, J = 4.4, 2H, CH2); 19F NMR δF, ppm (CD3OD): −188.62 (dt, J = 7.6 and 21.7, 4F, F-meta), −182.82 (dd, J = 7.7 and 21.5, 4F, F-meta), −179.06 (t, J = 21.7, 2F, F-para), −165.77 (dd, J = 7.7 and 21.5, 4F, F-ortho), and −163.75 (dd, J = 7.6 and 21.7, 4F, F-ortho); UV–vis (CH2Cl2) λmax, nm (logε): 412 (5.42), 506 (4.08), 582 (3.60), and 654 (2.92); HRMS (FAB+) m/z: calculated for C48H20F18N4O4 (M + H)+: 1059.1231, found: 1059.12498 (Fig. S3, S8, and S11 – ESI†).
The fourth fraction was identified as porphyrin P5 (40% yield), which had the following features: m.p. >300 °C; 1H NMR δH, ppm (CD3OD): 9.10 (broad s, 8H, H-β), 4.11 (t, J = 4.4, 2H, CH2), and 4.70 (t, J = 4.4, 2H, CH2); 19F NMR δF, ppm (CD3OD): −188.63 (dt, J = 7.3 and 21.3, 2F, F-meta), −182.84 (dd, J = 7.3 and 21.3, 6F, F-meta), −179.13 (t, J = 21.3, 1F, F-para), −165.75 (dd, J = 7.3 and 21.3, 6F, F-ortho), and −163.73 (dd, J = 7.4 and 21.3, 2F, F-ortho); UV–vis (CH2Cl2) λmax, nm (logε): 412 (5.30), 510 (4.19), 582 (3.65), and 654 (2.69). HRMS (FAB+) m/z: calculated for C50H25F17N4O6 (M + H)+: 1101.1536, found: 1101.1559 (Fig. S4, S9, and S11 – ESI†).
Finally, the fifth fraction was identified as porphyrin P6 (25% yield): m.p. >300 °C; 1H NMR δH, ppm (CD3OD): 9.10 (broad s, 8H, H-β), 4.11 (t, J = 4.4, 2H, CH2) , and 4.70 (t, J = 4.4, 2H, CH2); 19F NMR δF, ppm (CD3OD): −182.86 (dd, J = 7.5 and 21.5, 8F, F-meta) and −165.73 (dd, J = 7.4 and 21.5, 8F, F-ortho); UV–vis (CH2Cl2) λmax, nm (logε): 412 (4.90), 508 (4.20), 582 (3.69), and 654 (2.73); HRMS (FAB+) m/z: calculated for C52H31F16N4O8 (M + H)+: 1143.1842, found: 1143.1864 (Fig. S5, S10, and S11 – ESI†).
The above reaction yields were achieved after 24 h; when the reaction was performed for 8 h, the reaction yields were as follows: (7%), P2 (22%), P3 (11%), P4 (12%), P5 (40%), and P6 (3%).
The preparation of P6 was also optimized to 96% yield. For this purpose, the reaction was performed by reacting P5 (0.02 mmol) with an excess of ethylene glycol (12.5 mmol) and 0.21 mmol of NaH for 3 h at 80 °C (Fig. 1).
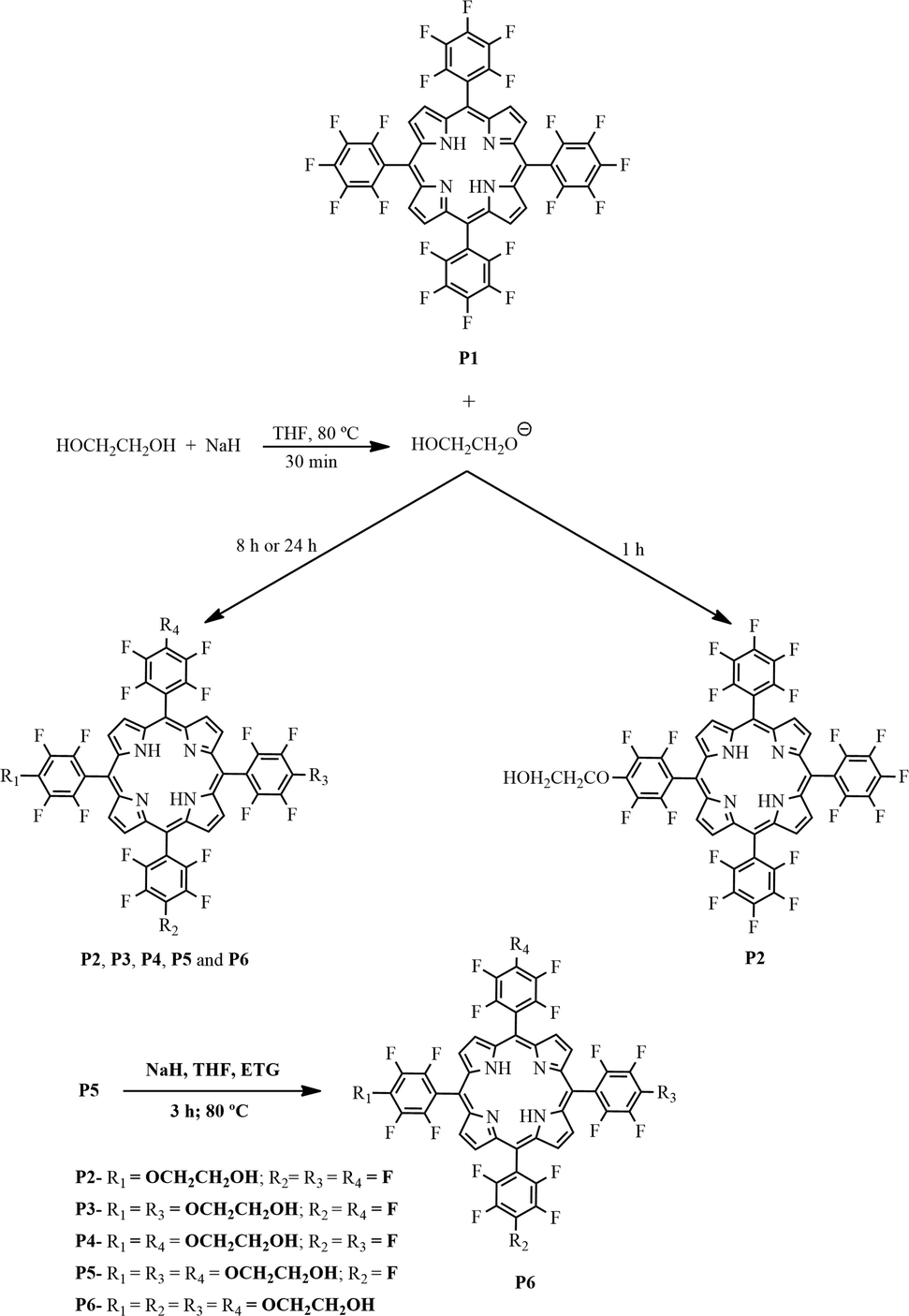 | ||
| Fig. 1 Schematic representation of the reactions of porphyrin P1 with ethylene glycol in order to obtain derivatives P2–P6. | ||
Synthesis of manganese and iron porphyrins (MPx)
The free-base porphyrins P2 and P6 were metallated with FeCl2 or Mn(CH3COO)2, by modifying the conventional methodology reported by Kobayashi, using 0.02 mmol of porphyrin dissolved in 15 mL of DMF and an excess of 5 times of the metal salt.61 For the metallation with manganese, two drops of pyridine were added to aid ligand deprotonation. This reaction was allowed to proceed under reflux conditions for 8 h (compounds MP9 and MP10). Concerning the metallation with iron, DMF was also used as the solvent with an excess of 5 times of the metal salt, and the reaction was carried out under reflux conditions for 24 h. To help with ligand deprotonation, sodium acetate (0.06 mmol) was also added to the reaction medium (compounds MP7 and MP8).After the metallation process, the solvents were removed under vacuum. The resulting solids were washed thoroughly with water to remove excess metal salts. The complexes were also purified by column chromatography using methanol as the eluent and silica as the stationary phase, the yields ranging from ca. 94 to 98%.
The resulting metalloporphyrins were designated MPx (where M = Fe3+ or Mn3+ and x = 7, 8, 9, or 10) (Fig. 2) and presented the following characteristics: FeP7 (C46H13F19FeN4O2) UV–vis (methanol) λmax, nm (logε): 406 (4.30), 502 (3.21), and 579 (3.10); MALDI (TOF/TOF) (m/z): 1070.01 [M + H]+, corresponding to the calculated formula C46H13F19FeN4O2. FeP8 (C52H28F16FeN4O8) UV–vis (CH2Cl2) λmax, nm (logε): 390 (4.56), 506 (3.55), and 572 (3.11); HRMS (FAB+) (m/z): 1196.09968 [M + H]+, corresponding to the calculated formula C52H18F16FeN4O8. MnP9 (C46H13F19MnN4O2) UV–vis (CH2Cl2) λmax, nm (logε): 456 (4.47), 500 (3.12), and 552 (3.36); HRMS (FAB+) (m/z): 1069.01101 [M + H]+, corresponding to the calculated formula C46H13F19MnN4O2. Anal. calc. for C48H16F19MnN4O4·2H2O: C, 49.5; H, 1.73; N, 4.81. Found: C, 49.5; H, 2.46; N, 5.17%. MnP10 (C52H28F16MnN4O8) UV–vis (CH2Cl2) λmax, nm (logε): 456 (4.38), 500 (3.09), and 552 (3.35); HRMS (FAB+) (m/z): 1195.10267 [M + H]+, corresponding to the calculated formula C52H18F16MnN4O8 (Fig. S13–S20 – ESI).
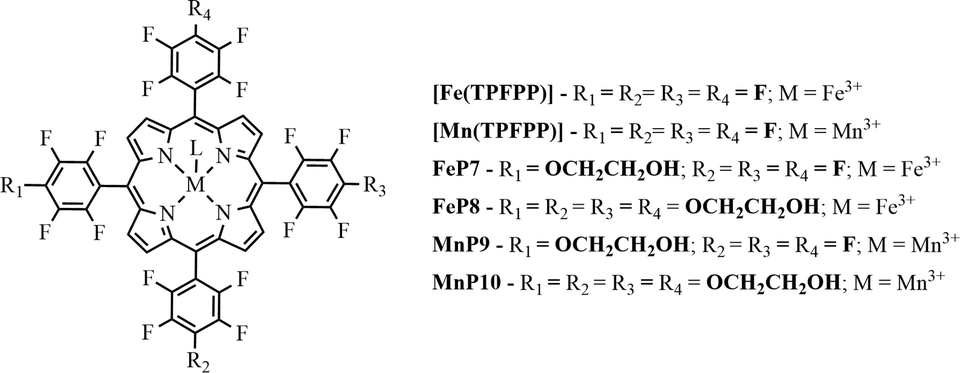 | ||
| Fig. 2 Schematic representation of iron(III) and manganese(III) porphyrins, where L represents the counter-ion (chloride or acetate). | ||
Immobilization of porphyrin derivatives MP2 and MP6 on different supports
:1) intercalated with nitrate anions was prepared using the co-precipitation methodology as described previously.26,59MPx–LDH (where M = Fe3+ or Mn3+ and x = 7, 8, 9, or 10) were prepared by dispersing around 500 mg of the support LDH in 10 mL of methanol and MPx (2.6 μmol (3.2 mg) of MnP9 and about 7.1 μmol (8.0 mg) of the other metalloporphyrins).26 This suspension was stirred for 5 h, and the resulting solid was filtered and exhaustively washed with methanol. The supernatant and the methanol from the washing process were analyzed by using UV–vis spectroscopy to quantify the MPx that was removed from the matrix by leaching during the washing process; the MPx loading was calculated for each solid. The final light brown solids MPx–LDH were dried at 55 °C for 48 h.
Catalytic oxidation reactions
Catalytic oxidation reactions were carried out in a 2 mL thermostatic glass reactor equipped with a magnetic stirrer placed inside a dark chamber. (Z)-Cyclooctene (previously purified on an alumina column) or cyclohexane oxidation by PhIO was accomplished in the presence of the tested catalyst. In a standard heterogeneous catalysis experiment, the different MPx-support catalysts and the oxidant (MPx/PhIO molar ratio = 1:50) were suspended in 400 μL of acetonitrile and degassed with argon for 15 min. The reaction started after the substrate was added to the reaction medium (MPx/substrate molar ratio = 1:5000); oxidation was carried out under magnetic stirring for 1 h. At the end of the reaction, sodium sulfite was introduced in the reaction mixture to eliminate excess PhIO. The reaction solution, along with the reaction products, was transferred to a 2 mL volumetric flask. The solid catalyst was washed several times with methanol and acetonitrile to extract any reaction product that might have remained adsorbed on it. The washing solutions were added to the previously separated reaction supernatant, and the content of these combined solutions was analyzed by gas chromatography, using high-purity (99.9%) undecane or 1-octanol (acetonitrile solution, 1.0 × 10−2 mol L−1) as the internal standard. Product yields were based on the amount of PhIO added to each reaction. Control reactions were also performed in the absence of MPx using the same methodology described above. A similar experimental procedure was followed for the homogeneous catalysis assays, but the solid catalyst washing process was unnecessary.
The solids that displayed better catalytic activity in the first use were recovered; washed with water, methanol, acetonitrile, and dichloromethane; dried; and reused in other catalytic cycles using (Z)-cyclooctene as substrate. The resulting washing solutions of all the recovered catalysts were analyzed by using UV–vis spectroscopy to evaluate whether MPx was leached from the support.
Characterization techniques
1H and 19F NMR spectra were recorded on a Bruker Avance 300 spectrometer at 300.13 and 282.38 MHz, respectively. Deuterated chloroform or methanol was used as the solvent; TMS (δ = 0 ppm) was the internal standard. Chemical shifts are reported in ppm (δ); coupling constants (J) are given in Hz. Mass spectra were acquired using a 4800 Proteomics Analyzer mass spectrometer (MALDI TOF/TOF); HRMS spectra were recorded on VG AutoSpec Q and M mass spectrometers (Vigo University).Electronic spectra (UV–vis) were obtained using a Cary–Varian or a Shimadzu UV-2501PC spectrophotometer in the 300–800 nm range. To characterize the solids by diffuse reflectance, a UV–vis Jasco V560 spectrophotometer was used.
Transmission Fourier Transform Infrared (FTIR) spectra were registered on a FT Mattson 7000 Galaxy spectrophotometer in the 400–4000 cm−1 range using KBr pellets. KBr was ground together with a small amount of the solid to be analyzed, and the spectra were collected with a resolution of 4 cm−1 and an accumulation of 64 scans.
For X-ray diffraction (XRD) measurements, self-oriented films were placed on neutral glass sample holders. The measurements were performed in the reflection mode using a Shimadzu XRD-6000 diffractometer operating at 40 kV and 40 mA (Cu-Kα radiation λ= 1.5444 Å) with a dwell time of 1° min−1.
Electron paramagnetic resonance (EPR) measurements of the powder materials were conducted on an EPR Bruker EMX microX spectrometer (frequency X, band 9.5 GHz) at room temperature and at 77 K (using liquid N2), using the perpendicular microwave polarization (perpendicular polarization CW-EPR).
The products from the catalytic oxidation reactions were quantified using the gas chromatographs Varian 3900 and Agilent 6850 (FID detector) equipped with a DB-5 type capillary column (J&W Scientific) or a DB-WAX type capillary column (J&W Scientific). Quantitative analysis was carried out by internal standard methodology.
Results and discussion
Synthesis of the free-base porphyrins P2–P6
Porphyrin P1 [H2(TPFPP)] is a suitable platform to obtain different porphyrin and metalloporphyrin derivatives. Nucleophiles can easily replace the fluorine atom in the p-position, affording the desired products in good yields.43The fluorine atoms' reactivity depends on the nucleophile. Sulfur-containing derivatives are more reactive than amines and alcohols (HS–CH2R > H2N–CH2R ~ HO–CH2R), so they require less drastic conditions.44
We adapted the reaction conditions used to prepare the free-base porphyrins P2–P6 (Fig. 1) from literature procedures.44,62 First, to improve ethylene glycol nucleophilicity, we generated the corresponding sodium salt by reacting this compound with sodium hydride in THF at room temperature. Then, we added porphyrin P1 to the resulting mixture and stirred it at 80 °C for 1, 8, or 24 h.
The yields of mono- (P2), di- (P3 and P4), tri- (P5), and tetra- (P6) substituted porphyrins (Fig. 1) depended on reaction time and NaH amount. Using the reaction time of 1 h and 8:1 NaH/porphyrin ratio, we were able to preferentially isolate the mono-substituted porphyrin P2 (61% yield). Increasing the reaction time to 24 h and using a 33:1 NaH/porphyrin ratio favored the formation of tri- and tetra-substituted porphyrins (P5, 40%; P6, 25%). After 8 h, we also isolated the tri-substituted porphyrin P5 in 40% yield, as well as porphyrins P2–P4 in yields ranging from ca. 11 to 22%; we obtained porphyrin P6 as a minor fraction (3%). As stated in the experimental part, we obtained the tetra-substituted porphyrin P6 in excellent yield (96%) when we conducted the last substitution on the tri-substituted porphyrin P5 in the presence of a large molar excess of ethylene glycol.
We identified the first isolated fraction as the mono-substituted porphyrin P2 using 1H and 19F NMR; this compound bore a single group derived from ethylene glycol, with an Rf (retention index) very close to that of the unsubstituted porphyrin P1. Fractions 2 and 3 corresponded to the di-substituted porphyrins P3 and P4; their Rf values were close. The fourth fraction corresponded to the tri-substituted porphyrin P5, whereas the fifth fraction, the most polar, consisted of the tetra-substituted porphyrin P6.
We numbered each of the porphyrins in accordance with their polarity; P1 was the least polar. We characterized porphyrins P2 to P6 (Fig. 1) by using UV–vis and FTIR spectroscopies, 1H and 19F NMR, and HRMS (FAB+) (Fig. S1–S12 – ESI†).
The UV–vis spectra of porphyrins P2–P6 did not significantly differ from the spectrum of the starting porphyrin P147 (Fig. S11 – ESI†). Therefore, substitution of the fluorine atom did not cause major changes in the free-base porphyrins. In other words, the different peripheral substituents did not alter the transition mode of the porphyrin molecules.63
P2 to P6 exhibited the typical FTIR bands of free-base porphyrins at 3342, 3122, and 2937 cm−1, due to NH, CH (phenyl), and CH (pyrrole) stretching, respectively; 1575 cm−1, related to symmetric angular deformation in the plane of the pyrrole ring NH; 1482 cm−1, corresponding to CH–R′–R′′ axial deformation; and 1372 cm−1, related to C–N axial deformation, among others. In addition, we also detected other bands characteristic of ethylene glycol binding to the porphyrin, namely at 1078 cm−1 (C–O–C symmetric axial deformation), 1152 cm−1 (C–O–C asymmetric axial deformation), 1457 cm−1 (CH deformation), and 3434 cm−1 (OH stretching)64 (Fig. S12 – ESI†).
Synthesis of manganese and iron porphyrins (MPx)
We metallated porphyrins P2 and P6 using the appropriate iron(III) and manganese(III) salts (Fig. 2). We employed UV–vis and FTIR spectroscopies, as well as HRMS (FAB+) and MALDI (TOF/TOF), to confirm the metallation process (Fig. S13–S16 – ESI†); we verified the MPx oxidation state using EPR (Fig. 3).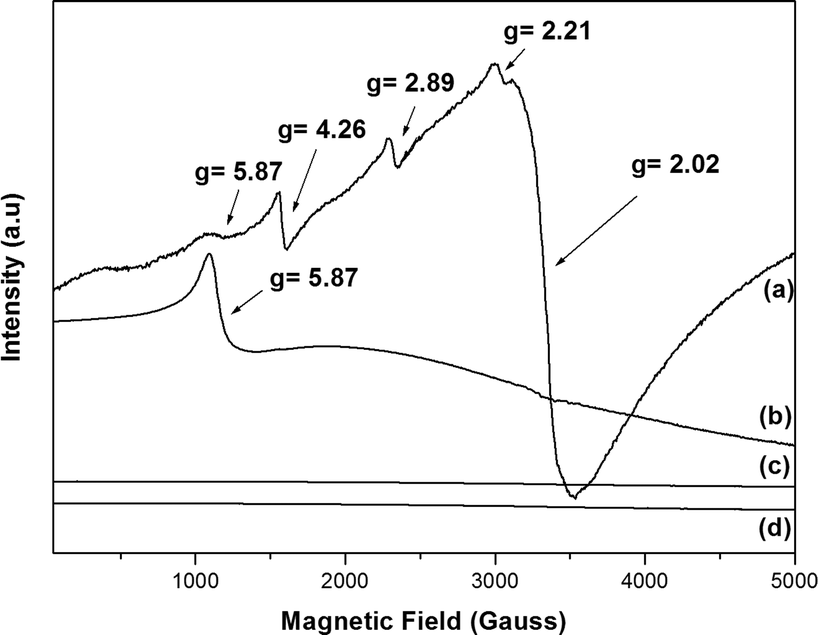 | ||
| Fig. 3 EPR spectra at room temperature: (a) FeP8, (b) FeP7, (c) MnP9, and (d) MnP10. | ||
The combination of five unpaired electrons in high-spin Fe3+ will result in three doublets, ±1/2, ±3/2, and ±5/2 (Kramer doublets). The extent to which these energy levels are filled depends on the separation field and the temperature.65 In a stronger symmetry tetragonal crystal field, the parameter field deployment is large; hence, one can detect the transitions only at ±1/2 (g⊥ = 6.0 and g∥ = 2.0). When the local symmetry changes from tetragonal to orthorhombic (less symmetric), the resulting distortion in the porphyrin plane distorts the associated levels, generating other signals. Systems with maximum distortion contain only one signal (g = 4.3). Two parameters can describe the separation field: D (axial separation) and E (rhombic separation); the ratio between D and E can range from 0 to 0.33 (maximum rhombohedral character), and the respective g values arise.66 Thus, in solution or in the solid state, at room temperature or at lower temperature, iron porphyrins generally present signals in the region of g⊥= 6.0 and g∥ = 2.0, which can be attributed to high-spin Fe(III) in axial symmetry and S = 5/2.66
The EPR spectra of complexes FeP7 and FeP8 (Fig. 3a and b) displayed the characteristic signal of high-spin Fe(III) in axial symmetry. Besides that, FeP8 presented low-intensity signals due to high-spin Fe(III) at g = 4.26 (Fig. 3a), which corresponds to high-spin Fe(III) in orthorhombic symmetry.67FeP8 also displayed other signals in the region of g = 2.0.
We estimated the D/E ratio using a rhombogram for Fe (S = 5/2)66 to verify whether all the signals appeared in the spectrum. We estimated the ratio to be 0.16; therefore, the signals should be detected at transition states ±1/2 and ±3/2, with the values of g = 8.3, g = 6.0, g = 2.9, and g = 2.0. Such ratio indicated that FeP8 had a rhombic distortion as compared with FeP7.
The influence of axial ligands and the presence of extra groups either in the macrocycle structure or in the axial ligand should be considered when analyzing macrocycle distortion. In our case, the chloride axial ligand in the iron porphyrin cannot distort the macrocycle and presents concomitant EPR signals, so the four OCH2CH2OH groups in FeP8 might explain the macrocycle distortion (orthorhombic symmetry) and the EPR signals.
The manganese porphyrin perpendicular microwave polarization X-band EPR spectra at room temperature and 77 K were similar, with no EPR signals (Fig. 3c and d), attesting that MnP9 and MnP10 are Mn(III) porphyrinates. The Mn(III) ion has four unpaired electrons and a d4 configuration with S = 2, typically featuring a pronounced Jahn–Teller distortion that results in a substantial spin-orbit coupling. As a result, mononuclear Mn(III) centers typically present no signals (EPR silent) in the X-band EPR technique under perpendicular microwave polarization.68
Immobilization of FeP7, FeP8, MnP9, and MnP10 on different supports
:1, containing nitrate as intercalated anion) through co-precipitation and used it without any further functionalization. We characterized the solids using powder X-ray diffraction and infrared spectroscopy; the results agreed with literature data.11–13,25,26,35,59 We immobilized MPx (MP7 to MP10) on the prepared LDH to obtain MPx–LDH. The way these neutral MPx anchor on the LDH support still requires elucidation, but one cannot exclude the interactions between the π-conjugated electron cloud of the macrocycle rings and the structure of highly hydroxylated layers11–13,25,26 (Fig. 4). Table 1 lists the percentages of MPx immobilized on the LDH.
 | ||
| Fig. 4 Proposed modes for MPx immobilization on LDH; mono-substituted MPx (MP7 and MP9) and tetra-substituted MPx (MP8 and MP10). | ||
| Solid | Percentage of immobilizationa | Loading (complex per mass of prepared solid) | |
|---|---|---|---|
| (mol g−1)b | % (m/m)c | ||
| a Immobilization percentage based on the mass of MPx used during MPx immobilization on LDH and on silica. b Mol of MPx per mass (g) of solid support. c Mass (g) of MPx per mass (g) of solid support. | |||
| FeP7–SGA | 100 | 3.2 × 10−6 | 0.36 |
| FeP7–LDH | 97 | 1.2 × 10−5 | 1.38 |
| FeP8–SGA | 100 | 3.1 × 10−6 | 0.38 |
| FeP8–LDH | 88 | 1.3 × 10−5 | 1.59 |
| MnP9–SGA | 100 | 2.8 × 10−6 | 0.34 |
| MnP9–LDH | 100 | 1.4 × 10−5 | 1.57 |
| MnP10–SGA | 100 | 2.8 × 10−6 | 0.35 |
| MnP10–LDH | 90 | 5.1 × 10−6 | 0.64 |
Powder X-ray diffraction is a powerful tool to monitor the intercalation of compounds into different layered compounds.11,12,69 The basal distances obtained before and after the immobilization process provide clues about the immobilization mode.12,25,26 For example, when the intercalation of the complex occurs in the LDH interlayer space, the basal spacing increases.12
The solids resulting from MPx immobilization on the LDH displayed an X-ray diffraction pattern similar to those obtained from the pure support (LDH) (Fig. 5). Diffraction peaks appeared at 10.90, 21.94, and 34.69 (in 2*θ), which correspond to the basal (00l) diffraction peaks. The basal distance of 8.9 Å, obtained from the higher order basal peak, is characteristic of hydrated nitrate anions intercalated between the LDH layers.70 This clearly shows that MPx did not intercalate between the layers, but they were adsorbed at the outer layered crystal surface or even became trapped in the voids of the agglomerated crystals (Fig. 4).
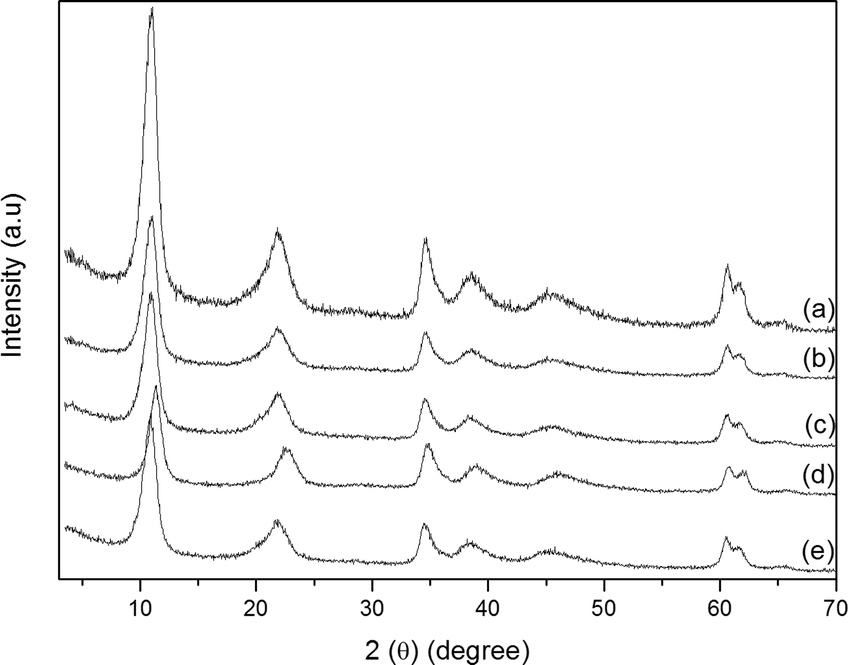 | ||
| Fig. 5 Powder X-ray diffraction patterns: (a) LDH, (b) FeP7–LDH, (c) FeP8–LDH, (d) MnP9–LDH, and (e) MnP10–LDH. | ||
We confirmed the presence of FePx and MnPx in the LDH by UV–vis analyses. We detected the typical Soret band of each MPx in the spectra of the solid samples (432, 418, 472, and 476 nm for FeP7–LDH, FeP8–LDH, MnP9–LDH, and MnP10–LDH, respectively, Fig. 6).
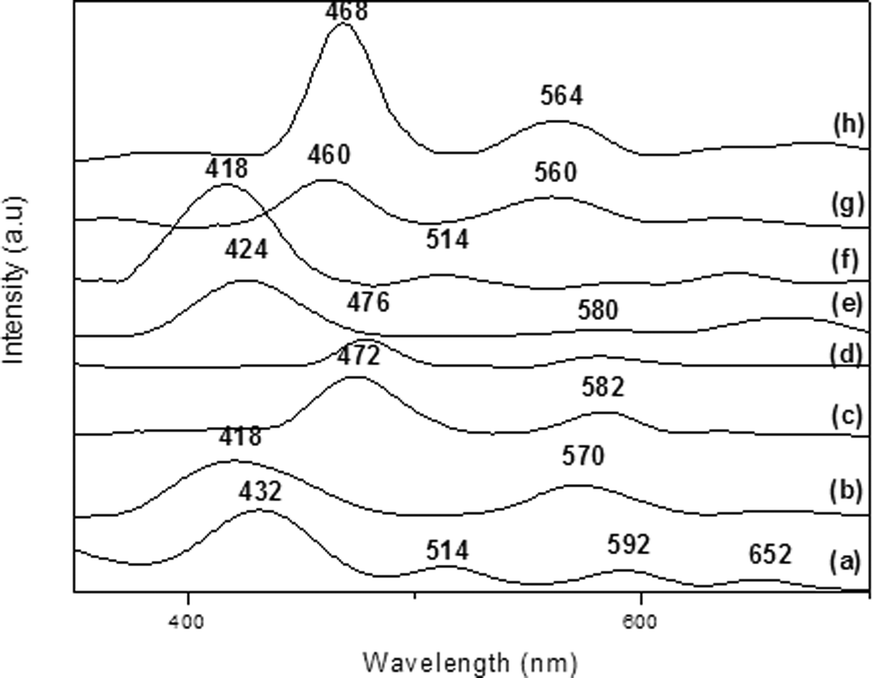 | ||
| Fig. 6 UV–vis spectra of (a) FeP7–LDH, (b) FeP8–LDH, (c) MnP9–LDH, (d) MnP10–LDH, (e) FeP7–SGA, (f) FeP8–SGA, (g) MnP9–SGA, and (h) MnP10–SGA. | ||
The spectrum of the raw LDH did not present bands in the region of 400 nm, so the bands in this region of the spectra of MPx–LDH can only be due to immobilized MPx. The structural distortion of the porphyrin ring upon metalloporphyrin immobilization on the rigid support tends to shift the electronic spectrum bands to higher wavelength. This behavior stems from the steric hindrance posed by the medium, which can substantially change the metalloporphyrin structure.34 When we compared the UV–vis spectra of the immobilized MPx–LDH (Fig. 6) with the spectra of the solid free MPx samples (Soret band at 418, 402, 460, and 460 nm for FeP7, FeP8, MnP9, and MnP10, respectively, figures not shown), we noted approximately 20 nm bathochromic shifts of the Soret band, depending on MPx. This indicated that immobilization significantly distorted the metalloporphyrin ring. These shifts can denote destabilization of the frontier orbital HOMO in relation to the LUMO in distorted systems, which could result from the physical and chemical interactions between MPx and the support surface.71 Metalloporphyrins can interact with the hydroxyl groups present on the LDH surfaces without significantly distorting the complex; on the other hand, metalloporphyrins can undergo distortion to maximize interaction with the support (Fig. 4). In fact, immobilization can greatly distort the porphyrin ring so that the a2u porphyrin orbital (HOMO orbital) approaches the eg orbital (LUMO), thereby reducing the total energy and shifting the band toward the red region of the spectrum.72
Compared with Fe, Mn perfectly fits the cavity of the porphyrin ring, so FePx tends to be more distorted. The metal size and its fitting into the porphyrin ring can influence the ring conformation and the stability of the complex.73
The interaction between MPx and the solid support may account for the shift and broadening of the bands in the spectra of MPx–LDH; indeed, this interaction may pose steric limitations on immobilized MPx.74 Interestingly, the immobilized tetra-substituted MPx (FeP8–LDH and MnP10–LDH) displayed a more red-shifted Soret band than FeP7 and MnP9, an effect that can be attributed to the MPx structure: FeP8 and MnP10 contain four extra substituents at the meso-phenyl groups, whereas and MnP9 have only one. As discussed before, these groups exert a larger steric effect on the porphyrin ring. To adjust the macrocycle structure to the LDH layers and to promote MPx immobilization, it is possible that the ring experiences some distortion, which shifts the Soret band. The larger the number of substituents bound to the porphyrin, the more red-shifted the Soret band becomes (Fig. 6).
Studies on the electronic spectroscopy and surface photochemistry of organic molecules adsorbed on silica gel have shown that the adsorption of organic molecules on silica generally results in red-shifted bands, a consequence of the interaction of π electrons (porphyrin) with the surface hydroxyls.75
In addition to the intense band in the region of 400 nm, characteristic metalloporphyrin bands appeared in the visible region of the MPx–LDH spectra (named Q bands).
The EPR technique also attested to the presence of the iron porphyrins in FeP7–LDH and FeP8–LDH: these solids displayed the characteristic Fe(III) EPR signals (figures not shown).
When one accomplishes metalloporphyrin immobilization on silica obtained by the sol–gel process, it is possible that the metallocomplex inserts into the three-dimensional structure voids or between the particles. The complex might also bind to the hydroxylated silica surface. The exact immobilization mode is difficult to determine, but different techniques can detect silica formation and the presence of each complex.
We analyzed the solids MPx–SGA (silica obtained by the sol–gel process under acid catalyzed conditions), as well as the control silica SGA (without MPx) using PXRD (figures not shown). All PXRD patterns exhibited the same profile: a typical halo in the region of 20 and 30° (in 2θ), corresponding to an amorphous structure.34,80
We also characterized MPx–SGA using FTIR (figures not shown). We detected the characteristic bands of the support in the region of 3420 cm−1, due to typical axial deformation of the surface Si–OH groups; at 1630 cm−1, assigned to the bending of physisorbed water molecules; and at 1100 cm−1, typical of the stretching of the 180-degree angle of the Si–O–Si groups of the four tetrahedral SiO2. We did not observe the FTIR bands related to MPx, probably because MPx concentration was low as compared with the amount of support (Table 1).
We confirmed the presence of FePx and MnPx in SGA by UV–vis spectroscopy (Fig. 6, spectra e–h); we detected the typical Soret band of each MPx in the spectra (424, 418, 460, and 468 nm for FeP7–SGA, FeP8–SGA, MnP9–SGA, and MnP10–SGA) (Fig. 6).
When we compared the UV–vis spectra of immobilized FePx–SGA with the spectra of the solid samples of free FePx, once again we noted that the Soret band shifted toward red. Hence, the FePx ring experiences some kind of distortion due to its confinement in the support. The shift verified that MnP10–SGA was smaller, suggesting that FePx underwent greater distortion in the silica network. As for MnP9–SGA, we did not notice any significant shifts, suggesting that it immobilizes on the support in a different fashion. In fact, Iamamoto and co-workers reported that such shift to wavelengths of lower energy upon metalloporphyrin immobilization is probably due to the putative distortion of the porphyrin ring; interaction of the macrocycle with the silica surface favors this distortion.81
The change in the UV–vis spectrum of metalloporphyrins immobilized on silica seems to depend on both the structure of the complex and the support. Vidoto et al.74 and Castro et al.34 observed a red-shifted Soret band when they immobilized β-halogenated73 and meso-substituted34 iron porphyrins on a silica matrix using the sol–gel process. Poltowicz et al. also observed that the UV–vis spectra of supported metalloporphyrins did not change with respect to the spectra of the corresponding metalloporphyrins in homogenous solution only when the ring did not undergo extensive distortion during the immobilization process.82
The EPR technique also helped us characterize FePx–SGA, namely the iron oxidation and spin state, and the possible distortions in the porphyrin ring due to immobilization. We obtained spectra similar to those observed for free FePx and FePx–LDH, suggesting that FePx immobilization on silica did not distort the ring considerably (figures not shown).
Catalytic oxidation reactions
We investigated the catalytic activities of non-immobilized MPx (homogeneous catalysis) and MPx supported on LDH or silica (MPx–LDH or MPx–SGA, heterogeneous catalysis) in (Z)-cyclooctene epoxidation (Tables 2 and 3, respectively). We also conducted recycling studies on the solids that performed better during heterogeneous catalysis (Table 4). Furthermore, we tested these catalysts in cyclohexane oxidation (Tables 5 and 6); cyclohexane is a more inert substrate, and metalloporphyrins catalyze its oxidation to two major products: cyclohexanol and cyclohexanone. For comparison purposes, we evaluated the homogeneous and heterogeneous catalysts under similar reaction conditions. We confirmed that the catalytic results achieved with MPx–LDH and MPx–SGA were really due to the immobilized MPx: control reactions carried out with the pure matrices of LDH and SGA (containing no MPx) did not give any oxidation products (in the case of cyclohexane) or furnished oxidation products in very low yields (for (Z)-cyclooctene).| Catalyst | Run | Epoxide yieldb (%) |
|---|---|---|
|
a The cyclooctene oxide yield was calculated on the basis of the amount of PhIO used in each reaction. The catalytic results represent an average of at least duplicate reactions. MP/PhIO/(Z)-cyclooctene molar ratio = 1:50:5000. Reaction time: 1 h.
b Cyclooctene oxide.
c Control reaction with no catalyst addition.
|
||
| [Fe(TPFPP)]Cl | 1 | 100 |
| FeP7 | 2 | 71 |
| FeP8 | 3 | 94 |
| [Mn(TPFPP)]Cl | 4 | 98 |
| MnP9 | 5 | 88 |
| MnP10 | 6 | 96 |
| PhIOc | 7 | 12 |
| Catalyst | Run | Epoxide yielda (%) |
|---|---|---|
|
a The yield of cyclooctene oxide was calculated on the basis of the amount of PhIO used in the reaction. The results represent an average of at least duplicate reactions. MP/PhIO/(Z)-cyclooctene molar ratio = 1:50:5000.
b Reaction time of 1 h.
c Reaction time of 3 h.
d Control reaction: the solid obtained by the sol–gel process in the absence of MPx was grounded, and the resulting powder was used as catalyst.
e Control reaction: the reaction was performed with LDH without MPx using the same mass as in the case of the catalytic reaction using MPx–LDH.
|
||
| FeP7–LDHb | 1 | 58 |
| FeP7–SGAb | 2 | 29 |
| FeP8–LDHb | 3 | 69 |
| FeP8–SGAb | 4 | 42 |
| MnP9–LDHb | 5 | 84 |
| MnP9–SGAb | 6 | 20 |
| MnP9–SGAc | 7 | 30 |
| MnP10–LDH | 8 | 71 |
| MnP10–SGAb | 9 | 15 |
| MnP10–SGAc | 10 | 32 |
| PhIO + SGAd | 11 | 12 |
| PhIO + LDHe | 12 | 10 |
| Catalyst | Run | Epoxide yielda (%) |
|---|---|---|
|
a The yield of cyclooctene oxide was calculated on the basis of the amount of PhIO used in the reaction. The results represent an average of at least duplicate reactions. MP/PhIO/(Z)-cyclooctene molar ratio = 1:50:5000. Reaction time: 1 h.
|
||
| FeP7–LDH first reuse | 1 | 55 |
| FeP7–LDH second reuse | 2 | 57 |
| FeP7–LDH third reuse | 3 | 54 |
| FeP8–LDH first reuse | 4 | 65 |
| FeP8–LDH second reuse | 5 | 70 |
| FeP8–LDH third reuse | 6 | 68 |
| MnP9–LDH first reuse | 7 | 74 |
| MnP9–LDH second reuse | 8 | 70 |
| MnP9–LDH third reuse | 9 | 68 |
| MnP10–LDH first reuse | 10 | 75 |
| MnP10–LDH second reuse | 11 | 74 |
| MnP10–LDH third reuse | 12 | 67 |
| Catalyst | Run | Alcohol yielda (%) | Ketone yielda (%) |
|---|---|---|---|
|
a The yield of cyclohexanol and cyclohexanone were calculated on the basis of the amount of PhIO used in the reaction. The results represent an average of at least duplicate reactions. MP/PhIO/cyclohexane molar ratio = 1:50:5000. Reaction time: 1 h.
b Control reaction, without catalyst.
|
|||
| [Fe(TPFPP)]Cl | 1 | 85 | 2.0 |
| FeP7 | 2 | 62 | Trace |
| FeP8 | 3 | 72 | Trace |
| [Mn(TPFPP)]Cl | 4 | 34 | 6.0 |
| MnP9 | 5 | 52 | Trace |
| MnP10 | 6 | 64 | Trace |
| PhIOb | 7 | Trace | Trace |
| Catalyst | Run | Yield (%)a | ||
|---|---|---|---|---|
| Alcohol | Ketone | |||
| 1 hb | 24 hb | 24 hc | ||
|
a The yields were calculated on the basis of the amount of PhIO used in the reaction. The results represent an average of at least duplicate reactions. MP/PhIO/cyclohexane molar ratio = 1:50:5000.
b Reaction time.
c No ketone was observed after 1 h of reaction.
d Control reaction, without catalyst.
|
||||
| FeP7–LDH | 1 | 1 | 15 | Trace |
| FeP8–LDH | 2 | 24 | 41 | Trace |
| MnP9–LDH | 3 | 5 | 16 | 3.8 |
| MnP10–LDH | 4 | 1 | 8 | Trace |
| PhIO + LDHd | 5 | — | — | — |
Usually, metalloporphyrins containing bulky or electronegative groups in the ortho-positions of the meso-substituents of the porphyrin ring give good results in the catalyzed oxidation reactions.5,6 This performance stems from the steric and electronic effects that the substituents exert on the porphyrin ring—the more electronegative the substituent, the longer the active catalytic species lifetime.5 Additionally, these bulky substituents may prevent the formation of inactive species (e.g., dimeric species) and the porphyrin ring auto-oxidative destruction. Together, these factors may explain the different MPx performances in (Z)-cyclooctene epoxidation (Table 2).
As expected, [Fe(TFPP)Cl] and [Mn(TFPP)Cl] (runs 1 and 4, respectively) furnished product yields as high as 98%.85,86 As for FeP7 and MnP9 (in which one fluorine atom is replaced with ethylene glycol, runs 2 and 5, respectively) and FeP8 and MnP10 (in which four fluorine atoms are substituted with ethylene glycol, runs 3 and 6, respectively), the epoxide yields decreased. Bearing in mind that electronegative groups increase the catalytically active metal-oxo species lifetime, the absence of these groups should reduce the catalytic activity.3 However, the epoxide yield decreased more markedly for the mono-substituted complexes as compared with the tetra-substituted ones. Therefore, the four ethylene glycol substituents in FeP8 and somehow inhibited the dimeric species formation and porphyrin ring auto-oxidative destruction. Moreover, the presence of bulkier substituents can affect ring symmetry, causing some distortion. Hence, substitution at the four p-positions of the phenyl rings distorted the porphyrin ring in FeP8 and to a larger extent than substitution at one p-position of the phenyl rings in FeP7 and MnP9. This fact may have influenced the formation and stabilization of the active catalytic oxo-species, resulting in lower epoxide yields for the mono-substituted complexes (FeP7 and MnP9).
When we used MPx–LDH (Table 3, runs 1, 3, 5, and 8) under the same reaction conditions (same reaction time), we achieved epoxide yields lower than or similar to those observed for homogeneous catalysis (Table 2, runs 2, 3, 5, and 6). The epoxide yields decreased by 13% for FeP7–LDH and 25% for FeP8–LDH and MnP10–LDH. Interestingly, MnP9–LDH was as effective as its homogeneous counterpart (84% vs. 88%, Table 3, run 5, and Table 2, run 5, respectively).
In general, bulky substituents seem to sterically hinder the approach between reactants and catalyst, thereby making the access of the oxidant and the substrate to the macrocycle metal center difficult and preventing the formation of the active catalytic oxo-species and the oxidation reaction products. The steric effect exerted by the substituents was even more evident in the case of immobilized MPx, as the catalytic results suggest. Furthermore, the mono- and tetra-substituted MPx behaved differently, suggesting distinct MPx immobilization modes on LDH; thus, the way MPx were immobilized on the matrix influenced their reactivity. The presence of four –OR groups probably favored the interaction of MnPx with the LDH support. Consequently, access of the substrate and oxidant to the catalytic center was more hindered in FeP8 and MnP10 (Table 4, runs 3 and 8) as compared with the mono-substituted FeP7 and MnP9 (Table 4, runs 1 and 5), leading to greater decline in product yields in the case of the tetra-substituted catalysts.
We also investigated the catalytic activity of MPx–SGA in (Z)-cyclooctene epoxidation (Table 3, runs 2, 4, 6, 7, 9, and 10). All the MPx–SGA afforded lower yields as compared with the parent MPx–LDH and MPx in homogeneous medium. We decided to carry out the epoxidation reactions for longer periods (3 h), and we verified that the product yield increased (Table 3, runs 7 and 10). This strongly suggests that the MnPx may have been trapped in the silica network, making the access of the oxidant to the metal center to form the active catalytic oxo-species difficult. In this sense, a longer reaction time should allow the reagents more time to interact.
Although the yields decreased for all the tested MPx–LDH and MPx–SGA as compared with MPx in homogeneous medium, immobilization offered a major advantage: it enabled catalyst recyclability. Indeed, UV–vis analyses did not detect any MPx leaching from the support after the first use or after the washing and drying processes. To investigate the recyclability of MPx–LDH, we reused them in a second (Z)-cyclooctene epoxidation reaction (Table 4). In general, all the reused MPx–LDH led to similar yields as compared with the corresponding fresh catalyst. Hence, MPx did not leach from the support LDH and enabled us to reuse MPx–LDH without loss of catalytic activity. The results obtained in the recycling reactions also indicated that MPx were resistant to the reaction conditions and interacted strongly with the support. We recovered MPx–LDH after the first reaction by simple filtration, washing, and drying. In fact, analysis of all the reaction mixtures after the catalytic reactions and the recovery procedure failed to display the characteristic MPx Soret band. After reuse, we recovered the MPx–LDH again and reused them in a third reaction; we did not detect any alterations in their activity. The reusability and stability of MPx–LDH point to a promising and economically viable process.
In accordance with the literature,3,5,7,84,85,87–93 [Fe(TPFPP)]Cl and [Mn(TPFPP)]Cl (homogeneous catalysis) catalyzed cyclohexane oxidation efficiently and selectively (runs 1 and 4; alcohol yield = 85 and 34%; ketone yield = 2.0 and 6.0%, respectively). Table 5 shows that the reaction yields for the modified iron porphyrins FeP7 and FeP8 were lower than the yields obtained for [Fe(TPFPP)]Cl. As discussed above in the case of (Z)-cyclooctene epoxidation, substitution of fluorine atoms by the –OR groups from the ethylene glycol moieties justifies this decrease.
For manganese porphyrins, we observed a reverse trend: the modified manganese porphyrins MnP9 and MnP10 afforded higher product yields than [Mn(TPFPP)]Cl. The solvent used in the catalytic studies—acetonitrile (ACN)—may have contributed to this result. We have verified that the best reaction medium for catalytic reactions using metalloporphyrins similar to [M(TPFPP)]Cl (M = Fe or Mn) is a mixture of solvents such as dichloromethane and acetonitrile 1:1 (v/v);12,13,26,35,93 this mixture solubilizes both the substrate and PhIO, while the catalyst is not always completely soluble in this medium. Some years ago, Iamamoto and co-workers92 reported that when they used dichloromethane (DCM) as the solvent, they achieved the best [Mn(TPFPP)]Cl catalytic performance in cyclohexane oxidation. However, when these authors used the same solvent for [Fe(TFPP)Cl], they verified competitive oxidation reactions between DCM and cyclohexane, which diminished the product yield. ACN can minimize competitive reactions between solvent and substrate, although in some cases it can also compete with cyclohexane for the catalytic active center, as it can be oxidized to HCN and formaldehyde.92 In the present work, we chose to use only ACN as solvent because it is more appropriate from an environmental standpoint and seems to affect the catalytic activity of the tested MPx to a lesser extent.
Table 5 also reveals that MPx bearing four –OR groups at the para-positions of the meso-phenyls of the macrocycle (more symmetric) furnished higher product yields as compared with MPx containing only one –OR group, a behavior we had already verified during (Z)-cyclooctene epoxidation. FePx provided better yields than MnPx,93 because recombination between the active catalytic species originated from FePx and cyclohexane was faster as compared with MnPx.93–95 Indeed, it is known that the intermediate species formed after the abstraction of a proton from the substrate by manganese porphyrin, (Mn(IV)(OH)Por)+, is more stable as compared with the intermediate species originated from the iron porphyrin, (Fe(IV)(OH)Por)+.95 In the generally accepted reaction pathway,96,97 the active catalytic species abstracts a hydrogen atom from the substrate, generating an alkyl radical and a hydroxo-metalloporphyrin in a cage solvent. Oxygen rebound preferably occurs within the cage, forming the alcohol. A more kinetically stable intermediate, as in the case of manganese porphyrins, results in recombination of the catalyst with the alkyl radical (radical species escapes from the vicinity of the intermediate species–solvent cage).94 In the case of the iron porphyrin, oxygen rebound is rapid.95
MPx immobilization on LDH elicited a drastic drop in the cyclohexanol yield as previously noted by some of us when we immobilized [Fe(TPFPP)]Cl on this same support.26 In the particular cases of FeP7–LDH, MnP9–LDH, and MnP10–LDH, the yields were less than 10% after 1 h of reaction (Table 6).
FeP8–LDH gave the best result after 1 h of reaction (Table 6, run 6, 24% alcohol yield). Although this yield was lower than that achieved with FeP8 in homogeneous catalysis (Table 5, run 1), this solid has the advantage of recyclability. Furthermore, FeP8–LDH catalyzed cyclohexane oxidation more effectively than Fe(TFPP)–LDH (alcohol yield = 7.0%),26 justifying the modification of P1 with ethylene glycol.
In general, under ideal reaction conditions, better yields are not expected upon increasing the reaction times when reactions are conducted in a homogeneous medium. We had previously optimized the reaction time used in all the catalytic experiments;12,24,26,34 in most cases, one hour of reaction was enough to obtain the best catalytic results. However, for heterogenized metalloporphyrins, increasing the reaction time can lead to better yields, mainly if the heterogenization process somehow blocks the access of the reactants to the metal center of the immobilized catalyst. The longer contact time between the solid catalyst, the substrate, and the oxidant can facilitate the access of both reactants to the active site, thus favoring catalysis.26,36,42,98,99 On the basis of our previous experience, we conducted the MPx–LDH-catalyzed cyclohexane oxidation reactions for 24 h (Table 6). The results show that all the MPx–LDH afforded better results after 24 h of reaction as compared with reaction time of 1 h. Once again, FeP8–LDH performed the best.
We did not detect any significant ketone formation. The exception was MnP9–LDH. Although a longer reaction time led to better yields, the reaction became less selective in this case. Over oxidation of alcohol might underlie ketone production.23,89
Conclusions
We successfully accomplished the chemical modification of the porphyrin [H2(TPFPP)] using ethylene glycol. We obtained five derivatives as a result of the substitution of the p-fluorine atoms on the meso-phenyl rings by an alkoxide from ethylene glycol. We characterized the obtained porphyrins using UV–vis and FTIR spectroscopies, and 1H and 19F NMR. We metallated the mono- and tetra-substituted derivatives with Fe(III) and Mn(III), which furnished stable metalloporphyrin complexes. We immobilized the resulting metalloporphyrins on two different supports: LDH and silica (obtained by the sol–gel methodology). Preliminary results demonstrated that these materials are catalytically active in the oxidation of (Z)-cyclooctene and cyclohexane by PhIO. Furthermore, we were able to reuse the LDH-immobilized metalloporphyrins in at least three additional cycles of (Z)-cyclooctene oxidation.Acknowledgements
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Araucária, Fundação da Universidade Federal do Paraná (FUNPAR), and Universidade Federal do Paraná (UFPR) for the financial support. Thanks are also due to Fundação para a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network, also supported with funds from FCT. Kelly A. D. F. Castro also thanks CAPES for the PhD sandwich scholarship (process 6883-10-9).Notes and references
- P. Battioni, J. F. Bartoli, D. Mansuy, Y. S. Byun and T. G. Traylor, J. Chem. Soc., Chem. Commun., 1992, 1051 RSC.
- A. Agarwala and D. Bandyopadhway, Chem. Commun., 2006, 4823 RSC.
- J. T. Groves, J. Inorg. Biochem., 2006, 100, 434 CrossRef CAS PubMed.
- R. Davydov, T. M. Makris, V. Kofman, D. E. Werst, S. G. Sligar and B. M. Hoffman, J. Am. Chem. Soc., 2001, 123, 1403 CrossRef CAS PubMed.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251 CrossRef CAS.
- K. S. Suslick, ed. K. Kadish, K. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, 1999 Search PubMed.
- A. J. Appleton, S. Evans and J. R. L. Smith, J. Chem. Soc., Perkin Trans. 2, 1995, 281 Search PubMed.
- M. E. Kosal and K. Suslick, J. Solid State Chem., 2000, 152, 87 CrossRef CAS.
- N. G. Giri and S. M. S. Chauhan, Catal. Commun., 2009, 10, 383 CrossRef CAS PubMed.
- J. H. Han, S.-K. Yoo, J. S. Seo, S. J. Hong, S. K. Kimb and C. Kim, Dalton Trans., 2005, 402 RSC.
- F. Wypych, A. Bail, M. Halma and S. Nakagaki, J. Catal., 2005, 234, 431 CrossRef CAS PubMed.
- M. Halma, K. A. D. F. Castro, C. Taviot-Gueho, V. Prévot, C. Forano, F. Wypych and S. Nakagaki, J. Catal., 2008, 257, 233 CrossRef CAS PubMed.
- S. Nakagaki, F. L. Benedito and F. Wpych, J. Mol. Catal. A: Chem., 2004, 217, 121 CrossRef CAS PubMed.
- S. Hirohara, M. Nishida, K. Sharyo, M. Obata, T. Ando and M. Tanihara, Bioorg. Med. Chem., 2010, 18, 1526 CrossRef CAS PubMed.
- N. M. M. Moura, F. Giuntini, M. A. F. Faustino, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva, E. M. Rakib, A. Hannioui, S. Abouricha, B. Röder and J. A. S. Cavaleiro, ARKIVOC, 2010, 24 CAS.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 136 CrossRef PubMed.
- D. K. Panda, F. S. Goodson, S. Ray, R. Lowell and S. Saha, Chem. Commun., 2012, 48, 8775 RSC.
- T. Malinski and Z. Taha, Nature, 1992, 358, 676 CrossRef CAS PubMed.
- D. Vlascici, E. Fagadar-Cosma, I. Popa, V. Chiriac and M. Gil-Agusti, Sensors, 2012, 12, 8193 CrossRef CAS PubMed.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed.
- B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS.
- J. R. Lindsay Smith, Y. Iamamoto and F. S. Vinhado, J. Mol. Catal. A: Chem., 2006, 252, 23 CrossRef CAS PubMed.
- G. M. Ucoski, K. A. D. F. Castro, K. J. Ciuffi, G. P. Ricci, J. A. Marques, F. S. Nunes and S. Nakagaki, Appl. Catal., A, 2011, 404, 120 CrossRef CAS PubMed.
- M. Halma, A. Bail, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2006, 243, 44 CrossRef CAS PubMed.
- K. A. D. F. Castro, A. Bail, P. B. Groszewicz, G. S. Machado, W. Schreiner and F. Wypych, Appl. Catal., A, 2010, 386, 51 CrossRef CAS PubMed.
- D. Mansuy, C. R. Chim., 2007, 10, 392 CrossRef CAS PubMed.
- B. Meunier, S. P. Visser and S. Shaik, Chem. Rev., 2004, 104, 3947 CrossRef CAS PubMed.
- M. M. Q. Simões, R. De Paula, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 589 CrossRef.
- S. M. G. Pires, M. M. Q. Simões, I. C. M. S. Santos, S. L. H. Rebelo, M. M. Pereira, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Appl. Catal., A, 2012, 439, 51 CrossRef PubMed.
- C. M. B. Neves, M. M. Q. Simões, I. C. M. S. Santos, F. M. J. Domingues, M. G. P. M. S. Neves, F. A. A. Paz, A. M. S. Silva and J. A. S. Cavaleiro, Tetrahedron Lett., 2011, 52, 2898 CrossRef CAS PubMed.
- S. L. H. Rebelo, A. R. Gonçalves, M. M. Pereira, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2006, 256, 321 CrossRef CAS PubMed.
- R. Paula, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2011, 345, 1 CrossRef PubMed.
- K. A. D. F. Castro, M. Halma, G. S. Machado, G. P. Ricci, G. M. Ucoski, K. J. Ciuffi and S. Nakagaki, J. Braz. Chem. Soc., 2010, 21, 1329 CrossRef CAS PubMed.
- S. Nakagaki, M. Halma, A. Bail, G. G. C. Arízaga and F. Wypych, J. Colloid Interface Sci., 2005, 281, 417 CrossRef CAS PubMed.
- M. Halma, A. Bail, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2006, 243, 44 CrossRef CAS PubMed.
- R. J. Abraham and I. Marsden, Tetrahedron, 1992, 48, 7489 CrossRef CAS.
- N. A. Stephenson and A. T. Bell, J. Am. Chem. Soc., 2005, 127, 8635 CrossRef CAS PubMed.
- E. H. Schaab, A. E. M. Crotti, Y. Iamamoto, M. J. Kato, L. V. C. Lotufo and N. P. Lopes, Biol. Pharm. Bull., 2010, 33, 912 CAS.
- F. R. Longo, M. G. Finarelli and J. B. Kim, J. Heterocycl. Chem., 1969, 6, 927 CrossRef CAS.
- C. K. Chang and F. Ebina, J. Chem. Soc., Chem. Commun., 1981, 778 RSC.
- W. Nam, H. J. Lee, S.-Y. Oha, C. Kimb and H. G. Jang, J. Inorg. Biochem., 2000, 80, 219 CrossRef CAS.
- J. I. T. Costa, A. C. Tomé, M. G. P. M. S. Neves and J. Cavaleiro, J. Porphyrins Phthalocyanines, 2011, 15, 1116 CrossRef CAS.
- J. Králova, T. Bríza, I. Moserova, B. Dolensky, P. Vasek, P. Pouckova, Z. Kejik, R. Klaplánek, P. Martázek, M. Dvorak and V. Král, J. Med. Chem., 2008, 51, 5964 CrossRef PubMed.
- P. Battioni, E. Cardin, M. Louloudi, B. Schollhorn, G. A. Spyroulias, D. Mansuy and T. G. Traylor, Chem. Commun., 1996, 2037 RSC.
- L. F. Pedrosa, M. C. Souza, M. A. F. Faustino, M. G. P. M. S. Neves, A. M. Silva, A. C. Tomé, V. F. Ferreira and J. A. S. Cavaleiro, Aust. J. Chem., 2011, 64, 939 CrossRef CAS.
- S. Silva, P. M. R. Pereira, P. Silva, F. A. A. Paz, M. A. F. Faustino, J. A. S. Cavaleiro and J. Tomé, Chem. Commun., 2012, 48, 3608 RSC.
- K. J. Ciuffi, H. C. Sacco, J. C. Biazzotto, E. A. Vidoto, O. R. Nascimento, C. A. P. Leite, O. A. Serra and Y. Iamamoto, J. Non-Cryst. Solids, 2000, 273, 100 CrossRef CAS.
- J. R. Lindsay Smith and Y. Iamamoto, J. Mol. Catal. A: Chem., 2006, 252, 23 CrossRef CAS PubMed.
- F. G. Doro, J. R. Lindsay Smith, A. G. Ferreira and M. D. Assis, J. Mol. Catal. A: Chem., 2000, 164, 97 CrossRef CAS.
- A. L. Faria, T. C. O. Mac Leod and M. D. Assis, Catal. Today, 2008, 863, 133 Search PubMed.
- A. T. Papacídero, L. A. Rocha, B. L. Caetano, E. Molina, H. C. Sacco, E. J. Nassar, Y. Martinelli, C. Mello, S. Nakagaki and K. J. Ciuffi, Colloids Surf., A, 2005, 275, 27 CrossRef PubMed.
- G. S. Machado, G. G. C. Arízaga, F. Wypych and S. Nakagaki, J. Mol. Catal., 2010, 274, 130 CrossRef CAS PubMed.
- G. S. Machado, F. Wypych and S. Nakagaki, J. Colloid Interface Sci., 2012, 377, 379 CrossRef CAS PubMed.
- W. Stober and A. Fink, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- E. L. Crepaldi and J. B. Valim, Quim. Nova, 1998, 21, 300 CrossRef CAS PubMed.
- V. Rives and M. A. Ullibarri, Coord. Chem. Rev., 1999, 181, 61 CrossRef CAS.
- J. C. Sharefkin and H. Saltzmann, Org. Synth., 1963, 43, 60 Search PubMed.
- J. Inacio, C. Taviot-Guého, C. Forano and J. P. Besse, Appl. Clay Sci., 2001, 18, 255 CrossRef CAS.
- A. M. R. Gonsalves, J. M. T. B. Varejão and M. M. Pereira, J. Heterocycl. Chem., 1991, 28, 635 CrossRef CAS.
- H. Kobayashi, T. Higuchi, Y. Kaizu, H. Osada and M. Aoki, Bull. Chem. Soc. Jpn., 1975, 48, 3137 CrossRef CAS.
- D. Samaroo, M. Vinodu, X. Chen and C. M. Drain, J. Comb. Chem., 2007, 9, 998 CrossRef CAS PubMed.
- W. Zheng, N. Shan, L. Yu and X. Wang, Dyes Pigm., 2008, 77, 153 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds parts A and B, Wiley Interscience Publication, 5th edn, 1997 Search PubMed.
- M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271 CrossRef CAS PubMed.
- R. Cammack and C. E. Cooper, Methods Enzymol., 1993, 227, 353 CAS.
- R. Cheng, P. Chen, P. Gau, C. Cheu and S. Peng, J. Am. Chem. Soc., 1997, 119, 2563 CrossRef CAS.
- M. T. Caudle, C. K. Mobley, L. M. Bafaro, R. Lobrutto, G. T. Yee and T. L. Groy, Inorg. Chem., 2004, 43, 506 CrossRef CAS PubMed.
- S. Nakagaki and F. Wypych, J. Colloid Interface Sci., 2007, 315, 142 CrossRef CAS PubMed.
- F. Cavani, F. Trifirb and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- H. Kameyama, H. Suzuki and A. Amano, Chem. Lett., 1988, 1117 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138 CrossRef CAS.
- M. J. Nappa and R. J. McKinney, Inorg. Chem., 1988, 27, 3740 CrossRef CAS.
- E. A. Vidoto, M. S. M. Moreira, F. S. Vinhado, K. J. Ciuffi, O. R. Nascimento and Y. Iamamoto, J. Non-Cryst. Solids, 2002, 304, 151 CrossRef.
- M. Espinosa, S. Pacheco and R. Rodriguez, J. Non-Cryst. Solids, 2007, 353, 2573 CrossRef CAS PubMed.
- H. C. Sacco, K. J. Ciuffi, J. C. Biazzotto, C. Mello, D. C. Oliveira, E. A. Vidoto, O. R. Nascimento, O. A. Serra and Y. Iamamoto, J. Non-Cryst. Solids, 2001, 284, 174 CrossRef CAS.
- M. A. G. Sánchez, S. R. Tello, R. Sosa and A. Campero, J. Sol-Gel Sci. Technol., 2006, 37, 93 CrossRef.
- M. Trytek, M. Madjan, A. Lipke and J. Fiedurek, J. Catal., 2012, 286, 193 CrossRef CAS PubMed.
- F. Figueira, J. A. S. Cavaleiro and J. P. C. Tomé, J. Porphyrins Phthalocyanines, 2011, 15, 517 CrossRef CAS.
- H. Tanaka, T. Yamada, S. Sugiyama, H. Shiradoti and R. Hino, J. Colloid Interface Sci., 2005, 286, 812 CrossRef CAS PubMed.
- L. B. Bolzon, H. R. Airoldi, F. B. Zanardi, J. C. Granado and Y. Iamamoto, Microporous Mesoporous Mater., 2013, 168, 37 CrossRef CAS PubMed.
- J. Poltowicz and J. Haber, J. Mol. Catal. A: Chem., 2004, 220, 43 CrossRef CAS PubMed.
- D. R. Leanord and J. R. Lindsay-Smith, J. Chem. Soc., Perkin Trans. 2, 1991, 25 RSC.
- P. Inchley, J. R. Lindsay-Smith and R. J. Lower, New J. Chem., 1989, 13, 669 CAS.
- E. C. Zampronio, M. C. A. F. Gotardo, M. D. Assis and H. P. Oliveira, Catal. Lett., 2005, 104, 53 CrossRef CAS PubMed.
- O. J. Lima, D. P. Aguirre, D. C. Oliveira, M. A. Silva, C. Mello, C. A. P. Leite, H. C. Sacco and K. J. Ciuffi, J. Mater. Chem., 2011, 11, 2476 RSC.
- G. R. Friedermann, M. Halma, K. A. D. F. Castro, F. L. Benedito, F. G. Doro, S. M. Drechsel, A. S. Mangrich, M. D. Assis and S. Nakagaki, Appl. Catal., A, 2006, 308, 172 CrossRef CAS PubMed.
- Y. Iamamoto, C. M. C. Prado, H. C. Sacco, K. J. Ciuffi, M. D. Assis, A. P. J. Maestrin, A. J. B. Melo, O. Baffa and O. R. Nascimento, J. Mol. Catal. A: Chem., 1997, 117, 259 CrossRef CAS.
- Y. Iamamoto, M. D. Assis, K. J. Ciuffi, H. C. Sacco, L. Iwamoto, A. J. B. Melo, O. R. Nascimento and C. M. C. Prado, J. Mol. Catal. A: Chem., 1996, 109, 189 CrossRef CAS.
- J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 6243 CrossRef CAS.
- T. G. Traylor and W. P. F. D. Bandyopadhhyay, J. Am. Chem. Soc., 1989, 111, 8009 CrossRef CAS.
- Y. Iamamoto, M. D. Assis, K. J. Ciuffi, C. M. C. Prado, B. Z. Prellwitz, M. Moraes, O. R. Nascimento and H. C. Sacco, J. Mol. Catal. A: Chem., 1997, 116, 365 CrossRef CAS.
- G. S. Machado, P. B. Groszewicz, K. A. D. F. Castro, F. Wypych and S. Nakagaki, J. Colloid Interface Sci., 2012, 374, 278 CrossRef CAS PubMed.
- J. A. Smegal and C. L. Hill, J. Am. Chem. Soc., 1983, 105, 3515 CrossRef CAS.
- J. T. Groves and D. V. Adhyam, J. Am. Chem. Soc., 1984, 106, 2177 CrossRef CAS.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884 CrossRef CAS.
- J. T. Groves, T. E. Nemo and R. S. Meyers, J. Am. Chem. Soc., 1979, 101, 1032 CrossRef CAS.
- M. Halma, K. A. D. F. Castro, V. Prévot, C. Forano, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2009, 310, 42 CrossRef CAS PubMed.
- S. Nakagaki, K. A. D. F. Castro, M. Halma, G. S. Machado, S. M. Drechsel and F. Wypych, J. Braz. Chem. Soc., 2006, 17, 1672 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR 1H and 19F, FTIR, HRMS and UV–vis of the free-base porphyrins P2–P6. UV–vis, MALDI (TOF/TOF) and HRMS of the metalloporphyrins. See DOI: 10.1039/c3cy00472d |
| This journal is © The Royal Society of Chemistry 2014 |